

ABSTRACT
The spleen is known as an important filter for blood-borne pathogens that are trapped by specialized macrophages in the marginal zone (MZ): the CD209+ MZ macrophages (MZMs) and the CD169+ marginal metallophilic macrophages (MMMs). Acute systemic infection strongly impacts MZ populations and the location of T and B lymphocytes. This phenomenon has been linked to reduced chemokine secretion by stromal cells. Brucella spp. are the causative agent of brucellosis, a widespread zoonotic disease. Here, we used Brucella melitensis infection as a model to investigate the impact of chronic stealth infection on splenic MZ macrophage populations. During the late phase of Brucella infection, we observed a loss of both MZMs and MMMs, with a durable disappearance of MZMs, leading to a reduction of the ability of the spleen to take up soluble antigens, beads, and unrelated bacteria. This effect appears to be selective as every other lymphoid and myeloid population analyzed increased during infection, which was also observed following Brucella abortus and Brucella suis infection. Comparison of wild-type and deficient mice suggested that MZ macrophage population loss is dependent on interferon gamma (IFN-γ) receptor but independent of T cells or tumor necrosis factor alpha receptor 1 (TNF-αR1) signaling pathways and is not correlated to an alteration of CCL19, CCL21, and CXCL13 chemokine mRNA expression. Our results suggest that MZ macrophage populations are particularly sensitive to persistent low-level IFN-γ-mediated inflammation and that Brucella infection could reduce the ability of the spleen to perform certain MZM- and MMM-dependent tasks, such as antigen delivery to lymphocytes and control of systemic infection.
KEYWORDS: Brucella melitensis, marginal zone macrophages, low-grade Th1 inflammation, brucellosis, infection, spleen
INTRODUCTION
The spleen is both the largest secondary lymphoid organ and the main filter of blood (for a general review, see reference 1). The white pulp, which contains T and B cell populations, is surrounded by the red pulp, a network of reticular fibers and fibroblasts rich in macrophages, where blood is filtered and old erythrocytes are removed. The marginal zone (MZ) located between the white pulp and the red pulp constitutes a specialized filtering area for blood content. The MZ contains specialized macrophage populations, various dendritic cell (DC) subsets, and MZ B cells. The marginal metallophilic macrophages (MMMs) form an inner ring between the MZ and white pulp, whereas MZ macrophages (MZMs) are situated at the outer boundary of the MZ, adjacent to the red pulp. Numerous reports document the filtering capacity of both MMMs and MZMs and their role in clearing pathogens from the systemic circulation, based on the expression of a large variety of pathogen receptor molecules (reviewed in references 1 and 2). MZMs express the C-type lectin CD209 (also known as SIGN-R1 and recognized by ER-TR9 monoclonal antibody [MAb]) and the type 1 scavenger receptor MARCO. CD209 binds to the capsular polysaccharide of Streptococcus pneumoniae (3) as well as to the polysaccharide dextran (4). MARCO binds to Staphylococcus aureus and Escherichia coli (5). Depletion experiments have demonstrated that MZMs are crucial for the entrapment of blood-borne pathogens such as lymphocytic choriomeningitis virus (LCMV) (6), Streptococcus pneumoniae (3), and Listeria monocytogenes (7). MMMs display CD169 (also known as Siglec-1 and recognized by MOMA-1 MAb [8, 9]) that recognizes sialylated lipopolysaccharides from Neisseria meningitidis (10) and Campylobacter jejuni (11) and binds to Trypanosoma cruzi (12). MZM populations also play a key role in antigen delivery to B cells (13) and CD4+ T cells (14) and regulate tolerance to apoptotic cells (15, 16).
Given the many functions of the MZ, interference of pathogens with the integrity and functionality of the MZ may render the host particularly vulnerable to secondary infections or affect self-tolerance and promote autoimmune diseases. Systemic infections with viruses (murine cytomegalovirus [MCMV] [17] and LCMV [18]), protozoa (Leishmania donovani [19] and Plasmodium berghei [20]), and bacteria (Salmonella enterica serovar Typhimurium [21]) have been reported to induce a dramatic alteration of MZ macrophage populations. Different mechanisms have been described depending on the model. During L. donovani infection, MZ alterations are reduced in the absence of tumor necrosis factor alpha (TNF-α) (19). In MCMV (17) and LCMV (18) models, loss of MZ macrophage populations is linked to alteration of CCL21 chemokine production by stromal cells. It has also been reported that mice presenting the mutation “paucity of lymph node T cells” (plt) display a deficiency in both CCL19 and CCL21 and have a reduced number of macrophages in the MZ (22). In addition, an in vitro study showed that MZMs can migrate in response to CCL19 and CCL21 (22). Taken together, these results suggest that CCL19/21 chemokines could be implicated in the location of MMMs and MZMs in homeostatic conditions.
Brucella (Alphaproteobacteria) bacteria are facultative intracellular Gram-negative coccobacilli that infect mammals and cause brucellosis, one of the most common bacterial zoonoses (reviewed in references 23 to 24). Due to its intracellular lifestyle, the weak agonist activity of its lipopolysaccharide, and the active suppression of the innate immune response by immuno-regulatory factors, Brucella establishes a stealth infection associated with low-level Th1 inflammatory reactions (reviewed in reference 25). In this work, our objective was to characterize the impact of chronic stealth Brucella melitensis infection on splenic MZ macrophage subsets in resistant C57BL/6 mice. Our results demonstrated that Brucella induces drastic, long-term, and selective alteration of splenic MZ macrophage populations.
RESULTS
Chronic Brucella infection causes selective alteration of spleen cell populations.
In order to characterize the impact of intraperitoneal (i.p.) Brucella melitensis infection on the spatial distribution and number of spleen cell populations, wild-type C57BL/6 mice were infected i.p. with 105 CFU of B. melitensis bacteria, and the spleens were harvested at 1, 5, 18, 28, and 50 days postinfection and analyzed. The bacterial load (Fig. 1A) peaked around day 5 in the spleens. Fluorescence microscopy analysis of frozen spleen sections from infected mice showed that splenomegaly was associated with significant recruitment of various cell types such as CD90+ T lymphocytes (see Fig. S1A in the supplemental material) and cells expressing high levels of Gr-1 (Gr-1high) cells (Fig. S1B). T lymphocytes, initially located in the T cell area of the white pulp, massively relocated to the red pulp, presumably to participate in granuloma formation induced by B. melitensis infection (26). The number of Gr-1high cells increased in the red pulp, where they formed dense aggregates and were frequently found to infiltrate the white pulp T cell area. Spleen cell counting and flow cytometry analysis of the major spleen cell populations were performed to characterize the kinetic evolution of the number of cell populations per spleen during the course of B. melitensis infection (Fig. 1B). We observed that all populations analyzed increased in number in response to infection. The numbers of inflammatory dendritic cells (CD11b+ CD11c+ Ly-6Chigh), B cells (CD45R/B220+ MHC-II+, where MHC-II is major histocompatibility complex class II), and both CD3+ CD4+ and CD3+ CD8+ T cells peak at 5 days. Red pulp macrophages (F4/80+ CD11c− Ly-6G−), neutrophils (F4/80− CD11c− Ly-6Ghigh), and classical dendritic cells (CD11c+ cells expressing low levels of Ly-6C [Ly-6Clow]) peaked later at 18 days. The late recruitment of neutrophils has been previously reported (27). It is surprising that cells of innate immunity are recruited later than those of adaptive immunity. We can hypothesize that this phenomenon is due to the stealthy nature (25) of Brucella.
FIG 1.

Brucella melitensis infection leads to increased numbers of lymphocytes and myeloid and granulocyte cells in the spleen. Wild-type C57BL/6 mice were infected intraperitoneally (i.p.) with a dose of 105 CFU of B. melitensis and sacrificed at the indicated times. The data represent the number of CFU per spleen (A) and the number of cells positive for the indicated cell markers (B) determined by combining the results of cell counting (using a Thoma cell counting chamber) and flow cytometry. These results are representative of at least two independent experiments (n = 8). Folli, follicular; infDC, inflammatory dendritic cells.
