

Abstract
Periodontitis is an oral chronic infection/inflammatory condition, identified as a source of mediators of inflammation into the blood circulation, which may contribute to exacerbate several diseases. There is increasing evidence that inflammation plays a key role in the pathophysiology of Alzheimer’s disease (AD). Although inflammation is present in both diseases, the exact mechanisms and crosslinks between periodontitis and AD are poorly understood. Therefore, this article aims to review possible comorbidity between periodontitis and AD. Here, the authors discuss the inflammatory aspects of periodontitis, how this oral condition produces a systemic inflammation and, finally, the contribution of this systemic inflammation for worsening neuroinflammation in the progression of AD.
Keywords: Alzheimer disease, amyloid beta-peptides, dementia, inflammation, neurodegenerative diseases, neurofibrillary tangles, periodontal diseases, periodontitis
Introduction
Life expectancy has increased considerably over the last three decades and the number of cases of dementia has expanded within aging population (Norton et al., 2014). Recent epidemiological projections indicate that the number of patients with dementia will more than triplicate by 2050 in comparison to 2010, and most of the cases of dementia are associated to Alzheimer’s disease (AD; Barnes and Yaffe, 2011; Norton et al., 2013, 2014).
Despite great efforts of researchers and clinicians, no effective disease-modifying drug has been approved for AD treatment to date (Norton et al., 2014; Bateman, 2015). Therefore, there is an increasing interest in identifying modifiable risk factors for dementia and AD, aiming to develop preventive strategies that could lower dementia prevalence over the next few years (Barnes and Yaffe, 2011; Norton et al., 2014; Deckers et al., 2015). The main identified preventable risk factors for AD worldwide are: low education attainment, smoking, physical inactivity, depression, midlife hypertension, diabetes mellitus and mild-life obesity (Norton et al., 2014). Taken together, these seven modifiable risk factors may contribute to 30%–50% of AD cases (Barnes and Yaffe, 2011; Norton et al., 2014).
Since the 1990s, it has been proposed that the brain innate immune response plays an important role in the development and progression of the neurodegeneration in AD (McGeer et al., 1990; McGeer and McGeer, 1995). Such response is characterized by the presence of activated microglia, the resident immune cells of the brain, which increase in cell density and undergoes changes in morphology and in the expression of surface antigens (Perry et al., 2007). Indeed, a number of clinical and experimental studies demonstrated the importance of systemic peripheral inflammation or infection as pivotal contributors to the pathophysiology of AD, supporting a bi-directional communication between brain and peripheral immune systems (Perry et al., 2007; Perry and Teeling, 2013). More recently, it has been proposed that peripheral inflammation/infection may be not just a contributor but indeed a key determinant of the cognitive decline associated to AD progression (Cunningham and Hennessy, 2015).
Interestingly, a considerable worldwide population is affected by periodontal diseases, that are oral infections that affect teeth supporting tissues (Dye, 2012; Eke et al., 2015; Oppermann et al., 2015). They can be classified as gingivitis, when the inflammation is localized in the gingival tissues, or it may assume a more severe destructive form, with the inflammatory process reaching deeper connective and bone tissue, causing bone and attachment loss, that may ultimately lead to tooth loss (Armitage, 1999; Kamer et al., 2008). This local inflammatory process may induce a systemic inflammatory state through mechanisms that include dissemination of pro-inflammatory cytokine and/or bacteria from the oral to extra-oral sites (Hajishengallis, 2015). Therefore, periodontitis could trigger and/or exacerbate an inflammatory condition especially in elderly subjects, leading to memory impairments, contributing to accelerate the progression of neurodegenerative diseases such as AD.
This review will discuss the findings that may clarify the influence of periodontitis on the magnitude of the neuroinflammatory status as well as to highlight experimental and clinical findings indicating a possible comorbidity between periodontitis and AD.
Role of Neuroinflammation in The Pathogenesis of Alzheimer’s Disease
The pathological hallmarks of AD include the synaptic loss and the presence of senile plaques and neurofibrillary tangles. The senile plaques are primarily composed of β-amyloid (Aβ) peptide, which is a 39–43 amino acidic peptide formed upon proteolytic processing, by β- and γ-secretases (Haass and De Strooper, 1999), of the larger amyloid precursor protein (APP), a ubiquitously expressed transmembrane glycoprotein. The Aβ cascade hypothesis in AD pathogenesis postulates that increased accumulation of Aβ appears to be related to a gradual synaptic loss and neuronal death finally leading to cognitive impairments (Selkoe, 1991; Hsiao et al., 1996; Hardy and Selkoe, 2002).
A contribution of neuroinflammation to the pathogenesis of AD has been pointed since complement factors surrounding senile plaques were observed in post mortem brain tissue from AD patients (Eikelenboom and Stam, 1982; McGeer and McGeer, 2013). Further support to this idea came from a number of epidemiological studies indicating that chronic use of nonsteroidal anti-inflammatory drugs reduces the risk of developing AD (McGeer et al., 1990; in ’t Veld et al., 2001; McGeer and McGeer, 2013), and that several inflammatory mediators are elevated in the brain and cerebrospinal fluid of AD patients (Heneka et al., 2015a). The traditional view postulates that Aβ deposits or oligomers trigger the recruitment and activation of microglia, which release inflammatory mediators that aggravate an already ongoing neurodegenerative process. However, some authors argue that neuroinflammation has a more central role to the pathogenesis of the disease than previously considered (Holmes, 2013; Heneka et al., 2015a,b).
