

ABSTRACT
RNase PH, encoded by the rph gene, is a 3′→5′ exoribonuclease that in E. coli participates primarily in the 3′ maturation of pre-tRNAs and the degradation of rRNA in stationary-phase cells. Interestingly, the routinely used laboratory strains of MG1655 and W3110 have naturally acquired the rph-1 allele, encoding a truncated catalytically inactive RNase PH protein which is widely assumed to be benign. Contrary to this assumption, we show that the rph-1-encoded Rph-1 protein inhibits RNase P-mediated 5′-end maturation of primary pre-tRNAs with leaders of <5 nucleotides in the absence of RppH, an RNA pyrophosphohydrolase. In contrast, RppH is not required for 5′-end maturation of endonucleolytically generated pre-tRNAs in the rph-1 strain and for any tRNAs in Δrph mutant or rph+ strains. We propose that the Rph-1 protein bound to the 3′ end of the substrate creates a steric hindrance that in the presence of a triphosphate at the 5′ end reduces the ability of RNase P to bind to the pre-tRNA.
IMPORTANCE In this paper, we demonstrate that the rph-1 mutation found in commonly used E. coli strains leads to the synthesis of a truncated functionally inactive RNase PH protein that interferes with the 5′-end maturation of specific tRNAs with short 5′ leaders by RNase P in the absence of RppH, an RNA pyrophosphohydrolase that converts primary 5′ triphosphates into 5′ monophosphates. The data presented indicate that the presence of the triphosphate interferes with RNase P binding to the pre-tRNA.
KEYWORDS: RNase P, RNase E, RNA pyrophosphohydrolase
INTRODUCTION
Escherichia coli contains 86 tRNAs that are processed both exo- and endonucleolytically from both mono- and polycistronic operons (1–4). E. coli has six 3′→5′ exoribonucleases that actively participate in the 3′ maturation of tRNAs: RNase T, RNase PH, RNase D, RNase BN, RNase II, and polynucleotide phosphorylase (PNPase) (5, 6). RNase T, encoded by rnt, and RNase PH, encoded by rph, are considered to be the major exoribonucleases involved in tRNA 3′-end maturation (7, 8). RNase T has been shown to precisely mature 3′ tRNA ends, unless it encounters C nucleotides immediately downstream of the CCA determinant (9). In that case, RNase PH completes the 3′-end maturation, as has been shown for the valU tRNA (5, 6). In addition, RNase PH has been shown to be required for the initiation of rRNA degradation under nutrient starvation (10, 11). Interestingly, many routinely used E. coli strains, such as MG1655 and W3110 and their derivatives, naturally contain the rph-1 allele, which arises from a GC base pair deletion near the 3′ end of the rph gene, resulting in a truncated catalytically inactive Rph-1 protein (12).
The 5′ leader sequences of the precursor tRNAs can vary from 2 to 55 nucleotides (nt) (13). Although there are multiple 3′→5′ exoribonucleases that can generate the mature 3′ termini of these tRNAs, RNase P is the only known enzyme implicated in the maturation of 5′ termini for all tRNAs. Bacterial RNase P is a ribozyme consisting of a highly conserved RNA (M1 encoded by rnpB), which is the catalytic subunit, and a small protein (C5 encoded by rnpA) responsible for binding the pre-tRNA 5′ leader sequence. Numerous studies have shown that the RNase P holoenzyme has a preference for certain nucleotides and leader lengths (the region upstream of +1 nucleotide of the mature tRNA sequence) at the 5′ termini of its tRNA substrates (13–22). It has been suggested that the affinity of the C5 protein subunit for the leader sequence is enhanced through its interaction at nt −5, −4, and −3 upstream of the cleavage site (16, 23). Consequently, pre-tRNAs with leaders of ≥5 nt will have stronger interactions than those with leaders of <5 nt (24). It is interesting to note that only 6 primary tRNA transcripts have predicted 5′ leader sequences of <5 nt (13).
Besides its role in the maturation of tRNA 5′ ends, RNase P, as well as RNase E, is required to separate a variety of polycistronic primary tRNA transcripts into pre-tRNAs (1–3, 5, 25), which are further processed to mature species that can be aminoacylated. In E. coli, endonucleolytic cleavages by RNase E and its paralog RNase G are significantly enhanced by the removal of the pyrophosphate from the 5′-terminal triphosphate of certain primary transcripts by the RNA pyrophosphohydrolase RppH, encoded by rppH (26, 27). RppH belongs to the nucleoside diphosphate X (Nudix) superfamily of hydrolases, of which there are 13 members in E. coli (29).
However, recent studies have demonstrated that the RNase E phosphate sensor domain, which interacts with the 5′ phosphate of a substrate (30), is not essential for its activity in vivo (31). Thus, RNase E can process some primary transcripts containing a 5′ triphosphate using “direct entry” at internal sites, bypassing the requirement for a monophosphate 5′ end (32, 33). In fact, RNase E, with or without its C-terminal scaffold domain, a region of the protein that facilitates the formation of the degradosome (34), has been shown to act on several primary tRNA precursors, such as argX, pheU, glyW, and leuZ (35).
With the exception of the results obtained with the leuX primary transcript (4), unlike with RNase E, nothing is known about whether RNase P activity is affected by the presence of a 5′ triphosphate. Accordingly, we carried out a comprehensive analysis of four polycistronic and 12 monocistronic tRNA transcripts with various 5′ leader lengths to determine if there was a requirement for RppH-dependent conversion of the 5′ triphosphate to a monophosphate for efficient tRNA maturation by RNase P. As a control, we also examined the maturation of several polycistronic transcripts that utilize RNase E to generate pre-tRNAs.
Here, we show that RNase P processing of primary tRNA transcripts with short (<5 nt) leader sequences is inhibited in an rph-1 mutant strain in the absence of RppH. The data suggest that this inhibition is due to a combination of the triphosphate group close to the RNase P cleavage site on the 5′ end, along with steric hindrance from the catalytically inactive Rph-1 protein bound at the 3′ end. Thus, tRNA processing proceeded normally in the presence of RppH or with a wild-type RNase PH protein, as well as with endonucleolytically generated pre-tRNAs from polycistronic tRNA transcripts. Additional support for the steric hindrance by Rph-1 protein came from the fact that 5′ processing by RNase P was normal in cells completely lacking RNase PH. Furthermore, the inhibition of RNase P activity was not observed if RNase E-mediated removal of the Rho-independent transcription terminator at the 3′ end was blocked.
RESULTS
The absence of RppH inhibits the 5′-end maturation of the pheU, pheV, and ileX tRNAs.
Conversion of the 5′ triphosphate on primary transcripts to a monophosphate by the RNA pyrophosphohydrolase encoded by rppH plays an important role in RNase E-dependent processing and decay of many mRNAs (26). Surprisingly, little is known about the ability of RNase P to process primary transcripts containing a 5′ triphosphate, considering that it is the only RNase known to be involved in the 5′ maturation of tRNAs. The one exception is the leuX transcript, which has a 22-nucleotide 5′ leader and is processed normally in the absence of RppH (4). Accordingly, we examined the processing of a number of monocistronic tRNA transcripts in a variety of ΔrppH mutants. Since it has been shown that the length of the 5′ leader of the tRNAs can influence their efficiency of processing by RNase P and, in turn, bacterial growth (13, 24), we included tRNAs with leader sequences ranging from 2 to 22 nt (Table 1). We tested the processing of the four asparagine precursors (asnT, asnU, asnV, and asnW), two proline precursors (proK and proL), two arginine precursors (argW and argU), two phenylalanine precursors (pheU and pheV), and the ileX tRNA precursor.
