

ABSTRACT
We previously found CC chemokine ligand 3 (CCL3) to be a potent effector of inflammation during otitis media (OM): exogenous CCL3 rescues the OM phenotype of tumor necrosis factor-deficient mice and the function of macrophages deficient in several innate immune molecules. To further delineate the role of CCL3 in OM, we evaluated middle ear (ME) responses of ccl3−/−mice to nontypeable Haemophilus influenzae (NTHi). CCL chemokine gene expression was evaluated in wild-type (WT) mice during the complete course of acute OM. OM was induced in ccl3−/− and WT mice, and infection and inflammation were monitored for 21 days. Phagocytosis and killing of NTHi by macrophages were evaluated by an in vitro assay. The nasopharyngeal bacterial load was assessed in naive animals of both strains. Many CCL genes showed increased expression levels during acute OM, with CCL3 being the most upregulated, at levels 600-fold higher than the baseline. ccl3−/− deletion compromised ME bacterial clearance and prolonged mucosal hyperplasia. ME recruitment of leukocytes was delayed but persisted far longer than in WT mice. These events were linked to a decrease in the macrophage capacity for NTHi phagocytosis and increased nasopharyngeal bacterial loads in ccl3−/− mice. The generalized impairment in inflammatory cell recruitment was associated with compensatory changes in the expression profiles of CCL2, CCL7, and CCL12. CCL3 plays a significant role in the clearance of infection and resolution of inflammation and contributes to mucosal host defense of the nasopharyngeal niche, a reservoir for ME and upper respiratory infections. Therapies based on CCL3 could prove useful in treating or preventing persistent disease.
KEYWORDS: chemokines, otitis media, nontypeable Haemophilus influenzae, innate immunity, nasopharyngeal culture, mucosal flora
INTRODUCTION
Otitis media (OM) is a primarily infectious disease that involves, in the majority of cases, pathogenic bacteria alone or in combination with a virus (1). The bacteria most frequently isolated from middle ear (ME) effusions during acute OM are Streptococcus pneumoniae, nontypeable Haemophilus influenzae (NTHi), and Moraxella catarrhalis (2). Children acquire these potential otopathogens, along with a broad variety of other microorganisms, as part of their normal nasopharyngeal flora early in infancy (3).
The pathogenesis of OM is considered multifactorial. Prior viral infection of the nasopharynx, significantly increased bacterial colonization of the nasopharynx (4) and/or cocolonization by different bacterial strains (4, 5), as well as dysfunction of the Eustachian tube leading to pressure differences between the nasopharynx and Eustachian tube (6) have all been associated with an elevated risk of OM. Immature or compromised immune status, allergy, and genetic predispositions (7) also contribute to OM susceptibility.
The fact that acute OM normally resolves in a few days, before cognate immunity can be effectively engaged, suggests that innate defenses are the primary means of recovery from OM. The host recognizes invading pathogens by innate immune receptors, including Toll-like receptors (TLRs) and nucleotide oligomerization domain (NOD)-like receptors. Pathogen molecule binding to these receptors initiates signaling cascades that result in a variety of antimicrobial responses. The TLRs that are the primary receptors for the recognition of NTHi (TLR1, TLR2, TLR4, TLR6, and TLR9 [8]) can signal via a myeloid differentiation primary response gene 88 (MyD88)-dependent pathway to induce the release of proinflammatory cytokines, including interleukin-1β (IL-1β), IL-6, IL-8, IL-12, and tumor necrosis factor alpha (TNF-α), and chemokines such as macrophage inflammatory protein 2 (CXCL2) and CC chemokine ligand 3 (CCL3)/macrophage inflammatory protein 1α (9, 10). Previous gene expression studies of the ME mucosa in mice after NTHi exposure demonstrated increased expression levels of TLR signaling molecules, especially during the initial stages of the inflammatory response, during acute OM (11, 12). Defects in TLR signaling are associated with an impaired immune response to NTHi-induced ME infection (13–15). In humans, polymorphisms in TLR genes are associated with increased susceptibility to ME infections (16, 17).
CCL3 belongs to the CC subfamily of the chemokine superfamily. CCL chemokines share the common biologic property of leukocyte chemoattraction. CCL3, because of its ability to attract and activate various inflammatory cells, including T cells, monocytes, eosinophils, and neutrophils, is an important determinant of inflammatory cell infiltration. This is mediated by interactions with its CC chemokine receptors (CCRs), CCR1 and CCR5 (18, 19), and is intimately involved in many inflammatory and infectious diseases (20).
Critical to the host defense of the ME against invading pathogens is the capacity of professional phagocytes, such as macrophages, to efficiently engulf and degrade microorganisms. Macrophages derived from TNF−/− and TLR2−/− mice have been shown in vitro to be defective in the phagocytosis/intracellular killing of NTHi and to express reduced levels of CCL3 (13). Moreover, stimulation with CCL3 was found to reverse the functional impairment of macrophages derived from TLR2−/−, MyD88−/−, and TNF−/− mice in previous studies (21). Furthermore, CCL3+/+ macrophages were shown to expand their phagocytic and killing activities after stimulation with CCL3 (13, 22). In addition to these in vitro effects, the administration of CCL3 in vivo, alone or in combination with TNF, prior to infection with NTHi restored the impaired immune response to OM in TNF−/− mice (13). Delayed recovery from OM was observed after blocking CCL3 activity with anti-CCL3 antibodies in vivo (13).
The above-mentioned findings make the CCL chemokines in general and CCL3 in particular an attractive target for further investigation in a well-established murine model of OM induced by NTHi infection of the ME (11). Our goal was to determine the extent to which CCL3, while sufficient to rescue innate immune defects, is also necessary for normal recovery from OM.
(Some of the data included in the manuscript were presented at the 9th Molecular Biology of Hearing and Deafness Conference in June 2013 at Stanford University [Palo Alto, CA].)
RESULTS
Regulation of CCL genes during acute OM.
Changes in the expression levels of the most highly regulated (peak increase of >20-fold) CCL chemokine genes over the course of an episode of acute NTHi-induced OM in WT mice are illustrated in Fig. 1. Data from all CCL genes and fold expression variability are provided in Table S1 in the supplemental material. Substantial changes in the expression levels of seven CCL genes were observed during OM. These changes fell into three categories: an increase early during OM (peaking 3 to 24 h after NTHi inoculation, e.g., CCL4 and CCL20), an increase later during OM (peaking 2 to 3 days after inoculation, e.g., CCL6, CCL7, and CCL12), or an increase during both time periods (e.g., CCL2 and CCL3). As shown in Fig. 1 and Table S1, CCL3 was by far the most highly regulated CCL gene. Expression peaked at a >600-fold increase 1 day after inoculation and increased >100-fold throughout the period from 3 h to 3 days, which corresponds to the most active ME inflammatory response. As can be seen from the range of fold change values presented in Table S1, the expression changes were replicated both in independent tissue samples and in the two chips of each sample. Mice injected with saline alone in the ME exhibited much smaller changes in the expression levels of CCL genes, as in almost all other regulated genes (11). For example, the peak fold change in CCL3 expression after saline injection was only 3% of that seen after NTHi injection.
