

Abstract
Significance: This review provides current overview of the emerging role of innate immunity in driving fibrosis, and preventing its resolution, in scleroderma (systemic sclerosis, SSc). Understanding the mechanisms of dysregulated innate immunity in fibrosis and SSc will provide opportunities for therapeutic interventions using novel agents and repurposed existing drugs.
Recent Advances: New insights from genomic and genetic studies implicate components of innate immune signaling such as pattern recognition receptors (PRRs), downstream signaling intermediates, and endogenous inhibitors, in fibrosis in SSc. Recent studies distinguish innate immune signaling in tissue-resident myofibroblasts and bone marrow–derived immune cells and define their roles in the development and persistence of tissue fibrosis.
Critical Issues: Activation of toll-like receptors (TLRs) and other PRR mechanisms occurs in resident nonimmune cells within injured tissue microenvironments. These cells respond to damage-associated molecular patterns (DAMPs), such as tenascin-C that are recognized as danger signals, and elicit matrix production, cytokine secretion, and myofibroblast transformation and survival. When these responses persist due to constitutive TLR activation or impaired termination by endogenous inhibitors, they interfere with fibrosis resolution. The genetic basis and molecular mechanisms of these phenomena in the context of fibrosis are under current investigation.
Future Directions: Precise delineation of the pathogenic DAMPs, their interaction with TLRs and other PRRs, the downstream signaling pathways and transcriptional events, and the fibroblast-specific regulation and function of endogenous inhibitors of innate immunity, will form the foundation for innovative targeted therapies to block fibrosis by reestablishing balanced innate immune signaling in fibroblasts.
Keywords: : fibrosis, systemic sclerosis, SSc, fibroblast, DAMP, TLR, fibronectin-EDA, tenascin-C, A20, TNFAIP3/A20

Swati Bhattacharyya, PhD

John Varga, MD
Scope and Significance
This review critically reviews emerging insights into the pathogenesis of fibrosis in systemic sclerosis (SSc). We discuss a novel paradigm implicating the innate immune system and its endogenous (sterile) triggers as key factors not only in initiating fibroblast activation but also in preventing reversion to a quiescent state. Thus, persistence of toll-like receptor (TLR)–mediated innate immune signaling in myofibroblasts and other mesenchymal resident cells appears to underlie the switch from a self-limited tissue repair response to persistent activation and nonresolving pathological fibrosis characteristic of SSc. Indeed, the relevance of this paradigm is strongly supported by genetic studies that identify numerous genes associated with SSc that are related to innate immune signaling and genomic studies revealing altered signatures in lesional skin and lungs reflecting innate immune activation. We particularly highlight data indicating that TLR signaling in resident stromal cells elicits overlapping but also distinct responses than in classical immune cells, with a primary commitment to rapid tissue repair and wound healing.
Translational Relevance
The emerging understanding of the regulation, mechanisms, and roles of stromal cell innate immune signaling opens new window into fibrosis research and provides exciting new opportunities for linking SSc-associated genetic factors to disease mechanisms. Moreover, these findings present numerous novel targets for therapeutically interrupting aberrant fibrotic signaling through innate immune receptors.
Clinical Relevance
Measuring the levels and distribution of pathogenic damage-associated molecular patterns (DAMPs) in target tissues and in the circulation might represent novel biomarkers for quantitating and monitoring fibrosis in SSc and for identifying patient subsets likely to benefit from interventions targeting the innate immune system. Defining the interaction of pathogenic DAMPs with their pattern recognition receptors (PRRs) in myofibroblasts can facilitate the design of small molecules and antibodies to selectively interrupt such interactions, while delineating downstream signaling pathways that lead to TLR-dependent fibrosis can present novel targets to selectively disrupt fibrotic signaling and speed fibrosis resolution without interfering with antimicrobial immunity.
Introduction
Organ fibrosis is the hallmark of SSc
SSc is a chronic autoimmune disease characterized by interstitial and perivascular fibrosis. In contrast to more common organ-based fibrotic diseases, fibrosis in SSc synchronously affects the skin and multiple internal organs. The disease occurs more commonly in women, has a worldwide distribution, and shows a complex multigenic inheritance pattern.1 There are no therapies effective in halting or resolving fibrosis. Current immunomodulatory treatments that are highly effective in rheumatoid arthritis and other inflammatory diseases generally show only modest and variable efficacy in SSc, and at best slow disease progression. Fibrosis, defined as aberrant accumulation of extracellular matrix (ECM) disrupting tissue structure and function, is the distinguishing hallmark of SSc. The fibrotic process in SSc is easily recognized in the skin, but it is fibrosis affecting the internal organs, most commonly the lungs, that contribute to the high mortality of SSc.2 In addition, fibrosis in the myocardium, gastrointestinal tract, tendons and muscles, and renal interstitium can also be frequent and prominent and contributes to disease burden and mortality.3
Vascular injury and evidence of cellular and humoral autoimmunity, including activated T and B cell, dendritic cells, and macrophages, are prominent in SSc, particularly in patients with early-stage disease. However, later stages of SSc are dominated by tissue fibrosis, and inflammatory cell infiltration in target organs is sparse. Understanding how the autoimmune/inflammatory, vascular, and fibrotic processes network in SSc to dictate disease evolution remains a major challenge in the field.4 Current thinking suggests a model for pathogenesis whereby exposure of genetically susceptible individuals to certain forms of environmental or toxic injuries elicits inflammation, which together with vascular injury, triggers a cascade leading to fibroblast activation and nonresolving fibrosis (Fig. 1).
Figure 1.
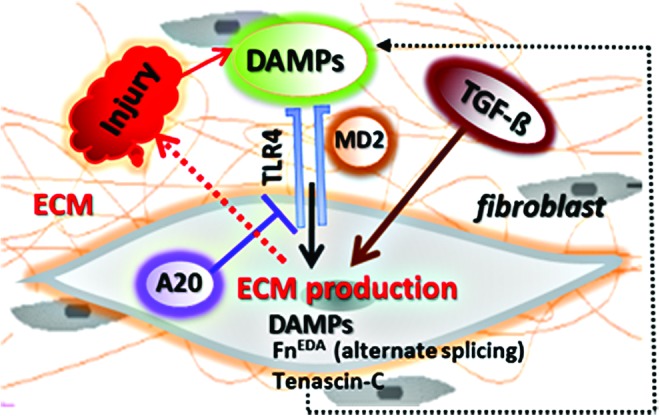
Vicious cycle of TLR4-driven fibrosis. Tissue damage from chronic injury causes local generation and accumulation of endogenous TLR4 ligands called “damage-associated molecular patterns (DAMPs)” such as fibronectin-EDA and tenascin-C. DAMPs in turn activate innate immune signaling in resident fibroblasts through TLR4. This results in enhanced matrix production and TGF-beta secretion, establishing a self-amplifying vicious cycle of fibrosis. Moreover, cell-intrinsic protective mechanisms that normally act as the brakes on fibroblast activation, such as A20/TNFAIP3, are hypofunctional in SSc, further aggravating unopposed TLR4 signaling and fibrogenesis. DAMP, damage-associated molecular pattern; TLR, toll-like receptor; TGF, transforming growth factor; SSc, systemic sclerosis. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Inflammatory signaling is tightly linked to fibrogenesis in SSc
Activation of tissue-resident quiescent fibroblasts and their in situ phenoconversion into biosynthetic and apoptosis-resistant myofibroblasts, as well as transition of various mesenchymal progenitor cell types (endothelial cells, pericytes, and adipose-derived progenitors) into myofibroblasts, accounts for the persistence of fibrosis, even in the absence of inflammation. The origins of fibrotic myofibroblasts, and the cues and mechanisms involved in their transdifferentiation and persistence, have been the subject of controversy, with data deriving largely from various animal models of organ fibrosis and differing among distinct types of organ fibrosis5; nevertheless, the fundamental pathogenic roles of transforming growth factor-β (TGF-β), Wnt/β-catenin, and cytokines, such as IL-13, oxidative stress, and biomechanical signals, across multiple types of organ fibrosis are supported by substantial experimental evidence.6