TABLE 1.
Effect of rph alleles on 5′-end maturation of tRNAs in ΔrppH mutant strains
The asparagine tRNA precursors are matured by a combination of RNase E, RNase P, and 3′→5′ exonucleases (2). The proline tRNA precursors are processed solely by RNase E and RNase P independent of any 3′→5′ exonucleolytic activity (36). Both the pheU and pheV transcripts are monocistronic with same mature tRNA sequence but contain differing Rho-independent transcription terminators that are removed by RNase E (2). They have predicted 5′ leaders of fewer than five nucleotides (2, 13, 37). The processing pathways for the argW and argU tRNAs are not known at this time.
The processing profiles of asnT, asnU, asnV, asnW, argU, argW, proK, and proL were identical in the rph-1 and ΔrppH rph-1 mutant strains (Table 1; also see Fig. S1 in the supplemental material). A single band of mature tRNA species in both strains suggested no effect of RppH on 5′-end processing of these tRNAs (Fig. S1). Since all of these tRNAs have 5′ leaders of 5 nt or longer (2, 13, 36), we tested pheU, pheV, and ileX tRNAs with predicted 5′ leader sequences of <5 nt (13, 37). Northern analysis of the pheU and pheV tRNAs yielded very different results (Fig. 1B). As expected, larger-than-mature tRNA species consistent with unprocessed 5′ ends were observed in the rnpA49 rph-1 mutant strain at the nonpermissive temperature (Fig. 1B, lane 3). However, a processing intermediate that was several nucleotides longer than the mature tRNA species was also detected in the ΔrppH rph-1 mutant strain compared to the rph-1 mutant strain (Fig. 1B, compare lanes 1 and 2). The processing profiles of rnpA49 rph-1 and ΔrppH rnpA49 rph-1 multiple mutants were experimentally identical (Fig. 1B, lanes 3 and 6). As shown previously (2), the full-length pheU and pheV transcripts that retained the Rho-independent transcription terminator appeared in the rne-1 rph-1 double mutant in addition to the mature species (lane 4), indicating a role of RNase E in the removal of the Rho-independent transcription terminator. Interestingly, the larger processing intermediate seen in the ΔrppH rph-1 double mutant (Fig. 1B, lane 2) was absent in the ΔrppH rne-1 rph-1 triple mutant (Fig. 1B, lane 5), suggesting that the retention of the Rho-independent transcription terminator prevented the formation of this product.
FIG 1.

Analysis of the processing of the pheU and pheV monocistronic transcripts in various genetic backgrounds. (A) Schematic presentation of the pheU/V transcripts. Predicted RNase E (E) (2) as well as RNase P (P) cleavage sites are shown above the cartoon. The position of the oligonucleotide probe (d) is shown below the cartoon. The diagram is not drawn to scale. (B) Northern blot of the phe transcripts using the oligonucleotide probe (d) that hybridizes to the mature sequences of both pheU and pheV. Northern analysis was conducted as described in Materials and Methods. The processing intermediates identified previously (2) are shown to the right of the image. The genotypes of the strains used are indicated above each lane. (C) Primer extension analysis of pheU and pheV transcripts. The two have identical coding sequences and sequences upstream of the 5′ termini. Primer extension analysis was conducted as described in Materials and Methods. The mature 5′ termini of both transcripts are indicated as M, and the transcription start sites are shown as M + 3 and M + 4, respectively. The nucleotide sequences corresponding to the autoradiogram are shown to the left. The transcription start sites M + 3 (G) and M + 4 (C) are underlined and the 5′ mature terminus M (G) is in bold. Unrelated lanes were removed from the image (dotted lines). (D) The nucleotide sequences upstream of the 5′ mature terminus G (bold), indicating the identified transcription start sites (bent arrows) and putative −10 and −35 sequences.
In order to determine if the larger products observed in the ΔrppH rph-1 and rnpA49 rph-1 double mutants had identical 5′ leader sequences, primer extension analysis was carried out (Fig. 1C). The intensities of the two primer extension products (M + 3 and M + 4) observed in the rnpA49 rph-1 double mutant were substantially reduced in the rph-1 mutant strain, suggesting their dependence on RNase P. More importantly, their levels were increased in the ΔrppH rph-1 mutant compared to the rph-1 mutant strain. Furthermore, a major primer extension product (M) observed in the rph-1 mutant strain was significantly reduced in the rnpA49 rph-1 mutant, but its level was higher in the ΔrppH rph-1 mutant strain.
The large increase of M + 3 and M + 4 bands in the rnpA49 rph-1 mutant strain, coupled with the significant reduction of the M band compared to the rph-1 mutant strain, indicated that M + 3 and M + 4 were the two primary transcription initiation sites and that M represented the mature 5′ end for pheU and pheV transcripts (Fig. 1D). In fact, near consensus −10 (5/6 nt) and −35 (5/6 nt) promoter sequences were identified upstream of M + 4 band (Fig. 1D). The increase in the levels of M + 3 and M + 4 bands in the ΔrppH rph-1 mutant strain was consistent with the inhibition of RNase P processing of the 5′ ends of the pheU and pheV pre-tRNAs. It should also be noted that in both the rph-1 and ΔrppH rph-1 mutant strains, approximately 15 to 20% of the mature pheU and pheV tRNAs contained an extra nucleotide at the 5′ mature terminus (M, Fig. 1C), indicating more than one RNase P cleavage site at the 5′ terminus. Two distinct 5′ mature termini have also been reported for the proL tRNA (36).
Since the inactivation of RppH inhibited the 5′-end processing of the pheU and pheV primary transcripts containing 3- to 4-nt 5′ leader sequences (Fig. 1), we also examined the ileX transcript, which has a predicted leader of 1 to 2 nucleotides (13, 37). In fact, Northern analysis showed that the putative mature species were approximately 1 or 2 nt longer in the ΔrppH rph-1 and rnpA49 rph-1 double mutants than in the rph-1 mutant strain, indicating retention of the 5′ leader sequence in the absence of RppH or RNase P, respectively (data not shown, see below).
Sequence analysis of the pheU, pheV, and ileX transcripts confirmed the reduced 5′-end processing in the absence of RppH.
To provide additional support for inefficient 5′-end processing of the pheU, pheV, and ileX transcripts in an ΔrppH rph-1 double mutant, we mapped both their 5′ and 3′ ends by sequencing the 5′-3′ junctions of the cDNAs derived from circularized tRNA transcripts (4, 25, 38). Steady-state RNA isolated from various strains was initially treated with tobacco acid pyrophosphatase (TAP) to convert all the 5′-triphosphate ends into monophosphates in order to facilitate the circularization. The differences in the 3′ downstream sequences of pheU and pheV following the encoded CCA determinant made it possible to distinguish between these two transcripts. As shown in Fig. 2, 24/28 (∼86%) clones had mature 5′ termini in the rph-1 mutant strain, while 4/28 (∼14%) had 2 to 4 unprocessed nucleotides at their 5′ termini. In contrast, 13/25 (∼52%) clones in the ΔrppH rph-1 and 23/24 (∼96%) clones in the rnpA49 rph-1 double mutants retained 2 to 4 unprocessed 5′ nucleotides. These data were consistent with the primer extension analysis (Fig. 1C).
FIG 2.