In striking contrast to other spleen cell populations, fluorescence microscopy analysis showed that the MZ macrophage populations decreased dramatically during the chronic phase of B. melitensis infection. Both the MMMs (Fig. 2A, CD169+ cells) and MZMs (Fig. 2B, CD209+ cells) had disappeared from the MZ of the mice at 18 days postinfection. This phenomenon is confirmed by quantitative PCR showing a significant decrease of CD169 and CD209 expression in the spleen at the same time (Fig. 2C).
FIG 2.

Loss of marginal zone macrophage populations following Brucella melitensis infection. Wild-type C57BL/6 mice were infected i.p. with a dose of 105 CFU of B. melitensis and sacrificed at 18 days postinfection. Thirty minutes before sacrifice, mice were injected i.p. with FITC-coupled dextran, as indicated in Materials and Methods. The panels represent the localization by immunofluorescence of CD45R/B200+ B cells and CD169+ and FITC-coupled dextran+ cells (A) and CD209+ and FITC-coupled Dextran+ cells (B) in the spleens of naive (control) and infected mice. The panels are color coded with the text for the antigen examined. r.p., red pulp; w.p., white pulp. (C) Wild-type C57BL/6 mice were infected i.p. with a dose of 105 CFU of B. melitensis and sacrificed at 18 days postinfection. Spleen cells were collected, and total RNA was extracted and analyzed by quantitative PCR. mRNA levels were normalized using β-actin RNA as the reference and compared with mRNA levels obtained under conditions without infection (control). The panels represent a comparison of CD169 and CD209 gene expression levels in naive (control) and 18-day-infected (18d Br) wild-type C57BL/6 mice. Significant differences between infected and control mice are marked with asterisks: *, P < 0.05; **, P < 0.01. The data are representative of at least two independent experiments (n = 5).
Brucella infection reduces the filtration capability of the splenic marginal zone.
As CD209 expressed by MZMs is known to bind to the dextran antigen (4), we injected fluorescein isothiocyanate (FITC)-coupled dextran i.p. into control mice and B. melitensis-infected mice at 18 days postinfection to examine MZM-dependent antigen uptake in the spleen. Fluorescence microscopy analysis showed that dextran staining colocalized closely with CD209 (Fig. 2B) in the control mice and was absent in the mice at 18 days postinfection, thus confirming the loss of MZMs and demonstrating that the ability of the spleen to take up circulating antigen is reduced during B. melitensis infection.
Fluorescent microspheres were taken up preferentially by CD209+ MZMs (28), as confirmed by the colocalization of fluorescent microspheres and CD209 in the spleens of control mice (Fig. 3A). In the absence of MZMs in the mice at 18 days post-B. melitensis infection, we observed that the density of fluorescent microspheres was reduced and that they were located mainly in the red pulp area (Fig. 3A and B for a low-magnification view).
FIG 3.

Reduction of the ability of the marginal zone to take up blood-borne fluorescent microspheres during chronic Brucella melitensis infection. Wild-type C57BL/6 mice were infected i.p. with a dose of 105 CFU of B. melitensis and sacrificed at 18 days postinfection. Thirty minutes before sacrifice, mice were injected intravenously with FITC-coupled fluorescent microspheres (Fluo-beads), as indicated in Materials and Methods. (A) Localization by immunofluorescence of CD45R/B200+ B cells, CD209+ MZMs, and Fluo-beads in the spleens of naive (control) and infected mice. (B) Localization of CD45R/B220+ and FITC-coupled bead-positive cells on a large spleen section. The panels are color coded with the text for the antigen examined. r.p., red pulp; w.p., white pulp. The data are representative of at least two independent experiments (n = 5).
Overall, the inability of the MZ to take up dextran antigens and microspheres suggests that the capture of blood-borne pathogens by the MZ could be reduced during chronic B. melitensis infection. In order to test this possibility, we injected 108 CFU of live green fluorescent protein (GFP)-expressing Salmonella enterica serovar Typhimurium (GFP-Salmonella) bacteria intravenously into control mice and mice at 18 days post-Brucella infection, and the spleens were harvested 20 min later for microscopic analysis. In the control mice, microscopic analysis of the spleen section confirmed the previously published data (29) which reported that S. Typhimurium is captured by both CD209+ MZMs and CD169+ MMMs (Fig. 4A), as well as by red pulp macrophages (data not shown). As expected, in the absence of both MZMs and MMMs in mice at 18 days post-Brucella infection, we observed a significant reduction of the ability of the spleen to capture GFP-Salmonella (Fig. 4B and C).
FIG 4.

Brucella melitensis infection reduces the capture of blood-borne bacteria by the spleen. Wild-type C57BL/6 mice were infected i.p. with a dose of 105 CFU of B. melitensis and sacrificed at 18 days postinfection. Twenty minutes before sacrifice, mice were injected intravenously with 108 CFU of live GFP-expressing Salmonella enterica serovar Typhimurium (GFP-Salmonella) bacteria, as indicated in Materials and Methods. (A and B) Localization by immunofluorescence of CD45R/B200+ B cells, GFP-Salmonella, and CD209+ MZMs or CD169+ MMMs in the spleens of naive (control) and infected mice. The panels are color coded with the text for the antigen examined. (C) The percentage of GFP-Salmonella-positive surface out of the total spleen surface for five spleens from control mice and mice at 18 days postinfection. This quantification was performed using the colocalization module of the AxioVision program (Zeiss). r.p., red pulp; w.p., white pulp. Significant differences between infected and control mice are marked with asterisks: **, P < 0.01. The data are representative of at least two independent experiments (n = 5).
Semiquantitative microscopic analysis of Brucella-induced MZ macrophage populations loss.
Previous studies (17–21) investigating the impact of infection on MZ macrophage populations have performed only qualitative analyses by comparing their distribution by immunohistology. This approach does not allow for a fine comparison between several experimental conditions. Unfortunately, MZ macrophage populations are present in low numbers and are notoriously very difficult to purify (30), rendering any quantitative analysis of these populations hazardous by flow cytometry. A similar problem was observed to quantify memory CD8 T cells in tissues (31). In addition, the functionality of MZ macrophage populations is strongly dependent on their localization in the MZ because the MZ constitutes anatomically a mandatory point of passage for host cells and pathogens carried by the blood. To overcome these technical problems, we developed a semiquantitative immunofluorescence microscopy approach to estimate in situ the number of both MZMs and MMMs surrounding the white pulp area. Briefly (see Materials and Methods and Fig. S2), images of spleen sections stained with MZM- and MMM-specific markers were processed using the NIS-Elements program (Nikon Instruments) to determine the percentage of specific signal per marginal zone. This produced a semiquantitative estimation of MZMs and MMMs found in the spleen and a comparison between control and infected mice. As shown in Fig. 5A, intraperitoneal B. melitensis infection induced a rapid and durable loss of both dextran-positive (dextran+) MZMs and CD169+ MMMs. Maximal loss was observed at 18 days for both populations. However, while the MMM numbers had recovered partially at 28 days, a major loss of MZMs persisted until 50 days postinfection. Representative images are shown in Fig. S3. Two weeks of antibiotic treatment starting at 18 days post-i.p. infection allowed for a partial recovery of both the MMMs and MZMs (Fig. 5A). Note that antibiotic treatment itself does not induce a detectable alteration of MZ macrophage populations in control mice (Fig. S4). In agreement with a previously published observation on the different turnover rates of the various splenic macrophage populations (32), reconstitution of the MZM population seems to be slower than that for the MMMs. We should point out that MZ cell loss is very selective as at the same time MZ B cells (MHC-II+ B220+ CD1d+) increased in number, as shown by flow cytometry analysis (Fig. 1C).
FIG 5.

Quantification of MZM and MMM loss induced by Brucella infection in the spleen. Wild-type C57BL/6 mice were infected i.p. with 105 CFU of live B. melitensis (A), injected i.p. with 5 × 107 or 105 CFU of heat-killed (HK) B. melitensis (B or C, respectively), or infected intranasally (i.n.) with 105 CFU of live B. melitensis (D). Mice were sacrificed at the indicated times. Spleens were harvested, fixed, frozen, and stained as indicated in Materials and Methods to detect MZM and MMM populations. The panels represent a semiquantitative estimation of the presence of MZMs and MMMs in the spleens of control and infected mice. Gray circles show the percentage of positive staining of individual white pulp areas from naive (control) or infected mice. Green and red circles show the mean of positive staining of at least 10 white pulp areas from one frozen spleen section and constitute the mean of positive staining per spleen. For each condition, at least seven frozen spleen sections from three individual mice were analyzed. The horizontal bar indicates the global median. The statistical analysis was performed on the mean of positive staining per spleen. These results are representative of at least two independent experiments (n = 8). Significant differences between infected and control mice are marked with asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001. The data in panel E represent the number of CFU per spleen from wild-type C57BL/6 mice infected i.p. or i.n., as indicated, with 105 CFU of live B. melitensis bacteria. These results are representative of at least two independent experiments (n = 5). ns, not significant.
