

Abstract
The past few years have witnessed a paradigm shift in molecular systematics from phylogenetic methods (using one or a few genes) to those that can be described as phylogenomics (phylogenetic inference with entire genomes). One approach that has recently emerged is phylo-transcriptomics (transcriptome-based phylogenetic inference). As in any phylogenetics experiment, accurate orthology inference is critical to phylo-transcriptomics. To date, most analyses have inferred orthology based either on pure sequence similarity or using gene-tree approaches. The use of conserved genome synteny in orthology detection has been relatively under-employed in phylogenetics, mainly due to the cost of sequencing genomes. While current trends focus on the quantity of genes included in an analysis, the use of synteny is likely to improve the quality of ortholog inference. In this study, we combine de novo transcriptome data and sequenced genomes from an economically important group of grass species, the tribe Paniceae, to make phylogenomic inferences. This method, which we call “genome-guided phylo-transcriptomics”, is compared to other recently published orthology inference pipelines, and benchmarked using a set of sequenced genomes from across the grasses. These comparisons provide a framework for future researchers to evaluate the costs and benefits of adding sequenced genomes to transcriptome data sets.
Introduction
Phylogenetic methods have undergone enormous changes over the past few years as the costs of next generation sequencing have declined. Where researchers once spent considerable time designing and testing PCR primers to sequence one or a few genes, it is now becoming common to sequence large numbers of genes, or even whole genomes, for phylogenomic analyses1–9. In an increasing number of cases, it is possible to build phylogenetic trees based on sequenced genomes, but even these are often re-sequenced or low coverage genomes1,10–20. For most groups of eukaryotic organisms, the costs of sequencing and assembling whole genomes remain prohibitive, limiting the applicability of whole genome sequencing for studies that sample large numbers of taxa. Whole genomes are also not generally necessary to allow phylogenomic methods to provide increased resolution of species relationships21. Reduced representation approaches, where part of the genome is excluded from sequencing, allow researchers to obtain sequence data for large numbers of nuclear genes across many species at a relatively low cost and have become increasingly common22–29.
The current study focuses on improving and testing the constraints of one of these approaches, transcriptome-based phylogenomics. Variations of this method have been applied to a range of organisms and scientific questions6,8,30–45. Transcriptome-based methods differ from other reduced representation approaches in the nature of the gene/transcript ascertainment bias that results. Transcriptomes produce a sampling of transcripts that are biased due to the biology of the organisms under study and the time point(s) and tissue(s) being sampled. Probe-based reduced representation methods on the other hand are biased by the methods used for discovering and choosing the probes. Transcriptome-based approaches to phylogenomics rely on sequencing RNA from multiple taxa at sufficient depth to enable de novo assembly of many (usually hundreds to thousands) of transcripts. The resulting transcripts are then used in phylogenetic analyses. The cost of sequencing transcriptomes is, of course, substantially less than that required for whole genomes. Transcriptome-based approaches also require less upfront time investment and a priori knowledge than probe hybridization/sequence capture-based methods. However, they require more bioinformatics time post-sequencing, the reason being that no probe design is required for transcriptome sequencing, but post-sequencing assembly is required. One key limitation of transcriptome-based methods is that they require access to fresh tissue or RNA, and therefore cannot be employed with, for example, museum collections. Conversely, one advantage of transcriptome-based methods is that the expression data can be used for additional biological analyses beyond phylogenetic inference, as RNA-Seq data is widely used to understand the evolution of gene expression46–49; of course, probe and hybridization-based methods can also be used for other types of functional exploration. Collecting and preserving RNA from fresh or frozen tissue has become routine in many laboratories50 and, at least in our hands, it is actually easier and less time consuming than DNA sequencing due to streamlined commercial kits (cited in materials and methods below) and the small quantities of RNA required for library preparation.
One area of rapid advancement in transcriptome-based phylogenomics (and most other phylogenomics approaches) is orthology determination. Once transcriptomes are generated and assembled, it is necessary to identify orthologous genes between the various transcriptomes; that is genes that are descended from a single gene copy present in the most recent common ancestor of the species being compared. To date, the most commonly used methods for orthology inference use sequence similarity-based methods like BLAST, often combined with the Markov Cluster (MCL) or other algorithms for differentiating between orthologs and paralogs. These platforms include OrthoMCL, HaMStR, OrthoFinder, and others51–55. While these methods have been useful for orthology inference, it is well established that sequence similarity can be misleading, and does not necessarily equate with orthology51,56,57. Some recent improvements on these methods start with all-by-all BLAST and/or MCL, but additionally use the topological features of phylogenetic trees to differentiate between orthologs and paralogs52,58–60. Two of the most advanced platforms for doing this are the Agalma and Yang & Smith pipelines2,3,61–63. These phylogenetically-informed methods have proven effective and become popular in large part because they are computationally tractable and because they require no a priori information about gene order (e.g., they do not require sequenced genomes). One of the major downsides to these methods is the use of an all-by-all BLAST step. Not only can sequence similarity searches be problematic for establishing orthology51, but when they are performed in an all-by-all framework they become extremely resource-intensive computationally. Some of these issues can be overcome through the phylogenetically informed approaches described above and the use of parallel computing, but many improvements remain to be made.
An alternative method for orthology inference that has been used in prokaryotes but received relatively little attention in eukaryotic phylogenetics is the use of gene synteny64–69. Synteny can be defined as the co-localization of the same gene at similar chromosomal positions across related taxa66,70. It has been compared to a street address system where, if one knows the physical location of a building, it is much easier to find that building than just looking for a building with specific features. Synteny-based orthology determination is then rooted in the assumption that orthologous genes will not only share sequence similarity, but will also reside in similar locations within the genomes of related species70. Synteny-based methods are widely employed in comparative genomics studies69,71–77. The omission of synteny-based approaches in most phylogenetic studies is likely due to the fact that syntenic analysis requires information on gene order in addition to gene sequence, and information on gene order is not captured by reduced representation methods, including amplification-based, probe-based and transcriptome-based datasets. Additionally, syntenic conservation levels vary depending on the phylogenetic proximity of the species being compared. However, for many phylogenetic clades, synteny is strongly conserved70,78,79, making it is possible to use syntenic data from a few genomes as an anchor for reduced representation data, an idea that has not yet been fully explored.
Here we describe the development and implementation of a method we call genome-guided phylo-transcriptomics. This method uses genome-derived syntenic orthologs to anchor transcripts for phylogenetic inference, and is here tested and applied in an economically and scientifically important group of grasses, the tribe Paniceae80–85. While the method still requires a BLAST step in which transcripts are mapped directly to reference genes that are known to be single-copy orthologs based on synteny, it bypasses the time consuming and error prone all-by-all BLAST and MCL algorithm steps commonly used in current phylo-transcriptomic methods51,56,57. Furthermore, by removing transcripts that map in multiple copies to the reference ortholog (see Materials and Methods section), one can avoid using BLAST to distinguish between paralogs and orthologs whose sequences are very similar. These are, of course, the sequences for which BLAST is most problematic51. We hypothesize that the use of a genome-guided method for orthology prediction will result in a greater percentage of “true” orthologs than those predicted by topology-based methods. This decrease in the signal-to-noise ratio in a data set could have serious impacts given the influence that even a single informative ortholog can have on a phylogenetic analysis86.