Genetic studies during the past decade have contributed to a better understanding of the immunity-fibrosis link in SSc pathogenesis. These studies have identified a substantial number of polymorphic loci associated with the risk of SSc.7 The most prominent of these genetic associations are within the major histocompatibility complex (MHC). The great majority of the non-MHC variants associated with SSc are related to immune cell activation and function, including T and B cell signaling, along with innate immunity, type I interferon, and toll-like receptor (TLR) signaling. It is remarkable that a majority of SSc-associated genetic loci are shared in common with other autoimmune diseases, particularly systemic lupus erythematosus.8 An especially intriguing genetic finding is the transethnic association of SSc with both synonymous and noncoding variants of TNFAIP3, which encodes the deubiquitinase enzyme A20, a prototypic intracellular inhibitor of TLR signaling.8–10 At the genomic level, pioneering transcriptome microarray studies carried out by the Whitfield laboratory demonstrated that ∼50% of skin biopsies from patients with diffuse cutaneous SSc show a very robust inflammatory gene signature that appears to be independent of disease duration.11 Many of the genes upregulated in these skin biopsies are related to the “interferon signature” and/or are involved in innate immunity and TLR signaling. It is noteworthy that inflammatory gene signatures are equally prominent in SSc skin biopsies from patients with both early- and late-stage disease, indicating that evidence of enhanced inflammatory gene expression in lesional tissue is not simply a function of disease stage.11 Finally, network analysis integrating SSc-associated genetic risk loci and differentially regulated genes in the skin underlines the intimate relationship between innate immunity fibrotic responses elicited by TGF-β and Wnt/β-catenin and ECM gene expression.12
Notwithstanding compelling genetic and genomic evidence pointing to the fundamental pathogenic role for inflammatory/autoimmune processes in SSc and the sometime dramatic beneficial clinical response seen following myeloablative therapy of SSc,13 the contribution of bone marrow–derived inflammatory cells to SSc fibrosis continues to be enigmatic and controversial, and biopsies from SSc patients with established disease are largely pauci-immune. Our laboratory has been interested in dissecting the cellular and molecular networks underlying self-sustaining fibroblast activity in SSc and the impact of inflammation in triggering and maintaining this process. Our recent studies have provided novel insights into fibrosis and revealed therapeutic targets for preventing fibrosis progression and promoting resolution. In particular, the results point to an unexpected function for innate immune signaling in fibroblasts triggered by endogenous DAMPs and mediated by TLRs. Despite substantial recent progress, however, the pathogenic roles, mechanisms, and regulation of DAMP-TLR signaling in the context of fibrosis and SSc remain inadequately defined. In this review, we present current understanding of the DAMP-TLR4 axis in fibroblasts and its pathogenic roles in unresolving fibrosis in SSc. We also discuss the application of these insights to the development of therapeutic and precision medicine approaches for targeting TLRs in fibrosis.
A Brief Overview of TLR Signaling
The first line of host defense is the innate immune system comprising a diverse set of PRRs. These receptors recognize both extrinsic pathogen-derived molecule patterns (PAMPs) from Gram-negative bacteria (lipopolysaccharide, LPS) or viral nucleic acids, as well as endogenous damage-associated molecule patterns (DAMPs) such as the ECM proteins tenascin-C and fibronectin-EDA that are generated within injured microenvironments.14 The best-characterized PRRs in addition to TLRs include nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), and the cGAS-STING pathway. Upon PAMP or DAMP recognition, PRRs stereotypically induce activation of NF-κB and interferon response factors (IRFs), resulting in the secretion of interferons and pro-inflammatory cytokines.15
The mammalian TLR family consists of 12 members, with each possessing a leucine-rich repeat (LRR) ligand-binding domain and a signaling Toll/Interleukin-1 (IL-1) receptor homology (TIR) domain. Some TLRs (TLR1, 2, 4, 5, 6, and 10) localize to the cell surface, while TLR3, 7, 8, 9, 11, and 13 are located within intracellular compartments.16 Binding of TLR ligand to the extracellular LRR region causes TLR dimerization and conformational changes of the intracellular TIR domains, and recruitment and activation of adaptor molecules MyD88, and/or TRIF, which then transduce signals. The MyD88-dependent signaling pathway recruits IRAKs and activates TRAF6 and the IκB kinase (IKK), leading to NF-κB activation. This pathway is negatively controlled by the deubiquitinase A20. The MyD88-independent TRIF signaling pathway culminates in the activation of both IRF3 and NF-κB. TLR4 is the only TLR that engages both MyD88 and TRIF for intracellular signaling.17
Endogenous TLR ligands
In response to LPS, the hallmark PAMP, TLR4 forms a complex with its coreceptor MD2. Structural studies show that five of the six LPS lipid chains bind to the hydrophobic pocket of MD2, while the remaining lipid chain associates directly with TLR4.18 There is an extensive, and growing, list of DAMPs that have been shown to serve as endogenous TLR4 ligands.19 These DAMPs, representing a diverse group of molecular responses to tissue damage, include single or double-stranded self-nucleic acids, fatty acids, serum amyloid A (SAA), ECM fragments, and alternatively-spliced “oncofetal” variants of normal ECM components. In contrast to LPS, DAMPs may not require the same coreceptors and accessory molecules to achieve signaling-competent TLR4 conformation. It is worth noting that many of the DAMPs were implicated as putative endogenous TLR4 ligands based on coimmunoprecipitation or functional cell-based assays in vitro, or using mutant mice deficient in TLRs or their adaptor proteins in vivo. No crystal structures of DAMP-TLR4 complex have been reported so far to confirm direct DAMP-TLR4 interactions or the requirements of specific coreceptors for the formation of active TLR signaling complexes.
One of the best studied endogenous TLR4 ligands linked to SSc fibrosis is the large modular glycoprotein tenascin-C.20,21 An intriguing recent study asked whether the TLR4-dependent signaling pathways and biological readouts elicited in macrophages by the tenascin-C fibrinogen-like globe (FBG) domain (Fig. 2) were similar to those elicited by LPS.22 Pursuing a comparative analysis of the signaling pathways and biological outcomes, the authors found that TLR4 activation elicited by LPS and FBG generated two distinct macrophage phenotypes, with only partially-overlapping sets of activation markers, secreted effector molecules, and phosphoproteomic profiles.22 FBG promoted a TLR4-dependent “profibrotic” macrophage phenotype, whereas LPS promoted a phenotype with enhanced matrix-degrading capacity. These observations provide evidence that different microenvironmental cues can elicit distinct biological responses through the same receptor. Whether TLR4 activation elicited by LPS versus DAMPs will generate similarly divergent responses in fibroblasts remains an important unanswered question with relevance to fibrosis.
Figure 2.

Fibronectin and tenascin-C modular structures. (A) Fibronectin exists as a protein dimer. Three regions of variable splicing occur along the length of the fibronectin. One or both of the “extra” type III modules EDA (between 7 and 8) and EDB (between 11 and 12) may be present in cellular fibronectin (FnEDA and FnEDB) and another connecting segment (CS, between the 14th and 15th type III module). EDA and EDB splicing is similar in all species, while that of the type III CS region is species specific. (B) Tenascin-C is a hexameric extracellular glycoprotein composed of multiple fibronectin TNIII repeats and a C-terminal FBG. Alternative splicing in TNIII (between 5 and 6 type III module) generates small and large tenascin-C isoforms. FBG, fibrinogen-like globe. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Altered TLR4 Signaling in SSc Fibrosis
In light of the genetic and genomic evidence suggesting a pathogenic role for dysregulated innate immunity and TLR activity in SSc, we undertook a series of studies focusing on the TLR ligand-receptor axis in patients with SSc and in animal models of disease. These studies demonstrated that levels of both TLR4, as well as certain endogenous TLR ligands, are elevated in skin and lung tissues from patients with SSc and correlate with clinical disease parameters23,24 (Fig. 3). For instance, tissue levels of TLR4 and its coreceptors MD2 and CD14 are elevated in SSc, and levels predict severity and/or progression of skin disease.23,24 Prominent TLR4 immunostaining was seen in skin and lung biopsies from patients with SSc, and TLR4 largely colocalized with myofibroblasts, as well as infiltrating macrophages and vascular cells within lesional tissue (Fig. 3A).23 Moreover, treatment of fibroblasts by LPS or by endogenous TLR4 ligands elicits potent fibrosis-related gene expression and phenoconversion into alpha smooth muscle actin–positive myofibroblasts (Fig. 4). Conversely, genetic targeting of TLR4 or its endogenous “damage-associated” ligands, or pharmacological disruption of signaling with drugs that block TLR4 or its coreceptor MD2, ameliorates progressive tissue fibrosis in various disease models.