FIG 1.
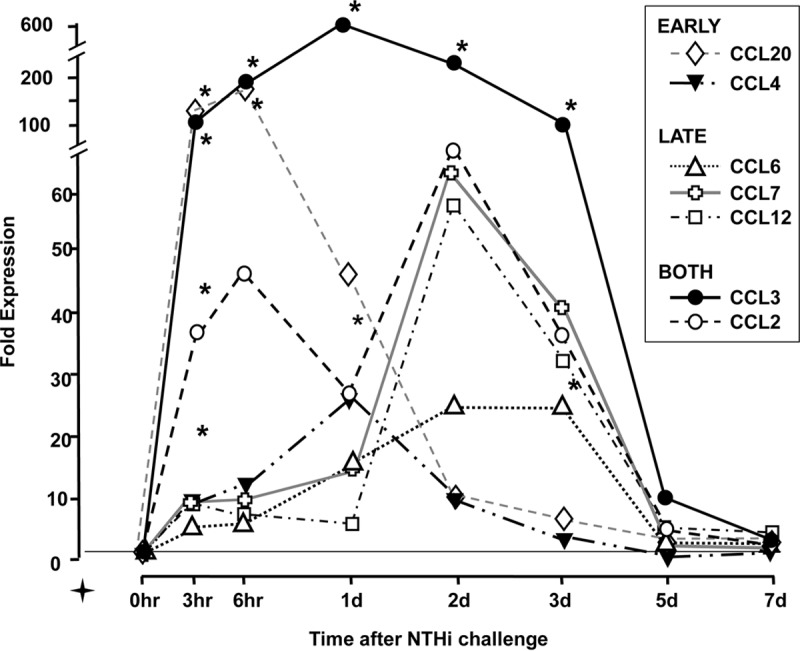
Regulation of CCL genes in the ME during the course of an episode of acute OM in mice. Data are from gene arrays using two independent groups of mice. CCL genes for which peak expression levels increased at least 20-fold are illustrated. Increased expression fell into three categories: (i) genes upregulated early (3 to 24 h) during OM, (ii) those upregulated late (2 to 3 days) during OM, and (iii) those upregulated both early and late. Results for all CCL genes and the variability of expression levels are presented in Table S1 in the supplemental material. CCL20 data were reported previously in a different form (14). Data analysis was performed by using VAMPIRE (41). For this and subsequent figures, * indicates a P value of <0.05, ** indicates a P value of <0.01, and *** indicates a P value of <0.001.
Mucosal hyperplasia in the absence of CCL3.
Hyperplasia of the ME mucosa is illustrated in Fig. 2A and B. When exposed to NTHi, WT mice exhibited characteristic, robust ME mucosal hyperplasia that reached a maximum on day 2 after inoculation. By day 7, the mucosa thickness was not different from preinfection values, although there was a nonsignificant tendency for increased thickness.
FIG 2.
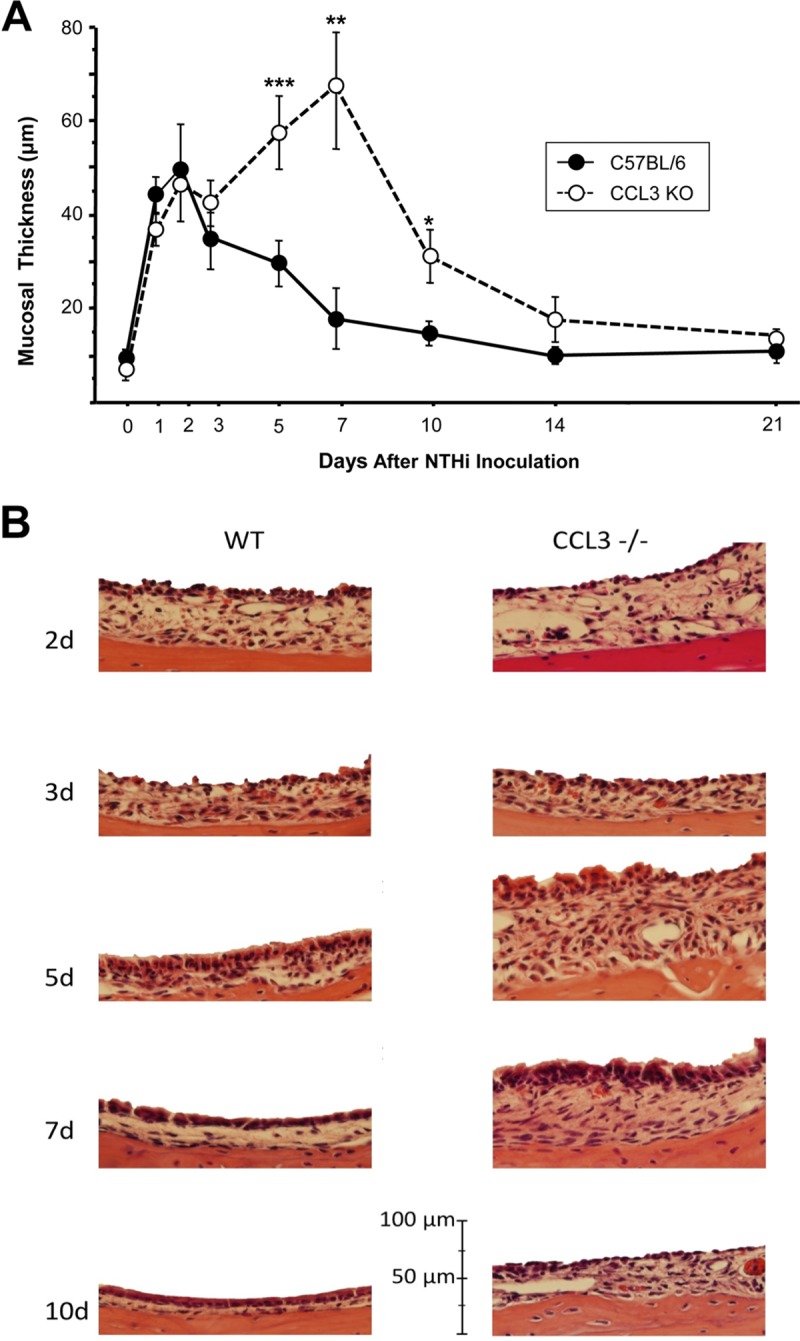
Mucosal response of the ME after NTHi challenge. (A) Time course of mucosal thicknesses in the MEs of CCL3-null versus WT mice after NTHi challenge. No significant differences in mucosal responses were noted during the first 3 days after NTHi infection. On day 5 (***, P < 0.001 by Fisher's exact t test), day 7 (**, P < 0.01), and day 10 (*, P < 0.05), ccl3−/− animals displayed significantly enhanced mucosal thickness (n = 6 to 7 MEs per time point and strain; bars and error bars represent means ± SEM). (B) Representative histological changes of the ME mucosa of ccl3−/− versus WT mice 2, 3, 5, 7, and 10 days after inoculation of NTHi in MEs (original magnification, ×40).