In addition to the Paniceae data set here generated, which covers 25–40 million years (m.y.) of evolutionary time, we applied the new method to a published dataset from the grape family (Vitaceae) to Arabidopsis thaliana which diverged 69–150 m.y. ago2,81,82,87,88. We also constructed and analyzed a data set from several publically available genomes from across the grasses (family Poaceae) and used it to benchmark the method’s reliability as compared to orthology inference based entirely on sequenced genomes. The three data sets were analyzed using both this genome-guided method as well as two recently published topology-based approaches for orthology inference with transcriptomes, the Agalma and Yang & Smith pipelines2,3,62.
Materials and Methods
Taxon Sampling and Plant Materials
Forty-five species from across the tribe Paniceae and outgroups were selected for RNA sequencing. Samples were obtained from the sources listed in Supplementary Table S1, with the majority of samples drawn from the USDA germplasm collection. Most samples were taken from the same plants as those used by Washburn et al.80 so results could be directly compared to the chloroplast phylogeny inferred in that study. Plants were grown and sampled in the greenhouse facilities at the University of Missouri, Columbia, MO and the Danforth Center, St. Louis, MO, with the exception of Neurachne alopecuroidea and Paraneurachne muelleri, for which RNA samples were obtained from Martha Ludwig, University of Western Australia. Leaf material was sampled from all plants and where possible, shoot, flower, and drought-stressed tissue samples were also taken with the hope of capturing a greater number of unique transcripts. RNA was extracted using the PureLink® RNA Mini Kit (Invitrogen, Carlsbad, CA, USA) or using Roche TriPure (Indianapolis, IN, USA), following the manufacturer’s instructions. The grape data set was obtained from NCBI. Details on its generation and record locators can be found in Wen, et al.87. The grass genomes and annotation were downloaded from Phytozome (phytozome.jgi.doe.gov) and included Zea mays 284 5b+ 89, Sorghum bicolor 255 v2.190, Setaria italica 312 v2.291, Oropetium thomaeum 386 v1.192, Oryza sativa 323 v7.093, and Brachypodium distachyon 283 v2.194.
Transcriptome Sequencing
Libraries were prepared using the TruSeq Stranded mRNA Sample Prep Kit (Illumina, Inc., San Diego, CA, USA) or the method described by Wang, et al.95 (See Supplementary Table S1). Sequencing was performed at the MU DNA Core facility on the campus of the University of Missouri and at Cornell University’s sequencing core facility, and was done on an Illumina Hiseq sequencer with 2 × 100 bp chemistry and six species per lane.
Sequence Processing
RNA-seq data were quality filtered following standard procedures96,97. Transcriptomes were assembled de novo using Trinity98–100 and processed as described in Yang and Smith (2013).
The sequenced genomes of S. bicolor and S. italica were used for syntenic ortholog determination because both are high quality and publically available, they represent an ingroup and outgroup taxa to the tribe Paniceae, and neither genome contains a recent whole genome duplication event90,91. Syntenic orthologs between S. bicolor and S. italica were infered using the SynMap tool in CoGe (https://genomevolution.org/CoGe/) with QuotaAlign set to filter out syntenic paralogous regions using a quota setting of 1:179,101. This insures that only genes which are present in a single copy and at the same syntenic location in both species are included. Protein sequences of the S. bicolor representative of the 1:1 S. italica/S. bicolor set were used as the reference sequence for the remainder of the analyses. The assembled tribe Paniceae transcripts (excluding outgroup transcriptomes) were then mapped to the S. bicolor reference orthologs using BLAST with a cutoff E-value of 0.00001 and 85% amino acid identity. When a given S. bicolor gene corrisponded to more than one transcript in a species, all transcripts maping to that S. bicolor gene where discarded to avoid the potential inclussion of paralogs. These sequences where then grouped into orthologous sets for each gene and a multiple alignment was created using mafft102,103. In this way, the use of all-by-all BLAST and the MCL algorithm are completely avoided. After further filtering with phyutility and several scripts from Yang and Smith2, concatenated trees, coalescence-based quartet summary species trees, and binned coalescence-based quartet summary trees were created using RAxML, ASTRAL, and binning followed by ASTRAL, respectivly104–107 (Fig. 1). To investigate syntenic block phylogenies, data from the genome-guided gene trees were grouped based on conserved syntenic blocks across the S. bicolor and S. italica genomes (again obtained from CoGe). Each transcript was mapped to its syntenic block and trees created using RAxML based on concatenated transcripts from each syntenic block. The same method was applied to the grape data set, except that the V. vinifera and Arabidopsis thaliana genomes where used and the E-value and protien identity cutoffs where lowered to 0.0001 and 75%, respectively, to account for the increased phylogenetic distances represented in the grape data set. Whole genome duplication events occuring in the ancestor of A. thaliana but not V. vinifera are excluded from the analysis because of the 1:1 setting in QuotaAlign. Scripts and instructions for the genome-guided method are available at: bitbucket.org/washjake/transcriptome_phylogeny_tools.
Figure 1.

Genome-guided phylo-transcriptomics workflow. Illustration of the workflow followed to produce the genome-guided phylogenies in this study.
Two gene tree topology-based approaches to orthology inference where also used for comparison: the Agalma pipeline (version 0.5.0) by Dunn, et al.3 and the Ya Yang pipeline2,61. As above, RAxML, ASTRAL, and binning combined with ASTRAL were used to infer phylogenies. Phylogenetic trees and other figures were generated using FigTree, Inkscape, and Vennerable in R108–111.
For the grass data used for benchmarking, several additional analyses were run. Single copy syntenic orthologs were found in a pairwise fashion between O. sativa and each of the other genomes using CoGe as described above. The likelihood of two non-orthologous genes evolving to have not only high sequence similarity, but also to be in the same physical location, and in single-copy across multiple species is incredibly small, giving us high confidence in orthologs inferred using this method. These orthologs where used to create a set of fully synteny-based, one-to-one orthologs across the grasses. While this set does not include all possible single-copy orthologs, it does include all of them for which we can have high confidence based on the available data and current methods, and is hence the best achievable ortholog set for the grasses at the current time. We refer to this as the benchmarking data set.
Each of the ortholog inference methods described above was then run using the transcriptomes generated by the genome sequencing projects referenced above. In this way, the transcripts could be followed by name through the pipelines (except for the Agalma method for which this could not be easily accomplished due to the way the pipeline is packaged). Ortholog sets derived from the genome-guided method and the Yang and Smith method were then compared to the benchmarking set to determine how many orthologs each method was able to find in common with the benchmark orthologs.