Figure 3.
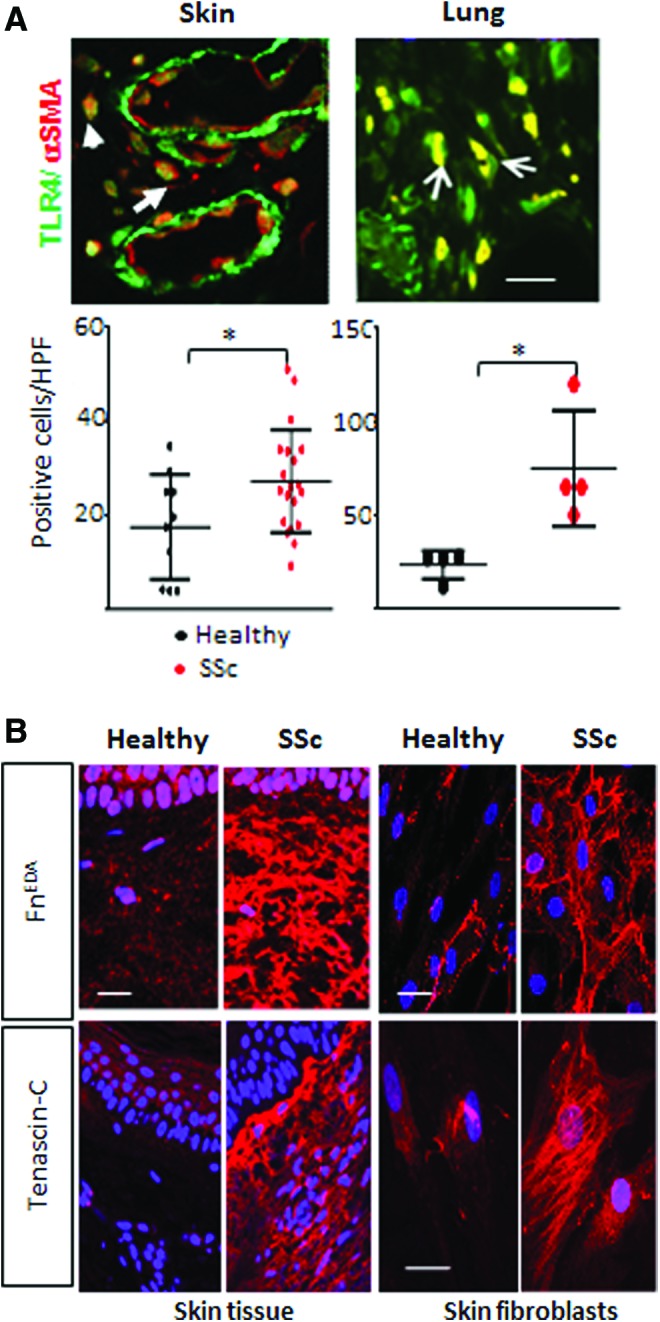
Elevated TLR4 and endogenous TLR4 ligand DAMP expression in SSc biopsies and fibroblasts. (A) Upper panels, immunofluorescence of skin and lung biopsies from SSc patients with pulmonary fibrosis. Yellow, colocalization of the two antibodies (white arrows). Scale bars: 50 μm. Bottom panels, quantitation of TLR4 staining. Each dot, number (mean) of immunopositive fibroblasts from four separate microscopic fields per biopsy (*p < 0.05). (B) Left panels, immunofluorescence of SSc and healthy control skin biopsies. Right panels, SSc fibroblasts showed increased FnEDA and tenascin-C production ex vivo compared to healthy controls. Scale bars: 50 μm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Figure 4.
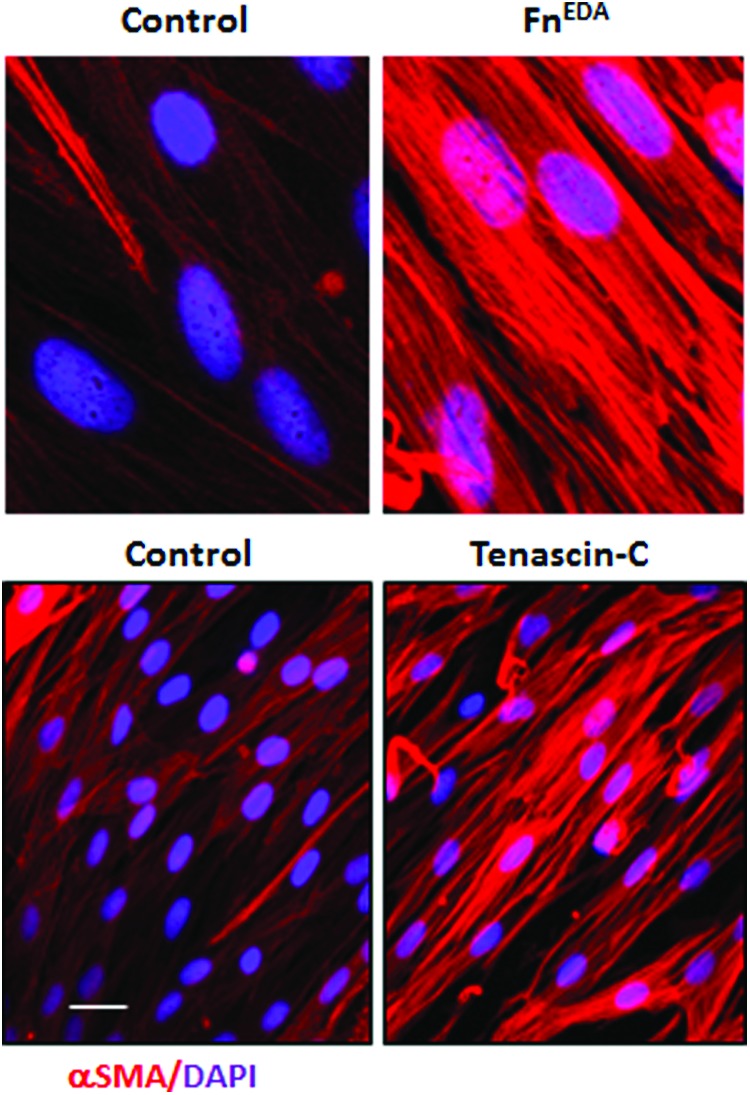
Endogenous TLR4 ligand DAMPs promote myofibroblast differentiation in normal fibroblasts. Human skin fibroblasts were incubated in medium with FnEDA or tenascin-C for 72 h. Immunofluorescence showed marked increase in myofibroblast differentiation compared to untreated control fibroblasts. Scale bar, 50 μm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Transcriptome analysis of SSc skin biopsies revealed substantial molecular heterogeneity among different patients.25,26 Cluster analysis reveals distinct subsets that are termed fibroproliferative, inflammatory, limited, and normal like. The inflammatory intrinsic subset, which accounts for a substantial portion of skin biopsies from patients with diffuse cutaneous SSc and was independent of disease duration, demonstrated upregulation of many genes involved in innate immunity. In contrast, the fibroproliferative, limited, and normal-like subsets did not show such associations.11 Relative paucity of immune cells within biopsies from patients with established SSc suggests that the inflammatory gene signatures might originate primarily from resident stromal cells. To explore this hypothesis, we generated an experimentally-derived “fibroblast TLR4 (LPS)-regulated gene signature” using normal skin fibroblasts transfected with TLR4.23,27 Comparison of this fibroblast TLR4 gene signature with those generated from human monocytes indicated only a partial overlap of differentially expressed genes in the two cell types (hypergeometric test; p = 0.02) (Table 1). The fibroblast TLR4 gene signature showed significant enrichment with pathways related to wound healing, matrix remodeling, and TGF-β signaling, revealing important cell type–specific differences between TLR responses in bone marrow–derived inflammatory versus stromal cells (Table 1). Most interestingly, we found that SSc skin biopsies displaying a strong fibroblast TLR4 gene signature mapped to the inflammatory intrinsic gene subset (Bhattacharyya S. and Varga J., unpublished). These findings suggest that elevated inflammatory gene expression in SSc skin biopsies, particularly in patients with late-stage disease, might originate from activated fibroblasts. From a translational perspective, a strong fibroblast TLR4 signature in skin biopsies might represent a predictive biomarker that identifies SSc patients with ongoing fibroblast activity; such patients might be potentially more responsive to therapies that block TLR signaling. A similar predictive strategy might also be relevant for SSc-associated interstitial lung disease (ILD). The leading cause of death in patients with SSc ILD is a frequent complication that can show sustained stability or rapid progression.1 Lung biopsies from patients with SSc-ILD show upregulation of genes related to TLR and TGF-β signaling, similar to lesional skin.27
Table 1.