Intraperitoneal inoculation of 5 × 107 CFU of heat-killed (HK) B. melitensis induced a less drastic loss of MMMs and MZMs (Fig. 5B) than i.p. inoculation of live B. melitensis, with a partial recovery of MMMs that was already observed at 12 days postinfection. This phenomenon is dose dependent as i.p. inoculation of 105 CFU of HK B. melitensis induces a lower loss of MMMs and MZMs (Fig. 5C) at 5 days postinfection. These results suggest that recognition by the immune system of B. melitensis-associated molecular patterns alone is able to induce MZ macrophage population loss and that the cell infection process is not absolutely required for this phenomenon to occur.
Finally, we observed that, like i.p. infection, intranasal (i.n.) infection with 105 CFU of B. melitensis induced significant MMM and MZM loss (Fig. 5D). Compared to i.p. infection, this phenomenon appears to be delayed and is detectable only from 18 days post-i.n. infection for MZMs, as expected based on delayed infection of the spleen in the i.n. infection model (Fig. 5E) (33). In order to generalize our observation to other pathogenic species of Brucella, we i.p. infected wild-type C57BL/6 mice with 105 CFU of Brucella abortus or Brucella suis. As with B. melitensis infection, we observed drastic MMM and MZM losses at 18 days postinfection (Fig. S5). Globally, these results demonstrate that MMM and MZM losses are also observed after B. abortus and B. suis i.p. infection and following a more natural route of B. melitensis infection.
The IFN-γ-dependent immune response controlling Brucella infection induces the loss of marginal zone macrophage populations.
B. melitensis is well known to induce the development of cytotoxic interferon gamma (IFN-γ)-producing CD4+ T (Th1) cells (34) that play a key role in the control of infection in the spleen (27, 35). However, IFN-γ production (36) and activated cytotoxic T cells (18, 37) have been described in several infection models to alter the cellular architecture of the spleen by affecting CCL21 production by stromal cells. Other inflammatory cytokines produced during B. melitensis infection, such as TNF-α, have also been linked with MZ alteration in a Leishmania donovani infection model (19). Therefore, we compared the Brucella-induced MZM and MMM losses in wild-type, interleukin-1 receptor deficient (IL-1R−/−), TNF-αR1−/−, IFN-γR−/−, and CD3−/− (T cell-deficient) C57BL/6 mice. The course of infection was similar in the wild-type and IL-1R−/− mice, but the TNF-αR1−/−, IFN-γR−/−, and CD3−/− mice displayed significantly enhanced CFU loads in the spleen at 18 days postinfection compared to levels in wild-type mice (Fig. 6A). Fluorescence microscopy analysis showed that IL-1R deficiency does not affect MZM and MMM loss (Fig. 6B). In contrast, IFN-γR deficiency allows for the maintenance of both MZM and MMM populations during B. melitensis infection (Fig. 6B and 7) or after HK B. melitensis injection (Fig. 6C). Surprisingly, we observed that neither neutralization of the TNF-α signaling pathway nor the complete absence of T cells in CD3−/− mice favored the retention of MMMs during B. melitensis infection (Fig. 6B and 7). Note that in the absence of lymphocytes, IFN-γ production is still carried out by natural killer T cells (CD3− NK1.1+ cells) during B. melitensis infection (35). In summary, these results suggest that IFN-γ plays a critical role in MZM and MMM loss but that TNF-α and T cells are dispensable for this phenomenon to occur. In our experimental model, as previously published (27), the frequency of IFN-γ-producing cells in the spleen peaks between 5 and 18 days postinfection, and IFN-γ-producing cells remain detectable until 28 days postinfection (Fig. S6). At 50 days postinfection, IFN-γ-producing cells are no longer detectable by flow cytometry without in vitro restimulation. Thus, the peak of IFN-γ in the spleen during i.p. Brucella infection correlates with the peak of the alteration of MZ macrophage populations.
FIG 6.

The IFN-γR signaling pathway is required for Brucella-mediated MZM and MMM loss. Wild-type (wt) and IL-1R−/−, TNF-αR1−/−, IFN-γR−/−, and CD3−/− C57BL/6 mice received 105 CFU of live B. melitensis bacteria (A and B) or 5 × 107 CFU of HK B. melitensis bacteria (C) by i.p. route and were sacrificed at the indicated times. (A) Number of CFU per spleen for each group of infected mice at 18 days (18d) postinfection. (B and C) A semiquantitative estimation of the presence of MZMs and MMMs in the spleens of naive (control) mice and mice at 18 days postinfection. White circles indicate the percentage of positive staining of individual white pulp areas from naive (control) or infected mice. Green and red circles show the mean of positive staining of at least 10 white pulps from one frozen spleen section and constitute the mean of positive staining per spleen. For each condition, at least seven frozen spleen sections from three individual mice were analyzed. The horizontal bar indicates the global median. The statistical analysis was performed on the mean of positive staining per spleen. These results are representative of at least two independent experiments. Significant differences between infected and control mice are marked with asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FIG 7.

Visualization of marginal zone macrophage populations in control and infected wild-type and deficient mice. Wild-type, TNF-αR1−/−, and IFN-γR−/− C57BL/6 mice were infected i.p. with a dose of 105 CFU of B. melitensis and sacrificed at the indicated time postinfection. The panels show the localization of CD45R/B220+ B cells, CD169+ cells (MMMs), and FITC-coupled dextran+ cells (MZMs) on spleen sections from PBS-treated mice (control) and mice at 18 days postinfection. The panels are color coded with the text for the antigen examined. r.p., red pulp; w.p., white pulp. These results are representative of at least two independent experiments (n = 6).
In agreement with previous studies (38, 39), we confirmed that the type IV secretion system (VirB) mutant strain of B. melitensis 16M appears to be significantly attenuated compared to the wild-type strain (Fig. 8A) and induces a lower frequency of IFN-γ-producing cells in the spleen (Fig. 8B). As expected, the VirB strain also induces reduced MMM and MZM loss compared to the level in the wild-type strain (Fig. 8C), demonstrating that the intensity of the alteration of MZ macrophage populations is partially dependent on the virulence of Brucella.
FIG 8.