Data Availability
The datasets generated during this study are available in the NCBI SRA repository under the identifiers noted in Supplemental Table S1.
Results
For species tree inference in the tribe Paniceae, the genome-guided method provided similar numbers of orthologous genes to both the Agalma and Yang & Smith methods at a 90% matrix occupancy cut-off (Table 1). However, for the full matrix runs, when any orthologous gene without all species represented was discarded, the genome-guided method returned fewer orthologs than the other two methods. This is probably due to the genome-guided method not using transcripts that map to the same ortholog. The genome-guided method however, produced more consistent tree topologies than the topology-based methods. For example, all species trees (concatenated, ASTRAL, binned, and with multiple matrix occupancies and taxonomic inclusion) built with the genome-guided orthology pipeline agreed in their subtribe level topologies. The topology-based methods on the other hand, occasionally produced conflicting subtribe-level topologies. In other words, the topology-based methods were more sensitive to perturbations in taxonomic inclusion (when a taxon is removed from the inference to test if the same general topology will result without it) than the genome-guided method. The genome-guided method was also many times faster than the topology-based methods (Table 2).
Table 1.
Total orthologs found in each method separated by matrix occupancy.
| Method | 8 spp | 90% | Full | ||
|---|---|---|---|---|---|
| Genome-guided | Genes | Total | 9,757 | 2,211 | 434 |
| Min | 5,389 | 1,963 | 434 | ||
| Amino Acids | Total | 4,182,364 | 835,229 | 144,503 | |
| Min | 1,775,925 | 669,215 | 128,896 | ||
| Agalma | Genes | Total | 11,563 | 2,308 | 555 |
| Min | 5,453 | 2,054 | 555 | ||
| Amino Acids | Total | 4,420,707 | 797,333 | 182,368 | |
| Min | 1,568,329 | 613,538 | 168,157 | ||
| Yang & Smith 1 to 1 | Genes | Total | 7,323 | 1,925 | 898 |
| Min | 3,685 | 1,781 | 898 | ||
| Amino Acids | Total | 2,408,802 | 789,203 | 361,901 | |
| Min | 1,129,993 | 628,190 | 310,283 | ||
| Yang & Smith MO | Genes | Total | 11,568 | 1,966 | 1,076 |
| Min | 6,417 | 1,879 | 1,076 | ||
| Amino Acids | Total | 4,362,686 | 857,857 | 456,597 | |
| Min | 2,009,430 | 687,942 | 380,988 |
Table 2.
Approximate run times in hours (hrs) for each orthology inference method based on a 16 CPU system.
| Synteny Step | BLAST Step | Alignment and Tree Building for Pruning | Total | |
|---|---|---|---|---|
| Genome-Guided | <1 | 6.7 | N/A | 7.7 |
| Agalma | N/A | 46.4 | 88.6 | 135.0 |
| Yang & Smith | N/A | 366.9 | 412.2 | 779.1 |
Figure 2 shows what we consider to be the best estimate of the Paniceae nuclear species tree, based on currently available data. This tree places Anthephorinae as direct sister to the MPC clade (subtribes Melinidinae, Panicinae, and Cenchrinae), which, although different from published chloroplast trees80,82,85, is consistent with the combined nuclear-chloroplast topology reported by Vicentini, et al.81.
Figure 2.

Genome-guided concatenation-based phylogeny of the tribe Paniceae. Phylogenetic tree of the tribe Paniceae (Poaceae) built using RAxML based on a concatenated matrix with 90% gene occupancy. Branches are labeled with maximum likelihood bootstrap values; unlabeled branches have values of 100.
As mentioned, the topology-based approaches (Yang & Smith and Agalma) generally resulted in the same tree topology as the genome-guided method (Fig. 3a). However, in some cases, depending on the taxon sampling included in the analysis, an alternative topology was obtained from these methods. This topology placed the subtribe Anthephorinae together with the Neurachninae and Sacciolipis lineages as sister to the MPC clade (Fig. 3b). Internode certainty (IC) scores for the main conflicting node in both the primary and secondary topologies were close to zero and in some cases even negative, suggesting high levels of gene tree incongruence1,112,113. Both the genome-guided method, and the topology-based methods included genes representative of each of the Sorghum bicolor chromosomes and the major Setaria italica scaffolds, indicating that the sampled genes from both methods came from across the entire genome (Supplementary Table S2).
Figure 3.

(a) Primary nuclear topology found using all methods, (b) Secondary nuclear topology, (c) Chloroplast topology based on Washburn, et al.80. (d) An ideogram of the Setaria italica chromosomes114 with conserved syntenic blocks between S. italica and Sorghum bicolor demarcated. Syntenic blocks are colored based on the phylogenetic patterns from a-c that each block supports. Gray indicates areas of the chromosomes not covered by our blocks. Asterisks below the blocks indicate significance level for pairwise Robinson-Foulds distance tests: ***0.001, **0.01, *0.05.
To further dissect the causes of gene tree incongruence within the Paniceae, the tree binning scripts described by Mirarab, et al.106 where used to separate groups of genes with distinct evolutionary histories. This method allows one to set a bootstrap significance threshold at which branches can be considered high confidence, and then compare large sets of gene trees for compatibility with each other. Different cut-off values were tested for this analysis, almost always (see exception below when a cut-off of 100 was used) resulting in several hundred distinct tree topologies that were incompatible with each other.
When a bootstrap cutoff value of 100 was used as the threshold, indicating that only gene tree branches with 100 percent bootstrap support were considered, the synteny-based data set still placed the trees into 18 unique topology groups (Supplementary Fig. S1). These eighteen topologies were then compared visually and examined for differences that could directly affect the relationship between the MPC clade and the subtribe Anthephorine. Of the eighteen (2211 total genes) topologies, eight topologies (981 total genes) showed strong support for the inclusion of Anthephorine within the MPC (as in the primary topology described above and shown in Figs 2 and 3b), five topologies (615 total genes) showed strong support for Anthephorine as sister to Neurachninae and Sacciolipis (as in the secondary topology described above and shown in Fig. 3b), and one topology (123 total genes) agreed with the chloroplast phylogeny from Washburn, et al.80 (Fig. 3c). The remaining four topologies (492 total genes) had low support for this area of the tree.
Another approach we developed to dissect gene tree incongruence consisted of building trees based on the combination of genes that share a similar physical location. A recent study was able to find likely introgression events using a non-overlapping window approach and constructing trees based on 1 Mb and 100 kb blocks of genes41. Because of the genome-guided approach, we were able to group genes into more biologically relevant blocks, namely blocks that are syntenically conserved between ingroup and outgroup taxa. The appearance of a block of genes sharing the same phylogeny, which differs from the species phylogeny, might suggest hybridization/introgression within a group as recently diverged as the Paniceae, but ILS could also produce these types of blocks.
