

SUMMARY
Nonhomologous end joining (NHEJ) must adapt to diverse end structures during repair of chromosome breaks. Here we investigate the mechanistic basis for this flexibility. DNA ends are aligned in a paired-end complex (PEC) by Ku, XLF, XRCC4, and DNA ligase IV (LIG4); we show by single-molecule analysis how terminal mispairs lead to mobilization of ends within PECs, and consequent sampling of more end-alignment configurations. This remodeling is essential for direct ligation of damaged and mispaired ends during cellular NHEJ, since remodeling and ligation of such ends both require a LIG4-specific structural motif, insert1. Insert1 is also required for PEC remodeling that enables nucleolytic processing when end structures block direct ligation. Accordingly, cells expressing LIG4 lacking insert1 are sensitive to ionizing radiation. Cellular NHEJ of diverse ends thus identifies the steps necessary for repair through LIG4-mediated sensing of differences in end structure, and consequent dynamic remodeling of aligned ends.
eTOC blurb
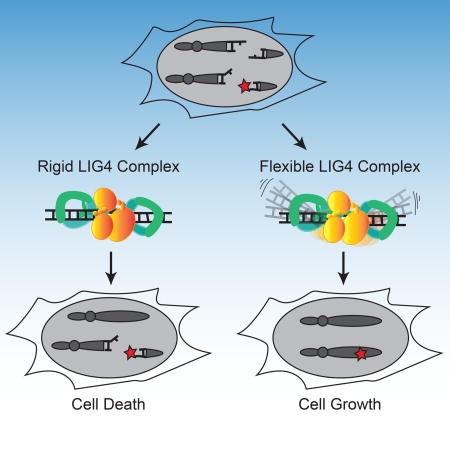
Conlin et al. show that when DNA double-strand breaks have terminal mispairs or damage, an unstructured loop unique to DNA ligase 4 allows for dynamic remodeling of the alignment and end-repair; the loop is also required for cellular resistance to ionizing radiation.
INTRODUCTION
DNA double-strand breaks (DSBs) are genomic lesions that play an important role in human health and disease. They are frequently generated by exogenous damaging agents (e.g. ionizing radiation) or as programmed intermediates in meiosis and V(D)J recombination (Mehta and Haber, 2014). The ends generated by these biological sources of chromosome breaks are often “complex,” with DNA helix-distorting nucleotide damage, mismatches, or chemical adducts that pose challenges to the ligases and polymerases needed for DSB repair (Breen and Murphy, 1995; Nitiss, 2009; Roth et al., 1992). This problem is especially relevant to the nonhomologous end joining (NHEJ) pathway since, unlike other DSB repair pathways, these complex ends are not extensively resected prior to synthetic steps (polymerase and ligase activity; Waters et al., 2014a).
Ligation is the only essential step in NHEJ, and is performed by one of the three mammalian ligases, DNA ligase IV (LIG4; Lieber et al., 1997). LIG4 is recruited to broken ends through participation in a complex of core NHEJ factors including XRCC4, the Ku 70/80 heterodimer (Ku; Nick McElhinny et al., 2000), and XLF. This NHEJ core complex is sufficient to physically link a pair of broken ends together, and can thus be termed the paired end complex, or PEC. The PEC is essential for repair of diverse end structures; for example, XLF is required both for stable PEC formation (Reid et al., 2015) and ligation of complex ends, but only modestly affects ligation of ends with complementary termini (Andres et al., 2007; Gu et al., 2007; Tsai et al., 2007). Recent physical analyses of PECs indicate that they are highly dynamic (Reid et al., 2015), and that both the flexibility and stability of PECs can be modulated by ligation-compatible DNA end chemistry (Reid et al., 2016). However, it is unclear how differences in end structure trigger these changes in dynamics, and whether these changes in dynamics impact cellular repair.
Here we address this problem by assessing the impact of diverse end structures on in vitro functional assays, single-molecule analyses of end-pairing dynamics, and cellular repair and survival. We show that mismatches near strand break termini trigger extensive PEC remodeling. Moreover, a separation-of-function mutation in LIG4 links this mispair-induced PEC remodeling to the sensing of these end structures by LIG4, and argues PEC remodeling is essential to the proficiency of cellular NHEJ in repairing these end structures.
RESULTS
LIG4WT is specialized to directly ligate mismatched or damaged ends
Activity of all three mammalian ligases requires the encircling of double stranded DNA at a strand break (Ellenberger and Tomkinson, 2008). Structural studies identified 6–10 amino acids inserted in LIG4 orthologs (residues 113–122 in human LIG4), relative to other eukaryotic ligases (Figure 1A; Supplementary Figure 1A; Ochi et al., 2013). This insert is located within the strand break-bound ligase on the opposite side of the double helix from the strand break and site of catalysis, suggesting a possible function specific to substrates with double helix-distorting mispairs or damage. Consistent with this idea, we purified LIG4 with this element specifically deleted (LIG4Δi) in a complex with XRCC4. We determined that insert1 had no significant impact on LIG4-XRCC4 intrinsic nick sealing activity (Supplementary Figure 1B), DNA binding (Supplementary Figure 1C), or ability to form a higher-order complex with NHEJ core factors Ku and XLF on DNA (Supplementary Figure 1D). In vitro NHEJ activity was also similar comparing LIG4Δi to LIG4WT when ends had complementary overhangs (Figure 1B; 5’ G:C, 3’ G:C). In contrast, when ends had mispairs or damage at strand-break termini, in vitro NHEJ activity using LIG4Δi was reduced 21-66-fold relative to LIG4WT (Figure 1B; 5’ GoxC, 3’ GxT, 3’ GxA). LIG4ΔI is thus specifically defective in supporting in vitro NHEJ when substrates have helix-distorting 8-oxoguanine (Go) damage or mispairs near strand termini.
Figure 1.