There were no significant differences in mucosal thicknesses between WT and ccl3−/− mice until day 5 (P < 0.001 by 2-way analysis of variance [ANOVA] and Fisher's exact t test with Bonferroni correction for multiple comparisons). The mucosa of ccl3−/− MEs reached its maximum thickness and difference from those of WT mice on day 7 (P < 0.01). Thereafter, mucosal hyperplasia in ccl3−/− mice decreased but remained significantly greater than that in WT mice through day 10 (P < 0.05).
Delayed and persistent leukocyte recruitment to the ME in the absence of CCL3.
In WT mice, the highest percentage of the ME lumen occupied by infiltrating leukocytes was observed on the first day after NTHi inoculation, and cells persisted in the ME until day 5. In the absence of CCL3, the initial recruitment of leukocytes was delayed. One day after NTHi instillation, the level of infiltration of inflammatory cells into the MEs of animals lacking CCL3 was significantly lower than that in WT mice (P < 0.05 by a Mann-Whitney U test), reaching the level seen in WT animals on day 2. However, on day 10 (<0.01) and day 14 (P < 0.05), it exceeded that in WT animals, and some ccl3−/− animals showed ME leukocytes even on day 21 (Fig. 3A).
FIG 3.

Inflammatory cells in the ME cavity after NTHi infection. (A) Percent area of the ME cavity occupied by leukocytes during OM in WT and ccl3−/− mice. The initial level of leukocyte infiltration on day 1 post-NTHi inoculation was significantly lower in CCL3−/− MEs (*, P < 0.05 by a Mann-Whitney U test), as was also observed on day 3. While the infiltration of both WT and ccl3−/− MEs declined on day 7, ccl3−/− MEs showed a significant increase on day 10. Thereafter, leukocytes were observed only in ccl3−/− MEs although not in every case. (B) Number of neutrophils in a standard area (×400 field) of ME infiltrates. Neutrophils were the primary cell type in ME infiltrates throughout the observation period. In the absence of CCL3, neutrophil infiltration peaked after 2 days following a slight delay. Numbers of neutrophils were significantly increased in the MEs of ccl3−/− animals on days 2, 7, 10, and 14 (*, P < 0.05; **, P < 0.01 by a Mann-Whitney U test) compared to those in WT animals and persisted in at least some MEs through day 21. HPF, high-power field. (C) Number of macrophages in a standard area of ME infiltrates. Macrophages were recruited to the ME by 2 days after NTHi infection of both WT and ccl3−/− animals. ccl3−/− mice displayed a prolonged presence of macrophages in MEs, with significantly increased numbers of macrophages on days 7 and 10 (*, P < 0.05 by a Mann-Whitney U test), and macrophages were still present in some ears on day 21 (n = 6 to 8 MEs per time point and strain; bars and error bars represent means ± SEM). It should be noted that since the measurements in panels B and C were taken from the largest cellular clusters in a given ear, the numbers do not reflect the total number of leukocytes in each ear. Rather, they reflect the density of the cell clusters and the proportions of neutrophils and macrophages present.
Neutrophils were the predominant cell type in the MEs of both mouse strains throughout the observation period. As shown in Fig. 3B, the maximum influx of neutrophils into WT MEs occurred on day 1, with a gradual decrease through day 5. In ccl3−/− MEs, consistent with the delayed total inflammatory cell recruitment, neutrophil counts reached a peak on day 2, with significantly increased numbers compared to those in WT mice (P < 0.05 by a Mann-Whitney U test). After this peak, the numbers of neutrophils declined in both groups, but the numbers of neutrophils peaked again and persisted longer in MEs of mice lacking CCL3, with significantly higher numbers on days 7 and 10 (P < 0.01) and day 14 (P < 0.05) and with some MEs exhibiting a small number even on day 21 (Fig. 3B).
Macrophages were observed in the ME cavities of both strains by day 2 after infection but in much smaller numbers than neutrophils. In WT mice, macrophage counts peaked on day 5 and were absent from the ME by day 7. In ccl3−/− mice, the early dynamics of macrophage infiltration were similar, but as with neutrophils, macrophages remained longer in the MEs. Macrophage counts were significantly higher than those in WT mice on day 7 (P < 0.01) and days 10 and 14 (P < 0.05), and some MEs retained macrophages on day 21 (Fig. 3C).
Bacterial clearance is delayed in ccl3−/− mice.
Next, we assessed ME bacterial clearance in WT and ccl3−/− mice (Fig. 4 and Table 1). As expected, MEs of neither CCL3-deficient nor WT mice showed bacterial colonies on day 0, prior to challenge with NTHi. For the first 3 days post-NTHi inoculation, MEs of both ccl3−/− and WT mice showed similarly robust culture positivity, between 63% and 100%. Thereafter, on day 5 postinoculation, 36% of ME cultures from WT mice were NTHi positive. This value decreased to 13% and 14% positivities on days 10 and 14, respectively. By day 21, no NTHi bacteria were recovered from WT mice.
FIG 4.
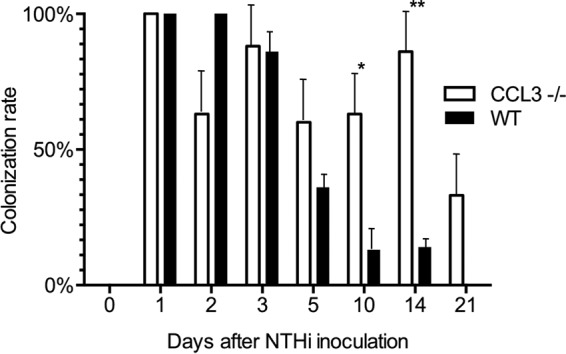
NTHi colonization rate as a percentage of culture-positive ME samples of ccl3−/− and WT mice. We found no significant differences in colonization rates of NTHi in the first 5 days post-NTHi inoculation. After day 5, colonization rates of NTHi diverged. ccl3−/− MEs were colonized by NTHi to a significantly higher degree on day 10 and 14 after NTHi inoculation (*, P < 0.05; **, P < 0.01 by a Fisher-Irwin t test for proportions). By day 21, no NTHi were cultured from WT mice. In contrast, NTHi remained present in the MEs of ccl3−/− animals throughout the observation period (n ≥ 6 MEs per time point and strain).
TABLE 1.
Recovery of NTHi from CCL3−/− and WT MEs, expressed as degree of culture plate colonizationa
| Day after NTHi instillation | Mean score for NTHi colonization per ME |
|
|---|---|---|
| CCL3−/− mice | WT mice | |
| 0 | 0 | 0 |
| 1 | 3.2 | 3.3 |
| 2 | 1.4 | 1.7 |
| 3 | 3 | 2.3 |
| 5 | 1.5 | 0.8 |
| 10 | 1.2 | 0.1 |
| 14 | 1.4 | 0.6 |
| 21 | 0.3 | 0 |
Bacterial colonization of MEs was evaluated by using a semiquantitative analysis of the presence of bacteria on culture plates. A 1-μl loop from the ME was sequentially streaked onto the four quadrants of each culture plate, where 0 indicates no CFU, 1 indicates one quadrant with CFU, 2 indicates two quadrants with CFU, 3 indicates three quadrants with CFU, and 4 indicates CFU in all four quadrants. Data represent means of results from at least 6 MEs per time point and strain.
