

SUMMARY
Association of aberrant glycosylation with melanoma progression is based mainly on analyses of cell lines. Here we present a systems-based study of glycomic changes and corresponding enzymes associated with melanoma metastasis in patient samples. Upregulation of core fucosylation (FUT8) and downregulation of α-1,2 fucosylation (FUT1, FUT2) were identified as features of metastatic melanoma. Using both in vitro and in vivo studies, we demonstrate FUT8 is a driver of melanoma metastasis which, when silenced, suppresses invasion and tumor dissemination. Glycoprotein targets of FUT8 were enriched in cell migration proteins including the adhesion molecule L1CAM. Core fucosylation impacted L1CAM cleavage and the ability of L1CAM to support melanoma invasion. FUT8 and its targets represent therapeutic targets in melanoma metastasis.
Keywords: primary melanoma, metastatic melanoma, glycosylation, lectin microarray, lectin array, core fucosylation, metastasis, glycomics
In Brief/eTOC Blurb
Using a systems-based approach to assess glycosylation in matched primary and metastatic melanoma samples, Agrawal et al. find increased core fucosylation mediated by FUT8 in metastatic melanoma. FUT8 facilitates invasion and tumor dissemination, in part due to reduced cleavage of core fucosylated L1CAM.
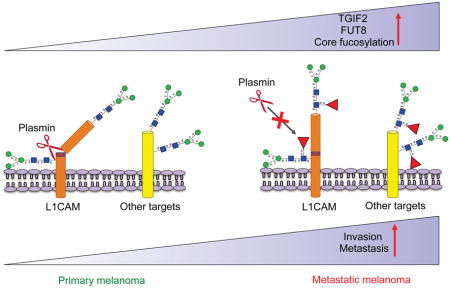
INTRODUCTION
The incidence of melanoma in the Western world doubles every 20 years (Giblin and Thomas, 2007). Despite recent approval of new therapies (i.e. BRAF, MEK and immune checkpoint inhibitors), metastatic melanoma remains, for the most part, an incurable disease. Identification of determinants of melanoma metastasis is a key step towards effectively controlling tumor progression. Aberrant glycosylation is often observed as a hallmark of cancer and is implicated in processes from cell signaling and communication to immune modulation and tumor invasion (Bird-Lieberman et al., 2012; Christiansen et al., 2014; Meany and Chan, 2011; Pinho and Reis, 2015; Stowell et al., 2015). Glycosylation is controlled by the actions of glycosyltranferases and glycosidases on glycoproteins and/or lipids. Differential expression of these enzymes in cancer produces glycoproteins with specific cancer-associated alterations in glycans. Aberrant glycosylation is not just a consequence of cancer, but also a driver of malignant phenotype, directly impacting key processes supporting tumor progression and metastasis including cell adhesion, motility, invasion, and immune evasion(Ciborowski and Finn, 2002; Gaziel-Sovran et al., 2011; Sackstein et al., 2008). Alterations in glycosyltransferase expression have been associated with both pro-metastatic and metastasis suppressor functions (Christiansen et al., 2014). Currently, few systematic studies of glycans in cancer exist to sort out these roles, none related to melanoma. In general, glycomic studies on cancer to date fall short of pinpointing enzymes responsible for the biosynthesis of observed aberrant glycan motifs and/or their relevant glycoprotein targets (Sugahara et al., 2012). Glycosylation remains an understudied portion of cancer biology due to issues including difficulties in glycan analysis and complex biosynthetic pathways (Agre et al., 2016; Rakus and Mahal, 2011).
Although there has been much interest in the role of glycosylation in melanoma metastasis, the vast majority of glycan analyses in melanoma has been limited to mouse models and tumor cell lines, where aberrant glycosylation has been linked to cancer progression (Christiansen et al., 2014; Laidler et al., 2006). The relevance of this data to melanoma patient samples is unknown, as there is little data on glycosylation in clinical cohorts, mainly due to the small size of primary melanomas and limitations in the analytical techniques used (Christiansen et al., 2014; Kinoshita et al., 2014; Rakus and Mahal, 2011). Previously, our group reported on the role of aberrant glycosylation in promoting melanoma cell invasion and immunosuppression (Gaziel-Sovran et al., 2011). We found that miR-30b/d, upregulated in a patient-derived signature associated with metastasis, drives tumor dissemination by targeting both GALNT7 and GALNT1, enzymes that are initiators of canonical O-linked glycan biosynthesis. This suggests that aberrant glycosylation can drive melanoma metastasis and points to the need for a more systematic study of glycosylation in clinical samples of melanoma.
RESULTS
Systematic dissection of the melanoma glycome and associated glycogenes in clinical samples reveals altered protein fucosylation linked to metastatic behavior
Although glycosylation has been studied in melanoma, these studies have focused on mouse models and tumor cell lines to the exclusion of clinical samples. The relevance of these studies to melanoma progression in patients is thus largely unknown (Christiansen et al., 2014). To examine the relationship between melanoma progression and altered glycosylation in a clinical cohort we utilized lectin microarrays (Figure 1A), our high-throughput technology for glycomic analysis (Bird-Lieberman et al., 2012; Pilobello et al., 2007). Our microarrays include 102 lectin and antibody probes, providing specific information on the glycan repertoire of cellular glycoproteins (Table S1) (Pilobello et al., 2013). We performed lectin microarray analysis on formalin-fixed paraffin-embedded (FFPE) patient-matched primary and metastasis tissues (n=17 patients; n=34 samples total). Samples were macrodissected to remove contamination of the melanoma sample by surrounding tissues such as skin. Glycomic profiles of the melanoma samples resolved into multiple clusters of primary and metastatic melanomas based on lectin binding (Figure S1A). A closer analysis of the data identified a subset of lectins whose binding differentiated between primary and metastatic melanoma (paired t test, p<0.05; Figure 1B). In general, metastatic tumors showed higher levels of poly/multiantennary-N-acetyl-lactosamine (polyLacNAc, bound by lectins: DSA, WGA), α-2,6 sialic acids (SNA and TJA-I), and core fucose (α-1,6 fucose; PSA, LcH) and lower levels of α-1,2 fucose structures (UEA-I, TJA-II and SNA-II) than the corresponding primary samples (Figure 1B). To gain insight into the biosynthetic underpinnings of these changes, we correlated our lectin microarray data with publicly available transcriptomic data including TCGA (Riker et al., 2008; Xu et al., 2008), focusing on glycogenes (glycosyltransferases) known to be involved in the synthesis of glycan epitopes bound by lectins discriminating between primary and metastatic melanoma (Figure 1C). Consistent with our observed glycan changes, metastatic melanomas displayed higher expression levels of N-glycan branching enzymes (MGAT2, MGAT4A), polylactosamine extension enzymes (B3GNT2), sialyltransferases (ST6GAL1, ST6GAL2) and the core fucosyltransferase FUT8 than primary melanomas, and lower levels of enzymes responsible for α-1,2 fucosylation (FUT1, FUT2) across multiple datasets (Figure 1C). Altered expression of these glycogenes would explain the changes in glycan structures associated with melanoma progression revealed by our glycomic analysis.