Another important updated concept is the fact that the central nervous system (CNS) is not an immune-isolated environment, since there is converging evidence of a bidirectional cross-talk between the brain and the peripheral immune system (Holmes, 2013; Perry and Teeling, 2013). The peripheral systemic inflammation may contribute not only to aggravate the progression of neurodegeneration in AD, but could also play a fundamental role in the development of the disease (Holmes, 2013; VanItallie, 2017). The main underlying mechanism is the “priming” of microglia, which postulates that microglia acquire a “primed” phenotype, ready to express a damaging pro-inflammatory response to further insults. Microglia priming could be initially triggered by a number of conditions such as: (i) aging; (ii) proteins associated to AD pathogenesis (e.g., Aβ, tau); and (iii) systemic inflammation (see Figure 1). Further events of systemic inflammation would switch primed microglia to an aggressive pro-inflammatory phenotype contributing to aggravate neuroinflammation and neurodegeneration (Perry and Teeling, 2013). Of note, at least five of the main preventable risk factors for AD, e.g., smoking, depression, hypertension, diabetes mellitus and obesity, have a common association with a systemic pro-inflammatory phenotype, giving further support to the hypothesis that systemic inflammation may play a fundamental role in the development and progression of AD (Holmes, 2013; VanItallie, 2017).
Figure 1.
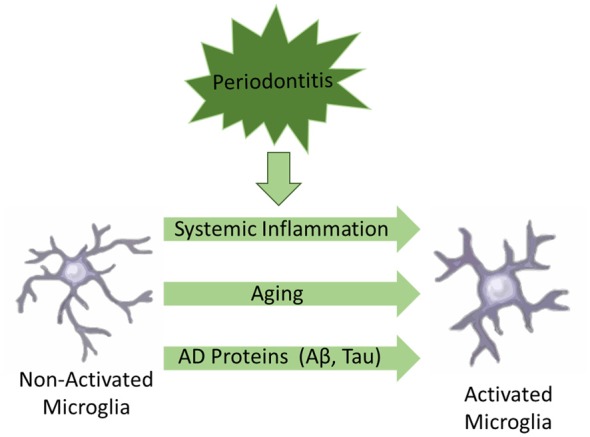
Schematic illustration of factors associated to microglial activation in Alzheimer’s disease (AD).
It is important to note that chronic activation of the complement system is also associated with AD (Fischer et al., 1995; Fonseca et al., 2016). Proteins associated to AD can activate complement and recruit activated glia (astrocytes and microglia) to the amyloid plaque, that secrete proinflammatory cytokines or other toxic mediators that could play a role in neuronal degeneration in AD (Fischer et al., 1995; Fonseca et al., 2016).
Preclinical studies have evaluated systemic inflammation by peripheral administration of lipopolysaccharide (LPS) in animals as a model of bacterial infection (Sly et al., 2001; Godbout et al., 2005). It has been observed that peripheral administration of LPS results in increased levels of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) in both periphery and the brain (Godbout et al., 2005; Teeling and Perry, 2009), alters the blood-brain barrier (BBB) transport of Aβ protein (Jaeger et al., 2009), increasing the brain levels of Aβ (Sly et al., 2001; Jaeger et al., 2009). This inflammatory response may predispose to neurodegenerative diseases (Godbout et al., 2005) or even amplify the ongoing neurodegenerative process (Sly et al., 2001). In aged laboratory animals, an exacerbated neuroinflammatory response is associated with sickness behavior including depressive-like behavior and cognitive deficits, which are also observed in AD (Godbout et al., 2005). In a prospective longitudinal study, it was observed that increased levels of serum TNF-α following that both acute and chronic systemic inflammation is associated with cognitive decline in AD patients (Holmes et al., 2009).
Some potential crosstalk sites between periphery and the brain are: vagal afferents; structures lacking the BBB such as the circumventricular organs; direct effects on vascular endothelial cells at the BBB; and entry of peripheral immune cells into the brain (Teeling and Perry, 2009; Holmes, 2013). Once in the brain, peripheral inflammatory signaling molecules stimulate microglia, which produce more pro-inflammatory cytokines. In young healthy brains, microglia activation is associated with a reparative inflammatory response, which suppresses the initial pro-inflammatory response. In contrast, in aged and/or diseased brains, where microglia has been already primed, an exacerbated inflammatory response takes place, which accelerates cognitive decline (Holmes, 2013; Cunningham and Hennessy, 2015).
Lastly, the regulatory mechanisms of entry and exit of immune cells from the CNS remain poorly understood (Louveau et al., 2015). The increasing knowledge of the brain lymphatic system may lead to a new point of view of this field in neuroimmunology and bring prospects on the etiology of neuroinflammatory and neurodegenerative diseases related with immune system dysfunctions (Berton et al., 2015). Dysfunctions of the meningeal lymphatic vessels may be the cause of a wide range of neurological disorders, in which altered immunity is a prevalent aspect, such as multiple sclerosis and AD (Akiyama et al., 2000; Berton et al., 2015).
Contribution of Periodontitis for Neuroinflammation and Diseases
Comorbidity between periodontal disease and AD has been reported in two front lines. The first line of evidence is that AD patients have greater impairment of oral health because of their progressive cognitive impairment, which would affect their oral hygiene habits (Kamer et al., 2008; Mancini et al., 2010; Gaur and Agnihotri, 2015). The second one is that uncontrolled periodontal disease could trigger or exacerbate neuroinflammatory phenomenon observed in AD (Kamer et al., 2008; Teixeira et al., 2014; Gaur and Agnihotri, 2015). However, it must be conceded that interventional studies reporting a direct association between periodontitis and AD are still missing.
Periodontitis is a chronic inflammatory disease, initiated by gram-negative bacteria that trigger host immuno-inflammatory response leading to tooth apparatus injury (Page and Kornman, 1997; Page, 1998; Watts et al., 2008; Gaur and Agnihotri, 2015). It is clinically characterized by bleeding on probing and clinical attachment loss (CAL; Armitage, 1999). The gums are usually swollen and discolored, dental calculus is frequently found on compromised teeth (Friedewald et al., 2009; Figure 2A), and tooth loss can result if left untreated or after unsatisfactory response to treatment (Page and Kornman, 1997; Figure 2B). Individuals with periodontitis are usually asymptomatic, except when acute processes occur, such as abscess and necrotizing periodontal diseases (Friedewald et al., 2009). Therefore, despite its high prevalence in the adult population of both developed (Eke et al., 2015) and developing countries (Oppermann et al., 2015), periodontitis is usually an unrecognized disease by both patients and health professionals.