In contrast to WT MEs, NTHi bacteria were isolated at a significantly higher percentage in ccl3−/− mice on day 10 (P < 0.05) and day 14 (P < 0.05 by a Fisher-Irwin test) following inoculation. Furthermore, NTHi persisted longer in MEs of CCL3 knockout (KO) animals and remained present in some ears even on day 21. Thus, the persistent inflammation seen in the MEs of ccl3−/− mice, as evidenced by mucosal hyperplasia and leukocyte infiltration, was associated with an impaired capacity to clear NTHi.
CCL gene expression in the absence of CCL3.
In many gene knockout studies, compensatory changes in the expression levels of genes with redundant functions are observed. We therefore compared the expression levels of CCL2, CCL7, and CCL12, which are expressed over approximately the same period as CCL3 in WT mice, in the MEs of WT and ccl3−/− mice during OM by quantitative PCR (qPCR). As shown in Fig. 5, CCL2 expression levels showed complex changes in ccl3−/− MEs. The CCL2 mRNA expression level was significantly lower 6 h and 3 days after NTHi inoculation, but was higher at 2 days, in the absence of CCL3. The CCL2 qPCR data for both genotypes exhibited the biphasic expression that was present in the gene array data for WT mice (Fig. 1). In contrast, CCL7 and CCL12 expression levels were significantly higher 6 h, 1 day, and 2 days after inoculation, but lower 3 days after inoculation, in ccl3−/− MEs. A biphasic response for these CCL mRNAs was also noted for both genotypes.
FIG 5.

Expression levels of CCL2, CCL7, and CCL12 mRNAs in the MEs of WT versus CCL3-null mice during OM. cDNAs were generated from 6 MEs at each time point. The 0-h time point denotes untreated mice. Results represent data from 2 replicates, with error bars denoting the 95% confidence intervals. Data points for which the confidence intervals do not overlap are significantly different between the two genotypes.
Bacterial phagocytosis by peritoneal macrophages is impaired, whereas intracellular killing is not affected, in the absence of CCL3.
ccl3−/− mice exhibited a reduced clearance of NTHi from the ME cavity despite the prolonged presence of macrophages in the ME lumen (Fig. 3C). We therefore hypothesized that macrophage function in mice lacking CCL3 is impaired. In in vitro assays, we observed significantly impaired phagocytic function in ccl3−/− macrophages compared to WT macrophages (P < 0.01 by a Mann-Whitney U test) (Fig. 6A). In contrast, the levels of intracellular killing of bacteria that were phagocytosed did not significantly differ between the two strains (nonsignificant by a Mann-Whitney U test) (Fig. 6B).
FIG 6.

Phagocytic and killing activities, assessed in peritoneal macrophages from ccl3−/− and WT mice. (A) Numbers of bacteria recovered from lysed macrophages after a 1-h ingestion period in culture and a 1-h exposure to gentamicin to kill extracellular bacteria (a measure of phagocytosis) were quantified by colony counting. Phagocytosis was significantly impaired in ccl3−/− macrophages (**, P < 0.01 as determined by a Mann-Whitney U test), compared to macrophages from WT mice. (B) Intracellular bacteria recovered 3 h after NTHi ingestion and gentamicin treatment as a percentage of the number observed at 1 h (a measure of intracellular killing). Bacterial killing by ccl3−/− macrophages and that by WT macrophages were similar (Mann-Whitney U test) (n = 6 per time point and strain; bars and error bars represent means ± SEM). ns, nonsignificant.
Higher bacterial load in the nasopharynx of CCL3−/− mice.
In order to determine whether the genetic loss of CCL3 is correlated with an increased bacterial load in the nasopharynx, we collected nasopharynx samples from both ccl3−/− and WT mice. The mean bacterial load of nasopharynx cultures obtained from ccl3−/− animals was significantly higher than that in cultures obtained from WT animals (P < 0.05 by a Mann-Whitney U test) (Fig. 7). In order to rule out general infection/hematogenous dissemination of bacteria, we collected blood samples from the same animals. Blood cultures were negative for all tested animals of both strains.
FIG 7.
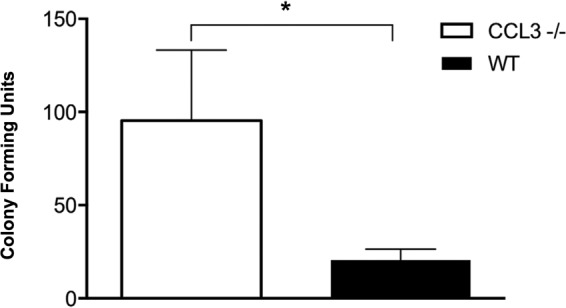
Presence of bacteria determined as CFU detected per microliter loop sample scraped from the nasopharynx of ccl3−/− versus WT mice. None of the mice tested had been previously infected with NTHi in the ME; thus, the CFU represent murine bacterial species. Significantly more bacteria were cultured from the nasopharynges of ccl3−/− mice than from those of WT animals (*, P < 0.05 as determined by a Mann-Whitney U test) (n = 6 to 8 per strain; bars and error bars represent means ± SEM).
DISCUSSION
The results of this study demonstrate that many CCL genes are regulated during acute OM, with CCL3 being the most highly expressed. Moreover, the lack of CCL3 has a significant effect upon the pathogenesis of and recovery from OM induced by NTHi. This effect includes a delayed early recruitment of leukocytes into the ME (Fig. 2). In addition, a longer duration of OM was evidenced by persistent ME mucosal hyperplasia and leukocyte infiltration (Fig. 2 and 3) and delayed bacterial clearance, with MEs showing infection even 21 days after ME inoculation (Fig. 4 and Table 1). This was associated with a reduced ability of CCL-null macrophages to phagocytose NTHi (Fig. 5) and increased bacterial colonization of the naive nasopharynx by murine bacterial pathogens (Fig. 6).
The CCL genes expressed in the ME included some that displayed biphasic regulation, with an initial peak in the first hours after NTHi inoculation and then a reduction at 24 h, followed by a second peak at 48 to 72 h. This was observed in both gene array and qPCR data (Fig. 1 and 2). This pattern was also noted more broadly in the complete transcriptome of acute OM (11). We can only speculate on the physiologic meaning of such responses. However, in the case of CCLs, they are produced in response to inflammation. Thus, the early peak in CCL expression may be related to the initial inflammatory response to bacteria. The later peak may be produced in response to inflammation induced by neutrophils and cytokines, both of which peak in the ME at around 24 h.
The delayed recruitment of neutrophils to CCL3-null MEs is consistent with the known role of this chemokine as a polymorphonuclear leukocyte (PMN) chemoattractant and activator (18–20, 23, 24). As at other sites, neutrophils are the first leukocytes to enter the ME in response to inflammation or infection, with macrophages entering 1 to 2 days later (25–27). Our data suggest that for the initial attraction of granulocytes to the ME, CCL3 plays a dominant role. The early expression of the ccl3 gene during NTHi-induced ME infection in WT animals (Fig. 1) is consistent with such a role. It would also appear that, at later stages of infection, other chemoattractants with redundant functions are able to compensate for the lack of CCL3. These chemoattractants include CCL7 and CCL12, which show enhanced expression levels in CCL3-null MEs. Other CCLs, the CXCL chemokines, which are broadly involved in neutrophil chemoattraction, and select cytokines may also participate.