Effect of LIG4 insert1 on NHEJ of complex ends in vitro (A) Structure of human LIG1 bound to DNA (1X9N; green), with inset emphasizing sequence and structural alignments of human LIG1 α helices 5–6 to human LIG3 (3L2P; blue) and human LIG4 (based on 3W1B; pink), with sequence and a modeled location of LIG4 insert1 (disordered in the 3W1B apoenzyme) in red. (B) Ku, XLF, and either XRCC4-LIG4WT (gray) or XRCC4-LIG4ΔI (orange) were incubated with substrates containing different complementary (5’ G:C, 3’ G:C) or non-complementary (5’ GoxC, 3’ GxT, 3’ GxA) overhangs as noted. Joining efficiency is expressed as a fraction of the total junctions recovered using the 5’ G:C substrate with NHEJ reactions containing LIG4WT. Ligation reactions were performed in triplicate and the mean joining efficiencies are shown, along with the fold difference between LIG4WT and LIG4ΔI for each substrate. Error bars represent the range of observed values for each set of experiments.
Ends with mispaired nucleotides are critical NHEJ substrates that arise during V(D)J recombination and after nucleolytic processing of radiation-induced breaks. They also presumably act as a model for ends with other sources of helical distortion, including nucleotide damage. To validate this inference, we measured in vitro NHEJ of ends with 8-oxoguanine (Figure 1B, 5’ GoxC), the most common form of oxidative base damage. NHEJ activity on this substrate was reduced over 50-fold with LIG4ΔI, which was comparable to the effect of a terminal G:A mispair. Therefore, insert1 is required for direct ligation of end structures with flanking helical distortions, whether the distortions are due to mispairs or nucleotide damage. To further explore the extent to which ligation of ends with terminal mispairs or damage is specific to wild-type LIG4, we generated a chimera (LIG3+4) with all three LIG4 catalytic sub-domains replaced with the equivalent sub-domains from mammalian wild-type LIG3 (Supplementary Figure 1E). Like LIG4ΔI, LIG3+4 physically associates with XRCC4 and was fully competent in Ku- and XLF-dependent ligation of ends with complementary overhangs. However, end joining with this chimera was even more sensitive than LIG4ΔI to terminal nucleotide damage (activity reduced more than 100-fold, relative to LIG4WT; Supplementary Figure 1E). This result consistent with the argument that LIG4 is unique amongst mammalian ligases in its ability to repair damaged termini. Additionally, the impact of LIG4ΔI on repair of damaged ends is less severe than that of the LIG3+4 chimera, suggesting that insert1 is not entirely responsible for the unique ability of LIG4 to tolerate mispairs and damage. We therefore sought to use the LIG4ΔI separation-of-function mutation to investigate both the mechanistic basis for the unique ability of NHEJ to tolerate helix-distorting mispairs or damage at the ligation step, as well as its significance to cellular double strand break repair.
Dynamic re-alignment of mismatched ends is required for their ligation
We previously described a single-molecule fluorescent resonance energy transfer (smFRET) assay that reports on pairing of DNA ends as mediated by a complex of Ku, XRCC4, LIG4, and XLF (Reid et al., 2015, 2016). These PECs are apparent as FRET pairs generated when a Cy3 labeled donor dsDNA fragment in solution stably associates with a Cy5 labeled acceptor dsDNA fragment immobilized on a surface (Figure 2A). In accord with the in vitro ligation assay, LIG4ΔI and LIG4WT similarly promote stable PEC formation when ends have complementary 4-nucleotide overhangs (G:C, Figure 2B). In contrast, pairing of overhangs with 3’ terminal G:T mismatches is significantly reduced when comparing LIG4ΔI to LIG4WT; this reduced pairing efficiency represents a diminished proportion of DNA ends associated in the PEC. The formation of PECs is even less efficient when termini have a bulkier purine:purine G:A mismatch, but is similarly inefficient for both LIG4WT and LIG4ΔI (Figure 2B). PECs thus form less efficiently with increasing terminal helical distortion, and PECs formed with LIG4ΔI are more sensitive to this challenge.
Figure 2.

Effect of complex end structures on pairing dynamics of single molecule complexes with LIG4WT or LIG4ΔI (A) smFRET NHEJ assay: (1) dsDNA with a Cy5 acceptor is tethered to a bitonylated PEG surface via a biotin-neutravadin linkage, (2) dsDNA with a Cy3 donor and NHEJ proteins (green) are added to the chamber, and (3) ends are paired and FRET is observed. (B) Quantitation of pairing efficiency of ends with complementary (G:C) or mismatched (GxT, GxA) overhangs by Ku, XLF, XRCC4 and either LIG4WT (gray) or LIG4ΔI (orange). Error bars represent standard error of the mean for 3 experiments. Means were assessed by two-way ANOVA as significantly different from control (LIG4WT on G:C substrate) with confidence p<0.001 (***). (C) Histograms of observed EFRET for PECs formed as in (B). (D) Representative smFRET trajectory for LIG4WT and LIG4ΔI PECs formed with GxT ends demonstrating altered transition frequency and FRET states (E) LIG4WT enables PECs to oscillate between high and low EFRET states in response to distortions, and this flexibility is essential for joining distorted breaks.
Changes in FRET efficiency (EFRET) reflect dynamic repositioning of DNA ends relative to each other within individual PECs (Reid et al. 2015, Reid et al. 2016). When using complementary ends (3’ G:C; Figure 2C), EFRET distributions were not significantly different when comparing PECs formed with LIG4WT (black line) vs. LIG4ΔI (orange line). LIG4ΔI PECs also had similar FRET distributions when ends had terminal mispairs (Figure 2C); importantly, LIG4WT PECs formed on ends with mispaired termini more often had lower EFRET (DNA labels located further apart; black lines for G:A and G:T mispairs, Figure 2C), and consequently overall wider distributions of EFRET (Figure 2C, S2A) when compared to paired temini (G:C). Ends with terminal distortions thus trigger PECs to sample a wider variety of end-alignment configurations to remain efficiently paired, but only when using LIG4WT.
Examination of smFRET trajectories from individual PECs also shows the transition frequency between FRET states increases when comparing LIG4WT and LIG4ΔI (Figure 2D, Supplementary Figure 2C). We quantified this difference by using autocorrelation of individual FRET trajectories to calculate the average transition times - “lag times” (τ) - between FRET states. For ends with G:T mismatches, these were approximately two fold lower for PECs formed with LIG4WT, compared to PECs formed with LIG4ΔI (Supplementary Figure 2B; Reid et al., 2016). These values are then used to calculate the relative stability of the DNA ends in the PECs (Supplementary Figure 2C). From these calculated end stabilities we can infer that LIG4WT PECs have lower energetic barriers in assuming new conformations, compared to LIG4ΔI PECs, but again only when ends have terminal mismatches.