Figure 2.

(A) Teeth from a chronic periodontal patient presented dental calculus, gingival recession and attachment loss. (B) Molar tooth showed in (A) extracted due to advanced periodontal disease involvement.
In periodontitis, the exacerbated host inflammatory response is associated with greater amount of tissue damage (Page and Kornman, 1997; Kamer et al., 2008). A systemic host immune-inflammatory response against periodontal pathogens is indicated by the presence of antibodies against periodontal pathogens, such as Porphyromonas gingivalis, Tannerella forsythia and Treponema denticola (Poole et al., 2013; Noble et al., 2014). Additionally, patients diagnosed with periodontitis present higher levels of inflammatory mediators in serum, such as C-reactive protein (CRP) in patients with chronic periodontitis (Bansal et al., 2014; Ardila and Guzmán, 2015), and leptin in patients with aggressive periodontitis (Shi et al., 2015). It has been suggested that the periodontal infection and the immuno-inflammatory response against periodontal pathogens may increase the host susceptibility to systemic diseases, including osteoporosis (Martelli et al., 2017), diabetes mellitus (Hanes and Krishna, 2010; Otomo-Corgel et al., 2012; Preshaw et al., 2012), cancer (Martelli et al., 2017), autoimmunity and cardiovascular disease (Page, 1998; Friedewald et al., 2009; Pejcic et al., 2011; Otomo-Corgel et al., 2012; Martelli et al., 2017), dementia (Pazos et al., 2016) and neurodegenerative diseases such as AD (Kamer et al., 2008, 2015; Rogers, 2008; Kubota et al., 2014; Gaur and Agnihotri, 2015; Ganesh et al., 2017; Sochocka et al., 2017a).
It is worth mentioning that a wrong clinical diagnosis of AD can generate other confounding factors. Vascular dementia, a subtype of dementia, such as AD (Appleton et al., 2017), may also be modulated indirectly by periodontitis, since periodontitis may be associated with clinical signs of atherosclerosis (Nakib et al., 2004; Yang et al., 2013; Etemadifar et al., 2015; Ahn et al., 2016), and atherosclerosis contribute to the development of vascular dementia (Appleton et al., 2017).
Periodontitis and Alzheimer’s Disease
This possible comorbity between periodontitis and AD has been indicated by clinical studies comparing the presence of periodontitis in individuals with and without AD (Stein et al., 2012; Martande et al., 2014; Noble et al., 2014; Cestari et al., 2016). One of these studies described an association between inflammatory cytokine levels in patients with AD and periodontitis, suggesting that periodontitis may be associated with onset, progression and aggravation of AD (Cestari et al., 2016). Similar results were described for serum IgG antibody levels to bacteria associated with periodontitis, observing an increasing incident AD onset/progression among participants with high serum antibody (Stein et al., 2012; Noble et al., 2014). When comparing periodontal health status, individuals with AD present a worsening of the condition with the progression of periodontitis, in which the probing depth and clinical attachment level, clinical parameters for periodontitis, were much higher in AD groups when compared to individuals without AD (Martande et al., 2014).
Periodontal pathogens and the immuno-inflammatory host response in periodontitis may affect the brain function, especially in more vulnerable elderly subjects, and may contribute to onset and progression of neurodegenerative disorders (Kamer et al., 2009). Some putative mechanisms that could explain how periodontitis may affect the homeostasis of the CNS have described by experimental studies and include: (i) translocation of bacteria into blood stream (bacteremia) or invasion into the brain via trigeminal nerve (e.g., Porphyromonas gingivalis); and (ii) production of pro-inflammatory cytokines that enter into the blood stream and act systemically or that reach the brain via peripheral nerves pathway (Gurav, 2014; Abbayya et al., 2015; Cerajewska et al., 2015; Gaur and Agnihotri, 2015; Olsen et al., 2016; Ganesh et al., 2017; Nezu et al., 2017; Sochocka et al., 2017a,b).
The first hypothesis relies on the fact that the microorganisms located on the dental biofilm can infiltrate in the brain by blood stream or peripheral nerves, mainly by trigeminal nerves (Riviere et al., 2002). Approximately 85% of the subgingival biofilm is composed by LPS-containing Gram-negative bacteria (Socransky and Haffajee, 2002). Such microorganisms and their immunogenic compounds in certain concentrations can trigger an inflammatory process in the CNS (Abbayya et al., 2015). This inflammatory process is a classic immune response similar in some aspects to that observed in AD, through TLR-2 and TLR-4 pathway, also related to cytokines interactions (including interleukins, TNF-α, transforming growth factor-β) and chemokines (monocyte chemotactic protein, IL-8, macrophage migration inhibitory factor and monokine induced by γ-interferon) released by neurons and glial cells. In addition, it is also observed increased production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) as well as complement system activation. The association of these factors triggers mechanisms of cell death and increases chronic inflammation already established by resident diseases or contribute to the development of new pathologies (Akiyama et al., 2000; Laflamme and Rivest, 2001; Gasque, 2004; Olson and Miller, 2004; Qin et al., 2005; Weller et al., 2009). Such hypothesis is supported by findings from independent research groups showing that peripheral infections can cross over the CNS (MacIntyre et al., 2003; Miklossy et al., 2006; Hammond et al., 2010). Of high interest, LPS has been linked to increased neuronal Aβ peptides levels and consequent disruption of BBB permeability and brain damage in animal models of AD (Lee et al., 2008; Jaeger et al., 2009).
Besides the inflammatory process generated by bacteria infection, some authors claim that there are subjects with “inflammatory traits” inherited which are similar to a vulnerability of the development of the neuroinflammatory diseases and the bacteria infection on the CNS may trigger the exaggerated innate immune response (van Exel et al., 2009; Singhrao et al., 2015; Olsen et al., 2016). Interestingly, periodontitis has been described to increase the gravity of AD in Down syndrome (DS) subjects (Kamer et al., 2016).