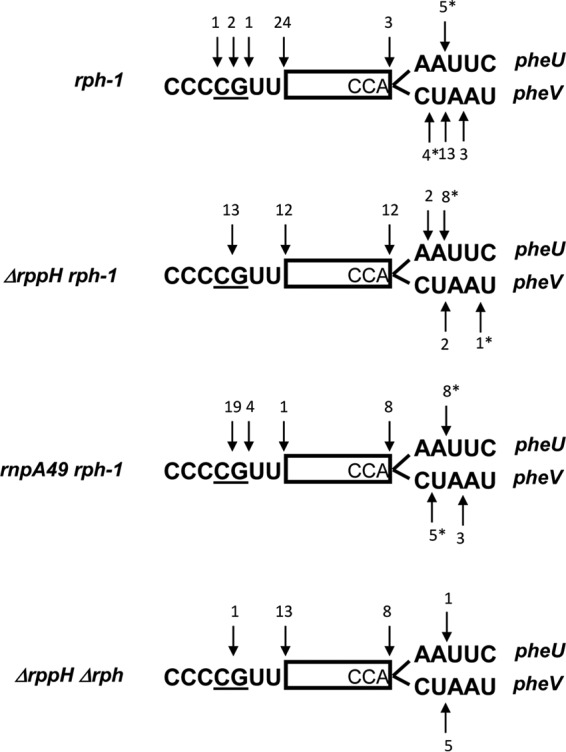
Identification of 5′ and 3′ termini of pheU and pheV transcripts by cDNA cloning and sequencing of circularized RNAs in various genetic backgrounds, as described in Materials and Methods. The mature sequences are identical for pheU and pheV and are represented as a rectangle. The 5′ upstream sequences are also identical for pheU and pheV, and the two transcription initiation sites, as determined in this study (Fig. 1C and D), are underlined. The 3′ sequences downstream of CCA are distinct for pheU and pheV and are shown in parallel. Each arrow (down or up) represents a terminus, as determined from the cDNA sequencing. Numbers above or below the arrow are numbers of 3′ or 5′ ends identified by sequencing. An asterisk indicates that some 3′ ends at a particular location contained untemplated poly(A) tails of 1 to 3 nt.
The analysis of ileX transcripts showed that only 9% of the transcripts had 2 extra nucleotides at the 5′ terminus in the rph-1 single mutant, compared to over ∼94% of the transcripts in both the ΔrppH rph-1 and rnpA49 rph-1 double mutants (Fig. S2).
Reduction in 5′-end maturation of the pheU, pheV, and ileX tRNAs in the ΔrppH mutant is specific to the rph-1 allele.
Since all derivatives of MG1655 and W3110 contain the naturally occurring rph-1 allele (12), we compared the processing of pheU and pheV transcripts in an rph-1 mutant with a strain carrying a complete deletion of the rph coding sequence (Δrph749::kan [39]). The profiles of the phe transcripts were identical in the two single mutants, showing an accumulation of processing intermediates of 1 or 2 nucleotides larger than the mature species (Fig. 3B, lanes 1 and 5), which disappeared in the strain carrying the wild-type rph gene (38) (Fig. 3B, lane 2), demonstrating that the Rph-1 protein was catalytically inactive. More importantly, the larger processing intermediates that accumulated in the ΔrppH rph-1 mutant strain were absent in the ΔrppH rph+ and ΔrppH Δrph mutant strains (Fig. 3B, lanes 3, 4, and 6). Cloning and sequencing of the 5′ and 3′ termini of the pheU and pheV transcripts isolated from an ΔrppH Δrph double mutant showed that 13/14 (∼93%) transcripts had mature 5′ termini, compared to 12/25 (∼48%) in the ΔrppH rph-1 double mutant (Fig. 2). A similar analysis of the ileX transcript showed that 8/18 (∼44%) transcripts had mature 5′ termini in the ΔrppH Δrph mutant strain compared to 1/19 (∼5%) in the ΔrppH rph-1 double mutant (Fig. S2).
FIG 3.
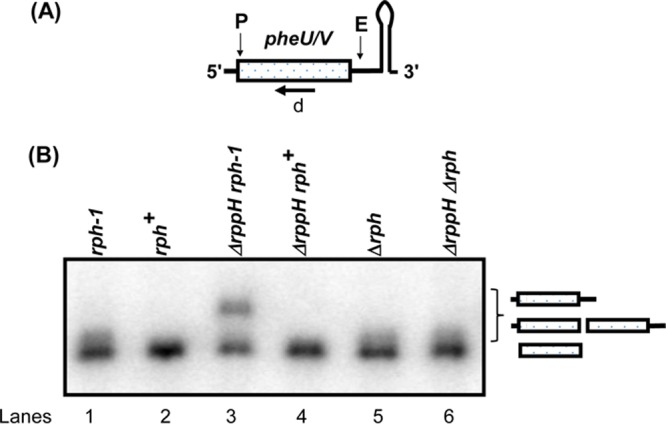
Analysis of the processing of the pheU and pheV monocistronic transcript in the presence and absence of RNase PH. (A) Schematic presentation of the phe transcripts is as described in Fig. 1A. (B) Northern blot of the phe transcripts using the oligonucleotide probe is as described in Fig. 1B. The genotypes of the strains used are indicated above each lane.
The sequencing analysis of the 3′ ends of pheU and pheV transcripts in the rph-1, ΔrppH rph-1, and rnpA49 rph-1 mutant strains showed that a large number of the transcripts retained 1 to 2 downstream nucleotides downstream of the CCA determinant (Fig. 2). This result was consistent with the heterogeneous bands observed during Northern analysis in the rph-1, Δrph, and ΔrppH Δrph mutants (Fig. 3B, lanes 1, 5, and 6). A similar analysis in an rph+ (wild-type) strain showed a much higher percentage of mature 3′ termini (Fig. 3B, lane 2, and data not shown), indicating that RNase PH played a significant role in the 3′-end maturation of the phe transcripts, following removal of the Rho-independent transcription terminator by RNase E (2). The involvement of RNase PH is likely mandated by the presence of a C residue immediately downstream of the CCA determinant in the pheV transcript (Fig. 2). Zuo and Deutscher (9) have shown that the presence of C residue among the nucleotides that need to be processed significantly inhibits the 3′→5′ exonuclease RNase T, the enzyme that normally is involved in the final maturation of the 3′ terminus. In addition, it should also be noted that a small fraction (between 8 and 14%) of the clones in the various genetic backgrounds had short poly(A) tails added to immature 3′ termini (Fig. 2, asterisks), in agreement with previously reported results (38).
The absence of RppH did not affect endonucleolytically processed pre-tRNA transcripts.
In order to determine if the effect of rph-1 allele was specific to primary transcripts with short leaders, such as pheU, pheV, and ileX, that have not undergone dephosphorylation, we also analyzed the processing of two well-characterized RNase E-dependent polycistronic tRNA transcripts, argX hisR leuT proM and glyW cysT leuZ, where pre-tRNAs are generated by initial endonucleolytic cleavages of the polycistronic transcripts (1, 2). Processing of the argX operon transcript was analyzed by Northern analysis (Fig. 4B) using either argX- or hisR-specific probes (a and b [Fig. 4A]). As expected, inactivation of RNase E in the rne-1 rph-1 double mutant led to the detection of various high-molecular-weight processing intermediates compared to the rph-1 control using the hisR-specific probe, which is a unique tRNA in the genome. (Fig. 4B, lanes 1 and 3). Consequently, the processed fraction (PF) of the mature hisR tRNA species was reduced in the rne-1 rph-1 mutant to 0.6 (lane 3), compared to 0.9 in the rph-1 mutant strain (lane 1). Inactivation of RNase P led to the expected accumulation of the hisR pre-tRNA that retained its 5′ leader sequences with a large reduction in the PF value (lane 2). These results were in agreement with previous reports showing that the argX hisR leuT proM primary transcript is initially processed by RNase E (1, 2). Almost identical hisR processing profiles and PF values between the rph-1 and ΔrppH rph-1 mutants and between the rne-1 rph-1 and ΔrppH rne-1 rph-1 mutant strains (Fig. 4B, compare lanes 1 versus 4 and 3 versus 5) suggested that the inactivation of RppH had no effect on the processing of either the argX hisR leuT proM primary transcript or the hisR pre-tRNAs.