Figure 1. Systematic dissection of the melanoma glycome and associated glycogenes in clinical samples reveals altered protein fucosylation linked to metastatic behavior.

(A) Schematic illustration of our systems biology approach to identify glycosylation enzymes involved in melanoma metastasis and their corresponding targets. (B) Heat map of lectin clustering of patient-matched primary and metastatic (MET) melanoma FFPE tissues (n=17 pairs) by ratiometric lectin microarray, p<0.05. Pink, log2(S/R) > log2(Smedian/Rmedian); blue, log2(Smedian/Rmedian) > log2(S/R). (C) Correlation of lectin data with glycogene expression. Fold change in selected lectin binding in melanoma metastasis vs. primary tissues using lectin microarray is shown. Fold change in expression of corresponding glycogenes is also shown, p<0.05. (D) Trans-well invasion assay on SkMel147 and WM3211 cells transfected with non-targeting control (NTC) or smart pool siRNAs (50 nM) against selected glycogenes (mean ± SD of 3 replicates), p<0.05. (E) N-linked α-1,6 core fucosylated glycan structure generated by FUT8 and recognized by LcH and PSA lectins. (F) α-1,2 terminal fucosylated glycan structure generated by FUT1 and FUT2 and recognized by UEA-I, TJA-II and SNA-II lectins. (G) Representative images of LcH and UEA-I lectin multiplex fluorescence microscopy of primary and metastatic melanomas (n=18 for LcH, n=19 for UEA-I). Biotinylated LcH- DY-647-Streptavidin (red), FITC-UEA-I (green) and DAPI stained sections, scale bar, 100 μm. Dot plots represent the average fluorescence intensity of 5 fields per image for each lectin. (H) Representative images of IHC staining with α-FUT8 antibody in 17 paired primary and metastatic melanoma show peri-nuclear staining pattern (FastRed counterstaining). IHC score was calculated combining the signal intensity and percentage of positive cells within the section. Dot plot shows distribution of FUT8 IHC score in primary and metastatic melanoma, scale bar, 10 μm. Two-tailed paired (B and H), and unpaired (C, D and G) t test. *p=0.01 to 0.05, **p=0.001 to 0.01. See also Figure S1.
We hypothesized that the glycosylation patterns observed in metastasis could contribute to melanoma aggressive behavior. Therefore, we assessed the effects of silencing identified glycogenes on cellular invasion, a biological property required by tumors for metastasis. We conducted a siRNA mini-screen for select glycosyltransferases identified as putative ‘pro-metastatic’ (MGAT2, MGAT4A, FUT8, ST6GAL1, ST6GAL2, B3GNT2, B3GNT3) or ‘anti-metastatic’ (FUT1, FUT2) factors based on lectin microarray or glycogene expression data. Studies were conducted in two melanoma cell lines (SkMel147 and WM3211) (Figure 1D, S1B-E). FUT8 knockdown consistently suppressed the invasion capacity of melanoma cells, while silencing of FUT1 or FUT2 had the opposite effect (Figure 1D). FUT8 is the only enzyme known to generate α-1,6 fucosylated structures on the core of N-glycans, the epitope recognized by the lectins LcH and PSA (Figure 1E). In contrast, FUT1 and FUT2 can generate α-1,2 fucosylated structures both at the termini of N-acetyllactosamine and on galactose linked with N-acetylgalactosamine, epitopes recognized by UEA-I and TJA-II/SNA-II lectins, respectively (Figure 1F). Data mining of publicly available transcriptomic datasets from five independent clinical cohorts, including TCGA (Kabbarah et al., 2010; Raskin et al., 2013; Riker et al., 2008; Xu et al., 2008), revealed consistently higher mRNA levels for FUT8 and lower levels for FUT1 and FUT2 in metastatic melanoma tissues in comparison to primary (Figure S1F). The remarkable consistency of these findings supports an antithetical role for the two forms of fucosylation (α-1,6 or core fucose: ‘pro-metastatic’, α-1,2 fucose: ‘anti-metastatic’) in clinical melanoma progression. We validated the differential presence of core fucosylated and α-1,2 fucosylated structures in a cohort of primary and metastatic samples using an orthogonal method, fluorescent lectin staining. In agreement with our lectin microarray results, metastatic sections displayed significantly higher LcH binding (p<0.01) and decreased UEA-I staining (p<0.05) (Figure 1G). Consistent with the observed increase in LcH binding, immunohistochemistry (IHC) analysis of the paired tissues confirmed higher FUT8 expression in metastatic tissues as evidenced by a perinuclear granular staining pattern (Figure 1H, S1G-H). In sum, our systems-based analysis identified the core fucosyltransferase FUT8, which has no known role in melanoma, as a modulator of in vitro cellular invasion and potential mediator of melanoma metastasis. We focused our functional and mechanistic studies on FUT8, as a candidate pro-metastatic gene, which represents a more suitable therapeutic target than fucosyltransferases FUT1 and FUT2.