The early recruitment of macrophages into the ME was unaffected by the lack of CCL3. Since CCL3 is known to chemoattract macrophages, this suggests that other chemokines predominantly regulate the entry of this cell type into the ME. As shown in Table S1 in the supplemental material, several other macrophage chemoattractants are upregulated in the ME both early and late during OM.
Our observation of delayed, but not decreased, inflammatory cell recruitment in CCL3-null mice (Fig. 3A) was unanticipated in view of the known role of CCL3 as a chemoattractant in the pathogenesis of various inflammatory diseases, its high level of expression during OM, and its broad chemoattractant profile. In contrast to our results, several other studies on infection at other sites in ccl3−/− mice reported a diminished infiltration of leukocytes (28–31). However, in accordance with our observations, inflammatory cell migration was not reduced in CCL3-null models of pulmonary infection (25) or periodontitis (32). Clearly, the context of CCL3 deletion can play a critical role in the null phenotype. This may well reflect the differing roles of redundant factors at various sites and under different inflammatory conditions.
The issue of functional redundancy between chemokines has been much discussed in recent literature. Chemokine receptors are promiscuous, as they are known to bind several different chemokines, and chemokines in turn can bind with high affinity to several different receptors. Thus, CCR1 and CCR5, the two receptors known to bind CCL3, are not selective for this chemokine. CCR5, for example, can also bind CCL4 and CCL5. Both of these chemokines are known to exert overlapping functions with CCL3. Other chemokines such as CCL2, CCL7, and CCL12 bind to different receptors but similarly recruit inflammatory cells, while cytokines such as macrophage migration inhibitory factor (MIF) can also be cooperatively involved in the trafficking of leukocytes (18, 33). Support for the possibility of redundancy comes from studies demonstrating enhanced expression levels of alternative chemokines as a mechanism of compensation for a genetic loss of CCL3 in other systems (27, 34). We evaluated the expression levels of CCL2, CCL7, and CCL12, regulated over the same time period as CCL3 during OM, in ccl3−/− mice. We detected significant differences in the expression levels of all three genes, with most expression levels being higher at intervals of 2 days or less after inoculation, and lower thereafter, than those in WT mice. These increases in expression levels are consistent with a compensatory response to CCL3 deletion. An exception to this was observed for CCL2, for which the expression level was lower at 6 h. This might reflect the lower number of neutrophils present early during OM, since this CCL is produced by PMNs, while CCL7 and CCL12 are not (18, 19, 24). Of course, other chemokines and cytokines that we did not assess may also help to compensate for CCL3 deletion.
Both neutrophils and macrophages persisted in ccl3−/− MEs longer than in WT MEs, in agreement with the results of our prior study (13) using antibody-mediated depletion of CCL3, but persisted for much longer periods in the genetic model. This persistence seems most likely to be due to the prolonged presence of NTHi, as has been observed with other defects in innate immunity (13–15). This could well be related to the phagocytic defect in ccl3−/− macrophages. However, it is possible that neutrophil bactericidal activity is also reduced in CCL3-null animals and that this contributes to persistence. The defect in macrophage NTHi phagocytosis due to the loss of CCL3 is consistent with data from a previous study of Pseudomonas aeruginosa uptake in ccl3−/− macrophages (22). Further evidence for the importance of CCL3 for macrophage function is provided by our previous finding that CCL3 was able to correct functional deficits in macrophages deficient in TLR2, MyD88, TNF (21), or CCL3 (22) (Fig. 5B). However, in contrast to our results, ccl3−/− macrophages displayed a defect in the intracellular killing of P. aeruginosa (22), while we saw no defect in the killing of NTHi. This could be related to the differential sensitivities of these bacterial species to the killing mechanism involved.
The OM phenotype observed in ccl3−/− animals is similar in many respects to that seen in mice genetically deficient for some TLR genes or TLR signaling genes. Mice with mutations in TLR2 (15), MyD88 (14), or TNF (13) are impaired in the clearance of NTHi from the ME, with a concomitant delay in mucosal recovery. However, our present study found that CCL3 deletion produces a somewhat milder OM phenotype. The deletion of these other genes caused a near-complete failure to resolve OM, as presented by a persistent inflammatory response with significant mucosal hyperplasia and the presence of bacteria in the ME at 21 or even 42 days postinoculation (13–15). Given the large number of other downstream molecules in innate immune pathways, including cytokines, other chemokines, and antimicrobials that are elicited by innate immune receptors, our findings are not surprising. In fact, given these many potentially redundant factors, the extent of the defect in recovery from OM caused by CCL3 deletion might be somewhat unexpected.
The ME would obviously not be predicted to be the only site affected by ccl3 gene deletion. The nasopharynx is the ecological niche for many commensal bacteria and potential respiratory pathogens. Ultimately, the colonization of the upper airways is a prerequisite for subsequent infection of the lower respiratory tract and ME (35, 36). Any imbalance of this complex microbiome, both qualitative and quantitative, may increase susceptibility to the spread of infection (3, 5). Since our nasopharyngeal cultures revealed that ccl3−/− mice were colonized with bacteria to a significantly higher degree than WT mice (Fig. 6), it seems likely that CCL3 is an important determinant of nasopharyngeal homeostasis. CCL3 may therefore be a determinant of resistance to ME infection beyond its direct role in the ME. However, it should be noted that despite the observation of increased bacterial colonization of the nasopharynx, the MEs of naive ccl3−/− mice were culture negative and showed no signs of inflammation such as increased mucosal thickness or the presence of leukocytes. Mice lacking MyD88 (14), TLR2 (15), or TNF (13) all displayed some signs of ME inflammation even in the absence of NTHi inoculation. Enhanced nasopharyngeal colonization has been cited as contributing to the etiology of OM (3). However, the lack of CCL3 alone does not appear to facilitate de novo ME infection from the nasopharynx.
With increased microbial antibiotic resistance resulting from the extensive use of antibiotics for the treatment of acute OM (37), there is an urgent need to develop novel therapeutic agents for otitis-prone children who suffer from recurrent or chronic ME disease. Promising results from the in vivo application of CCL3 in animals with OM recovery deficits (13) and the conclusions of our present study make this chemokine and its receptors interesting therapeutic targets.
MATERIALS AND METHODS
Animals.
For CCL gene expression studies, WT C57/WB F1 hybrid mice (gene array) or C57BL/6 mice (qPCR) were purchased from the Jackson Laboratory (Bar Harbor, ME). For the evaluation of OM without CCL3, CCL3-deficient (CCL3−/−) mice (B6.129P2-Ccl3tm1Unc/J) and age-matched WT mice (C57BL/6J) from the Jackson Laboratory were bred in our animal facility. Animals were housed under specific-pathogen-free conditions. All experiments were performed according to NIH guidelines following approval by the Institutional Animal Care and Use Committee of the Veterans Affairs Medical Center, San Diego, CA.
Bacteria.