Although the access of periodontal bacteria into the neural parenchyma may be a factor which contributes to the aggravation and acceleration of the AD progression, the neuroinflammatory mechanisms from a systemic inflammatory response triggered by periodontitis has been more consistent in the literature. As illustrated in Figure 3, several studies have shown a positive relationship between CRP blood levels and periodontitis (Ebersole et al., 1997; Fredriksson et al., 1999; Loos et al., 2000; Noack et al., 2001; Slade et al., 2003). CRP is a plasma protein that participate in the systemic response to inflammation and it is regulated by cytokines like IL-6, IL-1β and TNF-α, that trigger the production by hepatocytes (Craig et al., 2003), in which have been reported as a sensible marker of systemic inflammation (Barrientos et al., 2010). Additionally, increased levels of TNF-α in the systemic circulation of AD patients has been related to the presence of periodontopathogenic microorganisms (i.e., Aggregatibacter actinomycetemcomitans, Tannerella forsythia and Porphyromonas gingivalis) as well as the antibodies against such pathogens (Kamer et al., 2009; Olsen et al., 2016).
Figure 3.

Pathogenesis of AD and periodontal disease and their relationship. AB, antibody; Aβ, β amyloid protein; BoP, bleeding on probing; CAL, clinical attachment loss; CRP, C-reactive protein; IL, interleukin; MMP, matrix metalloproteinase; PG, prostaglandin; TNF-α, tumor necrosis factor-α.
Among the pathogens related to periodontitis, the P. gengivalis has been related to the levels of CRP found in the aged patients (Winning et al., 2015). Such relationship among this pathogen and the high levels of CRP clarify the positive correlation already described in a study, in which the patients with severe periodontitis have increased serum levels of CRP when compared with unaffected control population (Gomes-Filho et al., 2011). Besides, the level of CRP increases subsequently with the severity of the periodontal disease (Bansal et al., 2014).
In fact, a recent study showed a positive association between periodontal disease and brain Aβ load in humans (Kamer et al., 2015). These findings are consistent with previous animal studies data showing that peripheral inflammation/infections are sufficient to induce brain Aβ accumulation (Kamer et al., 2015). Moreover, the periodontal health status of individuals with AD deteriorates with disease progression and is closely related to their cognitive function (Martande et al., 2014; Sochocka et al., 2017b) and emotional disorders (Kiecolt-Glaser et al., 2002). Corroborating these findings, another study revealed a significant increase in the serum levels of TNF-α in patients with AD and chronic periodontitis in comparison to patients with AD and healthy periodontium (Farhad et al., 2014).
Abe et al. (2011) described increased levels of Aβ precursor protein (APP) in patients with chronic periodontitis. APP is recognized to play an important role in the pathophysiology of AD, increasing the accumulation of Aβ in the CNS (Otsuka et al., 1991) and can directly be modulated by NF-κB (Chami et al., 2015) and TNF-α (Keller et al., 2013), that are also elevated in periodontal diseases.
Considering the increasing recognition of periodontitis as an environmental modifiable factor for AD, recently it has been proposed that the adequate treatment or prevention of periodontal disease may represent a valuable strategy to prevent (or delay) AD development as well as to counteract AD progression (Kamer et al., 2016). However, it is also important to consider that mild systemic inflammatory response, as caused by periodontitis, before to an injury to CNS, may exert a neuroprotective effect in a rat model of ischemic stroke, minimizing the inflammatory response which usually occurs in response to stroke (Petcu et al., 2008).
Final Considerations
The findings reviewed here clearly pointed inflammation as an important role in both periodontitis and AD. Since periodontitis is a preventable and treatable factor, subjects diagnosed with periodontitis should be informed and treated in an effort to diminish the microbial challenge and the pro-inflammatory cytokines hyper-production, aiming to promote a better quality of life, especially in elderly period. More importantly, despite the existence of clinical studies indicating the comorbidity of periodontitis and AD and the identification of serum antibodies to periodontal pathogens in AD, there is no study showing clearly the causal link between periodontitis and AD.
Author Contributions
All authors wrote the manuscript. MTS graphed the illustration. All authors discussed and edited the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
FBT and FCM are supported by fellowships form the Brazilian Government/Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). RDP is supported by a research fellowship from CNPq. The authors would like to thank the CNPq and PROPESP—UFPA for providing financial support. The funders had no role in study design, literature review, decision to publish, or preparation of the manuscript.