FUT8 silencing decreases in vitro cell invasion and in vivo melanoma metastasis
To further investigate the role of core fucosylation in melanoma metastasis, we silenced FUT8 in the metastatic melanoma cell lines 113/6-4L (hereafter, 4L) and SkMel147, both of which express high endogenous levels of FUT8 (Figure S2A). In brief, 4L and SkMel147 cell lines were stably transduced with lentivirus carrying one of two independent short hairpin RNAs targeting FUT8 (FUT8 shA or shB) or a non-targeting control (shNTC). As expected, FUT8 knockdown, confirmed by both qRT-PCR, Western blot and IHC (Figure 2A,B, S2B), resulted in reduced core fucosylation (Figure 2C). FUT8 knockdown had no effect on cell proliferation (Figure 2D) but attenuated by ~60% the ability of both 4L and SkMel147 cells to invade through matrigel in a trans-well invasion assay (Figure 2E).
Figure 2. FUT8 silencing reduces melanoma cells invasiveness in vitro.

(A–B) FUT8 levels in 4L and SkMel147 cells stably expressing shRNA targeting FUT8 (FUT8 shA or FUT8 shB) or shNTC were assessed by real-time qPCR (A) and Western blotting (B). QPCR graph shows average relative expression normalized to GAPDH, 3 replicates per condition. QPCR data and Western blot images are representative of three independent experiments. (C) LcH fluorescence microscopy of 4L and SkMel147 cells transduced with FUT8 shA or FUT8 shB or shNTC. Right panel shows quantitation of fluorescent intensity of representative images. Data from 5 random areas of 2 biological replicates were averaged to generate graphs, scale bar, 100 μm. (D) Cell proliferation assay of 4L and SkMel147 cells transduced with FUT8 shA or FUT8 shB or shNTC and analyzed by Cell Titer Glo, 3 replicates per condition. Data shown is representative of two independent experiments. (E) Trans-well matrigel invasion by 4L and SkMel147 cells transduced with FUT8 shA or FUT8 shB or shNTC. Invading cells were quantified by counting the number of 4L and SkMel147 cells that invaded into the basal side of matrigel-coated trans-well inserts after 36 hr and 12 hr respectively, n=5 fields per replicate; 3 replicates per condition, data shown is representative of three independent experiments. Scale bar, 100 μm. *p=0.01 to 0.05. Data are presented as mean ± SD. Two-tailed unpaired t test. See also Figure S2.
We next determined whether aberrant FUT8 expression promotes melanoma aggressive behavior in vivo using a xenograft model of metastasis. We injected 4L cells stably transduced with lentiviral particles carrying a mCherry/luciferase reporter and one of the 2 independent shRNA against FUT8 (FUT8 shA and shB) or control (shNTC) into the flanks of immunocompromised NOD/Shi-scid/IL-2Rgamma null (NSG) mice, and monitored them for local tumor growth and metastasis (Figure 3A). All cell types started to form measurable tumors at primary implantation sites one week later. In concordance with our observations in vitro, there was no significant difference in tumor growth between shNTC and either FUT8 shA or FUT8 shB-transduced cells (Figure 3B). Lower FUT8 transcript (Figure 3C) and reduced core fucosylation levels (Figure S3) confirmed effective FUT8 knockdown by both shRNAs in tumors. After surgically resecting the lateral tumors (28 days post-injection), we monitored metastatic dissemination by bioluminescence imaging (BLI). Silencing FUT8 consistently inhibited the metastatic potential of 4L cells, shown by reduced luminescence in both lung and lymph nodes (Figure 3D–F). Forty-two days post-injection, we sacrificed the mice and dissected lungs and lymph nodes for histology (Figure 3G–I). Mice injected with FUT8 shA or FUT8 shB 4L cells exhibited significantly reduced lung tumor burden and total number of micrometastases per lung (p<0.05) (Figure 3G–I). Our findings provide compelling evidence that FUT8 silencing suppresses the ability of melanoma cells to colonize distal organs.
Figure 3. FUT8 silencing decreases in vivo melanoma metastasis.

(A) Schematic representation of the in vivo xenograft experiment. (B) Primary tumor growth of 4L cells transduced with FUT8 shA, FUT8 shB or shNTC following subcutaneous injection into NSG mice (n=9 mice per condition; ns: not significant). Data is presented as mean ± SD. (C) Real-time qPCR of FUT8 expression in flank tumors resected at day 25 post-injection. Data shown is representative of flank tumors from 2 mice, mean ± SD. (D) Representative images of whole-body in vivo BLI of mice injected with 4L cells transduced with FUT8 shA, FUT8 shB or shNTC at day 42 post-injection (17 days post-resection of lateral tumors). (E) Average radiance as measured by in vivo BLI at 27, 35 and 42 days post-injection of flank tumors (n=9 mice per condition). (F) Quantitation of BLI at day 42 (n=9). (G) Ex vivo brightfield (BF) and fluorescent (RFP) microscopic images of mouse lungs at termination of the experiment and corresponding H&E-stained sections, scale bar, 100 μm. White dotted ovals mark whole organ. (H) Average fluorescent intensity of macrometastases per lung. Fluorescence intensity of whole lung was measured (n=9/group). (I) Dot plots show the distribution of the number of metastases per section of lung (2 sections per lung). Two-tailed unpaired t test. *p=0.01 to 0.05 and **p=0.001 to 0.01. See also Figure S3.