Haemophilus influenzae strain 3655 (nontypeable, biotype II), originally isolated from the ME of an OM patient in St. Louis, Missouri, USA (kindly provided by Asa Melhus, Lund University, Sweden), was used at a concentration of 105 to 106 bacteria/ml to induce infection of the ME. Inocula were prepared as described previously (38, 39).
Surgery.
For gene expression studies, WT mice were used in groups of 20 per gene array. For the evaluation of OM in ccl3−/− mice, KO and WT C57BL/6J mice were divided into groups of at least 6 mice for each experimental time point (3 to 8 animals for histopathology and bacterial culture, respectively). All surgeries were performed under standard aseptic conditions. As described previously, all animals were deeply anesthetized, and approximately 5 μl of an NTHi inoculum was bilaterally injected into the ME via the surgically exposed ME bulla (39, 40). Following inoculation, the tympanic membranes were confirmed visually to be intact. Uninoculated animals (time 0 h) served as controls.
ME CCL gene expression.
ME mucosae were harvested from 40 deeply anesthetized WT mice 0, 3, and 6 h as well as 1, 2, 3, 5, and 7 days after inoculation of NTHi in saline. The tissues from 20 mice were pooled to generate two samples at each time point. Groups of control mice were injected with saline alone. The tissue was homogenized in TRIzol (Invitrogen, Carlsbad, CA), and total RNA was extracted. Total RNA quality was assessed by using the RNA 6000 Labchip kit on the Agilent 2100 bioanalyzer for the integrity of 18S and 28S rRNAs. The mRNA was reverse transcribed by using a T7 oligo(dT) primer and then in vitro transcribed by using T7 RNA polymerase to generate biotinylated cRNA probes that were hybridized to Affymetrix MU430 2.0 microarrays. Duplicate arrays were hybridized for each time point by using RNA from the two pools of mouse tissues to obtain independent biological replicates. Raw intensity data were median normalized, and statistical differences in gene transcript expression levels were evaluated by using variance-modeled posterior inference (VAMPIRE) (41). This program uses a Bayesian approach to identify altered genes. Statistical analysis by VAMPIRE requires two distinct steps: (i) modeling of the error structure of sample groups and (ii) significance testing with an a priori-defined significance threshold. VAMPIRE models the existing error structure to distinguish the signal from noise and identify the coefficients of expression-dependent and expression-independent variances. These models are then used to identify microarray features that are differentially expressed between treatment groups. This method allows the use of small numbers of replicates to evaluate gene expression across a continuum of conditions, down to two or even one array per condition if (as in the present study) many samples are pooled for each array (i.e., each array itself samples the mean value) and if multiple conditions are assessed. We compared mice inoculated with NTHi at each time point to uninoculated (0-h) controls to evaluate genes whose expression levels changed significantly over time following NTHi injection. Bonferroni multiple-testing correction (αBonf, <0.05) was applied to identify only those genes with the most robust changes. All CCL genes represented on the array were evaluated.
By using separate groups of three mice for each time point, the expression levels of CCL2, CCL7, and CCL12 mRNA were evaluated by qPCR, during the course of NTHi-induced OM in both WT and ccl3−/− mice. cDNA libraries were prepared from total RNA isolated from ME tissue and infiltrates by using the RNeasy kit (Qiagen, MD, USA). The RNA was reverse transcribed by using SuperScript III (Invitrogen, CA, USA) in a 25-μl reaction mixture, and a final 100 ng of the cDNA was used for each real-time quantitative PCR determination. QuantiTect primers (Qiagen, Hilden, Germany), specific for each of the chemokines CCL2, CCL3, CCL7, and CCL12, were used to probe the cDNA templates. Real-time qPCR was performed by using the StepOnePlus PCR cycler system (Applied Biosystems, Foster City, CA, USA). A 25-μl reaction mixture containing 12.5 μl 2× Sybr green PCR master mix (Applied Biosystems), 2.5 μl of the primer (10 μM), 5 μl cDNA, and 5 μl water (molecular biograde) was set up in duplicates per sample. PCR cycling conditions were 95°C for 10 min followed by 40 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 40 s. The relative expression level of each of the cytokines was normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by using the ΔΔCT method. Melt curves were used to ensure a single amplicon amplification. Results shown in Fig. 5 represent data from 2 replicates, with error bars denoting the 95% confidence intervals.
ME histology.
Mice used for histology were anesthetized and perfused intracardially with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). MEs were harvested at 1, 2, 3, 5, 7, 10, 14, and 21 days postinoculation; postfixed (4% PFA) overnight; decalcified (8% EDTA and 4% PFA) for 14 days; processed in paraffin; and then sectioned at 9 μm. Hematoxylin and eosin (H&E)-stained sections from standardized locations of the ME cavity were digitally recorded, and OM was quantified by using NIH Image-Pro (14, 40). Mucosal thickness was assessed at six standardized locations from each of 4 sections widely spaced throughout the ME (40). To provide an indirect measure of the number of infiltrating leukocytes, the area occupied by inflammatory cells on the section that displayed the maximum number of cells in the ME cavity, versus the total area of the cavity, was measured (40) to determine the percentage covered by leukocytes. To assess the density of cellular infiltrates and the proportions of neutrophils and macrophages comprising ME infiltrates, the two largest clusters of the cellular ME effusion for each ME were photographed (×400 magnification), and the number of cells of each type was counted. Histological measures were performed by two independent observers from sections blinded as to genotype, and the results were averaged to arrive at the final data values.
ME cultures.
The in vivo clearance of NTHi was assessed in 36 ccl3−/− mice and 38 WT mice. At least 6 ears were sampled at each time point. A 1-μl loop sample was obtained from the opened ME lumen 0 days (uninoculated control) and 1, 2, 3, 5, 10, 14, and 21 days after NTHi inoculation. Each loop was streaked over 4 successive quadrants of a chocolate agar plate (42). The plates were incubated for 24 h at 37°C prior to the evaluation of bacterial colonies. The colonization rate was calculated as the proportion of ME cultures positive for NTHi at each time point.
As described previously (14), a semiquantitative scoring system was used to further classify the degree of NTHi colonization per plate, with 0 indicating no CFU, 1 indicating CFU in one quadrant, 2 indicating CFU in two quadrants, 3 indicating CFU in three quadrants, and 4 indicating CFU in all four quadrants. The presence and identity of NTHi were verified by negative Gram staining and negative cultures on blood agar plates versus positive cultures on chocolate agar plates and bacitracin-incorporated chocolate blood agar (42).
Nasopharyngeal cultures.
The bacterial loads in the nasopharynges of 6 uninfected WT and 8 uninfected ccl3−/− mice were determined. A 1-μl sample was obtained by scraping the nasopharyngeal mucosa of deeply anesthetized mice with a sterile loop. The sample was spread directly onto chocolate agar and incubated overnight at 37°C. All CFU identified on the plates were quantified by manual counting in triplicate.
Blood cultures.
For the same mice (at least 6 per strain) used for nasopharyngeal cultures, bacteremia was monitored by culturing blood collected aseptically by cardiac puncture of anesthetized animals. A 1-μl loop sample was streaked directly onto chocolate agar and assessed as described above.