Many syntenic block phylogenies were inconclusive in that they yielded topologies that had little similarity to any of the previously described or published species trees. This seemed to be correlated with the number of genes in a syntenic block in that blocks with more genes generally (but not always) provided a resolved phylogeny that was similar in the placement of the subtribes Melinidinae, Panicinae, Cenchrinae, Anthephorinae, Neurachninae, and the Sacciolipis lineage to one of the three phylogenies in Fig. 3. When these blocks and their topologies were mapped to an ideogram114 of the S. italica chromosomes a striking patchwork of differing syntenic block histories was revealed (Fig. 3d). To further investigate whether or not syntenic blocks have distinct tree topologies we used Robinson-Foulds (RF) distances as implemented in the ETE Toolkit115. By computing pairwise RF distances for all genes in a given block we created a tree distribution for each of the blocks. We then took the complete set of gene trees (those from all blocks) and randomly re-assigned them to blocks eighty thousand times, each time computing the pairwise RF distance for each block. In this way, we created a simulated “random distribution” of trees for each block that could be used as the null distribution in a statistical test comparing the observed pair-wise RF distances in a block to the simulated distribution under the null hypothesis that all blocks share the same tree distribution. Of the 79 blocks, 15 had distributions that were significantly different than their respective simulated distributions at a significance level of alpha <0.001 (Fig. 3d). Hence, it appears that these blocks have a distribution of tree distances smaller than that expected. This observation implies that at least some local regions of the genome have similar evolutionary histories relative to the genome as a whole, either because of locally-coherent ILS or hybridization.
To further benchmark the Genome-Guided method here developed we applied it to two additional data sets. We used publically available sequenced grass genomes to compare our method with the Agalma and Yang and Smith methods. Syntenic relationships between the genomes were used to construct a list of high confidence single copy orthologs across the grasses. This list then served as a benchmark to which the orthologs predicted by each of the methods could be compared. Of course, this list does not contain all orthologs across the grasses, so it cannot tell us anything about the validity of ortholog combinations predicted for genes not found in the list. However, it can tell us when each ortholog prediction method correctly places orthologs in its list, and when it incorrectly identifies paralogs as orthologs. Therefore, we think that these comparisons are informative as to the reliability of different orthology assignment methods.
All three orthology detection pipelines and the synteny-derived benchmarking set generated the same tree topology, in agreement with previous phylogenetic studies, with high confidence82. Gene by gene comparisons between each method and the benchmarking set show that the genome-guided method recovers a much higher percentage of ortholog gene trees that agree, in terms of which genes are included, with the benchmarking set than either the Yang & Smith 1 to 1 or MO Methods (62%, 32%, and 31% respectively with a species cutoff of four and genes not found in the benchmarking set excluded. See Table 3 and Fig. 4). Because of the way Agalma is packaged, we were unable to modify its code to include it in this comparison, but it would likely perform similarly to the Yang & Smith method as it uses similar approaches. Beyond the orthologous genes in the benchmarking set, the Yang & Smith methods also include as many as 2,116 additional ortholog gene trees. These trees are based on genes which our direct synteny comparisons did not find (i.e., they did not have a detectable 1:1 orthologous relationship). In some cases, these are genes which the Yang & Smith method mapped in duplicate, for example, two S. italica genes mapped to one S. bicolor gene. In other cases, the expected 1:1 syntelog is present for some pairs of species but the O. sativa syntelogs is not present. These trees may or may not be based on correct orthology assignment, but because they are not in the benchmarking set they could not be evaluated here.
Table 3.
Grass (Poaceae) wide gene by gene comparisons of orthology detection methods to a benchmark set of orthologs derived entirely from syntenic relationships between sequenced genomes.
| Method | 4 species | 5 species | 6 species | ||
|---|---|---|---|---|---|
| Genome-Guided | All Trees Included | Trees Agreeing with Benchmark | 4,119 | 2,169 | 413 |
| Total Trees | 6,669 | 3,700 | 896 | ||
| Percent Trees in Agreement | 61.8% | 58.6% | 46.1% | ||
| Yang & Smith 1 to 1 | All Trees Included | Trees Agreeing with Benchmark | 1,936 | 1,741 | 1,370 |
| Total Trees | 7,933 | 6,989 | 5,171 | ||
| Percent Trees in Agreement | 24.4% | 24.9% | 26.5% | ||
| Excluding trees not in benchmark set | Trees Agreeing with Benchmark | 1,936 | 1,741 | 1,370 | |
| Total Trees | 6,088 | 5,417 | 4,320 | ||
| Percent Trees in Agreement | 31.8% | 32.1% | 31.7% | ||
| Yang & Smith MO | All Trees Included | Trees Agreeing with Benchmark | 2,000 | 1,795 | 1,404 |
| Total Trees | 8,619 | 7,560 | 5,464 | ||
| Percent Trees in Agreement | 23.2% | 23.7% | 25.7% | ||
| Excluding trees not in benchmark set | Trees Agreeing with Benchmark | 2,000 | 1,795 | 1,404 | |
| Total Trees | 6,503 | 5,757 | 4,516 | ||
| Percent Trees in Agreement | 30.8% | 31.2% | 31.1% |
Figure 4.
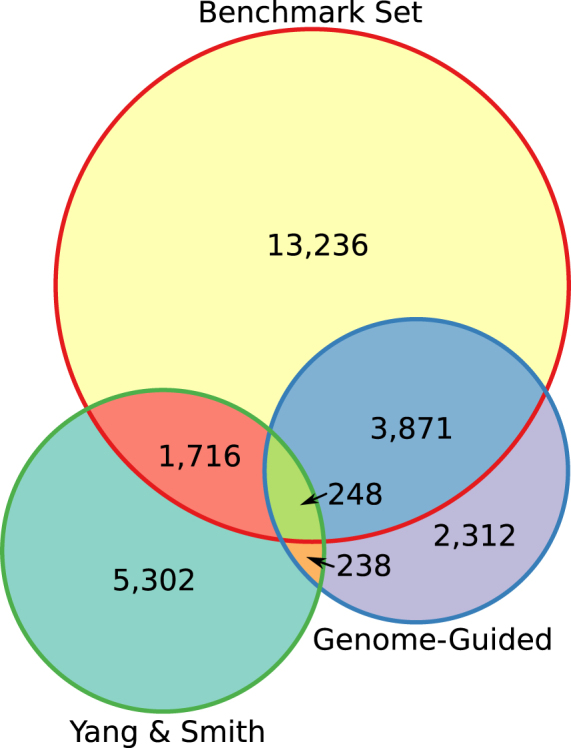
A Venn Diagram comparing the Poaceae gene sets derived from whole genomes, the genome-guided approach, and the Yang & Smith MO pipeline (the 1to1 pipeline is not shown because of large overlap with MO). Diagram created using Inkscape and the R package Vennerable108,109.