References
- Abbayya K., Puthanakar N. Y., Naduwinmani S., Chidambar Y. S. (2015). Association between periodontitis and Alzheimer’s disease. N. Am. J. Med. Sci 7, 241–246. 10.4103/1947-2714.159325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe D., Kubota T., Morozumi T., Shimizu T., Nakasone N., Itagaki M., et al. (2011). Altered gene expression in leukocyte transendothelial migration and cell communication pathways in periodontitis-affected gingival tissues. J. Periodont. Res. 46, 345–353. 10.1111/j.1600-0765.2011.01349.x [DOI] [PubMed] [Google Scholar]
- Ahn Y. B., Shin M. S., Han D. H., Sukhbaatar M., Kim M. S., Shin H. S., et al. (2016). Periodontitis is associated with the risk of subclinical atherosclerosis and peripheral arterial disease in Korean adults. Atherosclerosis 251, 311–318. 10.1016/j.atherosclerosis.2016.07.898 [DOI] [PubMed] [Google Scholar]
- Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G. M. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. 10.1016/S0197-4580(00)00124-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton J. P., Scutt P., Sprigg N., Bath P. M. (2017). Hypercholesterolaemia and vascular dementia. Clin. Sci. (Lond) 131, 1561–1578. 10.1042/CS20160382 [DOI] [PubMed] [Google Scholar]
- Ardila C. M., Guzmán I. C. (2015). Comparison of serum amyloid A protein and C-reactive protein levels as inflammatory markers in periodontitis. J. Periodontal Implant Sci. 45, 14–22. 10.5051/jpis.2015.45.1.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage G. C. (1999). Development of a classification system for periodontal diseases and conditions. Ann. Periodontol. 4, 1–6. 10.1902/annals.1999.4.1.1 [DOI] [PubMed] [Google Scholar]
- Bansal T., Dhruvakumar D., Pandey A. (2014). Comparative evaluation of C-reactive protein in peripheral blood of patients with healthy gingiva, gingivitis and chronic periodontitis: a clinical and particle-enhanced turbidimetric immuno-analysis. J. Indian Soc. Periodontol. 18, 739–743. 10.4103/0972-124X.147410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes D. E., Yaffe K. (2011). The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 10, 819–828. 10.1016/S1474-4422(11)70072-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos R. M., Frank M. G., Watkins L. R., Maier S. F. (2010). Memory impairments in healthy aging: role of aging-induced microglial sensitization. Aging Dis. 1, 212–231. [PMC free article] [PubMed] [Google Scholar]
- Bateman R. (2015). Alzheimer’s disease and other dementias: advances in 2014. Lancet Neurol. 14, 4–6. 10.1016/S1474-4422(14)70301-1 [DOI] [PubMed] [Google Scholar]
- Berton M., Lorette G., Baulieu F., Lagrue E., Blesson S., Cambazard F., et al. (2015). Generalized lymphedema associated with neurologic signs (GLANS) syndrome: a new entity? J. Am. Acad. Dermatol. 72, 333–339. 10.1016/j.jaad.2014.10.033 [DOI] [PubMed] [Google Scholar]
- Cerajewska T. L., Davies M., West N. X. (2015). Periodontitis: a potential risk factor for Alzheimer’s disease. Br. Dent. J. 218, 29–34. 10.1038/sj.bdj.2014.1137 [DOI] [PubMed] [Google Scholar]
- Cestari J. A., Fabri G. M., Kalil J., Nitrini R., Jacob-Filho W., Tesseroli de Siqueira J. T., et al. (2016). Oral infections and cytokine levels in patients with Alzheimer’s disease and mild cognitive impairment compared with controls. J. Alzheimers Dis. 54:845. 10.3233/JAD-169006 [DOI] [PubMed] [Google Scholar]
- Chami L., Buggia-Prévot V., Duplan E., Del Prete D., Chami M., Peyron J. F., et al. (2015). Nuclear factor-κB regulates βAPP and β- and γ-secretases differently at physiological and supraphysiological Aβ concentrations. J. Biol. Chem. 287, 24573–24584. 10.1074/jbc.A115.333054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R. G., Yip J. K., So M. K., Boylan R. J., Socransky S. S., Haffajee A. D. (2003). Relationship of destructive periodontal disease to the acute-phase response. J. Periodontol. 74, 1007–1016. 10.1902/jop.2003.74.7.1007 [DOI] [PubMed] [Google Scholar]
- Cunningham C., Hennessy E. (2015). Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res. Ther. 7:33. 10.1186/s13195-015-0117-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers J. W., Lobbestael J., van Wingen G. A., Kessels R. P., Arntz A., Egger J. I. (2015). The influence of stress on social cognition in patients with borderline personality disorder. Psychoneuroendocrinology 52, 119–129. 10.1016/j.psyneuen.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Dye B. A. (2012). Global periodontal disease epidemiology. Periodontol. 58, 10–25. 10.1111/j.1600-0757.2011.00413.x [DOI] [PubMed] [Google Scholar]
- Ebersole J. L., Machen R. L., Steffen M. J., Willmann D. E. (1997). Systemic acute-phase reactants, C-reactive protein and haptoglobin, in adult periodontitis. Clin. Exp. Immunol. 107, 347–352. 10.1111/j.1365-2249.1997.270-ce1162.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P., Stam F. C. (1982). Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 57, 239–242. 10.1007/bf00685397 [DOI] [PubMed] [Google Scholar]
- Eke P. I., Dye B. A., Wei L., Slade G. D., Thornton-Evans G. O., Borgnakke W. S., et al. (2015). Update on prevalence of periodontitis in adults in the united states: NHANES 2009 – 2012. J. Periodontol. 86, 611–622. 10.1902/jop.2015.140520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemadifar R., Konarizadeh S., Zarei A., Farshidi H., Sobhani A. (2015). Relationship between periodontal status and C-reactive protein and interleuckin-6 levels among atherosclerotic patients in Bandar Abbas, Iran in 2014. Electron Physician 7, 1010–1016. 10.14661/2015.1010-1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhad S. Z., Amini S., Khalilian A., Barekatain M., Mafi M., Barekatain M., et al. (2014). The effect of chronic periodontitis on serum levels of tumor necrosis factor-alpha in Alzheimer disease. Dent. Res. J. (Isfahan) 11, 549–552. [PMC free article] [PubMed] [Google Scholar]