Macrophage phagocytosis and killing.
Macrophage phagocytosis and bacterial killing were assessed by using an established in vitro assay (13, 14). Primary peritoneal macrophages were obtained from 6 ccl3−/− and 6 WT mice as described previously (13, 14) and seeded into 48-well plates at 5 × 105 cells per well in triplicate for each mouse and assay time point. Ten microliters of mid-exponential-phase NTHi bacteria (5 × 107 bacteria), a titer that does not saturate the cells, as demonstrated previously (13), was added to each well. After incubation for 1 h, extracellular bacteria were removed and killed by washing, followed by the addition of fresh Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS), macrophage colony-stimulating factor, and 50 μg/ml gentamicin. After the addition of gentamicin, incubation was continued for 1 or 3 h. The cells were then rinsed and lysed. Supernatants and cell lysates were plated onto chocolate agar in serial dilutions, incubated overnight at 37°C, and then evaluated by manually counting CFU. Six wells were used per time point and mouse strain. The recovery of bacteria after macrophage treatment with gentamicin for 1 h was used to represent phagocytosis. The ratio of the number of NTHi bacteria recovered after gentamicin treatment for 3 h compared to the number of bacteria recovered at 1 h posttreatment was considered to represent intracellular killing (14, 22). Percent bacterial killing was calculated as previously described (22), by using the following formula: % killing = 1 − (number of CFU in tested wells 3 h after cultivation with gentamicin/number of CFU in tested wells 1 h after cultivation) × 100.
Statistical analysis.
Statistical analyses for studies other than gene expression analyses were performed by using GraphPad Prism 6 software. Data are reported as means ± standard errors of the means (SEM). Differences were considered significant at a P value of <0.05. Two-way ANOVA with Bonferroni correction for multiple comparisons was performed on measures of mucosal thickness. ME colonization rates were evaluated statistically by the Fisher-Irwin test for proportions. The Mann-Whitney U test was employed for data lacking a normal distribution, as for analyses of ME inflammatory cells, CFU counts for the nasopharynx, and lysate NTHi counts for the phagocytosis assay. Data normality was evaluated by using the D'Agostino-Pearson omnibus test. Left and right ears in each mouse were considered to be independent of each other, as previously discussed in detail (40), and therefore were analyzed independently.
Supplementary Material
ACKNOWLEDGMENTS
We thank Eduardo Chavez for mouse colony maintenance and Julie Lightner for editorial help.
We certify that we have no conflicts of interest.
A.F.R., S.I.W., D.D., and B.N. conceived of and designed the experiments. D.D., B.N., K.P., J.H., and K.S. performed the experiments. D.D., J.H., A.K., and A.F.R. analyzed the data. D.D., A.K., and A.F.R. wrote the manuscript.
The research reported in this work was supported by the NIDCD of the National Institutes of Health under awards DC000129 to A.F.R., DC006279 to S.I.W., DC012595 to A.F.R., and DC014801 to A.K. and by the Research Service of the Veterans Administration under awards BX001205 and RX000977, both to A.F.R.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
A.F.R. is a cofounder of and stockholder in Otonomy, Inc., a company that develops slow-release drug formulations for the middle and inner ears. The company played no role in this research.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00148-17.
REFERENCES
- 1.Ruohola A, Meurman O, Nikkari S, Skottman T, Salmi A, Waris M, Osterback R, Eerola E, Allander T, Niesters H, Heikkinen T, Ruuskanen O. 2006. Microbiology of acute otitis media in children with tympanostomy tubes: prevalences of bacteria and viruses. Clin Infect Dis 43:1417–1422. doi: 10.1086/509332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casey JR, Kaur R, Friedel VC, Pichichero ME. 2013. Acute otitis media otopathogens during 2008 to 2010 in Rochester, New York. Pediatr Infect Dis J 32:805–809. doi: 10.1097/INF.0b013e31828d9acc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faden H, Duffy L, Wasielewski R, Wolf J, Krystofik D, Tung Y. 1997. Relationship between nasopharyngeal colonization and the development of otitis media in children. Tonawanda/Williamsville Pediatrics. J Infect Dis 175:1440–1445. doi: 10.1086/516477. [DOI] [PubMed] [Google Scholar]
- 4.Ruohola A, Pettigrew M, Lindholm L, Jalava J, Räisänen KS, Vainionpää R, Waris M, Tähtinen PA, Laine MK, Lahti E, Ruuskanen O, Huovinen P. 2013. Bacterial and viral interactions within the nasopharynx contribute to the risk of acute otitis media. J Infect 66:247–254. doi: 10.1016/j.jinf.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Revai K, Mamidi D, Chonmaitree T. 2008. Association of nasopharyngeal bacterial colonization during upper respiratory tract infection and the development of acute otitis media. Clin Infect Dis 46:e34–e37. doi: 10.1086/525856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bluestone CD. 1996. Pathogenesis of otitis media: role of Eustachian tube. Pediatr Infect Dis J 15:281–291. doi: 10.1097/00006454-199604000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Klein JO. 2000. The burden of otitis media. Vaccine 19(Suppl 1):S2–S8. doi: 10.1016/S0264-410X(00)00271-1. [DOI] [PubMed] [Google Scholar]
- 8.Shuto T, Xu H, Wang B, Han J, Kai H, Gu XX, Murphy TF, Lim DJ, Li JD. 2001. Activation of NF-kappa B by nontypeable Hemophilus [sic] influenzae is mediated by Toll-like receptor 2-TAK1-dependent NIK-IKK alpha/beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci U S A 98:8774–8779. doi: 10.1073/pnas.151236098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janeway CA, Medzhitov R. 2002. Innate immune recognition. Annu Rev Immunol 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 10.Takeda K, Kaisho T, Akira S. 2003. Toll-like receptors. Annu Rev Immunol 21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez M, Leichtle A, Pak K, Webster NJ, Wasserman SI, Ryan AF. 2015. The transcriptome of a complete episode of acute otitis media. BMC Genomics 16:259. doi: 10.1186/s12864-015-1475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacArthur CJ, Hausman F, Kempton JB, Choi D, Trune DR. 2013. Otitis media impacts hundreds of mouse middle and inner ear genes. PLoS One 8:e75213. doi: 10.1371/journal.pone.0075213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leichtle A, Hernandez M, Ebmeyer J, Yamasaki K, Lai Y, Radek K, Choung YH, Euteneuer S, Pak K, Gallo R, Wasserman SI, Ryan AF. 2010. CC chemokine ligand 3 overcomes the bacteriocidal and phagocytic defect of macrophages and hastens recovery from experimental otitis media in TNF−/− mice. J Immunol 184:3087–3097. doi: 10.4049/jimmunol.0901167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez M, Leichtle A, Pak K, Ebmeyer J, Euteneuer S, Obonyo M, Guiney DG, Webster NJ, Broide DH, Ryan AF, Wasserman SI. 2008. Myeloid differentiation primary response gene 88 is required for the resolution of otitis media. J Infect Dis 198:1862–1869. doi: 10.1086/593213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leichtle A, Hernandez M, Pak K, Yamasaki K, Cheng CF, Webster NJ, Ryan AF, Wasserman SI. 2009. TLR4-mediated induction of TLR2 signaling is critical in the pathogenesis and resolution of otitis media. Innate Immun 15:205–215. doi: 10.1177/1753425909103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emonts M, Veenhoven RH, Wiertsema SP, Houwing-Duistermaat JJ, Walraven V, de Groot R, Hermans PW, Sanders EA. 2007. Genetic polymorphisms in immunoresponse genes TNFA, IL6, IL10, and TLR4 are associated with recurrent acute otitis media. Pediatrics 120:814–823. doi: 10.1542/peds.2007-0524. [DOI] [PubMed] [Google Scholar]