We also analyzed previously published data from grape and its relatives in order to benchmark the method and explore the phylogenetic distance it is capable of spanning. This dataset was comprised of 17 species spanning from V. vinifera to A. thaliana, with the bulk of the species being found within the family Vitaceae (estimated age of ~90 m.y.)87. The grape data set performed similarly to the Paniceae dataset we generated in terms of the amount of time it took to perform the genome-guided method versus the other methods. The number of orthologs retrieved was much smaller for the genome-guided method than it was for the two topology-based methods (Supplementary Table S3). This dearth of orthologs is likely due to the simple fact that syntenic relationships are expected to break down as the evolutionary distance between two species increases. Grape and Arabidopsis likely diverged between 69–150 (m.y.) with most estimates around 100 m.y., while Sorghum and Seteria probably diverged between 25–40 m.y.81,82,88. Even with substantially fewer genes, the genome-guided method still predicted similar topologies to those of the other two methods and those previously published (Supplementary Fig. S2)2.
Discussion
Phylogenetic consistency, broadly defined as convergence on the “correct” tree topology with increasing data, is a well-established phylogenetic accuracy assessment criterion116–118. The Genome-guided method proposed here consistently inferred the same subspecies level tree topology regardless of the matrix occupancy used, the tree building approach applied, and the number of taxa included. The topology-based approaches also resulted in the same tree topology in most cases, however, when the number of taxa were reduced to 33 by removing species near the main areas of conflict (to test the robustness of the topology inference), the topology-based approaches no longer produced consistent results while the Genome-guided method continued to produce the same topology. In general, the inclusion of more taxa, which better represent the diversity of a group of organisms, will increase the accuracy of phylogenetic inference116,119,120. It then follows that the topology found by both genome-guided and topology-based methods, when all taxa were included, is likely to be the topological estimate nearest to the true species history. This implies that the genome-guided method should be able to infer that topology with less data than the topology-based methods require for similar confidence and accuracy. Additionally, the grass benchmarking data set comparisons indicate that, of the orthologs we know with high confidence, the genome-guided method predicts a higher percentage of them correctly than does the Yang & Smith method.
Orthology inference with the genome-guided pipeline is also many times faster than the topology-based methods and, except for the CoGe step, can be run efficiently on a standard desktop computer; something not possible with either of topology-based methods. This computational efficiency results from the fact that the genome-guided method does not require all-by-all BLAST or iterative tree pruning steps. The CoGe step is also very simple and straight-forward to run, as is the process of loading new genomes into the CoGe database. CoGe also has the capacity for uploading and analyzing private genomes without making them public and is exceptionally well documented.
A natural downside of the genome-guided method is the need for two genomes that span the taxonomic clade one is working with. While this approach could be used with only one genome or even a list of genes from a de novo transcriptome assembly, doing so negates its benefits and will increase the likelihood of including paralogs in the analysis. In these situations, topology-based methods are probably the best analysis choice. Because syntenic relationships often break down with increased phylogenetic distance there is likely a limit to the age of clades for which this method can be applied. Based on our experiments with the grape and Poaceae data sets, the method is functional up to at least 100 m.y. of divergence, and can likely be used successfully in any group for which a reasonable number of orthologs can be found between the ingroup and outgroup taxa. However, our experience indicates that syntenic conservation does not always correlate with divergence time, so the method’s utility will have to be evaluated on a case by case basis.
Based on both the binning analysis and the syntenic block trees, we conclude that the secondary topology, or at least the differential placement of the Anthephorine relative to the MPC, is not an artifact of the topology-based methods, but is supported by an appreciable number of genes regardless of the orthology determination method employed. The different topologies of these genes may result from either ILS or post-speciation hybridization, or both.
The small numbers of transcripts representing many of the syntenic blocks in Fig. 3, likely contributed to an inability to infer well supported phylogenies for some of the blocks. However, RF based topology distribution tests confirmed that tree topology distributions in at least certain areas of the genome are likely more similar for genes in a syntenic block than they are across the whole genome. This type of local-synteny analysis should become even more informative in future studies as more sequenced genomes are generated and included in phylogenetic inference. These types of analyses are also not limited to transcriptomic data but have the potential to add value to other data sets generated with probe/hybridization based data collection methods, as long as one or more sequenced genomes exist within the taxonomic group being studied.
The nuclear phylogeny of the Tribe Paniceae produced in this study is consistent with that produced in a previous study. However, that study was only able to sample one nuclear gene and because the inferred topology was incongruent with the many chloroplast phylogenies of the group, it was generally dismissed. This study demonstrates that in fact the nuclear phylogeny of the Paniceae is very different than the chloroplast one, and that those differences are not due to signals in one or a few genes, but are wide spread across the genome. This study also shows that while that original topology, based on only one nuclear gene is supported by many other genes, not all nuclear genes agree with it, and in fact a significant minority of the genes are incongruent with that topology.
The differences between the nuclear and chloroplast phylogenies shown here are critical to both basic and applied questions within the tribe Paniceae. For example, investigations within the tribe of the evolution of C4 photosynthesis, a trait with great economic importance, have focused on the MPC clade at the exclusion of the subtribe Anthephorinae80,82,121. Choices about resource investment, such as which genomes to sequence, have also been based almost exclusively on the chloroplast phylogeny122. Given our results, further resource investment in Paniceae (at least for the purpose of studying C4 photosynthesis) should be directed within the genus Sacciolepis or a close relative to it and the subtribe Anthephorinae. We suggest Sacciolepis indica as a model C3 species for further study as it is a close relative the MPCA clade in both chloroplast and nuclear phylogenies, has a genome size of approximately 523 Mb, and is easily self-pollinated80. An ideal Anthephorinae species for further investment is less clear, but Digitaria cuyabensis has an approximate genome size of 798 Mb making it a good candidate for genome sequencing80. Species within the Crabgrass complex, which includes several different species in the genus Digitaria, might also be good candidates for resource investment due to their economic importance as a noxious weed. Data from both nuclear and organellar genes allows for a more informed way to choose future genomes to sequence than simply using organellar data as was done in the past.
Electronic supplementary material
Acknowledgements
This work was supported by the National Science Foundation (DEB Award no. 1501406), the University of Missouri Research Board, the University of Missouri Mizzou Advantage, and the Sigma Xi Grants-in-Aid of Research Program. The authors thank Elizabeth Kellogg for species sampling advice, Antonis Rokas, Casey Dunn, Stephen A. Smith, Ya Yang, and James Pease for review and/or comments that greatly enhanced the manuscript.