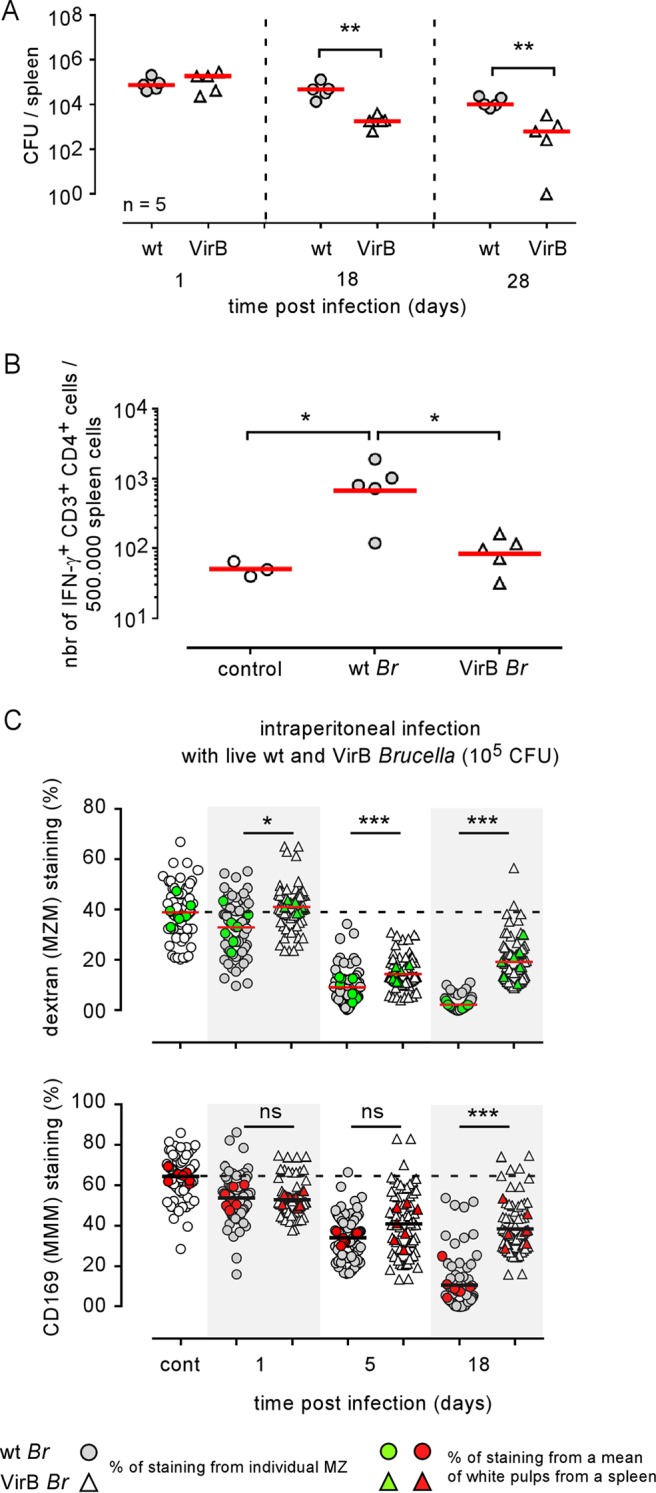
A VirB-deficient strain of Brucella melitensis induced reduced MZM and MMM loss. Wild-type (wt) C57BL/6 mice were infected i.p. with a dose of 105 CFU of wild-type and VirB B. melitensis in 500 μl of PBS or with PBS alone (control group) and sacrificed at the indicated time postinfection. (A) The number of CFU per spleen. (B) Frequency of IFN-γ-producing cells determined by flow cytometry. (C) Semiquantitative estimation of the presence of MZMs and MMMs in the spleens of control mice and mice at 1, 5, and 18 days postinfection. As indicated, white and gray symbols indicate the percentages of positive staining of individual white pulp areas from naive (control) or infected mice. Green and red symbols show the mean of positive staining of at least 10 white pulps from one frozen spleen section and constitute the mean of positive staining per spleen. For each condition, at least seven frozen spleen sections from three individual mice were analyzed. The horizontal bar indicates the global median. The statistical analysis was performed on the mean of positive staining per spleen. These results are representative of at least two independent experiments. Significant differences between infected and control mice are marked with asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Protection against MZM and MMM loss in IFN-γR−/− mice is not correlated with maintenance of CCL19, CCL21, and CXCL13 expression in the spleen.
In some acute bacterial and viral infection models (36), the splenic stromal cell-derived chemokines CCL21 and CXCL13 are transiently downregulated by a mechanism mainly controlled by IFN-γ-producing CD4+ T cells, leading to a dramatic alteration of the location of dendritic cells and lymphocytes. To determine the mechanism that protects MZMs and MMMs in B. melitensis-infected IFN-γR−/− mice, we analyzed the expression of CCL19, CCL21, and CXCL13 chemokines by quantitative PCR in control mice and mice at 18 days post-B. melitensis infection. Our results (Fig. 9) showed that chronic B. melitensis infection does not significantly modulate the expression of these chemokines in wild-type mice. In contrast, all of these chemokines were significantly downregulated in infected IFN-γR−/−, TNF-αR1−/−, and CD3−/− mice, thus demonstrating that in our experimental infection model their expression cannot be correlated with the loss or maintenance of MZ macrophage populations.
FIG 9.
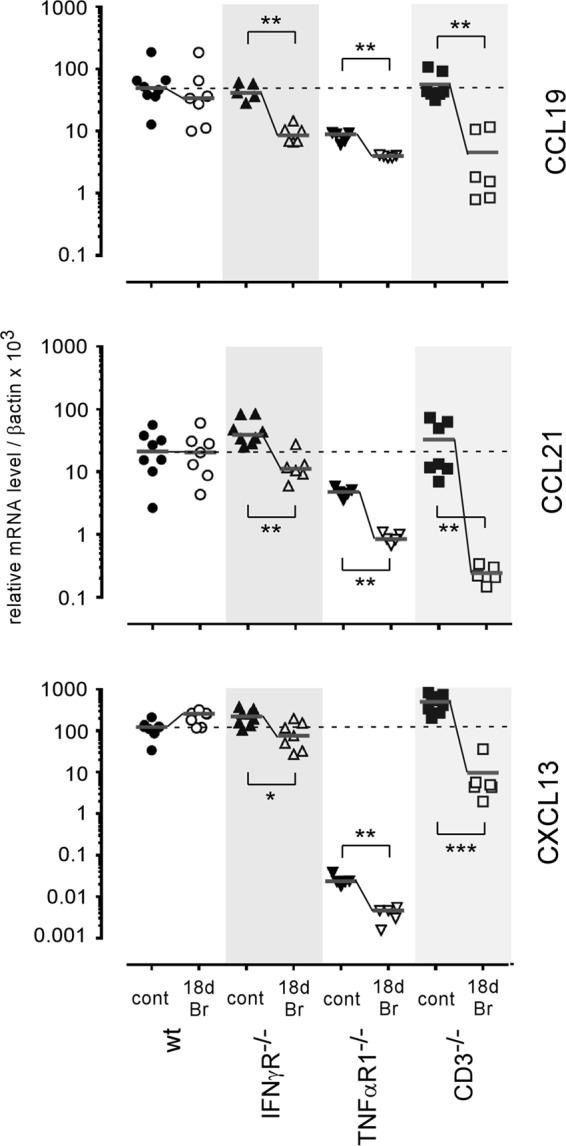
Impact of Brucella infection on CCL19, CCL21, and CXCL13 expression in the spleen. Wild-type, IFN-γR−/−, TNF-αR1−/−, and CD3−/− C57BL/6 mice were infected i.p. with a dose of 105 CFU of B. melitensis and sacrificed at 18 days postinfection. Spleen cells were collected, and total RNA was extracted and analyzed by quantitative PCR. mRNA levels were normalized using β-actin RNA as the reference and compared with mRNA levels obtained under conditions without infection (control). The panels represent a comparison of CCL19, CCL21, and CXCL13 gene expression levels in wild-type and deficient C57BL/6 mice in naive (control) mice and mice at 18 days postinfection. These results are representative of at least two independent experiments (n ≥ 6). Significant differences between infected and control mice are marked with asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
Although many studies have been performed, little real progress in our understanding of the Brucella-induced pathology has been reported recently. Whole-genome analysis demonstrated that Brucella does not display classic virulence factors that can directly harm eukaryotic cells, such as exotoxins, exoproteases, cytolysins, or other exoenzymes. All identified Brucella virulence factors play roles facilitating cell invasion and survival by subverting the innate cellular defense mechanisms (reviewed in reference 40) and are found in practically all Brucella species examined, independently of their pathogenicity for humans (41). Despite that, brucellosis can cause a debilitating chronic disease in humans, characterized by relapses of an undulant fever and lifelong severe complications, such as arthritis, meningitis, hepatitis, and endocarditis. We along with others have demonstrated that Brucella infection induces a protective (26, 35) but damaging (42) low-level IFN-γ-mediated inflammatory response in mice. A common characteristic of all localized Brucella-induced complications is the presence of inflammatory infiltrates in the lesions (43), suggesting that the incessant IFN-γ production generated by Brucella persistence could be the main source of the complications observed during brucellosis. Increasing evidence indicates that persistent low-level IFN-γ production can contribute to insulin resistance, type-2 diabetes, cardio-metabolic disease, autoimmune diseases (44), and cancer development (45).