PECs containing LIG4ΔI and mispaired ends are thus formed less efficiently (Figure 2B), and even when formed do not acquire the high degree of conformational plasticity observed when PECs are formed with LIG4WT (Figure 2C–D, Supplementary Figure 2D–E). We argue the inability of LIG4ΔI to allow for mispair-induced PEC remodeling accounts for its specific defect in direct ligation of such end structures (Figure 1B). There are also limits to the extent to which remodeling enables ligation, as even LIG4WT is inefficient in joining ends with bulky G:A mismatches (Figure 1B). PECs formed with paired termini favor a narrow distribution of high FRET end alignments that more closely resemble FRET distributions observed with products of ligation (Reid et al., 2015); these alignments thus likely directly juxtapose strand-break termini in anticipation of catalytic steps (“pre-catalytic”, Figure 2E). We attribute the LIG4WT-specific, insert1-dependent flexibility in accommodation of mispaired termini to a favoring of end alignments that both have lower FRET (more distally-located labels) and are more dynamic. These more dynamic and lower FRET PECs – “remodeling PECS” - may be catalytically incompetent, but allow for iterative attempts at the now transient (but occasionally catalytically competent) high-FRET intermediate (Figure 2E).
Cellular NHEJ of complex ends requires remodeling of the PEC
We next addressed whether the differences in PEC flexibility described above significantly impact cellular NHEJ. We employed scar-free gene targeting to exchange LIG4WT for LIG4ΔI alleles within the native LIG4 locus of a human cell line (Figure 3A). We independently generated two such cell lines (LIG4ΔI/ΔIa and LIG4ΔI/ΔIb), and confirmed they express only LIG4ΔI from endogenous loci (Methods; Supplementary Figure 3A–B). We then generated a cell line by another round of gene targeting where the LIG4 locus of LIG4ΔI/ΔIa was reverted back to wild type sequence (LIG4+r/+r), as a means of assessing the effects of possible off target mutations incurred in the original round of gene targeting (Figure 3A, Supplementary Figure 3A–B). Both LIG4ΔI/ΔI subclones acted equivalently in functional assays below. Similarly, results using parental wild type cells (LIG4+/+) matched those from the LIG4+r/+r reversion, confirming the differences observed in the LIG4ΔI/ΔI cells could be attributed to the 8 amino acids deletion.
Figure 3.

Effect of LIG4 insert1 on cellular joining of complex end structures (A) Cells were engineered to express LIG4ΔI from the native LIG4 locus by CRISPR/Cas9-based gene targeting, then the LIG4ΔI-a clone was reverted back to wild-type (LIG4+r/+r) by a second round of gene targeting (B–E) Substrates with varied end structures were introduced in the cell types described in (A). Joining efficiency was assessed by qPCR, and product structure by sequencing or diagnostic restriction digestion and defined as directly ligated or ligated after end processing as noted. Cellular NHEJ was assessed for (B) complementary ends, (C) ends with 3’ G:T terminal mismatches, (D) ends with 5’ terminal 8-oxoguanine (Go; product structures reported in Supplementary Figure 3D), and (E) ends with fully non-complementary overhangs (product structures reported in Supplementary Figure 3E). Error bars represent the standard error of the mean for 3 experiments. Means of linearized qPCR data and direct joining products were assessed by one-way ANOVA as significantly different from control (LIG4+/+ cells) with confidence p<0.001 (***) or not significantly different (ns).
DSB substrates with varied end structures were introduced into these cells, after which efficiency of repair was determined by qPCR and product structures were characterized by high-throughput sequencing. In accord with in vitro results, ends with complementary overhangs were efficiently joined almost entirely by direct ligation in both wild-type and LIG4ΔI/ΔI cells (Figure 3B). Also in accord with in vitro data, ends with terminal G:T mispairs were efficiently repaired by direct ligation (accounts for 60% of all repair) in both LIG4+/+ and LIG4+r/+r cells, while this class of product is rarely (<10%) seen in LIG4ΔI/ΔI clones (Figure 3C). Instead, repair in LIG4ΔI/ΔI cells typically requires re-alignment of overhangs and gap-repair synthesis prior to ligation of the now “sticky” end. This alternate pathway is sufficient to fully compensate for the inability of LIG4ΔI to directly ligate terminal mispairs, since overall joining efficiency was comparable for LIG4+/+ vs. LIG4ΔI/ΔI cells. Considering repair of ends with bulkier G:A mispairs, both wild-type and LIG4ΔI/ΔI cells rely on this compensating pathway (Supplementary Figure 3C), consistent with in vitro observations that neither LIG4WT nor LIG4ΔI can ligate this substrate in the absence of end processing. Repair of NHEJ substrates was severely reduced in LIG4−/− cells (Figure 3C; approximately 0.0005 products per cell), to the extent that we could not recover sufficient repair products to accurately assess product spectra.
Additional substrates were introduced into cells to assess whether barriers to mispair tolerance are routinely bypassed by cellular end processing. Similar to 3’ G:T mispairs, ends with 5’ Go terminal damage are primarily repaired by direct ligation in LIG4+/+ cells. Importantly, joining of 5’ GoxC in LIG4ΔI/ΔI cells is over 10-fold less efficient (Figure 3D), even though what little repair does occur is processing-dependent (Supplementary Figure 3D). We also investigated cellular NHEJ of end structures with entirely non-complementary overhangs (TTTT). Joining efficiency was again severely reduced in LIG4ΔI/ΔI cells, relative to wild-type cells (Figure 3D). For this substrate, the rare products recovered from LIG4ΔI/ΔI cells only subtly differed from wild-type controls in terms of junction structure (Supplementary Figure 3E). Thus, in contrast to previously tested substrates (Figure 3C), end processing was not sufficient to rescue repair of TTTT and 8-oxoguanine substrates in LIG4ΔI/ΔI cells. We initially linked LIG4WT PEC flexibility only to the ability of cellular NHEJ to directly ligate ends with terminal mispairs (Figures 1, 2, 3C); these latter results identify additional important contributions to cellular NHEJ associated with end-processing.