Biological processes designated by individual GO categories elicited by LPS in fibroblasts and macrophages
 |
(A) Confluent skin fibroblasts and macrophages treated with LPS for 24 h. At the end of incubation period, total RNA was harvested and processed for hybridizations to Illumina HumanHT-12 microarray chips. The significantly enriched GO categories elicited by LPS in fibroblasts and macrophages is shown. (B) Venn diagram showing numbers of differentially regulated genes in macrophages and fibroblasts. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
LPS, lipopolysaccharide.
We carried out a comparative analysis of SSc skin and lung transcriptomes to identify pathways shared between these two target organs. Pathways involved in positive and negative regulation of TLR signaling were most highly shared (Bhattacharyya S and Varga J; unpublished). The role of TLR4 in lung fibrosis appears to be complex and injury and cell type dependent, and the available evidence is contradictory. For instance, while bleomycin-induced lung fibrosis was ameliorated in TLR4-null and IRF5-null mice,28,29 a recent study demonstrated worse lung fibrosis and higher mortality in the absence of TLR4, attributed to failure of alveolar epithelial cell regeneration in the absence of TLR4.30,31 The findings suggest that epithelial cell TLR4 signaling might have a protective role during injury, and its absence results in worse fibrosis in organs such as the lung where postinjury regeneration is critically dependent of epithelial stem cell renewal. However, the conflicting data from these publications remain difficult to reconcile at the moment.
Mechanisms for the Profibrotic Responses Elicited By TLR4
In skin fibroblasts, TLR4 activation by LPS elicited global changes in gene expression.23 The response was dominated by genes involved in ECM remodeling, tissue repair, and wound healing, while changes in inflammatory genes were relatively modest. These results suggest distinct roles for TLR4 in inflammatory cells, where TLR4 serves to elicit a powerful inflammatory response designed to deal with invading microbial pathogens versus tissue-resident stromal/parenchymal cells, where TLR4 might have evolved primarily to promote robust repair of injured tissue.16,23 We found that TLR4 activation in explanted fibroblasts led to enhanced collagen synthesis and increased expression of genes involved in tissue remodeling and ECM homeostasis. Moreover, TLR4 dramatically enhanced the sensitivity of fibroblasts to the stimulatory effect of TGF-β. These profibrotic responses were abrogated by both genetic and pharmacological disruption of TLR4 signaling in vitro, and bleomycin-induced skin fibrosis was attenuated in mice harboring a mutated TLR4. Mechanistically, TLR4-dependent profibrotic fibroblast responses involved suppression of the endogenous TGF-β antagonist BAMBI (bone morphogenetic protein and activin membrane-bound inhibitor), resulting in unopposed TGF-β/Smad signaling.23,32 Moreover, levels of miR-29, a microRNA known to function as a negative regulator of fibrosis, were downregulated by both TLR4 activation and by TGF-β.23,33 Additional transcriptional mechanisms and epigenetic reprogramming underlying the persistent profibrotic TLR4 responses remain to be characterized.
Damps Acting as Endogenous TLR4 Ligands are Implicated in SSc Fibrosis
Sterile tissue injury results in the generation of DAMPs that enable cells to recognize and respond to danger.14 Persistent DAMP exposure however contributes to the pathogenesis of many inflammatory and autoimmune diseases. DAMPs encompass an extensive repertoire of molecules released by dying or damaged cells such as high mobility box 1 and heat shock proteins, self-nucleic acids, including single and double-stranded DNA, RNA, mitochondrial DNA, and nucleic acid-antibody complexes; SAA, fragments of ECM macromolecules, and “oncofetal” ECM macromolecule isoforms generated through alternative splicing. We performed an unbiased survey of putative endogenous TLR4 ligands detected in SSc skin biopsies.16 Using immunohistochemistry, we identified low molecular weight hyaluronic acid, alternately spliced fibronectin (FnEDA) isoform, and tenascin-C as most highly upregulated in the fibrotic skin.16,20,23,33 Normally, alternately spliced FnEDA and tenascin-C isoforms are detected in developing tissues during embryogenesis and subsequently decline. However, the ‘embryonic’ splicing pattern of these molecules is reestablished transiently during tissue repair and angiogenesis; in contrast, persistent re-expression of these isoforms is a hallmark of cancer and fibrosis.34,35
The Alternately-Spliced Fibronectin Isoform Acts as an Endogenous TLR4 Ligand with Potent Profibrotic Activity
Fibronectin is generated by alternate splicing from a 47-exon gene.34 Isoforms that include or exclude the EDA and EDB exons arise due to alternative splicing from a single fibronectin pre-mRNA. In adults, the protein occurs in two major forms: dimeric soluble plasma fibronectin (pFN), which lacks the EDA and EDB domains, and multimeric cellular fibronectin (cFN) that includes the EDA or EDB domains and is deposited within the ECM (Fig. 2). During wound healing, the EDA and EDB isoforms accumulate at the wound base.36 The inclusion or exclusion of EDA domain defines the ability of fibronectin to activate TLR4; recombinant EDA but not EDB can induce TLR4-dependent NF-kB signaling and cytokine synthesis.37–39
Since the EDA isoform of fibronectin had been previously shown to be required for the myofibroblast phenoconversion of normal fibroblasts,40 we sought to investigate its expression and regulation in SSc and its possible mechanisms of action in fibrosis. A series of studies revealed that FnEDA was selectively elevated in SSc skin biopsies, as well as in lesional tissues from mice with experimentally induced cutaneous fibrosis, and explanted SSc fibroblasts constitutively produced increased FnEDA ex vivo (Fig. 3B).33 Moreover, circulating FnEDA levels were elevated in patients with SSc. In subsequent studies we examined how FnEDA was regulated and how it affected normal fibroblasts. In normal fibroblasts, TGF-β induced an isoform-specific preferential upregulation of FnEDA.33 Serving as a bona fide endogenous TLR4 ligand, FnEDA elicited potent profibrotic responses, with enhanced synthesis of collagens and expression of the myofibroblast marker alpha smooth muscle actin. Moreover, similar profibrotic effects were elicited in fibroblasts cultured within a 3D human skin equivalent.33 Mice with genetic deletion of FnEDA showed no spontaneous phenotype, but were largely resistant to the induction of skin fibrosis.33 Other studies had shown that mice carrying a functionally hypomorphic TLR4 mutant23 or lacking TLR428 were similarly resistant to fibrosis. On the basis of these observations, we propose a model for persistent cutaneous fibrosis, where resident fibroblasts are chronically exposed to FnEDA acting as an endogenous TLR4 stimulus within the fibrotic microenvironment. In response, fibroblasts undergo TLR4-mediated activation and reprogramming, resulting in unopposed TGF-β signaling, enhanced profibrotic responses, and myofibroblast phenoconversion. Moreover, TLR4-dependent profibrotic responses in these cells include preferential production of the EDA isoform of fibronectin, along with other endogenous TLR4 ligands, which in turn drive further TLR4 activation. This potentially generates a cell-autonomous and inflammation-independent fibrosis amplification loop underlying persistent tissue fibrosis.