In this work, we analyzed the impact of chronic stealth Brucella infection on the specialized MZ macrophage populations of the spleen. Our results showed that Brucella infection leads to a drastic loss of both CD169+ MMMs and CD209+ MZMs. Similar results are observed during the course of B. abortus and B. suis infection. Alteration of MZM and MMM populations appears to be highly selective during brucellosis as all other lymphoid and myeloid populations analyzed increased in number during the course of infection. By comparing wild-type and IFN-γR−/−-infected mice, we demonstrated that Brucella-mediated loss of MMMs and MZMs is impaired in the absence of IFN-γ signaling, suggesting that MMM and MZM loss is a direct consequence of a sustained IFN-γ-dependent Th1 protective response. MZMs seem to be particularly sensitive to IFN-γ-mediated inflammation as this population had not recovered at 50 days postinfection in the absence of detectable IFN-γ production, which suggests that very low levels of IFN-γ are sufficient to induce MZM loss. We have previously observed (26, 46) that CD169+ and CD209+ spleen cells can be infected in vivo by B. melitensis. However, in our experimental model, injection of HK B. melitensis is also able to induce a dose-dependent transient loss of both MZMs and MMMs in the spleen, which suggests that the infection process is not crucial to the induction of MZ macrophage population loss. The microbial antigens implicated in this phenomenon remain to be identified. As Brucella-induced MMM and MZM loss is dependent on IFN-γ, we can hypothesize that all Brucella antigens able to induce IFN-γ production could participate in MMM and MZM loss. In vitro, spleen cells produce high IL-12 and IFN-γ counts when stimulated with HK Brucella or Brucella-derived DNA (47). In vivo, Toll-like receptor 4 (TLR4) and MyD88 deficiencies but not TLR2 and TLR9 deficiencies significantly reduce the frequency of IFN-γ-producing cells in spleens from infected mice (27), suggesting that Brucella-derived TLR4 ligands, but not Brucella-derived TLR2 and TLR9 ligands, could play a key role in MMM and MZM loss.
A study of Buiting et al. (48) reported that injection of 500 μg of HK Brucella bacteria in mice pretreated with liposome-encapsulated clodronate causes accelerated repopulation of the spleen with macrophage populations, including MZMs and MMMs. Administration of liposome-encapsulated clodronate is known to reduce inflammation in various experimental models (49). Therefore, these mice could display a reduced ability to produce IFN-γ in response to Brucella antigens compared to the ability of control mice. This difference could explain the apparent discrepancy between our results and the results of Buiting et al.
The structure of the spleen is supported by a network of stromal cells producing chemokines, such as CCL19, CCL21, and CXCL13 (50). Acute inflammatory reactions have been associated with a drastic reduction in the expression of these chemokines in the spleen and severe disorganization of T and B cell segregation in the white pulp (17, 18, 36). Loss of splenic MZ macrophage populations has also been reported in experimental infection models characterized by intense acute inflammation (17–20). In MCMV and LCMV infection models (17, 18), these alterations have been linked to a reduction in CCL19 and CCL21 chemokine expression. MCMV infection caused specific, but transient, transcriptional suppression of CCL21 (17). During LCMV infection, the virus preferentially infects CCL21-producing stroma cells that become the target of an intense T cell-mediated cytotoxic response (18). In contrast, in our experimental model, MZ alterations induced by B. melitensis culminated at 18 days postinfection, long after the peak of infection occurring in the spleen at 5 days. In infected wild-type mice, we did not observe a general perturbation of the T and B cell areas following B. melitensis infection. The B cell follicles were dense and normal in appearance, and while some T cells relocated to the red pulp area, they did not invade the B cell areas, suggesting that the balance of chemokines located at the boundaries of the B and T cell areas in the white pulp remained effective during B. melitensis infection. In keeping with this hypothesis, quantitative PCR did not reveal a significant decrease in the expression of stromal cell-derived chemokines known to regulate the location of B cells (CXCL13) and T cells (CCL19 and CCL21) during the chronic phase of B. melitensis infection in wild-type mice. However, the expression of these chemokines was significantly reduced in Brucella-infected IFN-γR−/− mice that displayed higher bacteria levels in the spleen but conserved the MZM and MMM populations. Other factors affecting MZ population maintenance have been reported previously. In the L. donovani infection model (19), TNF-α mediated MZM and MMM loss. However, the absence of TNF-α signaling did not impair MZM and MMM population disappearance in our model. B cells appeared to be crucial for MZM and MMM development and maintenance (51) as their absence in B cell-deficient mice was correlated with reduced counts of these cells. However, in our model, flow cytometry analysis showed that both follicular and MZ B cells remained present and even increased in number during B. melitensis infection, suggesting that they are not implicated in the Brucella-induced MZM and MMM loss in wild-type mice. Taken together, our results suggest that, in our chronic infection model, MZM and MMM loss is not correlated with reduced expression of CCL19, CCL21, and CXCL13, is not due to a B cell deficiency, and does not required TNF-α signaling.
A growing body of experimental and epidemiological data suggest that the efficiency of the host immune response to fight infectious microorganisms can be affected by past or chronic unrelated infection (reviewed in reference 52). MZMs and MMMs are highly specialized cells that play a key role in clearing pathogens from the systemic circulation (1–3, 6, 7). Our work demonstrated that low-level IFN-γ-mediated sustained inflammation generated by i.p. or i.n. B. melitensis infection leads to a drastic and durable loss of MZ macrophage populations in the spleens of infected mice. A direct consequence of the loss of MMMs and MZMs is the reduced ability of the spleen of B. melitensis-infected mice to capture blood-borne soluble antigens, microspheres, or unrelated bacteria, which suggests that Brucella persistence could affect the host's ability to control unrelated infections, to develop protective immunity following vaccination, or to maintain self-tolerance and prevent autoimmune disease. The occurrence of human brucellosis is high and probably underestimated in regions where Brucella is endemic in domestic animals (53). To our knowledge, there is no evidence that Brucella infection predisposes to secondary infection with unrelated pathogens. However, based on our work, it would be profitable and interesting to examine the impact of brucellosis on other infectious and noninfectious diseases in the regions where it is common. It has been recently reported (21) that, like infection with Brucella, chronic infection of mice with Salmonella enterica serovar Typhimurium induces a significant decline in the proportions of CD169+ MMMs, suggesting that MZ alteration by chronic bacterial infection could be a common occurrence.
On the whole, our results could improve the understanding of the complex consequences for the immune system of low-level Th1 inflammation associated with pathogen persistence and provide further insight about diseases that could arise within the context of chronic infection.
MATERIALS AND METHODS
Ethics statement.
The procedures in this study and handling of the mice complied with current European legislation (directive 86/609/EEC) and the corresponding Belgian law Arrêté royal relatif à la protection des animaux d'expérience du 6 avril 2010 publié le 14 mai 2010 (Royal Decree on the protection of experimental animals of April 6, 2010, published on May 14, 2010). The Animal Welfare Committee of the Université de Namur (Namur, Belgium) has reviewed and approved the complete protocol (permit number 05-058).
Mice and reagents.
Wild-type C57BL/6 mice were purchased from Harlan (Bicester, United Kingdom) and were used as the control. IFN-γR−/− C57BL/6 mice and TNF-αR1−/− C57BL/6 mice were acquired from C. De Trez (Vrije Universiteit Brussel, Belgium). CD3ε−/− and IL-1R−/− C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All wild-type and deficient C57BL/6 mice used in this study were bred in the Gosselies animal facility of the Université Libre de Bruxelles (Brussels, Belgium). We used wild-type strains of Brucella melitensis 16M, Brucella abortus 2308, and Brucella suis 1330 and a VirB-deficient strain of Brucella melitensis 16M. Construction of the VirB mutant has been previously described (54). All strains were grown in biosafety level III laboratory facilities of the Université de Namur (Namur, Belgium). Cultures were grown overnight with shaking at 37°C in 2YT medium (Luria-Bertani broth with a double quantity of yeast extract) and were washed twice in RPMI 1640 medium (Gibco Laboratories) (with centrifugation at 3,500 × g for 10 min) before inoculation of the mice. To prepare heat-killed (HK) B. melitensis, bacteria from an overnight liquid culture in 2YT medium were washed twice in phosphate-buffered saline (PBS) (with centrifugation at 3,500 × g for 10 min) before being heated at 80°C for 1 h. To confirm the killing, an aliquot was plated onto 2YT medium.
Mouse infection and bacterial count.