FIG 4.
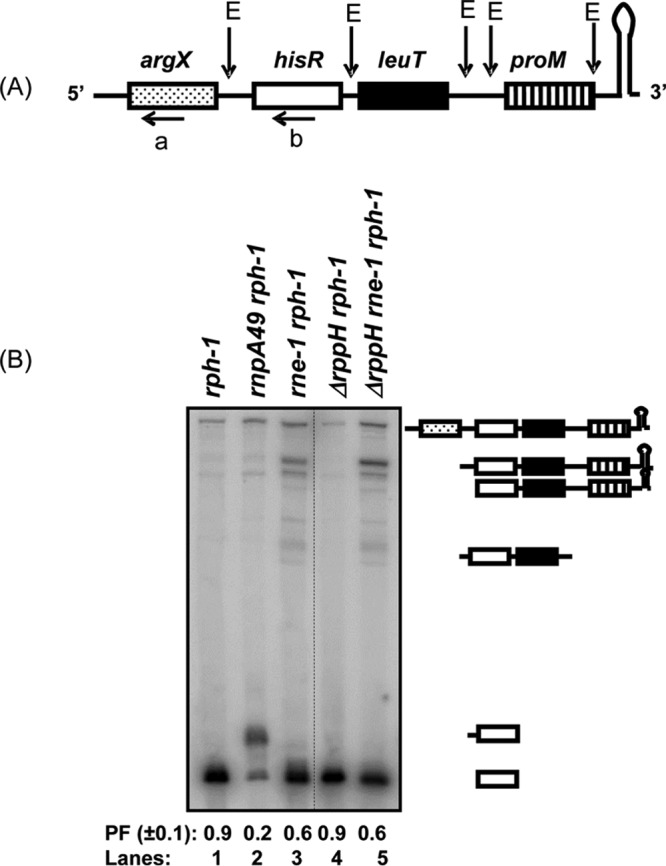
Analysis of the processing of the argX hisR leuT proM polycistronic transcript. (A) Schematic presentation of the argX hisR leuT proM transcript. Previously identified major RNase E cleavage sites (E) (1, 2, 25, 36) are shown above the cartoon. The positions of oligonucleotide probes (a and b) are shown below the respective tRNA. The diagram is not drawn to scale. (B) Northern blot of argX hisR leuT proM transcript probed with hisR-specific oligonucleotide probe (b). Northern analysis was conducted as described in Materials and Methods. All the major processing intermediates identified previously (1, 2) are shown to the right of the image. Processed fraction (PF) denotes the fraction of the mature tRNA relative to the total amount of unprocessed and processed tRNA. The genotypes of the strains used are indicated above each lane. Unrelated lanes were removed from the image (dotted line).
No effect of RppH in the processing of the argX operon transcript was consistent with previous reports, suggesting that RNase E can process certain polycistronic tRNA transcripts using a direct entry mechanism (31, 34, 35). If the argX operon transcripts were processed in vivo by RNase E via a direct entry mechanism, it would cleave 8 nt upstream of hisR (Fig. 4A) (25), generating an argX pre-tRNA with 5′-triphosphate leader sequence (13 nt) that would have to be processed by RNase P. However, an argX-specific probe (a, Fig. 4A) identified the mature argX tRNA only in the ΔrppH rph-1 double mutant, suggesting normal RNase P processing in the absence of RppH (Fig. S3 and Table 1).
We also analyzed glyW cysT leuZ, which is initially processed by RNase E to generate a cysT pre-tRNA with a 4-nucleotide leader sequence (2, 25) that has a 5′ monophosphate. While cysT was also processed normally in both the rph-1 and ΔrppH rph-1 mutant strains, a higher-molecular-weight cysT pre-tRNA consistent with unprocessed 5′ leader sequences was clearly present in the rnpA49 rph-1 double mutant (Fig. S3).
RNase P also plays a major role in the initial processing of a number of primary polycistronic tRNA transcripts into pre-tRNAs (3, 5, 25). In order to determine if the 5′-terminal triphosphate might affect RNase P processing of primary polycistronic tRNA transcripts, we analyzed the processing of the valV valW transcript (Fig. 5B). In agreement with a previous report (3), a significant accumulation of full-length valV valW transcripts was observed in the rnpA49 rph-1 double mutant (lane 2) when a Northern blot was probed with an oligonucleotide complementary to both valV and valW (c, Fig. 5A). There was no effect on valV valW transcript processing when RNase E was inactivated (lane 3). In addition, the comparable processing profiles observed in the rph-1 and ΔrppH rph-1 mutant strains and in the rnpA49 rph-1 and rnpA49 ΔrppH rph-1 mutants (compare lanes 1 versus 4 and 2 versus 5) suggested that RppH had no effect on the processing of the valV valW transcript by RNase P. Analysis of a second RNase P-dependent operon, leuQ leuP leuV, also showed normal RNase P processing of the transcript in the absence of RppH (data not shown).
FIG 5.
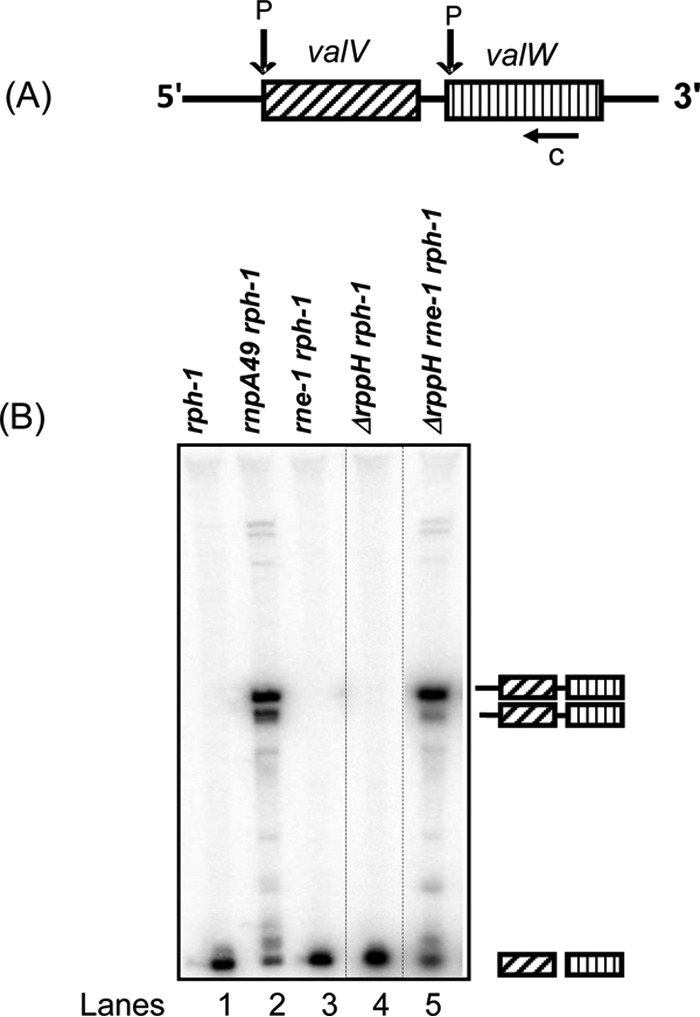
Analysis of the processing of the valV valW polycistronic transcript. (A) Schematic presentation of the valV valW transcript. Previously identified RNase P-processing sites (P) (3) are shown above the cartoon. The position of the oligonucleotide probe (c) is shown below the cartoon. The diagram is not drawn to scale. (B) Northern blot of the valV valW transcript probed with the oligonucleotide probe (c) that hybridizes to mature sequences of both valV and valW (3). Northern analysis was conducted as described in Materials and Methods. The processing intermediates identified previously (3) are shown to the right of the image. The genotypes of the strains used are indicated above each lane. Unrelated lanes were removed from the image (dotted lines).