In addition to its alternate splicing, another mechanism for fibronectin to become a signaling-competent DAMP has been reported. It was shown that exposure of fibroblasts to FNIII1c, a stable unfolded intermediate of FNIII1, can elicit TLR4-dependent inflammatory signaling and induce cytokine synthesis.41,42 This region of the fibronectin molecule is exposed through tensional forces within the rigid fibrotic microenvironment. These observations have clear implications for fibrosis, since fibrotic tissue is characterized by increased stiffness and mechanical forces.43 We speculate that in SSc, the stiff matrix of fibrotic skin and lungs might drive exposure of the EDA domain of fibronectin, which, combined with increased generation of the EDA isoform through alternate splicing, results in augmented FnEDA bioavailability and profibrotic activity as endogenous TLR4 ligands.
Tenascin-C accumulates within fibrotic lesions and drives TLR4-dependent profibrotic responses
Tenascin-C is a multifunctional hexameric ECM glycoprotein that undergoes extensive alternate splicing to generate multiple isoforms.35 Normally tenascin-C is under tight spatial and temporal regulation. Tenascin-C is prominent in tissues during embryogenesis, but undetectable in most healthy adult tissues, and only transiently re-expressed during wound healing and dynamic tissue remodeling.35 By contrast, persistent expression of the large isoform of tenascin-C is seen in nonhealing wounds, autoimmune diseases, and cancer.35,44 The first tenascin-C exon sequence isolated from human glioblastoma identified a clone with eight consecutive FNIII like repeats between FNIII 5 and 6, providing genetic evidence of alternative splicing (Fig. 2).35
Our unbiased survey identified tenascin-C as one of the most highly upregulated ECM proteins in SSc skin biopsies.16 Further studies revealed elevated tissue levels of tenascin-C in SSc skin and lung biopsies, as well as in circulation; moreover, serum levels showed positive correlation with the modified Rodnan skin score, a measure of skin fibrosis.20 The antibodies used in these studies detect the FNIII-B and FNIII-C epitopes of the large tenascin-C isoforms (Fig. 2B). Normal fibroblasts treated with TGF-β and PDGF preferentially synthesize the large (∼250 kDa) tenascin-C variant detected with an antibody against FN III-B domain, while SSc fibroblasts constitutively produce the large tenascin-C isoform (Fig. 3B).20
Recent studies provide insight into the physiological and pathological regulation of tenascin-C alternate splicing. The proto-oncogene serine/arginine-rich splicing factor 6 (SRSF6) is an essential regulator of tenascin-C alternate splicing.45 Transgenic mice overexpressing SRSF6 in collagen-producing cells spontaneously develop marked scleroderma-like skin hyperplasia.45 This was accompanied by aberrant alternative tenascin-C splicing and accumulation of the “large” tenascin-C isoform in the skin. By regulating alternative splicing of tenascin-C, SRSF6 thus has a powerful effect on skin homeostasis in mouse models. Remarkably, we recently observed that the expression of SRSF6 was highly elevated in SSc skin biopsies and levels correlated with tenascin-C accumulation (Bhattacharyya S, Roberson E, Varga J; unpublished). Moreover, RNA sequencing indicated increased abundance of alternatively spliced tenascin-C mRNA isoforms in SSc skin biopsies. While these intriguing observations implicate SRSF6 and alternate splicing of tenascin-C in the pathogenesis of skin fibrosis, further investigation of differential tenascin-C isoform expression and regulation in SSc and their role in pathogenesis are warranted.
Explanted skin fibroblasts isolated from SSc biopsies show constitutive tenascin-C production, indicating that tenascin-C accumulation in SSc might at least, in part, result from its cell-autonomous overproduction.20 In SSc patients, circulating and tissue levels of tenascin-C were upregulated in patients with both early- and late-stage disease, suggesting that persistent upregulation of tenascin-C might play a role in maintaining tissue fibrosis. It is noteworthy that tenascin-C levels showed a positive correlation with TLR4, as well as with IL-6, a readout for TLR4 signaling, in individual SSc skin biopsies.20 Treatment of skin fibroblasts with tenascin-C elicited TLR4-dependent profibrotic responses, including upregulation of TLR4 itself. Mice lacking tenascin-C were viable and fertile, but showed reduced skin and lung fibrosis in response to bleomycin. Moreover, lack of tenascin-C was associated with reduced hypodermal fibrosis in the Tsk/+ mouse. Moreover, bleomycin-induced skin fibrosis showed accelerated resolution in mice lacking tenascin-C. While the EDA isoform of fibronectin is a potent TLR4 agonist, it might be insufficient to compensate for the loss of tenascin-C in tenascin-C null mice, since its expression is reduced in lesional skin. We propose that reduced cutaneous TLR4 signaling in mice lacking tenascin-C accounts for attenuated fibrosis, and more importantly, accelerated resolution. Pathological tissue fibrosis in SSc might be similarly perpetuated through a TLR4-dependent fibrosis amplification loop driven by endogenous DAMPs that accumulate and persist within injured microenvironments. Since little is currently known about tenascin-C splicing in inflammation and tissue remodeling, it will be of great interest to determine if alternatively spliced tenascin-C isoforms, or the FBG domain of tenascin-C, are necessary and sufficient to activate fibroblast TLR4 signaling, and whether they can be targeted for antifibrotic therapy.
Multiple Mechanisms to Prevent Aberrant TLR4 Activation
To forestall injury resulting from aberrant innate immune activation, a number of cellular mechanisms that inhibit TLR signaling have evolved.46 These negative regulatory mechanisms include suppression of TLR4 expression; alternate splicing of TLR adaptors; the inhibitory surface molecule radioprotective 105 (RP105); ubiquitin ligase and deubiquitinase enzymes such as A20 that modulate the activity of key TLR signaling intermediates; transcriptional regulators; and microRNAs.46 Impaired negative regulation of TLR signaling results in unchecked TLR activation that might contribute to chronic inflammatory and fibrotic diseases.
In many cell types, RP105 forms a complex with the TLR coreceptor MD1, which then interacts directly with TLR4-MD2 and inhibits downstream TLR4 signaling. Mice lacking RP105 show enhanced sensitivity to arthritis.47 In addition, RP105-null mice develop exaggerated cardiac dysfunction, neointima formation upon injury, and aggravates vein graft disease while atherosclerosis is attenuated.48–51 Treatment of skin and lung fibroblasts with recombinant RP105 abrogated TLR4-dependent inflammatory, as well as profibrotic, responses (Bhattacharyya S, Varga J, unpublished data). Furthermore, bleomycin treatment of RP105-deficient mice resulted in accelerated skin and lung fibrosis (Bhattacharyya S, Varga J; unpublished), indicating that RP105 suppresses fibrotic signaling through attenuated TLR4-dependent fibroblast activation. The expression and function of RP105 in SSc remain to be investigated.
The level of cellular TLR4 expression regulates the sensitivity to TLR4 ligands. Not surprisingly, LPS is a potent stimulus for enhanced TLR4 expression, whereas proresolving mediators such as RvD2 suppress TLR4 expression, preventing sustained inflammatory responses.52 Both up- and downregulation of TLR4 involve microRNAs, including inhibitory miR146a.53 Whether miRNA-mediated modulation of the expression of TLR4, along with MD2, is involved in fibrosis and SSc, has not been explored.