Author Contributions
Material collection, experimental design, and/or library preparation were performed by J.W., J.S., S.Y., Z.Y., T.B., and M.L. Analysis was performed by J.W. with input from J.S., G.C., and J.P. Botanical identifications were done by J.W. and G.D. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
A correction to this article is available online at https://doi.org/10.1038/s41598-018-25620-4.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-13236-z.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
5/2/2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
- 1.Salichos L, Rokas A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature. 2013;497:327–331. doi: 10.1038/nature12130. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Smith SA. Orthology Inference in Nonmodel Organisms Using Transcriptomes and Low-Coverage Genomes: Improving Accuracy and Matrix Occupancy for Phylogenomics. Mol. Biol. Evol. 2014;31:3081–3092. doi: 10.1093/molbev/msu245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunn C, Howison M, Zapata F. Agalma: an automated phylogenomics workflow. BMC Bioinformatics. 2013;14:330. doi: 10.1186/1471-2105-14-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith SA, Moore MJ, Brown JW, Yang Y. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 2015;15:1–15. doi: 10.1186/s12862-015-0423-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misof B, et al. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014;346:763–767. doi: 10.1126/science.1257570. [DOI] [PubMed] [Google Scholar]
- 6.Burleigh JG, et al. Genome-Scale Phylogenetics: Inferring the Plant Tree of Life from 18,896 Gene Trees. Syst. Biol. 2011;60:117–125. doi: 10.1093/sysbio/syq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cibrián-Jaramillo A, et al. Using Phylogenomic Patterns and Gene Ontology to Identify Proteins of Importance in Plant Evolution. GBE. 2010;2:225–239. doi: 10.1093/gbe/evq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaux P-M, et al. Comparative Phylogenomics Uncovers the Impact of Symbiotic Associations on Host Genome Evolution. PLoS Genet. 2014;10:e1004487. doi: 10.1371/journal.pgen.1004487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee EK, et al. A Functional Phylogenomic View of the Seed Plants. PLoS Genet. 2011;7:e1002411. doi: 10.1371/journal.pgen.1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarvis ED, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–1331. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang G, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014;346:1311–1320. doi: 10.1126/science.1251385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamichhaney S, et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature. 2015;518:371–375. doi: 10.1038/nature14181. [DOI] [PubMed] [Google Scholar]
- 13.Malinsky M, et al. Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science. 2015;350:1493–1498. doi: 10.1126/science.aac9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orlando L, et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499:74–78. doi: 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- 15.Librado P, et al. Tracking the origins of Yakutian horses and the genetic basis for their fast adaptation to subarctic environments. Proc. Natl. Acad. Sci. USA. 2015;112:E6889–E6897. doi: 10.1073/pnas.1513696112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsagkogeorga G, Parker J, Stupka E, Cotton JA, Rossiter SJ. Phylogenomic Analyses Elucidate the Evolutionary Relationships of Bats. Curr. Biol. 2013;23:2262–2267. doi: 10.1016/j.cub.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Fontaine, M. C. et al. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science347, doi:10.1126/science.1258524 (2015). [DOI] [PMC free article] [PubMed]
- 18.Neafsey, D. E. et al. Highly evolvable malaria vectors: The genomes of 16 Anopheles mosquitoes. Science347, 10.1126/science.1258522 (2015). [DOI] [PMC free article] [PubMed]
- 19.Foote AD, et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 2015;47:272–275. doi: 10.1038/ng.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin Q, et al. The seahorse genome and the evolution of its specialized morphology. Nature. 2016;540:395–399. doi: 10.1038/nature20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemmon AR, Emme SA, Lemmon EM. Anchored Hybrid Enrichment for Massively High-Throughput Phylogenomics. Syst. Biol. 2012;61:727–744. doi: 10.1093/sysbio/sys049. [DOI] [PubMed] [Google Scholar]
- 22.Lemmon EM, Lemmon AR. High-Throughput Genomic Data in Systematics and Phylogenetics. Annu. Rev. Ecol., Evol. Syst. 2013;44:99–121. [Google Scholar]
- 23.Weitemier K, et al. Hyb-Seq: Combining Target Enrichment and Genome Skimming for Plant Phylogenomics. Appl. Plant Sci. 2014;2:1400042. doi: 10.3732/apps.1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmer EA, Wen J. Using nuclear gene data for plant phylogenetics: Progress and prospects II. Next-gen approaches. J. Syst. Evol. 2015;53:371–379. [Google Scholar]
- 25.Moyle RG, et al. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nature Communications. 2016;7:12709. doi: 10.1038/ncomms12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCormack JE, Tsai WLE, Faircloth BC. Sequence capture of ultraconserved elements from bird museum specimens. Molecular Ecology Resources. 2016;16:1189–1203. doi: 10.1111/1755-0998.12466. [DOI] [PubMed] [Google Scholar]
- 27.Schmickl R, et al. Phylogenetic marker development for target enrichment from transcriptome and genome skim data: the pipeline and its application in southern African Oxalis (Oxalidaceae) Molecular Ecology Resources. 2016;16:1124–1135. doi: 10.1111/1755-0998.12487. [DOI] [PubMed] [Google Scholar]
- 28.Glenn TC, Faircloth BC. Capturing Darwin’s dream. Molecular Ecology Resources. 2016;16:1051–1058. doi: 10.1111/1755-0998.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Budenhagen, C. et al. Anchored Phylogenomics of Angiosperms I: Assessing the Robustness of Phylogenetic Estimates. bioRxiv, doi:10.1101/086298 (2016).