Mice were injected intraperitoneally (i.p.) with 105 CFU of live Brucella melitensis, B. suis, or B. abortus or with 105 or 5 × 107 CFU of HK B. melitensis in 500 μl of PBS. For intranasal (i.n.) infection, mice were anesthetized with a mixture of xylazine (9 mg/kg) and ketamine (36 mg/kg) in PBS before being inoculated i.n. with a drop of 30 μl of PBS containing 105 CFU of live B. melitensis. Control animals were injected with the same volume of PBS. The infectious doses were validated by plating serial dilutions of the inocula. At selected time points to perform bacterial counts in the spleen, mice were sacrificed, and the spleens was harvested, crushed, and transferred to PBS–0.1% Triton X-100 (Sigma). We performed successive serial dilutions in RPMI medium to obtain the most accurate bacterial count and plated them on 2YT medium. The CFU were counted after 5 days of culture at 37°C.
Antibiotic treatment.
Antibiotic treatment was administered orally for 2 weeks. The treatment was a combination of rifampin (12 mg/kg) and streptomycin (450 mg/kg) prepared fresh daily and given in the drinking water. To ensure that the antibiotic treatment was effective, some mice from each group were sacrificed, and the CFU counts were evaluated in the spleen.
Flow cytometry analysis.
As described previously (55), spleens were harvested, cut into small pieces, and incubated for 30 min at 37°C with a mix of DNase I fraction IX (Sigma-Aldrich Chimie SARL, Lyon, France) (100 μg/ml) and 1.6 mg/ml of collagenase (400 U/ml). Spleen cells were washed and filtered and then incubated with saturating doses of purified 2.4G2 (anti-mouse Fc receptor; ATCC) in 200 μl of PBS, 0.2% bovine serum albumin (BSA), 0.02% NaN3 (fluorescence-activate cell sorting [FACS] buffer) for 20 min at 4°C to prevent antibody binding to the Fc receptor. Various fluorescent MAb combinations in FACS buffer were used to stain 3 × 106 to 5 × 106 cells, as follows: fluorescein (FITC)-coupled 145-2C11 (anti-CD3ε; BD Biosciences), FITC-coupled HL3 (anti-CD11c; BD Biosciences), phycoerythrin (PE)-coupled 53-6.7 (anti-CD8α; BD Biosciences), PE-coupled 1A8 (anti-Ly-6G; BD Biosciences), PE-coupled 1B1 (anti-CD1d; BD Biosciences), allophycocyanin (APC)-coupled M1/70 (anti-CD11b; BD Biosciences), APC-coupled RM4-5 (anti-CD4; BD Biosciences), APC-coupled RA3-6B2 (anti-CD45R/B220; BD Biosciences), APC-coupled BM8 (anti-F4/80; Abcam), biotin-coupled I-A/I-E (anti-MHC-II; BD Biosciences), biotin-coupled 1B1 (anti-CD1d; BD Biosciences), biotin-coupled AL-21 (anti-Ly-6C; BD Biosciences), and PE-coupled streptavidin (BD Biosciences). The cells were analyzed on a FACSCalibur flow cytometer. Dead cells and debris were eliminated from the analysis according to size and scatter.
Intracellular interferon gamma staining.
For intracellular interferon gamma staining, spleen cells were incubated after DNase and collagenase treatment for 4 h in RPMI 1640 medium and 10% fetal calf serum (FCS) with 1 ml/ml Golgi Stop (BD Pharmingen) at 37°C under 5% CO2. After cells were washed with FACS buffer and stained for cell surface markers, they were fixed in PBS–1% paraformaldehyde (PFA) for 15 to 20 min at 4°C. They were permeabilized for 30 min using a saponin-based buffer (103 Perm/Wash in FACS buffer; BD Pharmingen) and stained with allophycocyanin-coupled XMG1.2 (anti-IFN-γ; BD Biosciences). After final fixation in PBS–1% PFA, cells were analyzed on a FACSCalibur cytofluorometer. No signal was detectable with isotype controls.
Immunofluorescence microscopy.
Spleens were fixed for 2 h at room temperature in 2% paraformaldehyde (pH 7.4), washed in PBS, and incubated overnight at 4°C in a 20% PBS-sucrose solution. The tissues were then embedded in Tissue-Tek OCT compound (Sakura) and frozen in liquid nitrogen, and cryostat sections (thickness, 5 μm) were prepared. For the staining, tissue sections were rehydrated in PBS and incubated in a PBS solution containing 1% blocking reagent (Boeringer) (PBS–1% BR) for 20 min before being incubated overnight in PBS–1% BR containing the following MAbs or reagents: 4′,6′-diamidino-2-phenylindole (DAPI) for nucleic acid staining, Alexa Fluor 488 phalloidin (Molecular Probes) for actin staining to visualize the fibrous structure of the organ, and eFluor 570-coupled RA3-6B2 (anti-CD45R/B220; eBioscience), APC-coupled OX-7 (anti-CD90.1; eBioscience), Alexa Fluor 647-coupled BM8 (anti-F4/80; Abcam), Alexa Fluor 647-coupled HL3 (anti-CD11c; eBioscience), Alexa Fluor 647-coupled RB6-8C5 (anti-Gr-1; eBioscience), biotin-coupled MOMA1 (anti-marginal zone macrophages; BMA Biomedicals), and biotin-coupled ERTR9 (anti-SIGN-R1; BMA Biomedicals) to stain the cells of interest. Incubation with a streptavidin-coupled fluorochrome for 2 h was necessary for the biotin-coupled antibody (Ab): Alexa Fluor 350 or Alexa Fluor 647-streptavidin (Molecular Probes). Slides were mounted in Fluoro-Gel medium (Electron Microscopy Sciences, Hatfield, PA). Labeled tissue sections were visualized with an Axiovert M200 inverted microscope (Zeiss, Iena, Germany) equipped with a high-resolution monochrome camera (AxioCam HR, Zeiss). Images (1,384 by 1,036 pixels; 0.16 μm/pixel) were acquired sequentially for each fluorochrome with an A-Plan 10×/0.25, EC Plan-Neofluar 20×/0.50 Ph2 M27, and LD Plan-Neofluar 63×/0.75 Corr Ph2 dry objectives and recorded as eight-bit gray-level zvi files. The percentage of surface area associated with a signal was quantified using the colocalization module of the AxioVision program (Zeiss). At least three slides per organ were analyzed from three different animals, and the results are representative of two independent experiments.
Quantification of marginal zone macrophages in the spleen.
To quantify the number of both MZMs and MMMs around the white pulp area, frozen spleen sections were stained with specific markers (CD169, dextran-FITC, and CD209) (Fig. S1A in the supplemental material) as indicated previously. Entire tissue section surface areas were scanned automatically using a Zeiss Axiovert 200 fluorescent inverted microscope with a 20× objective and the MosaiX module. Within a same experiment, all images were acquired with the same parameters. Then, the images were processed with the NIS-Elements program (Nikon Instruments) to measure the percentage of positive staining per white pulp area. In practice, the experimenter drew an arbitrary line surrounding a white pulp in the marginal area (Fig. S1B), and the program calculated the total number of pixels per line and the number of pixels positive for the specific staining. This allowed us to calculate the percentage of positive staining per white pulp. Three arbitrary lines were drawn, and the percentage shown is the average of these measurements minus the background calculated from three measurements performed in a red pulp (negative) area. For each experimental condition, we analyzed at least 10 white pulp areas per frozen spleen section and at least seven frozen spleen sections from three individual mice.
Antigen, microsphere, and Salmonella capture assay.
To study the filtration capacity of the MZ, 200 μl of FITC-dextran (100 μg/ml, 500-kDa dextran; Molecular Probes), 200 μl of 0.05-μm Fluoresbrite YG (yellow green) microspheres (1.82 × 1013 beads/ml; Polysciences), or 108 CFU of live GFP-expressing Salmonella enterica serovar Typhimurium (GFP-Salmonella) bacteria containing the plasmid pFPV25.1-gfp in 200 µl of PBS were injected i.p., as indicated in the figure legends (56). S. Typhimurium was cultivated overnight with shaking at 37°C in Luria-Bertani broth and washed twice in RPMI 1640 medium (Gibco Laboratories) (centrifuged at 3,500 × g, 10 min) before it was injected intravenously into mice. Mice were sacrificed 20 min after injection. The spleen was harvested rapidly and treated for immunofluorescence microscopy as previously described.
RNA purification and real-time RT-PCR.