FUT8 silencing impairs the growth of established metastasis in vivo
Next we examined whether FUT8 is required for the maintenance or growth of existing metastasis using a model based on intracardiac instillation of tumor cells (Morsi et al., 2013). We generated 4L melanoma cells stably transduced with lentiviral particles carrying either a doxycycline (DOX)-inducible shRNA targeting FUT8 (pTRIPZ/FUT8 shC) or a scrambled control (pTRIPZ/shSCR) and a luciferase reporter vector constitutively expressing green fluorescent protein (GFP). This system couples DOX-regulated red fluorescent protein (RFP) expression to shRNA induction. In the presence of DOX, 4L FUT8 shC cells display decreased FUT8 mRNA and protein (Figure 4A) and reduced cell invasion in comparison to shSCR cells, without effects on cell proliferation (Figure 4B,C). We performed in vivo intracardiac injection of pTRIPZ/shSCR or pTRIPZ/FUT8 shC in NSG mice and assessed for metastatic dissemination by BLI (Figure 4D). Established metastases were detected 15 days post tumor cell instillation. At this point, mice within both groups (pTRIPZ/shSCR or pTRIPZ/FUT8 shC) were subdivided into two groups for +/−DOX feed (Figure 4D). DOX-fed mice injected with FUT8 shC-4L cells displayed significantly reduced metastatic burden in comparison to the corresponding −DOX mice or mice injected with shSCR (+/−DOX), as observed by BLI (p<0.05) (Figure 4E,F). Mice were sacrificed 27-days post injection, and ex vivo fluorescence imaging revealed a considerably lower metastatic burden in multiple organs (i.e. liver, brain, kidney) in the group of DOX-fed mice injected with 4L FUT8 shC cells (Figure 4G,H). The DOX-induced FUT8 shC-4L injected mice showed much smaller liver metastatic foci and fewer metastatic lesions in the kidneys and brains (data not shown). Moreover, the fraction of mice that developed brain metastases or kidney metastases at termination of the experiment was lower in the DOX-induced FUT8 shC group (3/9 for brain and 6/9 for kidney) than in the absence of DOX (8/9 for brain and 9/9 for kidney) (Figure 4H). These data demonstrate a requirement for FUT8 in metastatic colonization and supports FUT8 inhibition as a viable therapeutic strategy against advanced melanoma.
Figure 4. FUT8 silencing impairs the growth of established metastasis in vivo.

(A) Real-time qPCR and Western blot of FUT8 levels in 4L melanoma cells stably transduced with DOX inducible pTRIPZ-shSCR or pTRIPZ-FUT8 shC. QPCR graph shows average relative expression normalized to GAPDH, 3 replicates per condition. Data shown is representative of three independent experiments. (B) Cell proliferation assay of FUT8 shC or shSCR-transduced 4L cells in the presence (1 μg/ml) or absence of DOX. Cell growth was analyzed by Cell Titer Glo, 3 replicates per condition. Data shown is representative of two independent experiments. (C) Number of FUT8 shC or shSCR-transduced 4L cells that invaded into the basal side of matrigel-coated trans-well inserts after 36 hr. n=5 fields per replicate; 3 replicates per condition. Data shown is representative of three independent experiments. (D) Schematic representation of the in vivo metastasis assay with 4L melanoma cells stably transduced with DOX inducible pTRIPZ-shSCR or pTRIPZ-FUT8 shC instilled by intracardiac injection into NSG mice (n=4 mice per group in –DOX and + DOX shSCR, n=9 mice per group in –DOX and + DOX FUT8 shC). (E) Representative images of whole-body in vivo imaging 12 days post DOX treatment. (F) Average radiance after intracardiac injection of 4L cells followed over time. DOX treatment was started 16 days post injection. (G) Ex vivo fluorescent microscopic images of mouse liver, kidney and brain metastases at termination of the experiment. White dotted ovals mark organs. (H) Histogram shows the percentage of mice that developed brain or kidney metastases in each group of mice. Data are presented as mean ± SD in A, B and C. Two-tailed unpaired t test. *p=0.01 to 0.05, ns: not significant.
TGIF2 regulates FUT8 transcription
We sought to identify genomic or epigenetic changes or upstream transcriptional regulators of FUT8 potentially responsible for its upregulation in metastatic melanoma. Analysis of FUT8 mutations and amplifications showed a very low frequency of alterations in this gene (Figure S4A and data not shown). In addition, analysis of the methylation status showed that all the CpG islands in the FUT8 promoter are unmethylated in both primary and metastatic melanomas (β-value ~ 0; Figure S4B,C). We therefore turned our attention to transcriptional control. We identified a candidate list of transcriptional regulators based on both in silico analysis of transcription factors (TFs) predicted to bind the FUT8 promoter using the MatInspector software (Cartharius et al., 2005) and those shown to physically bind the FUT8 promoter in other cellular systems using the ChIP Enrichment Analysis (ChEA) database (Lachmann et al., 2010). From this list, we selected a subset of TFs whose expression correlated with FUT8 levels (p<0.2, data not shown) in multiple melanoma datasets including TCGA (Raskin et al., 2013; Riker et al., 2008; Xu et al., 2008) (TGIF2, SMAD3, NFKB1, SOX10, IRF1, FOXA2, RUNX1, SOX2). We examined the effect of silencing these TFs on FUT8 expression in 4L and SkMel147 melanoma cell lines (Figure 5A,B, S4D). Depletion of TGFβ-Induced Factor Homeobox 2 (TGIF2) consistently resulted in a ~4-fold reduction in FUT8 levels and concomitant loss of core fucosylation (Figure 5A–D). We next tested if TGIF2 silencing would recapitulate the effect of FUT8 suppression on melanoma cell invasion. Indeed, siTGIF2-transfected 4L melanoma cells exhibited a 5-fold reduction of in vitro cell invasion as compared to siNTC-treated cells (Figure 5E). To determine whether the regulation of FUT8 expression by TGIF2 is direct, we assessed the binding of TGIF2 to the FUT8 promoter (Yamaguchi et al., 2000), which is predicted to contain a consensus-binding site in exon 1 (Figure 5F). Chromatin immunoprecipitation (ChIP) of TGIF2 followed by quantitative PCR using three different sets of primers spanning the TGIF2 putative binding site in the vicinity of the FUT8 promoter showed a 6–10 fold enrichment in FUT8 signal over ChIP with non-specific IgG in 4L cells (Figure 5G). Meanwhile, negative controls, GAPDH, HPRT1, and primers spanning FUT8 distal regions at exons 5 and 8, showed no significant enrichment. TGIF2 binding to the FUT8 promoter was found to correlate with FUT8 expression, as low FUT8 expressing melanoma cell line SkMel85 showed no enrichment in TGIF2 ChIP compared to high FUT8 expressing SkMel147 and 4L cells (Figure S4E,F). To confirm the ability of TGIF2 to control FUT8 expression, we tested the effects of TGIF2 knockdown on luciferase activity driven by a FUT8 promoter construct encompassing the TGIF2 binding site in HEK293T cells. We observed a significant reduction in luciferase activity (p<0.05) (Figure 5H) upon TGIF2 silencing, with concomitant reduction of FUT8 mRNA levels measured by qPCR (Figure S4G). Moreover, TGIF2 and FUT8 mRNA levels positively correlate (R=0.57, p<0.0001) in patient tissues (Figure 5I), and multiple GEO datasets revealed higher TGIF2 expression in metastatic melanoma compared to primary (Figure S4H). Overall, our data reveal TGIF2 as a key regulator of FUT8 transcriptional induction in melanoma metastasis and an upstream contributor to melanoma aggressive behavior.