Role for PEC remodeling in guiding end processing choice during cellular NHEJ
We generated the substrate “EC1” (embedded complementarity 1) to further explore the relationship between PEC flexibility, cellular end processing, and ligation. EC1 has long (10-nt) non-complementary overhangs that can plausibly be aligned to juxtapose mispaired 3’OH:5’P termini, in anticipation of direct ligation (Figure 4A). Alternatively, EC1 can be re-aligned to pair complementary sequence embedded within the overhang, where unpaired tails are a presumptive substrate for nucleolytic end processing. These two alignments are readily distinguished by smFRET (Supplementary Figure 4A); PECs formed with a control substrate (with fully complementary 10nt overhangs) had low EFRET ranges, expected for EC1 alignments that juxtapose 3’OH:5’P termini (green lines), while PECs formed with 4-nt complementary overhangs had a clearly distinct population of high EFRET (blue lines) expected for EC1 alignments that pair embedded complementary sequence. Analysis of individual smFRET trajectories of PECs formed with EC1 and NHEJ core factors identified a much larger than typical fraction of transient complexes (lifetimes <5 seconds; Figure 4B). Transient PECs had two distinct populations of EFRET distributions, each roughly corresponding to the two alignment classes predicted above (Figure 4C). Long-lived PECs (persistent) favor only the high EFRET state, but sample both a wider range of alignment configurations (Figure 4C) and are more dynamic (have lower energetic barriers to transition; Supplementary Figure 4B) than PECs formed with complementary overhangs. To further address if the persistent PECs frequently involve pairing at embedded complementary sequence (as suggested by comparison to substrate standards; Figure 4C) we used a substrate where the complementary sequence was both reduced and re-located (“EC2”). As expected, PECs formed less efficiently with EC2 (Supplementary Figure 4C), and when formed had mostly lower EFRET (Supplementary Figure 4D). Importantly, LIG4ΔI was largely unable to form PECs with the EC1 substrate (Figure 4D), and the rare PECs that do form primarily have intermediate EFRET states that are inconsistent with either alignment (Supplementary Figure 4E). Therefore, only LIG4WT effectively promotes end-pairing of this substrate. Moreover, PECs formed with LIG4WT that juxtapose strand termini were “filtered out"; only the most plausibly productive alignments, i.e. those that could lead to ligation after nucleolytic end processing, were stable (Figure 4C).
Figure 4.

Effect of PEC flexibility on nucleolytic end processing (A) A substrate was designed with embedded complementarity (EC1) that can be aligned in a high-FRET conformation guided by base pairing (a presumptive nuclease substrate) or in a low-FRET conformation with juxtaposition of 5’ and 3’ termini. (B) Representative smFRET trajectories for transient (short lived; <30sec) and persistent (long lived; 30+ sec) PECs formed with the EC1 substrate (C) Histograms of EFRET of transient (top) or persistent (bottom) LIG4wt PECs formed on the EC1 substrate (black), compared to FRET standards with complementary overhangs either 4 nt (blue) or 10 nt (green) in length (D) Quantitation of pairing efficiency of EC1 substrate by LIG4WT (gray) or LIG4ΔI (orange). Error bars represent standard error of the mean for 3 experiments. Means were assessed for significance as in Figure 2B with confidence p<0.001 (***) or not significantly different (ns). (E) The EC1 substrate was transfected into cells and repair efficiency was quantitated by qPCR. Error bars represent standard error of the mean for 3 experiments. Linearized means were assessed for significance as in Figure 3B–E with confidence p<0.01 (**), p<0.001 (***) or not significantly different (ns).
We next assessed how the EC1 substrate was resolved by cellular NHEJ. Nearly all products (>99%) were indeed consistent with ligation after nuclease activity, with the dominant product guided by the alignment at embedded complementary sequence also favored in smFRET analysis (Supplementary Figure 4F). By comparison, direct ligation of EC1 accounted for less than 0.1% of all cellular repairs. Importantly, joining efficiency of this substrate was reduced over 60-fold in LIG4ΔI/ΔI cells, relative to LIG4+/+ cells (Figure 4E), even though LIG4ΔI/ΔI cells are fully proficient at ligating the inferred product of alignment-guided nuclease activity (a 4bp complementary overhang; Figure 3B). This result suggests that for this substrate, LIG4Δi fails to efficiently mediate repair because it is defective at an earlier step than ligation – specifically, stable accommodation of end-alignments required for nucleolytic end processing (Figure 4D).
Resistance to ionizing radiation requires tolerance of complex ends by LIG4
LIG4WT thus uniquely accommodates diverse end structures during end pairing. However, there is wide variation in how this flexibility impacts cellular NHEJ. Depending on the starting end structure it can be dispensable (Figure 3B, Supplementary Figure 3C), alter product spectra (Figure 3C), or can be critical for efficient repair (Figure 3D–E, Figure 4E). We therefore addressed the extent to which the inability of LIG4 to tolerate structural diversity impacts cell growth and survival after ionizing radiation. Using both standard colony forming assays and real-time imaging of cell growth, LIG4+/+ and LIG4+r/+r cells were similarly resistant to increasing dose of ionizing radiation. By comparison, LIG4ΔI/ΔI cells were radiosensitive to a degree intermediate between LIG4+/+ and LIG4−/− cells (Figure 5, Supplementary Figure 5), a result strikingly similar to joining efficiencies described for the majority of substrates with complex ends (Figures 3D–E, 4E). In contrast with ionizing radiation, LIG4ΔI/ΔI and LIG4+/+ cells are equally resistant to etoposide (Supplementary Figure 5). This is consistent with specific requirement for insert1 in repair of ends with mispairs or damage, since etoposide induced breaks can be processed by tyrosine phosphodiesterase 2 such that overhangs are undamaged and fully complementary (Gómez-Herreros et al., 2013). These results show that the ability of LIG4 to sense distortions facilitates cell survival following treatment with ionizing radiation.
Figure 5.

Effect of LIG4 insert1 on cellular sensitivity to ionizing radiation. Cells were exposed to indicated doses of X-rays and assessed for colony formation. Data represents the mean and standard deviation of 3 experiments. Mean surviving fractions were assessed by one-way ANOVA as significantly different from control (LIG4+/+ cells) independently for each dose with confidence p<0.05 (*), p<0.01 (**) or not significantly different (ns).