Spleen samples were collected and immediately frozen and stored at −80°C until processing. RNA was then extracted from total frozen tissue using a TriPure isolation reagent (Roche) according to the manufacturer's instructions. DNA contamination in the RNA samples was eliminated by incubation with DNase I (Fermentas). Reverse transcription (RT) and real-time PCRs were carried out using LightCycler 480 RNA master hydrolysis probes (one-step procedure) on a LightCycler 480 real-time PCR system (Roche Diagnostics). SYBR green PCR master mix kits (Roche) were used for quantification, and the specificity of the SYBR green assays was assessed by melting point analysis and gel electrophoresis. Results were normalized using the housekeeping gene β-actin. The primer sequences are presented in Table S1 in the supplemental materials.
Statistical analysis.
We used a (Wilcoxon-) Mann-Whitney test provided by the GraphPad Prism software to statistically analyze our results. Each group of deficient mice was individually compared to the wild-type mouse group. We also compared each group with each other and displayed the results when required. P values of <0.05 were considered to represent a significant difference.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Fonds National de la Recherche Scientifique (FRS-FNRS) (convention FRSM FNRS 3.4.600.06.F, Belgium) and the Communauté Française de Belgique (Action de Recherches Concertées 08/13–015) and by the Interuniversity Attraction Poles Programme initiated by the Belgian Science Policy Office. E.M. is a Research Associate from the FRS-FNRS (Belgium). A.M. holds Ph.D. grants from the FRIA (Belgium).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00115-17.
REFERENCES
- 1.Bronte V, Pittet MJ. 2013. The spleen in local and systemic regulation of immunity. Immunity 39:806–818. doi: 10.1016/j.immuni.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borges da Silva H, Fonseca R, Pereira RM, Cassado Ados A, Álvarez JM, D'Império Lima MR. 2015. Splenic macrophage subsets and their function during blood-borne infections. Front Immunol 6:480. doi: 10.3389/fimmu.2015.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang Y-S, Kim JY, Bruening SA, Pack M, Charalambous A, Pritsker A, Moran TM, Loeffler JM, Steinman RM, Park CG. 2004. The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc Natl Acad Sci U S A 101:215–220. doi: 10.1073/pnas.0307124101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang YS, Yamazaki S, Iyoda T, Pack M, Bruening SA, Kim JY, Takahara K, Inaba K, Steinman RM, Park CG. 2003. SIGN-R1, a novel C-type lectin expressed by marginal zone macrophages in spleen, mediates uptake of the polysaccharide dextran. Int Immunol 15:177–186. doi: 10.1093/intimm/dxg019. [DOI] [PubMed] [Google Scholar]
- 5.van der Laan LJ, Ea Döpp Haworth R, Pikkarainen T, Kangas M, Elomaa O, Dijkstra CD, Gordon S, Tryggvason K, Kraal G. 1999. Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J Immunol 162:939–947. [PubMed] [Google Scholar]
- 6.Seiler P, Aichele P, Odermatt B, Hengartner H, Zinkernagel RM, Schwendener RA. 1997. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol 27:2626–2633. doi: 10.1002/eji.1830271023. [DOI] [PubMed] [Google Scholar]
- 7.Aichele P, Zinke J, Grode L, Schwendener RA, Kaufmann SH, Seiler P. 2003. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J Immunol 171:1148–1155. doi: 10.4049/jimmunol.171.3.1148. [DOI] [PubMed] [Google Scholar]
- 8.Kraal G, Janse M. 1986. Marginal metallophilic cells of the mouse spleen identified by a monoclonal antibody. Immunology 58:665–669. [PMC free article] [PubMed] [Google Scholar]
- 9.Oetke C, Kraal G, Crocker PR. 2006. The antigen recognized by MOMA-I is sialoadhesin. Immunol Lett 106:96–98. doi: 10.1016/j.imlet.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Jones C, Virji M, Crocker PR. 2003. Recognition of sialylated meningococcal lipopolysaccharide by siglecs expressed on myeloid cells leads to enhanced bacterial uptake. Mol Microbiol 49:1213–1225. doi: 10.1046/j.1365-2958.2003.03634.x. [DOI] [PubMed] [Google Scholar]
- 11.Heikema AP, Bergman MP, Richards H, Crocker PR, Gilbert M, Samsom JN, van Wamel WJB, Endtz HP, van Belkum A. 2010. Characterization of the specific interaction between sialoadhesin and sialylated Campylobacter jejuni lipooligosaccharides. Infect Immun 78:3237–3246. doi: 10.1128/IAI.01273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monteiro VG, Lobato CS, Silva AR, Medina DV, de Oliveira MA, Seabra SH, de Souza W, DaMatta RA. 2005. Increased association of Trypanosoma cruzi with sialoadhesin positive mice macrophages. Parasitol Res 97:380–385. doi: 10.1007/s00436-005-1460-1. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Pomares L, Gordon S. 2012. CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol 33:66–70. doi: 10.1016/j.it.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Attanavanich K, Kearney JF. 2004. Marginal zone, but not follicular B cells, are potent activators of naive CD4 T cells. J Immunol 172:803–811. doi: 10.4049/jimmunol.172.2.803. [DOI] [PubMed] [Google Scholar]
- 15.Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, Chandler P, Tanaka M, Munn DH, Mellor AL, McGaha TL. 2014. Marginal zone CD169+ macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci 111:4215–4220. doi: 10.1073/pnas.1320924111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGaha TL, Chen Y, Ravishankar B, Van Rooijen N, Karlsson MCI. 2011. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood 117:5403–5412. doi: 10.1182/blood-2010-11-320028. [DOI] [PubMed] [Google Scholar]
- 17.Benedict CA, De Trez C, Schneider K, Ha S, Patterson G, Ware CF. 2006. Specific remodeling of splenic architecture by cytomegalovirus. PLoS Pathog 2:e16. doi: 10.1371/journal.ppat.0020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, Finke D, Luther SA, Junt T, Ludewig B. 2008. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat Immunol 9:667–675. doi: 10.1038/ni.1605. [DOI] [PubMed] [Google Scholar]
- 19.Engwerda CR, Ato M, Cotterell SE, Mynott TL, Tschannerl A, Gorak-Stolinska PM, Kaye PM. 2002. A role for tumor necrosis factor-alpha in remodeling the splenic marginal zone during Leishmania donovani infection. Am J Pathol 161:429–437. doi: 10.1016/S0002-9440(10)64199-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho LJM, Ferreira-da-Cruz MF, Daniel-Ribeiro CT, Pelajo-Machado M, Lenzi HL. 2007. Germinal center architecture disturbance during Plasmodium berghei ANKA infection in CBA mice. Malar J 6:59. doi: 10.1186/1475-2875-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosche KL, Aljasham AT, Kipfer JN, Piatkowski BT, Konjufca V. 2015. Infection with Salmonella enterica serovar Typhimurium leads to increased proportions of F4/80+ red pulp macrophages and decreased proportions of B and T lymphocytes in the spleen. PLoS One 10:e0130092. doi: 10.1371/journal.pone.0130092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ato M, Nakano H, Kakiuchi T, Kaye PM. 2004. Localization of marginal zone macrophages is regulated by C-C chemokine ligands 21/19. J Immunol 173:4815–4820. doi: 10.4049/jimmunol.173.8.4815. [DOI] [PubMed] [Google Scholar]
- 23.Godfroid J, Cloeckaert A, Liautard JP, Kohler S, Fretin D, Walravens K, Garin-Bastuji B, Letesson JJ. 2005. From the discovery of the Malta fever's agent to the discovery of a marine mammal reservoir, brucellosis has continuously been a re-emerging zoonosis. Vet Res 36:313–326. doi: 10.1051/vetres:2005003. [DOI] [PubMed] [Google Scholar]
- 24.Moreno E. 2014. Retrospective and prospective perspectives on zoonotic brucellosis. Front Microbiol 5:213. doi: 10.3389/fmicb.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martirosyan A, Gorvel J-P. 2013. Brucella evasion of adaptive immunity. Future Microbiol 8:147–154. doi: 10.2217/fmb.12.140. [DOI] [PubMed] [Google Scholar]