Figure 5. TGIF2 regulates FUT8 transcription.

Real-time qPCR of FUT8 mRNA levels after silencing of candidate transcription factors in 4L (A) and SkMel147 (B) cell lines. QPCR graph shows average relative expression normalized to GAPDH, 3 replicates per condition. Data shown is representative of two independent experiments. (C) Silencing of TGIF2 decreases FUT8, as shown by Western blot. (D) LcH fluorescence microscopy of 4L cells transfected with NTC or TGIF2 siRNA (50 nM). Lower panel shows quantitation of fluorescent intensity of representative images. Data from 5 random images of 2 biological replicates were averaged to generate graphs, scale bar, 100 μm. (E) Trans-well matrigel invasion assay on 4L cells transfected with NTC or TGIF2 siRNA, scale bar, 100 μm. n=5 fields per replicate; 3 replicates per condition. Data shown is representative of three independent experiments. (F) Schematic of the FUT8 promoter indicating the MatInspector-predicted binding site of TGIF2, the regions flanked by ChIP qPCR primers (Exon 1A, 1B and 1C) and the transcription start site. (G) TGIF2 ChIP qPCR of the FUT8 promoter at the indicated A, B and C regions in 4L cells. IgG, immunoglobulin. GAPDH, HPRT1, FUT8 exon 5 and FUT8 exon 8 served as negative controls for ChIP qPCR (mean ± SD of 3 replicates). (H) Promoter luciferase reporter assay to show pLS-FUT8 activity. Activity of the human FUT8 promoter was examined by co-transfection of the luciferase reporter construct pLS-FUT8 (100 ng) containing a 1000 bp long region of the human FUT8 promoter with NTC or TGIF2 siRNA (50 nM) in HEK293T cells. Relative luciferase activities were measured 24 hr after transfection (mean ± SD of 3 replicates). (I) Scatter plot shows positive co-relation of TGIF2 and FUT8 mRNA levels in a melanoma dataset, r= 0.57, p<0.0001 (Riker et al., 2008). Data are presented as mean ± SD in A, B, D and E. Two-tailed unpaired t test, *p=0.01 to 0.05 and **p=0.001 to 0.01. See also Figure S4.
Identification of core fucosylated glycoproteins in melanoma reveals regulators of invasion and metastasis
To identify core fucosylated proteins that could mediate the effects of FUT8 dysregulation in metastatic melanoma, we performed proteomic analysis of core-fucosylated membrane proteins. We prepared membrane extracts of three melanoma cell lines and enriched for core-fucosylated proteins using lectin chromatography with LcH (Figure 6A). Lectin blot analysis of affinity enriched protein fractions confirmed an enrichment of core fucosylated proteins (Figure S5A). Mass spectrometric analysis identified about 1100 proteins in each enriched fraction and a matrix of 471 overlapping proteins present in all three lines was created. After data filtration using median filtering and removal of contaminant proteins (known to be unglycosylated) such as ribosomal proteins, tubulins and heat shock proteins, 183, 221 and 152 candidate proteins were identified in the LcH enrichment of 4L, SkMel147 and MeWo membrane proteins respectively, 114 of which were common to all 3 cell lines (Figure 6B). Gene ontology analysis (DAVID) performed on the common proteins revealed enrichment in functional categories comprised of N-linked glycosylation, gangliosides, membrane and integrin (Figure S5B), and biological processes relevant to metastasis such as cell migration and locomotion control mechanisms (Figure 6C). Proteins identified as LcH bound that are involved in migration and adhesion include Neural cell adhesion molecule L1 (L1CAM), integrins-α4, 6 and V, integrin B1, ADAM10, laminin β1 (LAMB1) and γ1 (LAMC1), Pro-low-density lipoprotein receptor-related protein 1 (LRP1), cell surface glycoprotein MUC18 (MCAM), plexin-A1, plexin-B2 and neuropilin 2 (NRP2) (Table S2). To validate our proteomic analysis, we further examined the glycosylation state of L1CAM, NRP2, LAMB1, integrin B1 and ADAM10. LcH lectin enrichment followed by Western blot showed reduced L1CAM, NRP2, LAMB1, integrin B1 and ADAM10 levels in siFUT8- compared to siNTC-transfected 4L and SkMel147 (Figure 6D, S5C,D), consistent with lower core fucosylation on those proteins, while input show no differences in the expression of these proteins. Consistently, immunoprecipitation (IP) of L1CAM or NRP2 followed by LcH blot showed reduced LcH binding to L1CAM or NRP2 proteins in siFUT8- compared to siNTC-transfected 4L and SkMel147 (Figure 6E, S5E). The observed differences disappeared upon PNGase treatment, which removes all forms of N-linked glycosylation (Figure 6E). Western blots confirmed equal amount of IP input in each condition (Figure 6E, S5E). Thus, both proteins were confirmed as core fucosylated in line with our proteomic analysis.