Cell-autonomous regulation of inflammatory signaling involves the cytosolic ubiquitin-editing enzyme A20.54 In a variety of cells A20 controls both TLR-dependent and TLR-independent inflammatory responses. Genetic studies have uncovered consistent associations of A20 locus variants with a range of autoimmune and inflammatory diseases. Rare loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory syndrome.55 Altered A20 expression or function causes persistent inflammation and other pathologies, and functional A20 variants show robust association with disease susceptibility and severity in SSc. Consensus clustering of SSc-associated genetic variants with differentially expressed genes shows that subnetworks enriched for genes related to TLR pathways are connected to clusters enriched for genes associated with TGF-β and ECM remodeling, with A20 as a central hub within the network.56 We recently showed that in fibroblasts, A20 plays a fundamental role in limiting TGF-β activity and preventing excessive collagen production.57
Impaired A20 expression, regulation, or function due to genetic variants, epigenetic modulation, and/or environmental influences might contribute directly to the development or progression of fibrosis in SSc and represents a promising target for therapy. Treatment of fibroblasts with adiponectin, an adipocyte-derived peptide that is reduced in patients with SSc, blocked TLR-dependent fibrotic responses.58,59 Remarkably, adiponectin induces sustained upregulation of A20 in fibroblasts. Pharmacologic agents that induce A20 expression, such as vitamin E (γ-tocotrienol) and adiponectin, hold promise as antifibrotic therapies by restoring endogenous A20 expression, particularly when targeted in a tissue-restricted manner, such as by topical application.57,60,61 Connectivity mapping identified ikarugamycin and quercetin as A20-inducing agents with potential as anti-inflammatory drugs. In preclinical studies these compounds were shown to attenuate inflammatory responses by inducing A20 in cystic fibrosis airway epithelial cells.62
In the absence of A20, mice spontaneously develop sclerodermatous skin changes, including thickened dermis and loss of dermal white adipose tissue, and show premature lethality due to widespread inflammation.63 Deficiency of A20 in intestinal epithelial cells is associated with IBD, in myeloid cells causes joint inflammation, in dendritic cells causes colitis, in mast cells exacerbate inflammation,64,65 in B cells autoimmunity but only in old mice, and in vascular allografts aggravate severity of transplant arteriosclerosis.66–69 These observations highlight the diverse and cell type-, age-, and disease-specific functions of A20. The effect of fibroblast-specific A20 loss in fibrosis in vivo remains to be investigated.
TLR4 Signaling Represents a Potential Therapeutic Target in SSc
Currently, there are limited therapeutic options for patients with SSc. A major hurdle in the introduction of more effective treatments arises from disease heterogeneity—either in the form of differing disease subtypes or different rates of fibrosis progression between individual patients and even within the same patient between different organs (such as skin and lung). Application of a targeted precision medicine approach is therefore highly appropriate for SSc. For instance, we found that our experimentally derived “fibroblast TLR4 gene signature” was highly expressed in one patient subset only, mapping to the inflammatory intrinsic subset of SSc biopsies (Bhattacharyya S. and Varga J., unpublished data); this signature therefore might serve as a predictive biomarker identifying a subset of SSc patients who might be responders to anti-TLR4 therapy in future clinical trials.
As dysregulated TLR4 signaling underlies the pathology of diverse conditions, substantial effort has been committed to the design and development of selective inhibitors.70–73 A prime disease indication pursued for TLR4 inhibitors is sepsis. Eritoran (Eisai Co., Ltd.), a structural inhibitor of the lipid A portion of LPS, has been shown to inhibit TLR4 signaling in vitro and in vivo. TAK-242 (Takeda Pharmaceutical Company Limited) covalently binds to Cys747 of the TIR domain of TLR4, rendering TLR4 completely inactive. Both of these TLR4 inhibitors reached Phase III clinical trials for the treatment of septic shock, but unfortunately failed to demonstrate acceptable efficacy and safety.74–76 Notably, TAK-242 showed potent antifibrotic effects, preventing bleomycin-induced organ fibrosis (Bhattacharyya and Varga, unpublished observation) (Fig. 5).
Figure 5.
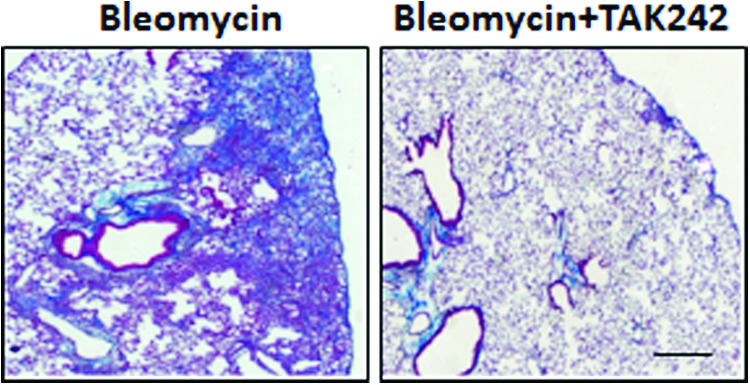
TAK-242 treatment ameliorates bleomycin-induced lung fibrosis. C57/BL6 mice were administered bleomycin s.c. synchronously with TAK242 daily by i.p. injections for 14 days, and lungs were collected 10 days following last injection. Trichrome stain showed reduced collagen with TAK-242 treatment. Scale bar, 10 μm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Recent studies implicating TLR4 and its coreceptor MD2 in SSc have focused attention of TLR4/MD2 as a novel therapeutic target.16 Since endogenous TLR4 ligand requires MD2 as a coreceptor for signaling and initiation of fibrotic responses,14,37 disrupting ligand-MD2-TLR4 complex formation represents a logical antifibrotic strategy. Based on this hypothesis, Yin and colleagues undertook extensive structure-activity-relationship studies to identify potent and selective drug candidates that disrupt TLR4-MD2 interactions. The most efficient compound in this series, T5342126, competes with MD2 for binding to TLR4 and disrupts LPS/TLR4-stimulated NF-kB-activity in vitro. Importantly, T5342126 showed potent antifibrotic effects, preventing stimulation of Type I collagen synthesis and myofibroblast differentiation induced in normal fibroblasts by microbial LPS or by EDA fibronectin or tenascin-C (Bhattacharyya S and Varga J; unpublished) Furthermore, the TLR4 inhibitor attenuated the constitutively activated phenotype of SSc fibroblasts. Treatment of mice with T5342126 not only prevented but also reversed organ fibrosis in multiple disease models (Bhattacharyya S, Yin H and Varga J unpublished). Additional agents targeting MD2 include caffeic acid phenethyl ester and rifampin, which prevent LPS-induced pro-inflammatory mediators, although their effects on fibrosis have not been examined.77,78 A viable therapeutic strategy for disrupting MD2-TLR4 complex formation with antibodies directed against TLR4 or the TLR4-MD2 complex showed promising results in preclinical studies.71
Selectively preventing TLR4 activation by disease-associated DAMPs represents a highly appealing innovative approach to TLR4 inhibition. By carefully choosing a target unique to the response to tissue damage and not to pathogen-mediated activation of the immune response may have the advantage of preserving an intact host response to infection. Several novel approaches for selectively targeting tenascin-C have been tried in cancer patients.79 The alternatively spliced domains of fibronectin and tenascin-C could be targeted using specific antibodies. For example, the F16 antibody targets the A1 domain of tenascin-C, whereas the 81C6 antibody recognizes domain D; the EDA domain of fibronectin is the target of F8 antibody.80 Alternately, as TLR4 activation involves the terminal FBG domain of tenascin-C (Fig. 3),21,81 selectively targeting the FBG domain using antibody represents another promising antifibrotic therapy. Further studies on preclinical and clinical data underlying therapeutic potential of targeting TLR4 DAMPs are warranted.
The precise mode of TLR4 interaction with DAMPs leading to active signaling remains to be elucidated. Although the fibronectin EDA domain has been shown to bind to cell surface TLR4,37 there are no data indicating whether tenascin-C binds directly to TLR4. Furthermore, the TLR4-dependent tenascin-C response in macrophages is not inhibited by MD2 blockade.21 In contrast, we observed that the MD2-TLR4 inhibitor T5342126 abrogated tenascin-C-induced profibrotic responses in fibroblasts, suggesting potential cell specificity. The identification of TLR4 coreceptors and their precise roles in DAMP-TLR4 signaling might help delineating the molecular events leading to ligand/cell-type specific pathological responses to DAMPs. A comparative analysis of coreceptors, kinases, and transcription factors involved in DAMP signaling versus PAMPs may highlight key differences that could hold promise to novel therapeutic approaches by selectively silencing DAMP-TLR4 signaling without compromising the host immune defense.