- 30.Barker MS, et al. Multiple Paleopolyploidizations during the Evolution of the Compositae Reveal Parallel Patterns of Duplicate Gene Retention after Millions of Years. Mol. Biol. Evol. 2008;25:2445–2455. doi: 10.1093/molbev/msn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cannon SB, et al. Multiple Polyploidy Events in the Early Radiation of Nodulating and Nonnodulating Legumes. Mol. Biol. Evol. 2015;32:193–210. doi: 10.1093/molbev/msu296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunn CW, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452:745–749. doi: 10.1038/nature06614. [DOI] [PubMed] [Google Scholar]
- 33.Edger PP, et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. USA. 2015;112:8362–8366. doi: 10.1073/pnas.1503926112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hittinger CT, Johnston M, Tossberg JT, Rokas A. Leveraging skewed transcript abundance by RNA-Seq to increase the genomic depth of the tree of life. Proc. Natl. Acad. Sci. USA. 2010;107:1476–1481. doi: 10.1073/pnas.0910449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKain MR, et al. Phylogenomic analysis of transcriptome data elucidates co-occurrence of a paleopolyploid event and the origin of bimodal karyotypes in Agavoideae (Asparagaceae) Am. J. Bot. 2012;99:397–406. doi: 10.3732/ajb.1100537. [DOI] [PubMed] [Google Scholar]
- 36.Sveinsson S, et al. Phylogenetic pinpointing of a paleopolyploidy event within the flax genus (Linum) using transcriptomics. Ann. Bot. 2014;113:753–761. doi: 10.1093/aob/mct306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wickett NJ, et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA. 2014;111:E4859–E4868. doi: 10.1073/pnas.1323926111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wickett NJ, et al. Transcriptomes of the Parasitic Plant Family Orobanchaceae Reveal Surprising Conservation of Chlorophyll Synthesis. Curr. Biol. 2011;21:2098–2104. doi: 10.1016/j.cub.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 39.Xi Z, Liu L, Rest JS, Davis CC. Coalescent versus Concatenation Methods and the Placement of Amborella as Sister to Water Lilies. Syst. Biol. 2014;63:919–932. doi: 10.1093/sysbio/syu055. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, et al. Comparative Transcriptome Analyses Reveal Core Parasitism Genes and Suggest Gene Duplication and Repurposing as Sources of Structural Novelty. Mol. Biol. Evol. 2015;32:767–790. doi: 10.1093/molbev/msu343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pease JB, Haak DC, Hahn MW, Moyle LC. Phylogenomics Reveals Three Sources of Adaptive Variation during a Rapid Radiation. PLoS Biol. 2016;14:e1002379. doi: 10.1371/journal.pbio.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barker MS, et al. Most Compositae (Asteraceae) are descendants of a paleohexaploid and all share a paleotetraploid ancestor with the Calyceraceae. Am. J. Bot. 2016;103:1203–1211. doi: 10.3732/ajb.1600113. [DOI] [PubMed] [Google Scholar]
- 43.McKain MR, et al. A Phylogenomic Assessment of Ancient Polyploidy and Genome Evolution across the Poales. GBE. 2016;8:1150–1164. doi: 10.1093/gbe/evw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei M, Dong D. Phylogenomic analyses of bat subordinal relationships based on transcriptome data. Scientific Reports. 2016;6:27726. doi: 10.1038/srep27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janouškovec J, et al. Major transitions in dinoflagellate evolution unveiled by phylotranscriptomics. Proc. Natl. Acad. Sci. USA. 2017;114:E171–E180. doi: 10.1073/pnas.1614842114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Todd EV, Black MA, Gemmell NJ. The power and promise of RNA-seq in ecology and evolution. Mol. Ecol. 2016;25:1224–1241. doi: 10.1111/mec.13526. [DOI] [PubMed] [Google Scholar]
- 47.Dunn CW, Luo X, Wu Z. Phylogenetic Analysis of Gene Expression. Integr. Comp. Biol. 2013;53:847–856. doi: 10.1093/icb/ict068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honaas LA, et al. Selecting Superior De Novo Transcriptome Assemblies: Lessons Learned by Leveraging the Best Plant Genome. PLoS ONE. 2016;11:e0146062. doi: 10.1371/journal.pone.0146062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conesa A, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:1–19. doi: 10.1186/s13059-016-1047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y, et al. An Efficient Field and Laboratory Workflow for Plant Phylotranscriptomic Projects. Appl. Plant Sci. 2017;5:1600128. doi: 10.3732/apps.1600128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith, S. A. & Pease, J. B. Heterogeneous molecular processes among the causes of how sequence similarity scores can fail to recapitulate phylogeny. Brief Bioinform, bbw034, 10.1093/bib/bbw034 (2016). [DOI] [PMC free article] [PubMed]
- 52.Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen F, Mackey AJ, Stoeckert JCJ, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ebersberger I, Strauss S, von Haeseler A. HaMStR: Profile hidden markov model based search for orthologs in ESTs. BMC Evol. Biol. 2009;9:157. doi: 10.1186/1471-2148-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Theobald DL. A formal test of the theory of universal common ancestry. Nature. 2010;465:219–222. doi: 10.1038/nature09014. [DOI] [PubMed] [Google Scholar]
- 57.Theobald DL. On universal common ancestry, sequence similarity, and phylogenetic structure: the sins of P-values and the virtues of Bayesian evidence. Biology Direct. 2011;6:60. doi: 10.1186/1745-6150-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duarte JM, et al. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 2010;10:1–18. doi: 10.1186/1471-2148-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Dongen, S. Graph Clustering by Flow Simulation. PhD thesis, University of Utrecht, (2000).
- 61.Yang Y, Smith S. Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genomics. 2013;14:328. doi: 10.1186/1471-2164-14-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Howison, M., Sinnott-Armstrong, N. A. & Dunn, C. W. BioLite, a lightweight bioinformatics framework with automated tracking of diagnostics and provenance in Proceedings of the 4th USENIX Workshop on the Theory and Practice of Provenance (2012).
- 63.Yang Y, et al. Dissecting Molecular Evolution in the Highly Diverse Plant Clade Caryophyllales Using Transcriptome Sequencing. Mol. Biol. Evol. 2015;32:2001–2014. doi: 10.1093/molbev/msv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prasanna AN, Mehra S. Comparative Phylogenomics of Pathogenic and Non-Pathogenic Mycobacterium. PLOS ONE. 2013;8:e71248. doi: 10.1371/journal.pone.0071248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Z, Wu M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Scientific Reports. 2015;5:7949. doi: 10.1038/srep07949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bekaert M, Conant GC. Copy Number Alterations among Mammalian Enzymes Cluster in the Metabolic Network. Mol. Biol. Evol. 2011;28:1111–1121. doi: 10.1093/molbev/msq296. [DOI] [PubMed] [Google Scholar]
- 67.Goodstadt L, Ponting CP. Phylogenetic Reconstruction of Orthology, Paralogy, and Conserved Synteny for Dog and Human. PLoS Comp. Biol. 2006;2:e133. doi: 10.1371/journal.pcbi.0020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wapinski I, Pfeffer A, Friedman N, Regev A. Automatic genome-wide reconstruction of phylogenetic gene trees. Bioinformatics. 2007;23:i549–i558. doi: 10.1093/bioinformatics/btm193. [DOI] [PubMed] [Google Scholar]