Figure 6. Identification of core fucosylated glycoproteins in melanoma reveals regulators of invasion and metastasis.

(A) Schematic illustration of the experimental approach showing affinity enrichment of core fucosylated proteins by LcH lectin affinity chromatography. (B) Number of proteins identified by mass spectrometry analysis of the LcH enriched fractions of 4L, SkMel147 and MeWo membrane proteins. (C) GO enrichment analysis (category of biological processes) of common core-fucosylated proteins in three cell lines. Also see Table S2. (D) LcH affinity chromatography of whole cell lysate of 4L cells transfected with NTC or FUT8 siRNA followed by Western blot with α-L1CAM or α-NRP2 antibody. Input shows no effect of FUT8 knockdown on L1CAM or NRP2 expression. (E) L1CAM and NRP2 immunoprecipitation from whole cell lysates of 4L cells transfected with NTC or FUT8 siRNA. Anti-L1CAM or anti NRP2 immunoprecipitates were treated with or without PNGase F and blotted with biotinylated LcH or α-L1CAM Ab1 (also referred as α-L1CAM) or α-L1CAM Ab2 or α-NRP2. L1CAM Ab1 preferentially recognizes glycosylated L1CAM while Ab2 preferentially recognizes non-glycosylated L1CAM. Experiments in D and E were performed in triplicates and representative images are shown. See also Figure S5.
L1CAM is a mediator of the pro-invasive effects of FUT8
L1CAM is a highly glycosylated protein known to regulate cell attachment, invasion and migration in several cancers (Altevogt et al., 2016; Valiente et al., 2014) and may promote melanoma progression (Meier et al., 2006). Thus, it is a plausible candidate as a mediator of the pro-invasive effects of FUT8. In keeping with the known impact of L1CAM on cell invasion, ectopic expression of L1CAM in melanoma cells expressing low (WM3248) to moderate (MeWo) L1CAM levels triggered a modest but significant increase in in vitro cell invasion (p<0.05) (Figure 7A,B; S6A–C). Concomitant FUT8 silencing, which resulted in reduced core fucosylated L1CAM, counteracted the pro-invasive effects of L1CAM over-expression (Figure 7A,B, S6A,B). These results suggest that core fucosylation is critical to L1CAM pro-invasive phenotype in melanoma.
Figure 7. L1CAM is a mediator of the pro-invasive effects of FUT8.

(A) Western blot of L1CAM or FUT8 in WM3248 cells stably overexpressing L1CAM or control vector and transfected with NTC or FUT8 siRNA. L1CAM IP on WM3248 cells lysates stably overexpressing L1CAM or control vector and transfected with NTC or FUT8 siRNA. Anti-L1CAM immunoprecipitates were blotted with biotinylated LcH or α-L1CAM Ab1. (B) Trans-well matrigel invasion assay on WM3248 melanoma cells stably overexpressing L1CAM or control vector and transfected with NTC or FUT8 siRNA. n=5 fields per replicate; 5 replicates per condition. Data shown is representative of three independent experiments. (C) Trans-well matrigel invasion by MeWo melanoma cells stably overexpressing FUT8 or control vector, scale bar, 100 μm. Western blot of FUT8 is also shown. n=5 fields per replicate; 5 replicates per condition. Data shown is representative of three independent experiments. (D) L1CAM IP on whole cell lysates of MeWo cells stably overexpressing FUT8 or control vector. Anti-L1CAM immunoprecipitates blotted with biotinylated LcH or α-L1CAM Ab1. (E) Western blot of FUT8 and L1CAM in MeWo cells stably overexpressing FUT8 or control vector and transfected with NTC or L1CAM siRNA. (F) Trans-well matrigel invasion by MeWo melanoma cells stably overexpressing FUT8 or control vector and transfected with NTC, L1CAM or FUT8 siRNA. n=5 fields per replicate; 5 replicates per condition. Data shown is representative of three independent experiments. (G) Western blot of L1CAM in MeWo cell line overexpressing FUT8 or control vector, and transfected with NTC or L1CAM siRNA. Samples were blotted with α-L1CAM Ab1. (H) Western blot of cleaved and full length L1CAM on SkMel147 cells transfected with NTC or FUT8 siRNA and treated with α2-antiplasmin. Cells were transfected with siRNAs for 48 hr then incubated with α2-antiplasmin (5 μg/ml) for 16 hr. L1CAM Ab3 preferentially recognizes cleaved L1CAM fragment while L1CAM Ab1 preferentially recognizes full length L1CAM. (I) Quantitation of cleaved L1CAM (~85 kDa) using ImageStudioLite software. Tubulin was used for normalization of loading (mean ± SD of 3 replicates). (J) Western blot of cleaved and full length L1CAM on lysates from MeWo cells stably overexpressing FUT8 or control vector and treated with plasmin. Cells were cultured for 24 hr then incubated with plasmin (3 μg/ml) for 24 hr. (K) Quantitation of cleaved L1CAM. Tubulin was used for normalization of loading (mean ± SD of 3 replicates). All experiments were performed in triplicates and representative images are shown. Data are presented as mean ± SD. Two-tailed unpaired t test. *p=0.01 to 0.05, **p=0.001 to 0.01 and *** p=0.0001 to 0.001, ns: not significant. See also Figure S6.
We next investigated whether FUT8 ectopic expression is able to promote melanoma cell invasion capacity and if core fucosylation of L1CAM contributes to that effect. FUT8 over-expression increased cell invasion 5-fold in a poorly invasive cell line (MeWo) and 2-fold in the highly invasive cell line 4L (Figure 7C, S6D). We confirmed that L1CAM exhibits increased core fucosylation in FUT8 over-expressing MeWo cells (Figure 7D). Silencing of L1CAM strongly counteracted the pro-invasive effects of FUT8 overexpression (Figure 7E,F; S6E–F). As expected, FUT8 pro-invasive effects were also ablated by FUT8 silencing (Figure 7F, S6F). Overall, our data support core-fucosylated L1CAM as a key mediator of FUT8 pro-metastatic effects.