Summary and Future Perspective
Emerging insights from in vitro experiments, experimental disease models using transgenic mice, and clinical information highlight a previously unsuspected pathogenic role of DAMP-TLR4 signaling in fibrosis. Activation of fibroblast TLR4 by disease-associated DAMPs appears to convert a controlled and self-limited tissue repair process into unresolving fibrosis. Transcriptome analyses highlight the association of aberrant TLR4 signaling with skin fibrosis in the “inflammatory” intrinsic gene subset of SSc, suggesting that patients mapping to this subset might be the optimal responders to therapy targeting the TLR4 axis. It is noteworthy that current drugs targeting TLR4, such as eritoran and TAK-242, failed to achieve their primary end point in septic shock clinical trials, highlighting an urgent need for new treatment strategies. Anti-TLR4 agents might also be considered as antifibrotic therapies in a drug “repurposing” strategy. We have highlighted promising approaches of targeting TLR4 signaling with small molecule TLR4 inhibitors. Alternatively, ablating the expression or function of individual DAMPs or blocking their recognition by TLR4 or restoring endogenous TLR inhibitors such as A20 or RP105 by pharmacologic agents might hold promise for antifibrotic therapies. Much remains to be learned regarding the unique and redundant functions of various TLR4 DAMPs, their specific coreceptors, negative regulators, and repertoire of molecular pathways. Moreover, it will be of great interest to investigate the genetic associations between A20 and specific SSc endophenotypes, as well as disease outcomes and response to therapy. In summary, this review highlighted an emerging novel paradigm implicating TLR4-DAMP signaling in persistent fibroblast activation underlying nonresolving fibrosis in SSc and possibly other forms of pathological fibrosis, appealing opportunities for therapy and novel approaches for identifying a homogeneous population of patients most likely to benefit from these therapies (Fig. 6).
Figure 6.
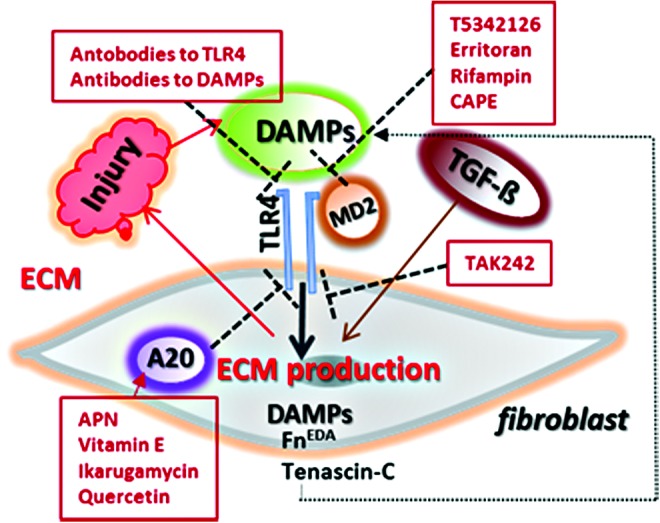
Distinct levels for blocking TLR4 signaling. Antibodies to either TLR4 or endogenous TLR4 ligands directly inhibit TLR4 signaling. Endotoxin antagonists or small molecule inhibitors of TLR4/MD2 interaction are potential therapeutic tools. TAK-242 through direct binding to the intracellular Cys747 residue of TLR4 blocks TLR4 signaling. Compounds such as adiponectin (APN), quercetin and Vitamin E inhibit TLR4 signaling by inducing endogenous A20, a negative regulator of TLR4. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Take-Home Messages.
• Triggering the innate immune system in nonclassical resident stromal cells elicits fibrotic responses that show only partial overlap with inflammatory responses elicited in classical immune cells
• Tenascin-C and other macromolecules are secreted within injured tissue and accumulate causing persistent fibrosis. In the fibrotic microenvironment, such “damage-associated molecular patterns” act as endogenous ligands for pattern recognition receptors such as TLRs expressed in myofibroblasts
• Persistent DAMP activation of myofibroblasts, coupled with impaired downregulation of innate immune signaling, underlies constitutive myofibroblast activation and failure of fibrosis resolution in SSc.
• Genomic and genetic surveys provide evidence for altered DAMP accumulation and innate immune system function in patients with SSc
• The novel paradigm of fibrosis persistence driven by sustained innate immune activation by DAMPs presents translational and clinical opportunities for the development of fibrosis biomarkers and treatments.
Abbreviations and Acronyms
- DAMP
damage-associated molecular pattern
- ECM
extracellular matrix
- FBG
fibrinogen-like globe
- ILD
interstitial lung disease
- IRF
interferon response factor
- LPS
lipopolysaccharide
- LRR
leucine-rich repeat
- MHC
major histocompatibility complex
- PAMP
pathogen-derived molecule pattern
- PRR
pattern recognition receptor
- SAA
serum amyloid A
- SSc
systemic sclerosis
- TGF-β
transforming growth factor-β
- TLR
toll-like receptor
Acknowledgments and Funding Sources
Supported by grants from the NIH (AR42309 and AR064925 to JV, GM101279 to HY), Arthritis Research UK Senior Fellowship 20003 (to KSM), and the Scleroderma Foundation (to SB).
Author Disclosure and Ghostwriting
The authors declare no competing interests in the studies discussed in this article. No ghostwriters were used to write this article.
About the Authors
Swati Bhattacharyya, PhD, is a Research Associate Professor of Rheumatology at Northwestern University, Feinberg School of Medicine. She is currently conducting research on the implication of innate immune signaling in fibrosis in scleroderma focusing specifically on stromal cell TLR4 signaling in injury and fibrosis. Kim S. Midwood, PhD, is a Professor of Matrix Biology in the Kennedy Institute of Rheumatology at Oxford University. Hang Yin, PhD, is an Associate Professor at University of Colorado, Boulder. John Varga, MD, is a John and Nancy Hughes Professor of Rheumatology and Director of the Northwestern Scleroderma Program at the Feinberg School of Medicine. His studies focus on the pathogenesis of scleroderma, with the goal of identifying and validating novel targets for antifibrotic therapy.