- Fischer B., Schmoll H., Riederer P., Bauer J., Platt D., Popa-Wagner A. (1995). Complement C1q and C3 mRNA expression in the frontal cortex of Alzheimer’s patients. J. Mol. Med. 73, 465–471. 10.1007/bf00202265 [DOI] [PubMed] [Google Scholar]
- Fonseca M. I., Chu S., Pierce A. L., Brubaker W. D., Hauhart R. E., Mastroeni D., et al. (2016). Analysis of the putative role of CR1 in Alzheimer’s disease: genetic association, expression and function. PLoS One 11:e0149792. 10.1371/journal.pone.0149792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson M. I., Figueredo C. M., Gustafsson A., Bergström K. G., Asman B. E. (1999). Effect of periodontitis and smoking on blood leukocytes and acute-phase proteins. J. Periodontol. 70, 1355–1360. 10.1902/jop.1999.70.11.1355 [DOI] [PubMed] [Google Scholar]
- Friedewald V. E., Kornman K. S., Beck J. D., Genco R., Goldfine A., Libby P., et al. (2009). The american journal of cardiology and journal of periodontology editors’ consensus: periodontitis and atherosclerotic cardiovascular disease. Am. J. Cardiol. 104, 59–68. 10.1016/j.amjcard.2009.05.002 [DOI] [PubMed] [Google Scholar]
- Ganesh P., Karthikeyan R., Muthukumaraswamy A., Anand J. (2017). A potential role of periodontal inflammation in Alzheimer’s disease: a review. Oral Health Prev. Dent. 15, 7–12. 10.3290/j.ohpd.a37708 [DOI] [PubMed] [Google Scholar]
- Gasque P. (2004). Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 41, 1089–1098. 10.1016/j.molimm.2004.06.011 [DOI] [PubMed] [Google Scholar]
- Gaur S., Agnihotri R. (2015). Alzheimer’s disease and chronic periodontitis: is there an association? Geriatr. Gerontol. Int. 15, 391–404. 10.1111/ggi.12425 [DOI] [PubMed] [Google Scholar]
- Godbout J. P., Chen J., Abraham J., Richwine A. F., Berg B. M., Kelley K. W., et al. (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 19, 1329–1331. 10.1096/fj.05-3776fje [DOI] [PubMed] [Google Scholar]
- Gomes-Filho I. S., Freitas Coelho J. M., da Cruz S. S., Passos J. S., Teixeira de Freitas C. O., Aragão Farias N. S. (2011). Chronic periodontitis and C-reactive protein levels. J. Periodontol. 82, 969–978. 10.1902/jop.2010.100511 [DOI] [PubMed] [Google Scholar]
- Gurav A. N. (2014). Alzheimer’s disease and periodontitis—an elusive link. Rev. Assoc. Med. Bras. (1992) 60, 173–180. 10.1590/1806-9282.60.02.015 [DOI] [PubMed] [Google Scholar]
- Haass C., De Strooper B. (1999). The presenilins in Alzheimer’s disease—proteolysis holds the key. Science 286, 916–919. 10.1126/science.286.5441.916 [DOI] [PubMed] [Google Scholar]
- Hajishengallis G. (2015). Periodontitis: from microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 15, 30–44. 10.1038/nri3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C. J., Hallock L. R., Howanski R. J., Appelt D. M., Little C. S., Balin B. J. (2010). Immunohistological detection of Chlamydia pneumoniae in the Alzheimer’s disease brain. BMC Neurosci. 11:121. 10.1186/1471-2202-11-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes P. J., Krishna R. (2010). Characteristics of inflammation common to both diabetes and periodontitis: are predictive diagnosis and targeted preventive measures possible? EPMA J. 1, 101–116. 10.1007/s13167-010-0016-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., et al. (2015a). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T., Golenbock D. T., Latz E. (2015b). Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236. 10.1038/ni.3102 [DOI] [PubMed] [Google Scholar]
- Holmes C. (2013). Review: systemic inflammation and Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 39, 51–68. 10.1111/j.1365-2990.2012.01307.x [DOI] [PubMed] [Google Scholar]
- Holmes C., Cunningham C., Zotova E., Woolford J., Dean C., Kerr S., et al. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774. 10.1212/WNL.0b013e3181b6bb95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., et al. (1996). Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. 10.1126/science.274.5284.99 [DOI] [PubMed] [Google Scholar]
- in ’t Veld B. A., Ruitenberg A., Hofman A., Launer L. J., van Duijn C. M., Stijnen T., et al. (2001). Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 345, 1515–1521. 10.1056/NEJMoa010178 [DOI] [PubMed] [Google Scholar]
- Jaeger L. B., Dohgu S., Sultana R., Lynch J. L., Owen J. B., Erickson M. A., et al. (2009). Lipopolysaccharide alters the blood-brain barrier transport of amyloid β protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 23, 507–517. 10.1016/j.bbi.2009.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer A. R., Craig R. G., Dasanayake A. P., Brys M., Glodzik-Sobanska L., de Leon M. J. (2008). Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimers Dement. 4, 242–250. 10.1016/j.jalz.2007.08.004 [DOI] [PubMed] [Google Scholar]
- Kamer A. R., Craig R. G., Pirraglia E., Dasanayake A. P., Norman R. G., Boylan R. J., et al. (2009). TNF-α and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J. Neuroimmunol. 216, 92–97. 10.1016/j.jneuroim.2009.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer A. R., Fortea J. O., Videla S., Mayoral A., Janal M., Carmona-Iragui M. (2016). Periodontal disease’s contribution to Alzheimer’s disease progression in Down syndrome. Alzheimers Dement. (Amst) 2, 49–57. 10.1016/j.dadm.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer A. R., Pirraglia E., Tsui W., Rusinek H., Vallabhajosula S., Mosconi L., et al. (2015). Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging 36, 627–633. 10.1016/j.neurobiolaging.2014.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C. W., Schmitz M., Münz C., Lünemann J. D., Schmidt J. (2013). TNF-α upregulates macroautophagic processing of APP/β-amyloid in a human rhabdomyosarcoma cell line. J. Neurol. Sci. 325, 103–107. 10.1016/j.jns.2012.12.011 [DOI] [PubMed] [Google Scholar]
- Kiecolt-Glaser J. K., McGuire L., Robles T. F., Glaser R. (2002). Emotions, morbidity and mortality: new perspectives from psychoneuroimmunology. Annu. Rev. Psychol. 53, 83–107. 10.1146/annurev.psych.53.100901.135217 [DOI] [PubMed] [Google Scholar]