- 69.Lechner M, et al. Orthology Detection Combining Clustering and Synteny for Very Large Datasets. PLOS ONE. 2014;9:e105015. doi: 10.1371/journal.pone.0105015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang H, et al. Synteny and Collinearity in Plant Genomes. Science. 2008;320:486–488. doi: 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- 71.Schnable JC, Freeling M, Lyons E. Genome-wide analysis of syntenic gene deletion in the grasses. Genome Biol Evol. 2012;4:265–277. doi: 10.1093/gbe/evs009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schnable JC, Springer NM, Freeling M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA. 2011;108:4069–4074. doi: 10.1073/pnas.1101368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schnable JC, Wang X, Pires JC, Freeling M. Escape from preferential retention following repeated whole genome duplications in plants. Front. Plant Sci. 2012;3:94. doi: 10.3389/fpls.2012.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cannon SB, Young ND. OrthoParaMap: Distinguishing orthologs from paralogs by integrating comparative genome data and gene phylogenies. BMC Bioinformatics. 2003;4:35. doi: 10.1186/1471-2105-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fu Z, et al. MSOAR: A High-Throughput Ortholog Assignment System Based on Genome Rearrangement. J. Comput. Biol. 2007;14:1160–1175. doi: 10.1089/cmb.2007.0048. [DOI] [PubMed] [Google Scholar]
- 76.Han MV, Hahn MW. Identifying Parent-Daughter Relationships Among Duplicated Genes. Pacific Symposium on Biocomputing. 2009;14:114–115. [PubMed] [Google Scholar]
- 77.Jun J, Mandoiu II, Nelson CE. Identification of mammalian orthologs using local synteny. BMC Genomics. 2009;10:630. doi: 10.1186/1471-2164-10-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lyons E, Freeling M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. 2008;53:661–673. doi: 10.1111/j.1365-313X.2007.03326.x. [DOI] [PubMed] [Google Scholar]
- 79.Lyons E, Pedersen B, Kane J, Freeling M. The Value of Nonmodel Genomes and an Example Using SynMap Within CoGe to Dissect the Hexaploidy that Predates the Rosids. Tropical Plant Biol. 2008;1:181–190. [Google Scholar]
- 80.Washburn JD, Schnable JC, Davidse G, Pires JC. Phylogeny and photosynthesis of the grass tribe Paniceae. Am. J. Bot. 2015;102:1493–1505. doi: 10.3732/ajb.1500222. [DOI] [PubMed] [Google Scholar]
- 81.Vicentini A, Barber JC, Aliscioni SS, Giussani LM, Kellogg EA. The age of the grasses and clusters of origins of C4 photosynthesis. Global Change Biol. 2008;14:2963–2977. [Google Scholar]
- 82.Grass Phylogeny Working Group II New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 2012;193:304–312. doi: 10.1111/j.1469-8137.2011.03972.x. [DOI] [PubMed] [Google Scholar]
- 83.Washburn JD, Bird KA, Conant GC, Pires JC. Convergent Evolution and the Origin of Complex Phenotypes in the Age of Systems Biology. Int. J. Plant Sci. 2016;177:305–318. [Google Scholar]
- 84.Spriggs EL, Christin P-A, Edwards EJ. C4 Photosynthesis Promoted Species Diversification during the Miocene Grassland Expansion. PLoS ONE. 2014;9:e97722. doi: 10.1371/journal.pone.0097722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burke SV, et al. Evolutionary relationships in Panicoid grasses based on plastome phylogenomics (Panicoideae; Poaceae) BMC Plant Biol. 2016;16:1–11. doi: 10.1186/s12870-016-0823-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brown, J. M. & Thomson, R. C. Bayes factors unmask highly variable information content, bias, and extreme influence in phylogenomic analyses. Syst. Biol., syw101, 10.1093/sysbio/syw101 (2016). [DOI] [PubMed]
- 87.Wen J, et al. Transcriptome Sequences Resolve Deep Relationships of the Grape Family. PLOS ONE. 2013;8:e74394. doi: 10.1371/journal.pone.0074394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stevens, P. F. Angiosperm Phylogeny Website, http://www.mobot.org/MOBOT/research/APweb/ (2017).
- 89.Schnable PS, et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 90.Paterson AH, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 91.Bennetzen JL, et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012;30:555–561. doi: 10.1038/nbt.2196. [DOI] [PubMed] [Google Scholar]
- 92.VanBuren R, et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature. 2015;527:508–511. doi: 10.1038/nature15714. [DOI] [PubMed] [Google Scholar]
- 93.Ouyang S, et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 2007;35:D883–D887. doi: 10.1093/nar/gkl976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.The International Brachypodium Initiative Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 95.Wang L, et al. A Low-Cost Library Construction Protocol and Data Analysis Pipeline for Illumina-Based Strand-Specific Multiplex RNA-Seq. PLoS ONE. 2011;6:e26426. doi: 10.1371/journal.pone.0026426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Babraham Bioinformatics. FastQC A quality control tool for high throughput sequence data., http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2015)
- 97.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protocols. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Henschel, R. et al. In Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the campus and beyond 1–8 (ACM, Chicago, Illinois, USA, 2012).
- 101.Tang H, et al. Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinformatics. 2011;12:1–11. doi: 10.1186/1471-2105-12-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Katoh K, Misawa K, Kuma Ki, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 105.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mirarab S, Bayzid MS, Boussau B, Warnow T. Statistical binning enables an accurate coalescent-based estimation of the avian tree. Science. 2014;346:1250463. doi: 10.1126/science.1250463. [DOI] [PubMed] [Google Scholar]
- 107.Mirarab S, et al. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics. 2014;30:i541–i548. doi: 10.1093/bioinformatics/btu462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Swinton, J. Vennerable, Venn and Euler area-proportional diagrams. https://github.com/js229/Vennerable (2011).
- 109.Inkscape’s Contributors Inkscape. The Inkscape Project, Oregon, USA. https://inkscape.org/en/ (2017).
- 110.Rambaut, A. FigTree. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, UK. http://tree.bio.ed.ac.uk/software/figtree/ (2014).
- 111.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.r-project.org/ (2015).
- 112.Salichos L, Stamatakis A, Rokas A. Novel Information Theory-Based Measures for Quantifying Incongruence among Phylogenetic Trees. Mol. Biol. Evol. 2014;31:1261–1271. doi: 10.1093/molbev/msu061. [DOI] [PubMed] [Google Scholar]
- 113.Kobert K, Salichos L, Rokas A, Stamatakis A. Computing the Internode Certainty and related measures from partial gene trees. Mol. Biol. Evol. 2016;33:1606–1617. doi: 10.1093/molbev/msw040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.National Center for Biotechnology Information. Genome Assembly and Annotation report, Setaria italica (foxtail millet), http://www.ncbi.nlm.nih.gov/genome/10982?genome_assembly_id=276542 (2017).
- 115.Huerta-Cepas J, Serra F, Bork P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016;33:1635–1638. doi: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nabhan AR, Sarkar IN. The impact of taxon sampling on phylogenetic inference: a review of two decades of controversy. Briefings in Bioinformatics. 2011;13:122–134. doi: 10.1093/bib/bbr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hillis DM. Approaches for Assessing Phylogenetic Accuracy. Syst. Biol. 1995;44:3–16. [Google Scholar]
- 118.Huelsenbeck JP. Performance of Phylogenetic Methods in Simulation. Syst. Biol. 1995;44:17–48. [Google Scholar]
- 119.Hillis DM, Pollock DD, McGuire JA, Zwickl DJ. Is Sparse Taxon Sampling a Problem for Phylogenetic Inference? Syst. Biol. 2003;52:124–126. doi: 10.1080/10635150390132911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Havird JC, Miyamoto MM. The importance of taxon sampling in genomic studies: An example from the cyclooxygenases of teleost fishes. Mol. Phylogen. Evol. 2010;56:451–455. doi: 10.1016/j.ympev.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 121.Washburn, J. D. et al. Ancestral Reconstruction and C3 Bundle Sheath Transcript Abundance in the Paniceae Grasses Indicate the Foundations for all Three Biochemical C4 Sub-Types Were Likely Present in the Most Recent Ancestor. bioRxiv, 10.1101/162644 (2017).
- 122.Studer AJ, et al. The draft genome of the C3 panicoid grass species Dichanthelium oligosanthes. Genome Biol. 2016;17:223. doi: 10.1186/s13059-016-1080-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available in the NCBI SRA repository under the identifiers noted in Supplemental Table S1.