DISCUSSION
Repair by NHEJ implicitly requires the pairing together of broken chromosome ends. A complex of Ku, XRCC4, DNA Ligase IV, and XLF (paired end complex, or PEC) is necessary and sufficient for this purpose (Reid et al., 2015); we describe here dynamic changes in this complex that are triggered by differences in end structure, and show that this response is essential for efficient cellular repair.
Mechanistic basis for repair of complex ends by NHEJ
Ends with complementary (“sticky”) overhangs are aligned efficiently and with little mobility, to the extent that pairing EFRET more closely resemble the narrow distributions observed in products of ligation (Reid et al., 2015), relative to other end structures tested here. We suggest these PECs describe “pre-catalytic” end alignments, where strand break termini are directly juxtaposed in anticipation of ligase-mediated catalytic steps (Figure 6). In contrast, ends with helix distorting mispairs or damage near strand termini – complex ends - induce the sampling of a much wider variety of alignment configurations, most or all of which no longer juxtapose strand termini.
Figure 6.

Sensing of differences in end structure by LIG4 guides repair. Model for LIG4-dependent remodeling of paired end complexes in response to complex ends.
We use a LIG4 separation of function mutation (LIG4ΔI) to identify an essential role for this second, more dynamic “remodeling” class of PECs in cellular NHEJ for the repair of complex ends. We show LIG4ΔI is specifically unable to accommodate PEC remodeling in response to complex ends. As a consequence, PECs formed with LIG4ΔI are unable to directly ligate such substrates, but are also – with rare exception (Figure 3C) – unable to couple ligation to end processing when end complexity is sufficient to block direct ligation.
Notably, the exceptions are restricted to contexts where alignment-directed synthesis generates a fully complementary 6-nt overhang, a substrate expected to be especially permissive for the ligation step.
By comparison, insert1 is dispensable for the XLF-, XRCC4-, and Ku-dependent alignment of ends with complementary overhangs, as well as catalytic activity on this conventional ligase substrate. Moreover, a chimeric ligase with all three LIG4 catalytic subdomains replaced with LIG3 counterparts is equally effective in ligation of “sticky” ends (and is similarly stimulated by Ku and XLF), but is even less able to repair complex ends. Prior work emphasized the importance of a variety of NHEJ factors, including PAXX (Ochi et al., 2015; Xing et al., 2015), end processing factors (Chang et al., 2016; Ma et al., 2002; Pryor et al., 2015), and especially the end-bridging filament of XRCC4-XLF (Andres et al., 2012; Gu et al., 2007; Roy et al., 2015) in repairing complex breaks. Indeed, we previously reported that these filaments form on bleomycin-induced DSBs and orchestrate their repair (Reid et al., 2015). Here we identify a critical role for specialization of LIG4 catalytic subdomains in repair of complex ends, and show this role is attributable to insert1-dependent PEC remodeling.
How does insert1 contribute to PEC remodeling? The three subdomains of eukaryotic ligases are extended in the absence of DNA (“open” conformation), and engage substrates by forming a ring around double stranded DNA (“closed” conformation; Cotner-Gohara et al., 2010; Nair et al., 2007; Pascal et al., 2004). In the closed conformation, the central catalytic subdomain is bound to strand break termini while insert1 is located in the N-terminal subdomain on the opposite side of the double helix (Ochi et al., 2013). Though not resolved in current apo-enzyme crystal structures, its location suggests that insert1 helps LIG4 maintain a closed conformation, either by stabilizing the ring-closing interactions between N and C terminal catalytic subdomains or by interacting with DNA (Ochi et al., 2012). We suggest stable end-pairing is dependent on LIG4 maintaining a closed conformation, even if LIG4 can directly interact with only the 5’ phosphate side of a strand break (“half-site” binding). LIG4ΔI instead transitions to an open conformation in this context (like conventional ligases), which leads to failure of end pairing.
Significance of LIG4 sensing complex ends
Prior work indicates that LIG4 has functions in NHEJ distinct from the ligation step, most clearly in promoting end pairing (Budman et al., 2007; Cottarel et al., 2013; Davis et al., 2008; Reid et al., 2015, 2016). Data presented here identify a much more sophisticated function. Differences in how LIG4 catalytic domains interact with different end structures trigger dramatic changes in the dynamics of the entire paired-end complex – i.e. including Ku, XRCC4, and XLF paired ends – and these altered dynamics determine the steps taken to complete repair. This role is distinct from critical LIG4 roles in catalysis and end pairing, since both of the latter functions are fully intact in PECs formed with LIG4ΔI. LIG4 can thus be identified as the PEC “sensor,” helping tailor the path to repair as is appropriate to end structure, possibly even to the extent that how LIG4 interacts with aligned ends may dictate the identity of the end processing factor that next engages the end.
Inhibitors of LIG4 are being explored for their potential to sensitize tumors to radiation therapy (Tomkinson et al., 2013). Here we identify a role of LIG4 that is specific to the ability of cells to repair complex damage, identify a structural element required for this role, then show deletion of this element leads to cellular sensitivity to ionizing radiation. Since this structural element is unique to LIG4 and required for radioresistance, it presents a promising therapeutic target, as it is less likely to engender the off-target effects observed with current LIG4 inhibitors (Greco et al., 2016).
METHODS
DSB Substrates
DSB substrates were made by ligating the 15–30 bp double stranded oligonucleotide “caps” described in Table S1 to a 285-bp PCR-generated common DNA “core” segment that had been digested with BsaI to generate appropriate sticky ends. Substrates were purified with the QiaQuick PCR purification kit (Qiagen), 5’ phosphorylated with T4 polynucleotide kinase (NEB), and substrate assembly validated by gel electrophoresis.