- Kubota T., Maruyama S., Abe D., Tomita T., Morozumi T., Nakasone N. (2014). Amyloid β (A4) precursor protein expression in human periodontitis-affected gingival tissues. Arch. Oral Biol. 59, 586–594. 10.1016/j.archoralbio.2014.03.004 [DOI] [PubMed] [Google Scholar]
- Laflamme N., Rivest S. (2001). Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 15, 155–163. 10.1096/fj.00-0339com [DOI] [PubMed] [Google Scholar]
- Lee J. W., Lee Y. K., Yuk D. Y., Choi D. Y., Ban S. B., Oh K. W., et al. (2008). Neuroinflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of β-amyloid generation. J. Neuroinflammation 5:37. 10.1186/1742-2094-5-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos B. G., Craandijk J., Hoek F. J., Wertheim-van Dillen P. M., van der Velden U. (2000). Elevation of systemic markers related to cardiovascular diseases in the peripheral blood of periodontitis patients. J. Periodontol. 71, 1528–1534. 10.1902/jop.2000.71.10.1528 [DOI] [PubMed] [Google Scholar]
- Louveau A., Smirnov I., Keyes T. J., Eccles J. D., Rouhani S. J., Peske J. D. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. 10.1038/nature14432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIntyre A., Abramov R., Hammond C. J., Hudson A. P., Arking E. J., Little C. S., et al. (2003). Chlamydia pneumoniae infection promotes the transmigration of monocytes through human brain endothelial cells. J. Neurosci. Res. 71, 740–750. 10.1002/jnr.10519 [DOI] [PubMed] [Google Scholar]
- Mancini M., Grappasonni I., Scuri S., Amenta F. (2010). Oral health in Alzheimer’s disease: a review. Curr. Alzheimer Res. 7, 368–373. 10.2174/156720510791162359 [DOI] [PubMed] [Google Scholar]
- Martande S. S., Pradeep A. R., Singh S. P., Kumari M., Suke D. K., Raju A. P., et al. (2014). Periodontal health condition in patients with Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 29, 498–502. 10.1177/1533317514549650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli M. L., Brandi M. L., Martelli M., Nobili P., Medico E., Martelli F. (2017). Periodontal disease and women’s health. Curr. Med. Res. Opin. 24, 1005–1015. 10.1080/03007995.2017.1297928 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., McGeer E. G. (1995). The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 21, 195–218. 10.1016/0165-0173(95)00011-9 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., McGeer E. G. (2013). The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 126, 479–497. 10.1007/s00401-013-1177-7 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., McGeer E., Rogers J., Sibley J. (1990). Anti-inflammatory drugs and Alzheimer disease. Lancet 335:1037. 10.1016/0140-6736(90)91101-f [DOI] [PubMed] [Google Scholar]
- Miklossy J., Kis A., Radenovic A., Miller L., Forro L., Martins R., et al. (2006). β-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol. Aging 27, 228–236. 10.1016/j.neurobiolaging.2005.01.018 [DOI] [PubMed] [Google Scholar]
- Nakib S. A., Pankow J. S., Beck J. D., Offenbacher S., Evans G. W., Desvarieux M., et al. (2004). Periodontitis and coronary artery calcification: the Atherosclerosis Risk in Communities (ARIC) study. J. Periodontol. 75, 505–510. 10.1902/jop.2004.75.4.505 [DOI] [PubMed] [Google Scholar]
- Nezu A., Kubota T., Maruyama S., Nagata M., Nohno K., Morozumi T., et al. (2017). Expression of neprilysin in periodontitis-affected gingival tissues. Arch. Oral Biol. 79, 35–41. 10.1016/j.archoralbio.2017.03.003 [DOI] [PubMed] [Google Scholar]
- Noack B., Genco R. J., Trevisan M., Grossi S., Zambon J. J., De Nardin E. (2001). Periodontal infections contribute to elevated systemic C-reactive protein level. J. Periodontol. 72, 1221–1227. 10.1902/jop.2000.72.9.1221 [DOI] [PubMed] [Google Scholar]
- Noble J. M., Scarmeas N., Celenti R. S., Elkind M. S., Wright C. B., Schupf N., et al. (2014). Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS One 9:e114959. 10.1371/journal.pone.0114959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton S., Matthews F. E., Barnes D. E., Yaffe K., Brayne C. (2014). Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 13, 788–794. 10.1016/S1474-4422(14)70136-X [DOI] [PubMed] [Google Scholar]
- Norton S., Matthews F. E., Brayne C. (2013). A commentary on studies presenting projections of the future prevalence of dementia. BMC Public Health 13:1. 10.1186/1471-2458-13-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson J. K., Miller S. D. (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 173, 3916–3924. 10.4049/jimmunol.173.6.3916 [DOI] [PubMed] [Google Scholar]
- Olsen I., Taubman M. A., Singhrao S. K. (2016). Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis and Alzheimer’s disease. J. Oral Microbiol. 22:33029. 10.3402/jom.v8.33029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann R. V., Haas A. N., Rösing C. K., Susin C. (2015). Epidemiology of periodontal diseases in adults from Latin America. Periodontol. 2000 67, 13–33. 10.1111/prd.12061 [DOI] [PubMed] [Google Scholar]
- Otomo-Corgel J., Pucher J. J., Rethman M. P., Reynolds M. A. (2012). State of the science: chronic periodontitis and systemic health. J. Evid. Based Dent. Pract. 12, 20–28. 10.1016/S1532-3382(12)70006-4 [DOI] [PubMed] [Google Scholar]
- Otsuka N., Tomonaga M., Ikeda K. (1991). Rapid appearance of β-amyloid precursor protein immunoreactivity in damaged axons and reactive glial cells in rat brain following needle stab injury. Brain Res. 568, 335–338. 10.1016/0006-8993(91)91422-w [DOI] [PubMed] [Google Scholar]
- Page R. C. (1998). The pathobiology of periodontal diseases may affect systemic diseases: inversion of a paradigm. Ann. Periodontol. 3, 108–120. 10.1902/annals.1998.3.1.108 [DOI] [PubMed] [Google Scholar]
- Page R. C., Kornman K. S. (1997). The pathogenesis of human periodontitis: an introduction. Periodontol. 2000 14, 9–11. 10.1111/j.1600-0757.1997.tb00189.x [DOI] [PubMed] [Google Scholar]