Glycosylation can affect protein folding, activation and the access of proteases to cleavage sites (Cheng et al., 2015; Sola and Griebenow, 2009). L1CAM is cleaved by both ADAM10 and plasmin (Maretzky et al., 2005). In particular, cleavage of L1CAM by plasmin inhibits its ability to mediate cell invasion and metastatic outgrowth (Valiente et al., 2014). We hypothesized that altered core fucosylation might alter L1CAM cleavage by proteases, impacting melanoma invasive behavior. Indeed, we observed that MeWo cells overexpressing FUT8 show lower levels of a ~85 kDa L1CAM product when compared to controls (Figure 7G). This cleavage fragment disappears upon L1CAM knockdown, confirming it as a L1CAM specific product. Silencing of FUT8 in SkMel147 cells (expressing high FUT8 levels) results in increased levels of the ~85 kDa L1CAM product compared to siNTC (Figure 7H,I). Treatment of SkMel147 with a plasmin inhibitor, α-2 antiplasmin, resulted in reduced ~85 kDa L1CAM fragment (Figure 7H–I). Silencing of plasminogen activator (PLAT), which converts plasminogen into plasmin, reduced the levels of this ~85 kDa L1CAM fragment in SkMel147 cells (Figure S6G,H), confirming this fragment as a plasmin cleavage product. To further support that core fucosylation inhibits L1CAM cleavage, we performed in vitro plasmin treatment in control and FUT8-overexpressing MeWo cells. In presence of plasmin a significant increase in cleaved L1CAM (~85 kDa) was observed in control cells while in FUT8 overexpressing MeWo cells L1CAM was protected against cleavage (Figure 7J,K). Overall, our results suggest that core fucosylation is critical to L1CAM function through modulation of plasmin cleavage and that L1CAM is a major mediator of the pro-metastatic role played by core fucosylation (i.e. FUT8) in melanoma.
DISCUSSION
Although it has long been speculated that glycosylation plays important roles in melanoma progression, there are no comprehensive glycomic analyses of clinical samples to support this assertion (Christiansen et al., 2014). The biosynthesis of glycans is an intricate process requiring the coordinated action of multiple glycosyltransferases and glycosidases to synthesize discrete structures (Cummings and Pierce, 2014; Moremen et al., 2012). There is currently little understanding of how glycan biosynthesis is controlled (Nairn et al., 2012) or which enzymes are important in the biosynthesis of specific epitopes underlying clinical pathologies, hindering the development of glycosylation-based therapeutic strategies (Dalziel et al., 2014; Dube and Bertozzi, 2005). Here we have taken a systematic, multi-disciplinary approach to identify glycomic changes and the underlying glycosyltransferases associated with melanoma metastasis in patient samples. Our analysis revealed α-1,6 fucosylation, mediated by the FUT8, is functionally important for melanoma invasion and provided insight into how core fucosylation impacts melanoma metastasis.
Glycomic analysis of clinical melanoma has long been inhibited by the small quantities of sample afforded by primary melanoma (Christiansen et al., 2014). Lectin microarray analysis requires as little as 50 μg of FFPE tissue, enabling a systematic analysis of glycosylation in clinical samples. Our analysis revealed strong discrimination between primary melanomas and metastases based on glycosylation, with several glycans (α-2,6 sialic acid, poly/multiantennary LacNAc) following predictions from model systems (Kinoshita et al., 2014; Pinho and Reis, 2015). However, the changes in fucosylation were surprising. In recent work it was reported that transcription factor ATF2 promotes melanoma metastasis by suppressing global fucosylation (Lau et al., 2015). While we observed a loss of α-1,2 fucosylation (UEA-I, TJA-II), we saw a marked increase in core fucosylation (α-1,6 fucose, LcH, PSA), arguing that the effects of fucose on metastasis are linkage-dependent. In the ATF2 study, UEA-I, specific for α-1,2 fucosylation, was used as a global marker for fucose, and loss of UEA-I binding correlated with lower survival rates (Lau et al., 2015). This data is consistent with our lectin microarray analysis in which binding to UEA-I is lost with progression to metastasis.
Mapping of candidate glycogenes from clinical transcriptomic datasets onto our lectin microarray data provided further evidence for the linkage-specific effects of fucosylation in melanoma, identifying FUT8, the sole enzyme responsible for core fucosylation, as upregulated in metastatic cohorts, and FUT1 and FUT2, enzymes that create α-1,2 fucosylated epitopes, as high in primary melanoma (Kanehisa and Goto, 2000; Nairn et al., 2012). Lectin histochemistry on patient samples confirmed increased core fucose and loss of α-1,2 fucose in metastatic samples. In line with this data, we identified FUT8 as a positive regulator of, and FUT1 and FUT2, as potential suppressors of invasion. Although data in a mouse melanoma line and the recent work on ATF2 support a role for α-1,2 fucose in suppressing the metastatic potential of melanoma (Gorelik et al., 1997; Lau et al., 2015), there is no known role for core fucosylation in melanoma progression, and the importance of these enzymes to clinical melanoma is unknown. Our results support the value of a systems-based approach to identify specific glycogenes (and corresponding enzyme and glycan changes) that contribute to melanoma metastatic behavior.