DNA Constructs and Protein Purification
Constructs for expression after baculovirus delivery of human Ku, XLF, XRCC4, and LIG4WT into Hi-5 insect cells have been previously described (Nick McElhinny et al., 2005; Roberts et al., 2010). LIG4ΔI was generated by modifying LIG4WT as noted in Figure 1A and validated by sequencing. The LIG3+4 chimera was generated by replacing amino acids 1–638 of LIG4WT with a fusion of amino acids 170–862 of human LIG3 to the linker (GGGGS)x3 (Genewiz). Cell pellets were extracted, lysed by sonification, and purified by sequential chromatography on Histrap and MonoQ columns (GE Biosciences). Figure 1A structures were prepared in Pymol and include hLIG1 (1X9N; Pascal et al., 2004), hLIG3 (3L2P; Cotner-Gohara et al., 2010), and hLIG4 (3W1B; Ochi et al., 2013), with disordered insert1 modeled by the SWISSmodel server (Biasini et al., 2014).
In vitro Joining Assays
NHEJ reactions were initiated by incubating 2 nM DSB substrates, 25 nM Ku, 40 nM XLF, and 40 nM XRCC4-LIG4 in a buffer with 25 mM Tris pH 7.5, 100 uM EDTA, 1 mM DTT, 5 mM MgCl, 100 uM ATP, 150 mM KCl, 8.5% polyethelene glycol 3000, and 100 ng of supercoiled plasmid DNA. Reactions were carried out for 10 minutes at 37 C and stopped with 0.1% SDS and 20 mM EDTA. Repair products were purified by phenol-chlorform extraction and recovery was measured by real-time PCR (qPCR) using a QuantStudio 6 system (Applied Biosciences), primers that amplify head-to-tail junctions (Table S1), and VeriQuest SYBR Green master mix (Affymetrix). The relative numbers of molecules recovered were quantified by a well characterized qPCR assay (Pryor et al., 2015; Waters et al., 2014b).
For nick sealing assays, a 5’Cy5 labeled, nicked 41 bp substrate was generated by annealing three oligonucleotides (Table S1). 5 nM substrate was incubated with XRCC4-LIG4 at 37 C and products were characterized by denaturing polyacrylamide gel electrophoresis (PAGE). Wild-type XRCC4-LIG4 was titrated to determine that 0.5 nM ligase (1:10 enzyme:substrate) generates sub-saturating (19–21%) amounts of nick sealing in 10 minutes, after which reactions were carried out under these conditions in triplicate to generate data presented in Figure S1B. Reaction velocity was determined by quantifying band intensities using ImageJ software.
Electrophoretic Mobility Shift Assay (EMSA)
Substrates for EMSA were generated by annealing oligonucleotides (Table S1) to produce a Cy5 labeled 15 bp substrate to assess DSB end binding (Supplementary Figure 1C), as well as a Cy5 labeled, 60 bp substrate to assess complex formation (Supplementary Figure 1D). To assess intrinsic end binding, the 15 bp substrate was incubated at 10 nM with 125, 250, or 500 nM XRCC4-LIG4. For complex formation, the 60 bp substrate was incubated at 10 nM with 2 nM Ku, 40 nM XLF, and 40 nM XRCC4-LIG4. These samples were incubated for 20 min on ice in EMSA buffer (50 mM NaCl, 75 mM KCl, 25 mM Tris pH 8, 13% Glycerol, and 0.015% X100). Samples were run on 4% (Supplementary Figure 1C) or 6% (Supplementary Figure 1D) polyacrylamide gels in 0.5x TBE buffer and imaged using a Typhoon (GE).
smFRET Assays
SmFRET assays and analysis were performed as described previously (Reid et al., 2015, 2016). Briefly, NHEJ reactions composed of 50 nM Ku, LX, XLF gloxy (0.5 mg/mL glucose oxidase and 0.4 µg/mL catylase), and 1 nM dsDNA were added stepwise to NEB4 (20 mM pH 7.5 TricAc, 50 mM KAc, 10 mM MgAc) supplemented with 0.8% glucose, ~5 mM Trolox, 1 mg/mL BSA, and 2 mM DTT. The reaction was immediately flowed into an imaging chamber that had been prepared with surface dsDNA (~250 pM). Movies consisting of 1000 frames (33Hz) were acquired for analysis of PECs. Trajectory analysis, histogram assembly, and autocorrelation of PECs were performed in Matlab (Reid et al. 2016). Oligonucleotides used in smFRET experiments are detailed in Table S1.
Cell Lines
LIG4−/− cells were generated from parental HCT116 human colorectal cancer cells by conventional gene targeting and were the gift of Dr. Eric Hendrickson (Oh et al., 2013). We generated additional variants of the parental cells by CRISPR/Cas9 gene targeting. We introduced by electroporation plasmids to express Cas9 (Addgene 44758; Shen et al., 2013; 5 µg) and an sgRNA (Addgene 51133; Shen et al., 2014; 5 µg; guide sequence described in Table S1) that targets insert1-encoding sequence from wild-type LIG4, as well as a gene-targeting donor plasmid. The donor plasmid was engineered such that it contains 1.1kb of sequence identical to the LIG4 gene except as modified such that gene targeting ablates the sgRNA target site, generates the LIG4ΔI mutation as described in Figure 1A, and introduces synonymous mutations that result in a BsmFI site used for screening. The native LIG4 sequence in this region and resulting LIG4ΔI alleles are described in Supplementary Figure 3 and Table S1. Targeted puromycin resistant clones were identified by amplification of the insert1 region using primers specific to the native locus (i.e. originate outside of donor sequence identity). Two independently generated clones, LIG4ΔI/ΔI a and b, were produced that possessed only targeted alleles after sequencing (Supplementary Figure 3A). To generate LIG4+r/+r reverted cells we repeated gene targeting, but started with LIG4ΔI/ΔI a cells and used an sgRNA specific for the LIG4ΔI allele (Table S1) as well as a gene-targeting donor with wild-type sequence in this region. We verified LIG4 expression in all of these cell lines using standard western blot techniques (Supplementary Figure 3B) and antibodies against human LIG4 (Serotec cat no. AHP554) and human Ku70 (Abcam cat no. ab62820). All 5 cell lines were cultured in McCoy’s 5A medium (Corning) with 10% fetal bovine serum (Sigma) and determined to be free of mycoplasma contamination by PCR (Uphoff and Drexler, 1999); we additionally employed a third party to validate the absence of mycoplasma by an alternate method for a randomly selected cell line (Hoechst staining; Battaglia et al., 1980).