RNase PH, RppH, and RNase P have no physical interactions.
The data presented in Fig. 3 and Table 1 suggested that RppH is required for efficient 5′ processing of specific tRNA transcripts (pheU, pheV, and ileX) by RNase P in the rph-1 genetic background. Western blot analysis demonstrated that the Rph-1 protein was only ∼2 to 3 kDa smaller in size and was produced at significantly lower levels than the wild-type RNase PH (Fig. S4, lanes 1 to 4). No band was detected in the Δrph mutant strain (Fig. S4, lanes 5 and 6). This result was in contrast to previous minicell protein expression experiments showing the truncated Rph-1 protein expression almost at a comparable level to the wild-type protein (12).
While the Rph-1 protein had no enzymatic activity (12) (Fig. 3, compare lanes 1, 2, 4, and 5), the expression of rph-1 allele from a low-copy-number plasmid (pNWK18, rph-1 Cmr) in either the rph+ or Δrph genetic background led to the inhibition of RNase P processing of pheU and pheV tRNA 5′ ends (data not shown), indicating a dominant negative effect of the rph-1 allele. To determine if there were any physical interactions among RNase PH and RppH and/or RNase PH and RNase P, we carried out immunoprecipitation experiments using polyclonal antibodies against either RNase PH, RppH, or the protein subunit of RNase P. In these experiments, highly specific polyclonal RNase PH antibodies (40) were used for the initial pulldown experiments. Subsequently, the precipitated material was tested for the presence of RppH, RNase P, or RNase PH by Western blotting. No physical interactions were detected under the experimental conditions we employed (data not shown).
Strains with rph-1 allele have a growth defect.
Based on the inefficient 5′-end maturation of certain primary tRNAs in the rph-1 mutant genetic background, we speculated that these mutants would manifest a growth defect. A comparison of the growth rates of rph+, ΔrppH, rph-1, and ΔrppH rph-1 mutant strains yielded generation times of 25 ± 2, 26 ± 2, 30 ± 2, and 32 ± 3 min, respectively, suggesting a small but consistent growth difference between the rph+ and rph-1 mutant strains.
DISCUSSION
The RNase PH plays an important role in tRNA 3′-end maturation (5, 6) and the initiation of rRNA degradation during starvation (10, 11). Strikingly, the routinely used E. coli strains MG1655 and W3110, and their derivatives, have long been known to contain a naturally acquired rph-1 mutation that results in a truncated but inactive RNase PH protein (12). Here, we have shown that that rph-1 allele inhibits 5′-end maturation of certain primary tRNA transcripts in the absence of the RNA pyrophosphohydrolase, RppH, mimicking the phenotype observed with rnpA49 allele at nonpermissive temperature. However, the inhibition of 5′-end processing by RNase P was limited only to primary tRNA transcripts having leaders of <5 nt (Fig. 1 to 3 and Table 1) and not to the endonucleolytically processed pre-tRNAs (Fig. 4 and 5; also see Fig. S3 in the supplemental material), which have 5′-phosphomonoester termini.
The Rph-1 protein (Fig. S4) had no detectable 3′→5′ exonucleolytic activity (Fig. 3B, lanes 1, 2, and 5) and did not physically interact with either RppH or RNase P under the conditions tested (data not shown). Thus, the complete loss of the inhibition of RNase P activity on the pheU, pheV, and ileX transcripts in the presence of the wild-type rph allele or when the rph gene was completely deleted (Fig. 3B, lanes 2, 4, and 6) indicated some type of physical interference by the Rph-1 protein with RNase P. Furthermore, the fact that the 5′ ends of endonucleolytically processed pre-tRNAs were processed normally in the ΔrppH rph-1 double mutant strongly suggested that the presence of a 5′ triphosphate on the primary transcript in the absence of RppH somehow interfered with RNase P binding (Fig. 6). The normal processing of the pheU and pheV transcripts in the rph-1 single mutant when RppH was also present suggested that the Rph-1 protein did not interfere with the conversion of a 5′-triphosphate end to a 5′ monophosphate end (Fig. 1 and 3).
FIG 6.

Modulation of RNase P processing of specific pre-tRNAs by Rph-1 protein. A RNase P holoenzyme-pre-tRNA complex is formed prior to RNase P cleavage at the mature 5′ end of the tRNA (41). (A) The RNase P cleavage is normal in a strain containing either wild-type RNase PH or no RNase PH and for tRNAs with 5′ leader sequence larger than 5 nucleotides. (B) Binding of the Rph-1 protein to the 3′ terminus, even though it is functionally inactive, would likely block the ability of RNase P to interact with CC of CCA (43, 55). The presence of a triphosphate at the 5′ end of a short leader sequence closer to the RNase P cleavage site would also interfere with the interaction of C5 protein with single-stranded leader sequence. (C) Conversion of 5′ triphosphate to monophosphate by RppH is sufficient for C5 protein to restore RNase P binding and cleavage at the 5′ mature terminus. (D) Presence of a longer 3′ trailer sequence, such as the Rho-independent transcription terminator, in the absence of the endonucleolytic cleavage, would exclude Rph-1 protein binding to CCA. This would result in normal interaction of RNase P with CC of CCA and cleavage at the 5′ mature terminus. This hypothesis would also explain why the loss of RNase P activity on the pheU and pheV substrates in the ΔrppH rph-1 double mutant is not as severe as seen in the rnpA49 rph-1 double mutant.
Based on the recent work of Luciano et al. (28), in which they demonstrated that a significant number of mRNAs contain 5′ diphosphates, we hypothesize that a 5′-terminal diphosphate or triphosphate affects the processing of pre-tRNAs by RNase P with short leader sequences, based on its proximity to the processing site. It has previously been shown that the catalytic efficiency of the RNase P holoenzyme depends on interaction of the M1 RNA subunit with CC of CCA at the 3′ terminus and C5 protein subunit with the 5′ leader sequence of a pre-tRNA substrate (41) (Fig. 6A). An interaction of the central cleft of the RNase P protein with the −4 to −8 region upstream of the RNase P cleavage site in the single-stranded 5′ leader sequence has been shown by photo-cross-linking and primer extension analysis (16). Thus, the presence of a triphosphate or diphosphate at the 5′ end of a short leader region may prevent a strong interaction with C5 protein (Fig. 6B). Endonucleolytically or RppH processed pre-tRNAs containing a 5′ monophosphate end would not interfere with RNase P binding (Fig. 6C). The tRNAs with longer 5′ leaders, such as leuX and the four asn tRNAs, would not also be subjected to this type of interference (Fig. 6A).