- 17.Lee YC, Kim C, Shim JS, Byun JY, Park MS, Cha CI, Kim YI, Lee JW, Yeo SG. 2008. Toll-like receptors 2 and 4 and their mutations in patients with otitis media and middle ear effusion. Clin Exp Otorhinolaryngol 1:189–195. doi: 10.3342/ceo.2008.1.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rollins BJ. 1997. Chemokines. Blood 90:909–928. [PubMed] [Google Scholar]
- 19.Luster AD. 1998. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med 338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 20.Maurer M, von Stebut E. 2004. Macrophage inflammatory protein-1. Int J Biochem Cell Biol 36:1882–1886. doi: 10.1016/j.biocel.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Leichtle A, Yamasaki K, Euteneuer S, Wasserman SI, Wollenberg B, Ryan AF. 2009. Impaired antibacterial function is restored via CCL3. Otolaryngol Head Neck Surg 141:82–83. [Google Scholar]
- 22.Takahashi H, Tashiro T, Miyazaki M, Kobayashi M, Pollard RB, Suzuki F. 2002. An essential role of macrophage inflammatory protein 1alpha/CCL3 on the expression of host's innate immunities against infectious complications. J Leukoc Biol 72:1190–1197. [PubMed] [Google Scholar]
- 23.Ramos CD, Canetti C, Souto JT, Silva JS, Hogaboam CM, Ferreira SH, Cunha FQ. 2005. MIP-1a[CCL3] acting on the CCR1 receptor mediates neutrophil migration in immune inflammation via sequential release of TNF-a and LTB4. J Leukoc Biol 78:167–177. doi: 10.1189/jlb.0404237. [DOI] [PubMed] [Google Scholar]
- 24.Xue ML, Thakur A, Cole N, Lloyd A, Stapleton F, Wakefield D, Willcox MD. 2007. A critical role for CCL2 and CCL3 chemokines in the regulation of polymorphonuclear neutrophils recruitment during corneal infection in mice. Immunol Cell Biol 85:525–531. doi: 10.1038/sj.icb.7100082. [DOI] [PubMed] [Google Scholar]
- 25.Lindell DM, Standiford TJ, Mancuso P, Leshen ZJ, Huffnagle GB. 2001. Macrophage inflammatory protein 1alpha/CCL3 is required for clearance of an acute Klebsiella pneumoniae pulmonary infection. Infect Immun 69:6364–6369. doi: 10.1128/IAI.69.10.6364-6369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eming SA, Krieg T, Davidson JM. 2007. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 127:514–525. doi: 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- 27.Janeway CA Jr, Travers P, Walport M, Shlomchik. 2001. Immunobiology: the immune system in health and disease, 5th ed. Garland Science, New York, NY: http://www.ncbi.nlm.nih.gov/books/NBK27122/. [Google Scholar]
- 28.Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, Smithies O. 1995. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science 269:1583–1585. doi: 10.1126/science.7667639. [DOI] [PubMed] [Google Scholar]
- 29.Lu P, Li L, Wu Y, Mukaida N, Zhang X. 2008. Essential contribution of CCL3 to alkali-induced corneal neovascularization by regulating vascular endothelial growth factor production by macrophages. Mol Vis 14:1614–1622. http://www.molvis.org/molvis/v14/a191. [PMC free article] [PubMed] [Google Scholar]
- 30.Heinrichs D, Berres M, Nellen A, Fischer P, Scholten D, Trautwein C, Wasmuth HE, Sahin H. 2013. The chemokine CCL3 promotes experimental liver fibrosis in mice. PLoS One 8:e66106. doi: 10.1371/journal.pone.0066106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X, Walton W, Cook DN, Hua X, Tilley S, Haskell CA, Horuk R, Blackstock AW, Kirby SL. 2011. The chemokine, CCL3, and its receptor, CCR1, mediate thoracic radiation-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 45:127–135. doi: 10.1165/rcmb.2010-0265OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Repeke CE, Ferreira SB, Claudino M, Silveira EM, de Assis GF, Avila-Campos MJ, Silva JS, Garlet GP. 2010. Evidences of the cooperative role of the chemokines CCL3, CCL4 and CCL5 and its receptors CCR1+ and CCR5+ in RANKL+ cell migration throughout experimental periodontitis in mice. Bone 46:1122–1130. doi: 10.1016/j.bone.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 33.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, McColl SR, Hickey MJ. 2006. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol 177:8072–8079. doi: 10.4049/jimmunol.177.11.8072. [DOI] [PubMed] [Google Scholar]
- 34.Yanaba K, Mukaida N, Matsushima K, Murphy PM, Takehara K, Sato S. 2004. Role of CC chemokine receptors 1 and 5 and CCL3/macrophage inflammatory protein-1alpha in the cutaneous Arthus reaction: possible attenuation of their inhibitory effects by compensatory chemokine production. Eur J Immunol 34:3553–3561. doi: 10.1002/eji.200425426. [DOI] [PubMed] [Google Scholar]
- 35.von Eiff C, Becker K, Machka K, Stammer H, Peters G. 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study group. N Engl J Med 344:11–16. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- 36.Bogaert D, De Groot R, Hermans PW. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4:144–154. doi: 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 37.Grijalva CG, Nuorti JP, Griffin MR. 2009. Antibiotic prescription rates for acute respiratory tract infections in US ambulatory settings. JAMA 302:758–766. doi: 10.1001/jama.2009.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melhus A, Hermansson A, Prellner K. 1994. Nontypeable and encapsulated Haemophilus influenzae yield different clinical courses of experimental otitis media. Acta Otolaryngol 114:289–294. doi: 10.3109/00016489409126058. [DOI] [PubMed] [Google Scholar]
- 39.Melhus A, Ryan AF. 2003. A mouse model for acute otitis media. APMIS 111:989–994. doi: 10.1034/j.1600-0463.2003.1111012.x. [DOI] [PubMed] [Google Scholar]
- 40.Ebmeyer J, Furukawa M, Pak K, Ebmeyer U, Sudhoff H, Broide D, Ryan AF, Wasserman S. 2005. Role of mast cells in otitis media. J Allergy Clin Immunol 116:1129–1135. doi: 10.1016/j.jaci.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 41.Hsiao A, Ideker T, Olefsky JM, Subramaniam S. 2005. VAMPIRE microarray suite: a Web-based platform for the interpretation of gene expression data. Nucleic Acids Res 33:W627–W632. doi: 10.1093/nar/gki443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH (ed). 1995. Manual of clinical microbiology, 7th ed. ASM Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.