Cellular NHEJ Assays
Extrachromosomal DNA substrates described above (20 ng) were electroporated into 2×105 cells with pMAX-GFP plasmid (600 ng) at 1350 V in one 30 ms pulse in 10 µL (Neon, Invitrogen). Transfected cells were incubated for 30 minutes in antibiotic-free McCoy’s 5A media with 10% fetal bovine serum. Cellular repair products were harvested using a QIAamp DNA mini kit (Qiagen). Each electroporation was reproduced in triplicate from 3 independent preparations of cells. Repair efficiency was quantified by qPCR as described above for in vitro joining assays.
Repair product structures were determined by restriction digest for the 8-oxoguanine (2-Amino-7,9-dihydro-1H-purine-6,8-dione; Go) substrate, and by high-throughput sequencing for all other substrates. For the Go substrate, harvested repair products were amplified with Cy5-labeled primers (Table S1) and digested with BstZ17I (New England Biolabs; recognizes transversion mutation after amplification of Go) and BamHI (New England Biolabs; recognizes accurately amplified Go) to identify directly ligated products. The intensities of digested and undigested bands were quantified using ImageJ.
To determine repair product structures of all other substrates, sequencing libraries were prepared by PCR amplification of repair products with primers containing 6-nucleotide indices on their 5’ ends (Table S1). Amplified DNA (40 ng per library) was pooled into groups of 8–12 libraries, 5’ phosphorylated, and treated with Klenow exo-(NEB) to add dA to the 3’ termini. Ends were ligated to adapters for paired-end sequencing (Illumina). Pooled libraries were purified from 3% agarose gels to remove unligated adapters using the QIAquick gel extraction kit (Qiagen). Recovered samples were amplified for an additional 9 cycles using enrichment primers (Illumina). Products were again purified using Ampure XP beads (Beckman Coulter). 27.27 ng from each of the 11 pools was combined (for a total of 300 ng of sample), supplemented with PhiX174 (40% final concentration) and submitted to the UNC high throughput sequencing facility for a 2×75-bp MiSeq run (Illumina). Genomics Workbench was used to remove PhiX174 DNA, merge read pairs, de-index libraries, and remove low quality sequences (CLC-Bio). Remaining sequences were analyzed using Microsoft Excel.
Colony Formation and Cell Growth Assays
For colony formation assays, seeding densities were determined independently for each dose and cell line such that 50–150 colonies would be produced per 10 cm dish. Cells were plated on 10 cm dishes in fresh McCoy’s 5A media with 10% fetal bovine serum, incubated for 4 hours, and then irradiated with indicated doses of X-rays using a RS 2000 irradiator (Rad Source Technologies). Colonies formed after 14 days were stained with a solution of crystal violet (0.5%) and glutaraldehyde (6%). Colonies were manually counted on three plates per dose and cell line. The surviving fraction of LIG4−/− cells treated with 3 Gy of X-rays was much less than 10-3, and was excluded from analysis because the resulting faint, small colonies could not be reliably discriminated above background debris staining.
For live cell imaging, 2000 cells were plated into 96-well plates in triplicate for each dose and cell line. After overnight incubation, cells were irradiated or treated with etoposide and placed into the IncuCyte live cell imager (Essen biosciences). Four 215 mm2 images were taken per well at 10x objective every 4 hours for a total of 120 hours. The confluence of each image was determined by generating a confluence mask with IncuCyte software (Essen biosciences).
Statistical Analysis
For all experiments, means were tested for significance against a control (e.g. LIG4WT, LIG4+/+ cells) using two-tailed t-tests for single comparisons, one-way ANOVA for multiple comparisons, and two-way ANOVA for comparisons with multiple variables. Dunnett’s correction for testing multiple hypotheses was applied as necessary. For each experiment, the value and definition of n, the representation of error bars, the specific tests used, the specific control tested, and the determination of statistical significance are described in the figure legends.
Supplementary Material
Highlights.
Mobility of DNA ends aligned by NHEJ factors increases when ends are mispaired.
Increased end mobility requires insert1, a motif unique to the NHEJ ligase (LIG4).
End mobilization is also essential for cellular repair of damaged and mispaired ends.
This mechanism explains the remarkable flexibility of NHEJ in repair of diverse ends.
Acknowledgments
M. C. was supported by F31CA203156 and T32GM007092. M. C., D. Ramsden, and G. S. were supported by CA084442. E. R. and D. Reid were supported by GM108118. M. R. L., H. H. C., and G. W. were supported by CA100504 and GM118009. We thank Dr. Eric Hendrickson for providing LIG4+/+ and LIG4−/− cell lines used in this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
M. C., D. Reid, E. R., and D. Ramsden prepared the manuscript and designed the experiments described therein. Experiments were performed by M. C., D. Reid, G. S., M. R. L., H. H. C. and G. W. Data were analyzed by M. C., D. Reid, E. R., and D. Ramsden.
We declare no conflicts of interest.