References
- 1.Allanore Y, et al. Systemic sclerosis. Nat Rev Dis Primers 2015;1:15002. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol 2012;8:42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009;360:1989–2003 [DOI] [PubMed] [Google Scholar]
- 4.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 2007;117:557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 2013;304:C216–C225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis—a lethal component of systemic sclerosis. Nat Rev Rheumatol 2014;10:390–402 [DOI] [PubMed] [Google Scholar]
- 7.Martin JE, et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum Mol Genet 2013;22:4021–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayes MD, et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Hum Genet 2014;94:47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koumakis E, et al. Brief report: candidate gene study in systemic sclerosis identifies a rare and functional variant of the TNFAIP3 locus as a risk factor for polyautoimmunity. Arthritis Rheum 2012;64:2746–2752 [DOI] [PubMed] [Google Scholar]
- 10.Korman BD, Criswell LA. Recent advances in the genetics of systemic sclerosis: toward biological and clinical significance. Curr Rheumatol Rep 2015;17:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson ME, et al. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PloS One 2015;10:e0114017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahoney JM, et al. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput Biol 2015;11:e1004005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sullivan KM, Shah A, Sarantopoulos S, Furst DE. Review: hematopoietic Stem Cell Transplantation for Scleroderma: effective Immunomodulatory Therapy for Patients With Pulmonary Involvement. Arthritis Rheumatol 2016;68:2361–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010;2010 pii: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010;11:373–384 [DOI] [PubMed] [Google Scholar]
- 16.Bhattacharyya S, Varga J. Emerging roles of innate immune signaling and toll-like receptors in fibrosis and systemic sclerosis. Curr Rheumatol Rep 2015;17:474. [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011;34:637–650 [DOI] [PubMed] [Google Scholar]
- 18.Park BS, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009;458:1191–1195 [DOI] [PubMed] [Google Scholar]
- 19.Bryant CE, et al. Advances in Toll-like receptor biology: modes of activation by diverse stimuli. Critic Rev Biochem Mol Biol 2015;50:359–379 [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharyya S, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun 2016;7:11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Midwood K, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 2009;15:774–780 [DOI] [PubMed] [Google Scholar]
- 22.Piccinini AM, Zuliani-Alvarez L, Lim JM, Midwood KS. Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Sci Signal 2016;9:ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharyya S, et al. Toll-like receptor 4 signaling augments transforming growth factor-beta responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. Am J Pathol 2013;182:192–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stifano G, et al. Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther 2014;16:R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milano A, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PloS One 2008;3:e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pendergrass SA, et al. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol 2012;132:1363–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christmann RB, et al. Association of Interferon- and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol 2014;66:714–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi T, et al. Amelioration of tissue fibrosis by toll-like receptor 4 knockout in murine models of systemic sclerosis. Arthritis Rheumatol 2015;67:254–265 [DOI] [PubMed] [Google Scholar]
- 29.Saigusa R, et al. Multifaceted contribution of the TLR4-activated IRF5 transcription factor in systemic sclerosis. Proc Natl Acad Sci U S A 2015;112:15136–15141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang J, et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med 2016;22:1285–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang HZ, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol 2012;180:275–292 [DOI] [PubMed] [Google Scholar]
- 32.Seki E, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–1332 [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharyya S, et al. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med 2014;6:232ra250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White ES, Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011;63:538–546 [DOI] [PubMed] [Google Scholar]
- 35.Giblin SP, Midwood KS. Tenascin-C: form versus function. Cell Adhes Migration 2015;9:48–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ffrench-Constant C, Van de Water L, Dvorak HF, Hynes RO. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol 1989;109:903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okamura Y, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 2001;276:10229–10233 [DOI] [PubMed] [Google Scholar]
- 38.Gondokaryono SP, et al. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J Leukoc Biol 2007;82:657–665 [DOI] [PubMed] [Google Scholar]
- 39.Lefebvre JS, et al. Extra domain A of fibronectin primes leukotriene biosynthesis and stimulates neutrophil migration through activation of Toll-like receptor 4. Arthritis Rheum 2011;63:1527–1533 [DOI] [PubMed] [Google Scholar]
- 40.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 2003;200:500–503 [DOI] [PubMed] [Google Scholar]
- 41.Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: synergism between the III-1 and EDA domains. PloS One 2014;9:e102974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You R, Zheng M, McKeown-Longo PJ. The first type III repeat in fibronectin activates an inflammatory pathway in dermal fibroblasts. J Biol Chem 2010;285:36255–36259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinz B, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 2012;180:1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Udalova IA, Ruhmann M, Thomson SJ, Midwood KS. Expression and immune function of tenascin-C. Critic Rev Immunol 2011;31:115–145 [DOI] [PubMed] [Google Scholar]
- 45.Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol 2014;21:189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol 2012;33:449–458 [DOI] [PubMed] [Google Scholar]
- 47.Tada Y, et al. Toll-like receptor homolog RP105 modulates the antigen-presenting cell function and regulates the development of collagen-induced arthritis. Arthritis Res Ther 2008;10:R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Louwe MC, et al. RP105 deficiency aggravates cardiac dysfunction after myocardial infarction in mice. Int J Cardiol 2014;176:788–793 [DOI] [PubMed] [Google Scholar]
- 49.Karper JC, et al. TLR accessory molecule RP105 (CD180) is involved in post-interventional vascular remodeling and soluble RP105 modulates neointima formation. PloS One 2013;8:e67923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wezel A, et al. Deficiency of the TLR4 analogue RP105 aggravates vein graft disease by inducing a pro-inflammatory response. Sci Rep 2016;6:24248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wezel A, et al. RP105 deficiency attenuates early atherosclerosis via decreased monocyte influx in a CCR2 dependent manner. Atherosclerosis 2015;238:132–139 [DOI] [PubMed] [Google Scholar]
- 52.Croasdell A, Sime PJ, Phipps RP. Resolvin D2 decreases TLR4 expression to mediate resolution in human monocytes. Faseb J 2016;30:3181–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He X, Jing Z, Cheng G. MicroRNAs: new regulators of Toll-like receptor signalling pathways. BioMed Res Int 2014;2014:945169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 2012;12:774–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Q, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet 2016;48:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson ME, Pioli PA, Whitfield ML. Gene expression profiling offers insights into the role of innate immune signaling in SSc. Semin Immunopathol 2015;37:501–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhattacharyya S, Wang W, Graham LV, Varga J. A20 suppresses canonical Smad-dependent fibroblast activation: novel function for an endogenous inflammatory modulator. Arthritis Res Ther 2016;18:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fang F, et al. The adipokine adiponectin has potent anti-fibrotic effects mediated via adenosine monophosphate-activated protein kinase: novel target for fibrosis therapy. Arthritis Res Ther 2012;14:R229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lakota K, et al. Levels of adiponectin, a marker for PPAR-gamma activity, correlate with skin fibrosis in systemic sclerosis: potential utility as biomarker? Arthritis Res Ther 2012;14:R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Park NY, Jang Y, Ma A, Jiang Q. Vitamin E gamma-Tocotrienol Inhibits Cytokine-Stimulated NF-kappaB Activation by Induction of Anti-Inflammatory A20 via Stress Adaptive Response Due to Modulation of Sphingolipids. J Immunol 2015;195:126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim Y, et al. Suppression of NLRP3 inflammasome by gamma-tocotrienol ameliorates type 2 diabetes. J Lipid Res 2016;57:66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malcomson B, et al. Connectivity mapping (ssCMap) to predict A20-inducing drugs and their antiinflammatory action in cystic fibrosis. Proc Natl Acad Sci U S A 2016;113:E3725–E3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000;289:2350–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moll HP, et al. A20 Haploinsufficiency Aggravates Transplant Arteriosclerosis in Mouse Vascular Allografts: implications for Clinical Transplantation. Transplantation 2016;100:e106–e116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferran C. Preface. The multiple therapeutic targets of A20. Adv Exp Med Biol 2014;809:ix–x [PubMed] [Google Scholar]
- 66.Matmati M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet 2011;43:908–912 [DOI] [PubMed] [Google Scholar]
- 67.Hammer GE, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol 2011;12:1184–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hovelmeyer N, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Euro J Immunol 2011;41:595–601 [DOI] [PubMed] [Google Scholar]
- 69.Heger K, et al. A20-deficient mast cells exacerbate inflammatory responses in vivo. PLoS Biol 2014;12:e1001762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hennessy EJ, Parker AE, O'Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Revi Drug Discov 2010;9:293–307 [DOI] [PubMed] [Google Scholar]
- 71.Savva A, Roger T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol 2013;4:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li J, Wang X, Zhang F, Yin H. Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol Ther 2013;138:441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang X, Smith C, Yin H. Targeting Toll-like receptors with small molecule agents. Chem Soc Rev 2013;42:4859–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rice TW, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Critic Care Med 2010;38:1685–1694 [DOI] [PubMed] [Google Scholar]
- 75.Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 2011;79:34–41 [DOI] [PubMed] [Google Scholar]
- 76.Opal SM, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 2013;309:1154–1162 [DOI] [PubMed] [Google Scholar]
- 77.Kim SY, et al. Suppression of Toll-like receptor 4 activation by caffeic acid phenethyl ester is mediated by interference of LPS binding to MD2. Br J Pharmacol 2013;168:1933–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, et al. Rifampin inhibits Toll-like receptor 4 signaling by targeting myeloid differentiation protein 2 and attenuates neuropathic pain. Faseb J 2013;27:2713–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Midwood K, Orend G. Special issue of Cell Adhesion & Migration on Tenascins: defining their role in tissue homeostasis and cancer. Cell Adhes Migration 2015;9:1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–777 [DOI] [PubMed] [Google Scholar]
- 81.Maqbool A, et al. Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. World J Cardiol 2016;8:340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]