- 26.Copin R, Vitry M-A, Hanot Mambres D, Machelart A, De Trez C, Vanderwinden J-M, Magez S, Akira S, Ryffel B, Carlier Y, Letesson J-J, Muraille E. 2012. In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog 8:e1002575. doi: 10.1371/journal.ppat.1002575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Copin R, De Baetselier P, Carlier Y, Letesson J-J, Muraille E. 2007. MyD88-dependent activation of B220-CD11b+ LY-6C+ dendritic cells during Brucella melitensis infection. J Immunol 178:5182–5191. doi: 10.4049/jimmunol.178.8.5182. [DOI] [PubMed] [Google Scholar]
- 28.Aoshi T, Carrero JA, Konjufca V, Koide Y, Unanue ER, Miller MJ. 2009. The cellular niche of Listeria monocytogenes infection changes rapidly in the spleen. Eur J Immunol 39:417–425. doi: 10.1002/eji.200838718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salcedo S, Noursadeghi M, Cohen J, Holden D. 2001. Intracellular replication of Salmonella typhimurium strain in specific subsets of splenic macrophages in vivo. Cell Microbiol 3:587–597. doi: 10.1046/j.1462-5822.2001.00137.x. [DOI] [PubMed] [Google Scholar]
- 30.Backer R, Schwandt T, Greuter M, Oosting M, Jüngerkes F, Tüting T, Boon L, O'Toole T, Kraal G, Limmer A, den Haan JMM. 2010. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc Natl Acad Sci 107:216–221. doi: 10.1073/pnas.0909541107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, Masopust D. 2015. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161:737–749. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Rooijen N, Kors N, Kraal G. 1989. Macrophage subset repopulation in the spleen: differential kinetics after liposome-mediated elimination. J Leukoc Biol 45:97–104. [DOI] [PubMed] [Google Scholar]
- 33.Hanot Mambres D, Machelart A, Potemberg G, De Trez C, Ryffel B, Letesson J-J, Muraille E. 2016. Identification of immune effectors essential to the control of primary and secondary intranasal infection with Brucella melitensis in mice. J Immunol 196:3780–3793. doi: 10.4049/jimmunol.1502265. [DOI] [PubMed] [Google Scholar]
- 34.Martirosyan A, Von Bargen K, Arce Gorvel V, Zhao W, Hanniffy S, Bonnardel J, Méresse S, Gorvel JP. 2013. In vivo identification and characterization of CD4+ cytotoxic T cells induced by virulent Brucella abortus infection. PLoS One 8:e82508. doi: 10.1371/journal.pone.0082508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitry M-A, De Trez C, Goriely S, Dumoutier L, Akira S, Ryffel B, Carlier Y, Letesson J-J, Muraille E. 2012. Crucial role of gamma interferon-producing CD4+ Th1 cells but dispensable function of CD8+ T cell, B cell, Th2, and Th17 responses in the control of Brucella melitensis infection in mice. Infect Immun 80:4271–4280. doi: 10.1128/IAI.00761-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller SN, Hosiawa-Meagher KA, Konieczny BT, Sullivan BM, Bachmann MF, Locksley RM, Ahmed R, Matloubian M. 2007. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science 317:670–674. doi: 10.1126/science.1144830. [DOI] [PubMed] [Google Scholar]
- 37.Matter MS, Hilmenyuk T, Claus C, Marone R, Schürch C, Tinguely M, Terracciano L, Luther SA, Ochsenbein AF. 2011. Destruction of lymphoid organ architecture and hepatitis caused by CD4+ T cells. PLoS One 6:e24772. doi: 10.1371/journal.pone.0024772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.den Hartigh AB, Sun Y, Sondervan D, Heuvelmans N, Reinders MO, Ficht TA. 2004. Differential requirements for VirB1 and VirB2 during Brucella abortus infection. Infect Immun 72:5143–5149. doi: 10.1128/IAI.72.9.5143-5149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rolan HG, Tsolis M. 2008. Inactivation of the type IV secretion system reduces the Th1 polarization of the immune response to Brucella abortus infection. Infect Immun 76:3207–3213. doi: 10.1128/IAI.00203-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chain PS, Comerci DJ, Tolmasky ME, Larimer FW, Malfatti SA, Vergez LM, Aguero F, Land ML, Ugalde RA, Garcia E. 2005. Whole-genome analyses of speciation events in pathogenic brucellae. Infect Immun 73:8353–8361. doi: 10.1128/IAI.73.12.8353-8361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Audic S, Lescot M, Claverie J-M, Scholz HC. 2009. Brucella microti: the genome sequence of an emerging pathogen. BMC Genomics 10:352. doi: 10.1186/1471-2164-10-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corsetti PP, de Almeida LA, Carvalho NB, Azevedo V, Silva TM, Teixeira HC, Faria AC, Oliveira SC. 2013. Lack of endogenous IL-10 enhances production of proinflammatory cytokines and leads to Brucella abortus clearance in mice. PLoS One 8:e74729. doi: 10.1371/journal.pone.0074729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baldi PC, Giambartolomei GH. 2013. Immunopathology of Brucella infection. Recent Pat Antiinfect Drug Discov 8:18–26. doi: 10.2174/1574891X11308010005. [DOI] [PubMed] [Google Scholar]
- 44.Minihane AM, Vinoy S, Russell WR, Baka A, Roche HM, Tuohy KM, Teeling JL, Blaak EE, Fenech M, Vauzour D, McArdle HJ, Kremer BHA, Sterkman L, Vafeiadou K, Benedetti MM, Williams CM, Calder PC. 2015. Low-grade inflammation, diet composition and health: current research evidence and its translation. Br J Nutr 114:999–1012. doi: 10.1017/S0007114515002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiba T, Marusawa H, Ushijima T. 2012. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 143:550–563. doi: 10.1053/j.gastro.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 46.Hanot Mambres D, Machelart A, Vanderwinden JM, De Trez C, Ryffel B, Letesson JJ, Muraille E. 2015. In situ characterization of splenic Brucella melitensis reservoir cells during the chronic phase of infection in susceptible mice. PLoS One 10:e0137835. doi: 10.1371/journal.pone.0137835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang L, Krieg AM, Eller N. 1999. Induction and regulation of Th1-inducing cytokines by bacterial DNA, lipopolysaccharide, and heat-inactivated bacteria. Infect Immun 67:6257–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buiting AM, De Rover Z, Van Rooijen N. 1995. Brucella abortus causes an accelerated repopulation of the spleen and liver of mice by macrophages after their liposome-mediated depletion. J Med Microbiol 42:133–140. doi: 10.1099/00222615-42-2-133. [DOI] [PubMed] [Google Scholar]
- 49.van Rooijen N, van Kesteren-Hendrikx E. 2002. Clodronate liposomes: perspectives in research and therapeutics. J Liposome Res 12:81–94. doi: 10.1081/LPR-120004780. [DOI] [PubMed] [Google Scholar]
- 50.Mueller SN, Germain RN. 2009. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol 9:618–629. doi: 10.1038/nri2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nolte MA, Arens R, Kraus M, van Oers MHJ, Kraal G, van Lier RAW, Mebius RE. 2004. B Cells are crucial for both development and maintenance of the splenic marginal zone. J Immunol 172:3620–3627. doi: 10.4049/jimmunol.172.6.3620. [DOI] [PubMed] [Google Scholar]
- 52.Muraille E. 2015. The unspecific side of acquired immunity against infectious disease: causes and consequences. Front Microbiol 6:1525. doi: 10.3389/fmicb.2015.01525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. 2006. The new global map of human brucellosis. Lancet Infect Dis 6:91–99. doi: 10.1016/S1473-3099(06)70382-6. [DOI] [PubMed] [Google Scholar]
- 54.Nijskens C, Copin R, De Bolle X, Letesson JJ. 2008. Intracellular rescuing of a B. melitensis 16M virB mutant by coinfection with a wild type strain. Microb Pathog 45:134–141. doi: 10.1016/j.micpath.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 55.Vitry M-A, Hanot Mambres D, De Trez C, Akira S, Ryffel B, Letesson J-J, Muraille E. 2014. Humoral immunity and CD4+ Th1 cells are both necessary for a fully protective immune response upon secondary infection with Brucella melitensis. J Immunol 192:3740–3752. doi: 10.4049/jimmunol.1302561. [DOI] [PubMed] [Google Scholar]
- 56.Valdivia RH, a Hromockyj E, Monack D, Ramakrishnan L, Falkow S. 1996. Applications for green fluorescent protein (GFP) in the study of host-pathogen interactions. Gene 173:47–52. doi: 10.1016/0378-1119(95)00706-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.