References
- Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. Crystal Structure of Human XLF: A Twist in Nonhomologous DNA End-Joining. Mol. Cell. 2007;28:1093–1101. doi: 10.1016/j.molcel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012;40:1868–1878. doi: 10.1093/nar/gks022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia MF, Balducci L, Finocchiaro M. Use of Hoechst 33258 fluorochrome for detection of mycoplasma contamination in cell cultures: development of a technique based on simultaneous fixation and staining. Boll. Ist. Sieroter. Milan. 1980;59:155–158. [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- Budman J, Kim SA, Chu G. Processing of DNA for nonhomologous end-joining is controlled by kinase activity and XRCC4/ligase IV. J. Biol. Chem. 2007;282:11950–11959. doi: 10.1074/jbc.M610058200. [DOI] [PubMed] [Google Scholar]
- Chang HHY, Watanabe G, Gerodimos CA, Ochi T, Blundell TL, Jackson SP, Lieber MR. Different DNA End Configurations Dictate Which NHEJ Components Are Most Important for Joining Efficiency. J. Biol. Chem. 2016;291:24377–24389. doi: 10.1074/jbc.M116.752329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotner-Gohara E, Kim I-K, Hammel M, Tainer JA, Tomkinson AE, Ellenberger T. Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states. Biochemistry. 2010;49:6165–6176. doi: 10.1021/bi100503w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottarel J, Frit P, Bombarde O, Salles B, Négrel A, Bernard S, Jeggo PA, Lieber MR, Modesti M, Calsou P. A noncatalytic function of the ligation complex during nonhomologous end joining. J. Cell Biol. 2013;200:173–186. doi: 10.1083/jcb.201203128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BJ, Havener JM, Ramsden DA. End-bridging is required for pol to efficiently promote repair of noncomplementary ends by nonhomologous end joining. Nucleic Acids Res. 2008;36:3085–3094. doi: 10.1093/nar/gkn164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberger T, Tomkinson AE. Eukaryotic DNA Ligases: Structural and Functional Insights. Annu. Rev. Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Herreros F, Romero-Granados R, Zeng Z, lvarez-Quiló A, Quintero C, Ju L, Umans L, Vermeire L, Huylebroeck D, Caldecott KW, et al. TDP2–Dependent Non-Homologous End-Joining Protects against Topoisomerase II-Induced DNA Breaks and Genome Instability in Cells and In Vivo TDP2-Dependent Non-Homologous End-Joining Protects against Topoisomerase II-Induced DNA Breaks and Genome Instability in Cells and. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TGW, Walter JC, Loparo JJ. Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Mol. Cell. 2016;61:850–858. doi: 10.1016/j.molcel.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco GE, Matsumoto Y, Brooks RC, Lu Z, Lieber MR, Tomkinson AE. SCR7 is neither a selective nor a potent inhibitor of human DNA ligase IV. DNA Repair (Amst) 2016;43:18–23. doi: 10.1016/j.dnarep.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lu H, Tsai AG, Schwarz K, Lieber MR. Single-stranded DNA ligation and XLF-stimulated incompatible DNA end ligation by the XRCC4-DNA ligase IV complex: influence of terminal DNA sequence. Nucleic Acids Res. 2007;35:5755–5762. doi: 10.1093/nar/gkm579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, Wilson TE, Grawunder U. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair PA, Nandakumar J, Smith P, Odell M, Lima CD, Shuman S. Structural basis for nick recognition by a minimal pluripotent DNA ligase. Nat. Struct. Mol. Biol. 2007;14:770–778. doi: 10.1038/nsmb1266. [DOI] [PubMed] [Google Scholar]
- Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol. Cell. Biol. 2000;20:2996–3003. doi: 10.1128/mcb.20.9.2996-3003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Havener JM, Garcia-Diaz M, Juárez R, Bebenek K, Kee BL, Blanco L, Kunkel TA, Ramsden DA. A Gradient of Template Dependence Defines Distinct Biological Roles for Family X Polymerases in Nonhomologous End Joining. Mol. Cell. 2005;19:357–366. doi: 10.1016/j.molcel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Wu Q, Chirgadze DY, Grossmann JG, Bolanos-Garcia VM, Blundell TL. Structural insights into the role of domain flexibility in human DNA ligase IV. Structure. 2012;20:1212–1222. doi: 10.1016/j.str.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Gu X, Blundell TL. Structure of the Catalytic Region of DNA Ligase IV in Complex with an Artemis Fragment Sheds Light on Double-Strand Break Repair. Structure. 2013;21:672–679. doi: 10.1016/j.str.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, Travers J, Wu Q, Draviam VM, Robinson CV, et al. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science (80-.) 2015;347:185–188. doi: 10.1126/science.1261971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Wang Y, Zimbric J, Hendrickson EA. Human LIGIV is synthetically lethal with the loss of Rad54B-dependent recombination and is required for certain chromosome fusion events induced by telomere dysfunction. Nucleic Acids Res. 2013;41:1734–1749. doi: 10.1093/nar/gks1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- Pryor JM, Waters CA, Aza A, Asagoshi K, Strom C, Mieczkowski PA, Blanco L, Ramsden DA. Essential role for polymerase specialization in cellular nonhomologous end joining. Proc. Natl. Acad. Sci. 2015;112:E4537–E4545. doi: 10.1073/pnas.1505805112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid DA, Keegan S, Leo-Macias A, Watanabe G, Strande NT, Chang HH, Oksuz BA, Fenyo D, Lieber MR, Ramsden DA, et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc. Natl. Acad. Sci. 2015;112:E2575–E2584. doi: 10.1073/pnas.1420115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid DA, Conlin MP, Yin Y, Chang HH, Watanabe G, Lieber MR, Ramsden DA, Rothenberg E. Bridging of double-stranded breaks by the nonhomologous end-joining ligation complex is modulated by DNA end chemistry. Nucleic Acids Res. 2016;45:1872–1878. doi: 10.1093/nar/gkw1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, Hasty P, Ramsden DA. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature. 2010;464:1214–1217. doi: 10.1038/nature08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DB, Menetski JP, Nakajima PB, Bosma MJ, Gellert M. V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell. 1992;70:983–991. doi: 10.1016/0092-8674(92)90248-b. [DOI] [PubMed] [Google Scholar]
- Roy S, de Melo AJ, Xu Y, Tadi SK, Négrel A, Hendrickson E, Modesti M, Meek K. XRCC4/XLF Interaction Is Variably Required for DNA Repair and Is Not Required for Ligase IV Stimulation. Mol. Cell. Biol. 2015;35:3017–3028. doi: 10.1128/MCB.01503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, Zhang X, Zhang P, Huang X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013;23:720–723. doi: 10.1038/cr.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- Tomkinson AE, Howes TRL, Wiest NE. DNA ligases as therapeutic targets. Transl. Cancer Res. 2013;2 [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, Kim SA, Chu G. Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7851–7856. doi: 10.1073/pnas.0702620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff CC, Drexler HG. Detection of mycoplasma contaminations in cell cultures by PCR analysis. Hum. Cell. 1999;12:229–236. [PubMed] [Google Scholar]
- Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA. Nonhomologous end joining: a good solution for bad ends. DNA Repair (Amst) 2014a;17:39–51. doi: 10.1016/j.dnarep.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters Ca, Strande NT, Pryor JM, Strom CN, Mieczkowski P, Burkhalter MD, Oh S, Qaqish BF, Moore DT, Hendrickson EA, et al. The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nat. Commun. 2014b;5:4286. doi: 10.1038/ncomms5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, Ning S, Yan Z, Li W, Wang Q, et al. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat. Commun. 2015;6:6233. doi: 10.1038/ncomms7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.