- Pazos P., Leira Y., Domínguez C., Pías-Peleteiro J. M., Blanco J., Aldrey J. M. (2016). Association between periodontal disease and dementia: a literature review. Neurologia [Epub ahead of print]. 10.1016/j.nrl.2016.07.013 [DOI] [PubMed] [Google Scholar]
- Pejcic A., Kesic L. J., Milasin J. (2011). C-reactive protein as a systemic marker of inflammation in periodontitis. Eur. J. Clin. Microbiol. Infect. Dis. 30, 407–414. 10.1007/s10096-010-1101-1 [DOI] [PubMed] [Google Scholar]
- Perry V. H., Cunningham C., Holmes C. (2007). Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 7, 161–167. 10.1038/nri2015 [DOI] [PubMed] [Google Scholar]
- Perry V. H., Teeling J. (2013). Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 35, 601–612. 10.1007/s00281-013-0382-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petcu E. B., Kocher T., Kuhr A., Buga A. M., Klöting I., Herndon J. G., et al. (2008). Mild systemic inflammation has a neuroprotective effect after stroke in rats. Curr. Neurovasc. Res. 5, 214–223. 10.2174/156720208786413424 [DOI] [PubMed] [Google Scholar]
- Poole S., Singhrao S. K., Kesavalu L., Curtis M. A., Crean S. (2013). Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimers Dis. 36, 665–677. 10.3233/JAD-121918 [DOI] [PubMed] [Google Scholar]
- Preshaw P. M., Alba A. L., Herrera D., Jepsen S., Konstantinidis A., Makrilakis K., et al. (2012). Periodontitis and diabetes: a two-way relationship. Diabetologia 55, 21–31. 10.1007/s00125-011-2342-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Li G., Qian X., Liu Y., Wu X., Liu B., et al. (2005). Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 52, 78–84. 10.1002/glia.20225 [DOI] [PubMed] [Google Scholar]
- Riviere G. R., Riviere K. H., Smith K. S. (2002). Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 17, 113–118. 10.1046/j.0902-0055.2001.00100.x [DOI] [PubMed] [Google Scholar]
- Rogers J. (2008). The inflammatory response in Alzheimer’s disease. J. Periodontol. 79, 1535–1543. 10.1902/jop.2008.080171 [DOI] [PubMed] [Google Scholar]
- Selkoe D. (1991). The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498. 10.1016/0896-6273(91)90052-2 [DOI] [PubMed] [Google Scholar]
- Shi D., Liu Y. Y., Li W., Zhang X., Sun X. J., Xu L., et al. (2015). Association between plasma leptin level and systemic inflammatory markers in patients with aggressive periodontitis. Chin. Med. J. (Engl) 128, 528–532. 10.4103/0366-6999.151110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhrao S. K., Harding A., Poole S., Kesavalu L., Crean S. (2015). Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediators Inflamm. 2015:137357. 10.1155/2015/137357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade G. D., Ghezzi E. M., Heiss G., Beck J. D., Riche E., Offenbacher S. (2003). Relationship between periodontal disease and C-reactive protein among adults in the Atherosclerosis Risk in Communities study. Arch. Intern. Med. 163, 1172–1179. 10.1016/j.accreview.2003.08.039 [DOI] [PubMed] [Google Scholar]
- Sly L. M., Krzesicki R. F., Brashler J. R., Buhl A. E., McKinley D. D., Carter D. B., et al. (2001). Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res. Bull. 56, 581–588. 10.1016/s0361-9230(01)00730-4 [DOI] [PubMed] [Google Scholar]
- Sochocka M., Sobczyński M., Sender-Janeczek A., Zwolińska K., Błachowicz O., Tomczyk T., et al. (2017a). Association between periodontal health status and cognitive abilities. The role of cytokine profile and systemic inflammation. Curr. Alzheimer Res. [Epub ahead of print]. 10.2174/1567205014666170316163340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochocka M., Zwolińska K., Leszek J. (2017b). The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol. 15, 996–1009. 10.1007/978-3-662-03248-0_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socransky S. S., Haffajee A. D. (2002). Dental biofilms: difficult therapeutic targets. Periodontol. 2000 28, 12–55. 10.1034/j.1600-0757.2002.280102.x [DOI] [PubMed] [Google Scholar]
- Stein P., Steffen M. J., Smith C., Jicha G., Ebersole J. L., Abner E., et al. (2012). Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement. 8, 196–203. 10.1016/j.jalz.2011.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling J. L., Perry V. H. (2009). Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience 158, 1062–1073. 10.1016/j.neuroscience.2008.07.031 [DOI] [PubMed] [Google Scholar]
- Teixeira F. B., Pereira Fernandes Lde M., Noronha P. A., dos Santos M. A., Gomes-Leal W., Maia C. S. F., et al. (2014). Masticatory deficiency as a risk factor for cognitive dysfunction. Int. J. Med. Sci. 11, 209–214. 10.7150/ijms.6801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Exel E., Eikelenboom P., Comijs H., Frölich M., Smit J. H., Stek M. L., et al. (2009). Vascular factors and markers of inflammation in offspring with a parental history of late-onset Alzheimer disease. Arch. Gen. Psychiatry 66, 1263–1570. 10.1001/archgenpsychiatry.2009.146 [DOI] [PubMed] [Google Scholar]
- VanItallie T. B. (2017). Alzheimer’s disease: innate immunity gone awry? Metabolism 69S, S41–S49. 10.1016/j.metabol.2017.01.014 [DOI] [PubMed] [Google Scholar]
- Watts A., Crimmins E. M., Gatz M. (2008). Inflammation as a potential mediator for the association between periodontal disease and Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 4, 865–876. 10.2147/ndt.s3610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller R. O., Boche D., Nicoll J. A. (2009). Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 118, 87–102. 10.1007/s00401-009-0498-z [DOI] [PubMed] [Google Scholar]
- Winning L., Patterson C. C., Cullen K. M., Stevenson K. A., Lundy F. T., Kee F., et al. (2015). The association between subgingival periodontal pathogens and systemic inflammation. J. Clin. Periodontol. 42, 799–806. 10.1111/jcpe.12450 [DOI] [PubMed] [Google Scholar]
- Yang J., Feng L., Ren J., Wu G., Chen S., Zhou Q., et al. (2013). Correlation between the severity of periodontitis and coronary artery stenosis in a Chinese population. Aust. Dent. J. 58, 333–338. 10.1111/adj.12087 [DOI] [PubMed] [Google Scholar]