Furthermore, an interaction between the two cytosine residues of the CCA determinant at the 3′ terminus of the pre-tRNA and two conserved G residues in the L15 loop of the M1 RNA (42) has also been shown to increase RNase P substrate affinity, selection of the correct cleavage site, and catalysis (43). It should be noted that the presence of a C nucleotide immediately downstream of the CCA determinant in the pheV transcripts (Fig. 2) indicates that RNase PH is actually required for the efficient processing of this particular tRNA. Thus, although catalytically inactive, it is likely that the Rph-1 protein still binds to the 3′ termini of some pre-tRNA substrates, preventing the M1 RNA from interacting efficiently with CC of CCA (Fig. 6B). The suppression of the RNase P processing defect in an rph deletion strain (Fig. 3, lane 6) is consistent with this hypothesis. Furthermore, the fact that RNase P inhibition was also abolished in the presence of a wild-type RNase PH protein (Fig. 3, lane 4) suggested that the Rph-1 protein binds more tightly to potential substrates than the wild-type protein. Thus, by blocking the removal of the Rho-independent transcript terminator by inactivating RNase E (Fig. 1, lane 5), the Rph-1 was prevented from binding close to the CCA determinant (Fig. 6D). Even if the Rph-1 protein does bind to the Rho-independent transcription terminator, which is unlikely, the extra nucleotides associated with the terminator would alter its physical relationship to the 5′ terminus, thus allowing RNase P to bind.
The normal processing of argX and glyW polycistronic transcripts by RNase E in the absence of RppH to generate the pre-tRNAs (Fig. 4 and data not shown) was consistent with various studies (31, 33–35), suggesting an internal entry mechanism for RNase E, which bypasses the requirement of a 5′-monophosphate end for certain substrates (32). Alternatively, E. coli has 13 Nudix family members, one of which could possibly also encode an enzyme that can specifically catalyze the conversion of a 5′-terminal triphosphate to monophosphate on primary tRNA transcripts. This scenario is probably unlikely, considering that the interaction between the RNase E sensor domain and the 5′ phosphate of a tRNA substrate has been shown to be dispensable (34), suggesting an efficient alternative mechanism for substrate identification by RNase E. Overall, the presence of a 5′ triphosphate did not inhibit the removal of Rho-independent transcription terminators by RNase E from either mono- or polycistronic transcripts (Fig. 1 and 4).
Finally, the apparent dominant negative effect of the rph-1 allele raises some interesting questions regarding the MG1655 and W3110 genetic backgrounds, which are some of the most widely used E. coli K-12 strains. For example, although the ΔrppH rph-1 double mutant had only a small growth defect, inactivation of the rppH gene in either an rnpA49 rph-1 or rne-1 rph-1 mutant strain significantly exacerbated the lethality associated with the rnpA49 or rne-1 allele (data not shown). Thus, it should be noted that when analyzing RNA metabolism in E. coli, the use of point mutations can sometimes lead to results that may not accurately reflect the actual role of a particular RNase in the cell. As shown here, the phenotypes of point mutations should be checked against those observed with a true deletion allele, particularly, since we have noticed other discrepancies between true deletions and deletion/insertion mutations (B. K. Mohanty and S. R. Kushner, unpublished data).
MATERIALS AND METHODS
Bacterial strains and plasmids.
The E. coli strains used in this study were all derived from MG1693 (rph-1 thyA715) (E. coli Genetic Stock Center, Yale University) and are listed in Table 2. SK10153 (an rph+ derivative) (38), SK2525 (rnpA49 rph-1) (2), and SK5665 (rne-1 rph-1) (44) have been previously described. The rne-1 and rnpA49 alleles encode temperature-sensitive RNase E and RNase P proteins, respectively, which are unable to support cell viability at 44°C (44–46). The rne-1 allele retains residual RNase E activity on some substrates at the nonpermissive temperature (25).
TABLE 2.
Bacterial strains used in this work
| Strain | Genotypea | Reference or source |
|---|---|---|
| JW2798 | ΔrppH754::kan rph-1 | 54 |
| JW3618 | Δrph749::kan rph-1 | 54 |
| MG1693 | thyA715 rph-1 | E. coli Genetic Stock Center |
| SK10153 | thyA715 rph+ | 38 |
| SK2525 | rnpA49 thyA715 rph-1 rbsD296::Tn10 Tcr | 2 |
| SK4390 | ΔrppH754::kan thyA715 rph-1 | 4 |
| SK4394 | ΔrppH754::kan rne-1 thyA715 rph-1 | This study |
| SK4395 | ΔrppH754::kan rnpA49 thyA715 rph-1 rbsD296::Tn10 Kmr Tcr | 4 |
| SK5665 | rne-1 thyA715 rph-1 | 44 |
| SK10751 | Δrph749 thyA715 | This study |
| SK10752 | Δrph749 ΔrppH754::kan thyA715 | This study |
Tcr, tetracycline resistant; Kmr, kanamycin resistant.
For this study, a P1 lysate grown on JW2798 (ΔrppH754::kan; Keio Collection, Japan) (54) was used to transduce MG1693, SK2525, and SK5665 to construct SK4390 (ΔrppH754::kan rph-1), SK4395 (ΔrppH754::kan rnpA49 rph-1), and SK4394 (ΔrppH754::kan rne-1 rph-1), respectively. SK10751 (Δrph749 rph-1) was constructed by transduction using a P1 lysate grown on JW3618 (Δrph749::kan; Keio Collection, Japan) and MG1693 as a recipient, followed by removal of the kanamycin cassette, as described previously (47). SK10752 (Δrph749 ΔrppH754::kan rph-1) was constructed by transducing P1 lysate grown on JW2798 (ΔrppH754::kan) into SK10751.
Plasmid pNWK18 (rph-1 Cmr [Cmr, chloramphenicol resistant]) was constructed by cloning the rph-1 coding sequence containing its own promoter into a Cmr derivative of the 6- to 8-copy vector pSBK29 (48) using the PstI-BamHI sites. A PCR fragment containing the rph-1 coding sequence was amplified using the primer pair RPH-PST and RPH-1291 using Q5 high-fidelity DNA polymerase (NEB). The plasmid pNWK18 was sequenced to confirm the presence of the rph-1 allele.
Growth of bacterial strains and isolation of total RNA.
Bacterial strains were grown with shaking at 37°C in Luria broth supplemented with thymine (50 μg/ml) and kanamycin (25 μg/ml) (when appropriate). Cell densities were measured every 30 min using a Klett meter (no. 42 green filter), and the cultures were maintained in mid-exponential phase (80 Klett units) by diluting with fresh prewarmed medium. The generation times were calculated by plotting the Klett values on a graph after adjusting to reflect the appropriate dilution factors. For total RNA isolation, cells were grown until a cell density of 20 Klett units above the background (∼5.0 × 107 cells/ml). All cultures were then shifted to 44°C for 2 h to inactivate temperature-sensitive ribonucleases and maintained at 80 Klett units above background by diluting, if necessary, with fresh prewarmed medium.
Unless otherwise noted, RNA was extracted using the RNAsnap method (49). RNA was quantified using a NanoDrop 2000c (Thermo Scientific) apparatus. Five hundred nanograms of each RNA sample was run on a 1% agarose-Tris-acetate-EDTA gel and visualized with ethidium bromide to ensure satisfactory quality for further analysis. RNA to be used in primer extensions, reverse transcription-PCR (RT-PCR) cloning, and sequencing experiments was further treated with DNase I using the DNA-free kit (Ambion) to remove any contaminating DNA. Subsequently, the treated samples were quantified using the NanoDrop 2000c.
Northern analysis.
Northern analyses were performed as previously described previously (50). Five micrograms of total RNA was run on either 6% or 8% polyacrylamide-8.3 M urea gels and transferred to a positively charged nylon membrane (Nytran SPC; Whatman) for 2.5 h at 20 V, followed 45 min at 40 V. Northern blots were probed with 32P-5′-end-labeled oligonucleotides (51) specific to the mature sequence of each tRNA being tested. The probe sequences are available on request. The blot was then scanned with a PhosphorImager (Storm 840; GE Healthcare), and the data were quantified using the ImageQuant TL software (GE Healthcare).
Primer extension analysis.
Primer extension analysis of the various tRNA transcripts was carried out as previously described (3). The sequences were analyzed on a 6% PAGE gels containing 8 M urea.
RT-PCR cloning and sequencing of 5′-3′-ligated transcripts.
The 5′ and 3′ ends of the pheU, pheV, and ileX transcripts were determined by cloning and sequencing the RT-PCR products obtained from 5′→3′ self-ligated circular RNAs, according to the methods previously described (25). The 5′-3′ junctions of the cDNAs were amplified with pairs of gene-specific primers using Q5 high-fidelity DNA polymerase (NEB) and cloned into pWSK29 (52) for sequencing. All DNA sequencing was carried out using the MWG sequencing service.
Primers and probes.
The sequences of all primers and oligonucleotide probes used in the study are available upon request.
Immunoprecipitation analysis.
Strains were grown at 37°C with shaking at 255 rpm to Klett 80 in Luria broth supplemented with thymine (50 μg/ml) and antibiotic, where appropriate. Cell cultures (50 ml) were collected for protein isolation and quantified as described previously (53). Immunoprecipitations were performed at 4°C by mixing 1 mg of total protein and RNase PH antibody cross-linked to 25 μl of Pierce protein A/G magnetic beads (Thermo Scientific) for 1 h in a shaker. The protein-bead complexes were washed three times with radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8], 1.0% Nonidet P-40, 0.5% deoxycholate [DOC], 0.1% SDS). The immunocomplexes were subsequently eluted from the beads in 40 μl of the SDS-PAGE loading buffer after heating at 55°C for 15 min. Immunoprecipitated samples were separated on 12% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon TM-P; Millipore) using a Bio-Rad Mini-Protean 3 electrophoretic apparatus. The membranes were probed with either RNase PH (1:10,000 dilution), RppH (1:5,000 dilution), or RNase P (1:2,500 dilution) antibodies using the ECL Plus Western blotting detection kit (GE Healthcare), as per the manufacturer's instructions. The RNase PH, RppH, and RNase P antibodies were raised against RNase PH-, RppH-, and RnpA-specific peptides, respectively, by GenScript USA, Inc., NJ.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a grant from the National Institutes of Health (grant GM081544) to S.R.K.
We thank V. F. Maples for valuable technical assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00301-17.
REFERENCES
- 1.Li Z, Deutscher MP. 2002. RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA 8:97–109. doi: 10.1017/S1355838202014929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ow MC, Kushner SR. 2002. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev 16:1102–1115. doi: 10.1101/gad.983502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohanty BK, Kushner SR. 2007. Ribonuclease P processes polycistronic tRNA transcripts in Escherichia coli independent of ribonuclease E. Nucleic Acids Res 35:7614–7625. doi: 10.1093/nar/gkm917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohanty BK, Kushner SR. 2010. Processing of the Escherichia coli leuX tRNA transcript, encoding tRNALeu5, requires either the 3′→5′ exoribonuclease polynucleotide phosphorylase or RNase P to remove the Rho-independent transcription terminator. Nucleic Acids Res 38:597–607. doi: 10.1093/nar/gkp997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agrawal A, Mohanty BK, Kushner SR. 2014. Processing of the seven valine tRNAs in Escherichia coli involves novel features of RNase P. Nucleic Acids Res 42:11166–11179. doi: 10.1093/nar/gku758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z, Deutscher MP. 1996. Maturation pathways for E. coli tRNA precursors: a random multienzyme process in vivo. Cell 86:503–512. doi: 10.1016/S0092-8674(00)80123-3. [DOI] [PubMed] [Google Scholar]
- 7.Kelly KO, Reuven NB, Li Z, Deutscher MP. 1992. RNase PH is essential for tRNA processing and viability in RNase-deficient Escherichia coli cells. J Biol Chem 267:16015–16018. [PubMed] [Google Scholar]
- 8.Deutscher MP. 1993. Ribonuclease multiplicity, diversity and complexity. J Biol Chem 268:13011–13014. [PubMed] [Google Scholar]
- 9.Zuo Y, Deutscher MP. 2002. The physiological role of RNase T can be explained by its unusual substrate specificity. J Biol Chem 277:29654–29661. doi: 10.1074/jbc.M204252200. [DOI] [PubMed] [Google Scholar]
- 10.Sulthana S, Basturea GN, Deutscher MP. 2016. Elucidation of pathways of ribosomal RNA degradation: an essential role for RNase E. RNA 22:1163–1171. doi: 10.1261/rna.056275.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sulthana S, Quesada E, Deutscher MP. 2017. RNase II regulates RNase PH and is essential for cell survival during starvation and stationary phase. RNA 23:1456–1464. doi: 10.1261/rna.060558.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen KF. 1993. The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol 175:3401–3407. doi: 10.1128/jb.175.11.3401-3407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fredrik Pettersson BM, Ardell DH, Kirsebom LA. 2005. The length of the 5′ leader of Escherichia coli tRNA precursors influences bacterial growth. J Mol Biol 351:9–15. doi: 10.1016/j.jmb.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Kazantsev AV, Krivenko AA, Pace NR. 2009. Mapping metal-binding sites in the catalytic domain of bacterial RNase P RNA. RNA 15:266–276. doi: 10.1261/rna.1331809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rueda D, Hsieh J, Day-Storms JJ, Fierke CA, Walter NG. 2005. The 5′ leader of precursor tRNAAsp bound to the Bacillus subtilis RNase P holoenzyme has an extended conformation. Biochemistry 44:16130–16139. doi: 10.1021/bi0519093. [DOI] [PubMed] [Google Scholar]
- 16.Niranjanakumari S, Stams T, Crary SM, Christianson DW, Fierke CA. 1998. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc Natl Acad Sci U S A 95:15212–15217. doi: 10.1073/pnas.95.26.15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerrier-Takada C, Altman S. 1993. A physical assay for and kinetic analysis of the interactions between M1 RNA and tRNA precursor substrates. Biochemistry 32:7152–7161. doi: 10.1021/bi00079a012. [DOI] [PubMed] [Google Scholar]
- 18.Brannvall M, Mattsson JG, Svard SG, Kirsebom LA. 1998. RNase P RNA structure and cleavage reflect the primary structure of tRNA genes. J Mol Biol 283:771–783. doi: 10.1006/jmbi.1998.2135. [DOI] [PubMed] [Google Scholar]
- 19.McClain WH, Lai LB, Gopalan V. 2010. Trials, travails and triumphs: an account of RNA catalysis in RNase P. J Mol Biol 397:627–646. doi: 10.1016/j.jmb.2010.01.038. [DOI] [PubMed] [Google Scholar]
- 20.Li H. 2007. Complexes of tRNA and maturation enzymes: shaping up for translation. Curr Opin Struct Biol 17:293–301. doi: 10.1016/j.sbi.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Hartmann RK, Gossringer M, Spath B, Fischer S, Marchfelder A. 2009. The making of tRNAs and more—RNase P and tRNase Z. Prog Mol Biol Transl Sci 85:319–368. doi: 10.1016/S0079-6603(08)00808-8. [DOI] [PubMed] [Google Scholar]
- 22.Koutmou KS, Zahler NH, Kurz JC, Campbell FE, Harris ME, Fierke CA. 2010. Protein-precursor tRNA contact leads to sequence-specific recognition of 5′ leaders by bacterial ribonuclease P. J Mol Biol 396:195–208. doi: 10.1016/j.jmb.2009.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai HY, Masquida B, Biswas R, Westhof E, Gopalan V. 2003. Molecular modeling of the three-dimensional structure of the bacterial RNase P holoenzyme. J Mol Biol 325:661–675. doi: 10.1016/S0022-2836(02)01267-6. [DOI] [PubMed] [Google Scholar]
- 24.Crary SM, Niranjanakumari S, Fierke CA. 1998. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry 37:9409–9416. doi: 10.1021/bi980613c. [DOI] [PubMed] [Google Scholar]
- 25.Mohanty BK, Kushner SR. 2008. Rho-independent transcription terminators inhibit RNase P processing of the secG leuU and metT tRNA polycistronic transcripts in Escherichia coli. Nucleic Acids Res 36:364–375. doi: 10.1093/nar/gkm991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deana A, Celesnik H, Belasco JG. 2008. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451:355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- 27.Richards J, Liu Q, Pellegrini O, Celesnik H, Yao S, Bechhofer DH, Condon C, Belasco JG. 2011. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol Cell 43:940–949. doi: 10.1016/j.molcel.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luciano DJ, Vasilyev N, Richards J, Serganov A, Belasco JG. 2017. A novel RNA phosphorylation state enables 5′ end-dependent degradation in Escherichia coli. Mol Cell 67:44–54. doi: 10.1016/j.molcel.2017.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLennan AG. 2006. The Nudix hydrolase superfamily. Cell Mol Life Sci 63:123–143. doi: 10.1007/s00018-005-5386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Callaghan AJ, Marcaida MJ, Stead JA, McDowall KJ, Scott WG, Luisi BF. 2005. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 437:1187–1191. doi: 10.1038/nature04084. [DOI] [PubMed] [Google Scholar]
- 31.Garrey SM, Blech M, Riffell JL, Hankins JS, Stickney LM, Diver M, Hsu YH, Kunanithy V, Mackie GA. 2009. Substrate binding and active site residues in RNases E and G: role of the 5′-sensor. J Biol Chem 284:31843–31850. doi: 10.1074/jbc.M109.063263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker KE, Mackie GA. 2003. Ectopic RNase E sites promote bypass of 5′-end-dependent mRNA decay in Escherichia coli. Mol Microbiol 47:75–88. doi: 10.1046/j.1365-2958.2003.03292.x. [DOI] [PubMed] [Google Scholar]
- 33.Clarke JE, Kime L, Romero AD, McDowall KJ. 2014. Direct entry by RNase E is a major pathway for the degradation and processing of RNA in Escherichia coli. Nucleic Acids Res 42:11733–11751. doi: 10.1093/nar/gku808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garrey SM, Mackie GA. 2011. Roles of the 5′-phosphate sensor domain in RNase E. Mol Microbiol 80:1613–1624. doi: 10.1111/j.1365-2958.2011.07670.x. [DOI] [PubMed] [Google Scholar]
- 35.Kime L, Clarke JE, Romero AD, Grasby JA, McDowall KJ. 2014. Adjacent single-stranded regions mediate processing of tRNA precursors by RNase E direct entry. Nucleic Acids Res 42:4577–4589. doi: 10.1093/nar/gkt1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohanty BK, Petree JR, Kushner SR. 2016. Endonucleolytic cleavages by RNase E generate the mature 3′ termini of the three proline tRNAs in Escherichia coli. Nucleic Acids Res 44:6350–6362. doi: 10.1093/nar/gkw517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muniz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T, Kaipa P, Spaulding A, Pacheco J, Latendresse M, Fulcher C, Sarker M, Shearer AG, Mackie A, Paulsen I, Gunsalus RP, Karp PD. 2011. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res 39:D583–D590. doi: 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohanty BK, Maples VF, Kushner SR. 2012. Polyadenylation helps regulate functional tRNA levels in Escherichia coli. Nucleic Acids Res 40:4589–4603. doi: 10.1093/nar/gks006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baba T, Mori H. 2008. The construction of systematic in-frame, single-gene knockout mutant collection in Escherichia coli K-12. Methods Mol Biol 416:171–181. doi: 10.1007/978-1-59745-321-9_11. [DOI] [PubMed] [Google Scholar]
- 40.Mohanty BK, Kushner SR. 2013. Deregulation of poly(A) polymerase I in Escherichia coli inhibits protein synthesis and leads to cell death. Nucleic Acids Res 41:1757–1766. doi: 10.1093/nar/gks1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mondragón A. 2013. Structural studies of RNase P. Annu Rev Biophys 42:537–557. doi: 10.1146/annurev-biophys-083012-130406. [DOI] [PubMed] [Google Scholar]
- 42.Kirsebom LA, Svard SG. 1994. Base pairing between Escherichia coli RNase P RNA and its substrate. EMBO J 13:4870–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wegscheid B, Hartmann RK. 2006. The precursor tRNA 3′-CCA interaction with Escherichia coli RNase P RNA is essential for catalysis by RNase P in vivo. RNA 12:2135–2148. doi: 10.1261/rna.188306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arraiano CM, Yancey SD, Kushner SR. 1988. Stabilization of discrete mRNA breakdown products in ams pnp rnb multiple mutants of Escherichia coli K-12. J Bacteriol 170:4625–4633. doi: 10.1128/jb.170.10.4625-4633.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schedl P, Primakoff P. 1973. Mutants of Escherichia coli thermosensitive for the synthesis of transfer RNA. Proc Natl Acad Sci U S A 70:2091–2095. doi: 10.1073/pnas.70.7.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ono M, Kuwano M. 1979. A conditional lethal mutation in an Escherichia coli strain with a longer chemical lifetime of mRNA. J Mol Biol 129:343–357. doi: 10.1016/0022-2836(79)90500-X. [DOI] [PubMed] [Google Scholar]
- 47.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perwez T, Hami D, Maples VF, Min Z, Wang BC, Kushner SR. 2008. Intragenic suppressors of temperature-sensitive rne mutations lead to the dissociation of RNase E activity on mRNA and tRNA substrates in Escherichia coli. Nucleic Acids Res 36:5306–5318. doi: 10.1093/nar/gkn476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stead MB, Agarwal A, Bowden KD, Nasir R, Meagher RB, Mohanty BK, Kushner SR. 2012. RNAsnap: a rapid, quantitative, and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res 40:e156. doi: 10.1093/nar/gks680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohanty BK, Kushner SR. 2014. In vivo analysis of polyadenylation in prokaryotes. Methods Mol Biol 1125:229–249. doi: 10.1007/978-1-62703-971-0_19. [DOI] [PubMed] [Google Scholar]
- 51.Mohanty BK, Giladi H, Maples VF, Kushner SR. 2008. Analysis of RNA decay, processing, and polyadenylation in Escherichia coli and other prokaryotes. Methods Enzymol 447:3–29. doi: 10.1016/S0076-6879(08)02201-5. [DOI] [PubMed] [Google Scholar]
- 52.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 53.Mohanty BK, Maples VF, Kushner SR. 2004. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli. Mol Microbiol 54:905–920. doi: 10.1111/j.1365-2958.2004.04337.x. [DOI] [PubMed] [Google Scholar]
- 54.Baba T, Huan HC, Datsenko K, Wanner BL, Mori H. 2008. The applications of systematic in-frame, single-gene knockout mutant collection of Escherichia coli K-12. Methods Mol Biol 416:183–194. doi: 10.1007/978-1-59745-321-9_12. [DOI] [PubMed] [Google Scholar]
- 55.Wegscheid B, Hartmann RK. 2007. In vivo and in vitro investigation of bacterial type B RNase P interaction with tRNA 3′-CCA. Nucleic Acids Res 35:2060–2073. doi: 10.1093/nar/gkm005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.