We focused our functional and mechanistic studies on FUT8, as a candidate pro-metastatic gene and potential therapeutic target. Core fucosylation is elevated in liver and ovarian cancerous tissues in comparison to normal (Abbott et al., 2008; Mehta et al., 2016), and linked with EGFR dimerization and activation, TGF-β-activation, and HGF and EGF signaling pathways (Gu et al., 2013; Liu et al., 2011; Wang et al., 2015). However, a role for this enzyme in melanoma and the specific core fucosylated glycoproteins underlying its effects are unknown. FUT8 silencing in highly metastatic melanoma cell lines reduced their invasive capacity without affecting proliferation. This is in contrast to work in both non-small cell lung (Chen et al., 2013) and hepatocellular carcinoma (Wang et al., 2015), where FUT8 silencing was anti-proliferative. Silencing FUT8 reduced metastatic dissemination of melanoma cells to the lungs and inhibited the growth of pre-seeded metastases in liver, brain and kidney, suggesting core fucosylation may be required for the adaptations that disseminated cancer cells undergo to survive in ‘foreign’ tissues. FUT8 inhibition of pre-established metastases seems independent of any growth advantage conferred by FUT8, as FUT8 silencing did not impact growth of primary melanoma tumors. Together our data point towards metastatic-specific promoting effects of FUT8 and provide compelling evidence that FUT8 silencing inhibits the processes underlying melanoma metastasis in vivo.
We identified TGIF2 as a positive regulator of FUT8. This is in agreement with previous evidence that TGIF2 inhibition by miR-34a blocks bone metastasis and melanoma invasive growth (Krzeszinski et al., 2014; Yamazaki et al., 2012). FUT8 has also been described as a miR-34a target (Bernardi et al., 2013; Cheng et al., 2016) opening the possibility that miR-34a controls FUT8 both directly and indirectly, via TGIF2 suppression. Multiple datasets of melanoma patient samples support TGIF2 upregulation in metastasis highlighting the clinical relevance of the TGIF2-FUT8 axis.
Core fucosylation is found on a myriad of proteins including adhesion molecules, receptors, and inflammatory molecules (Ohtsubo and Marth, 2006). Glycoproteomic analysis of metastatic melanoma cell lines identified 114 common core-fucosylated proteins, with enrichment in proteins involved in cell invasion and migration. These proteins included NRP2, a cell surface receptor promoting extravasation and metastasis (Karpanen et al., 2006), multiple integrins, which play well known roles in metastasis (Desgrosellier and Cheresh, 2010), and the neural cell adhesion molecule L1CAM. Overexpression of L1CAM contributes to the metastatic cascade in several cancers, including ovarian and pancreatic cancers (Altevogt et al., 2016). L1CAM transcripts are not upregulated in metastasis in melanoma datasets; however, L1CAM activity or interaction with other receptors or growth factors might be altered by core fucosylation. FUT8 conferred increased invasive capacity to the poorly invasive MeWo and moderately invasive 4L cell lines. Remarkably, this phenotype was mostly rescued by silencing of L1CAM. Taken together, the data suggest that core fucosylation enhances the ability of L1CAM to promote metastasis, altering either L1CAM activity or interactions with its counterparts (Kiefel et al., 2010).
L1CAM is a 220 kDa glycoprotein and its cleavage by proteases like ADAM10 and plasmin results in 32 and 85 kDa cellular fragments, respectively (Kiefel et al., 2012; Silletti et al., 2000). Cleavage by plasmin impairs L1CAM ability to promote vascular co-option and metastatic outgrowth (Valiente et al., 2014), while ADAM10 cleavage renders a soluble L1CAM fragment important for invasion or migration of cancer cells (Altevogt et al., 2016). We observed lower amounts of cleaved ~85 kDa L1CAM in FUT8 overexpressing cells and increased cleaved L1CAM upon FUT8 silencing, suggesting that core fucosylation impairs L1CAM cleavage and providing a rationale for the prometastatic behavior of highly fucosylated L1CAM. Our mass spectrometry analyses identified a L1CAM glycopeptide 842GYNVTYWR849 in close proximity to the plasmin cleavage site of L1CAM (851GSQRKHSKRHIHKDHV866). The molecular weight of the detected L1CAM cleavage product matched that of plasmin cleavage (Silletti et al., 2000). Treatment of melanoma cells with plasmin or α2-antiplasmin increased or decreased its levels, respectively, and plasmin-mediated cleavage of L1CAM was inhibited in FUT8 overexpressing cells. The ability of uncleaved L1CAM to co-opt the existing vasculature at distal organs could explain why FUT8 is required for the growth of established metastases without affecting intrinsic cell proliferation. Our data argues that manipulating the glycosylation status of L1CAM may provide a way to increase its cleavage and impair metastasis.
Despite remarkable advances that have led to FDA approval of new compounds, outcomes for metastatic melanoma patients remain poor. Lack of response, initial response followed by relapse with treatment-refractory disease and adverse side effects are common occurrences (Lo and Fisher, 2014; Zaretsky et al., 2016). Therefore, there is a clear need to identify new targets and develop innovative drugs that target molecules essential for metastatic melanoma. Our study highlights the therapeutic potential of targeting FUT8 to treat metastatic melanoma and other advanced tumors. Exemplifying the value of glycosylation enzymes as druggable targets are both glycosyltransferase (Zavesca for Gaucher’s disease (Dwek et al., 2002; Gloster and Vocadlo, 2012)) and glycosidase inhibitors (zanamivir (Relenza) and oseltamivir (Tamiflu) for influenza (Dalziel et al., 2014), migalastat for Fabry’s disease (Gloster and Vocadlo, 2012)), currently in use or in advanced clinical trials. Our work provides a rationale for the future design of small molecule inhibitors against FUT8, to prevent or treat established metastasis.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-L1CAM antibody [UJ127.11] L1CAM Ab1 | Abcam | ab20148 |
| Anti-L1CAM antibody [EPR18750] L1CAM Ab2 | Abcam | ab208155 |
| Anti-L1CAM antibody [2C2] L1CAM Ab3 | Abcam | ab24345 |
| Anti-GAPDH antibody [6C5] | Abcam | ab8245 |
| Anti-Neuropilin-2 antibody (C-9) | Santa Cruz | sc13117 |
| Anti-Laminin beta 1 antibody [LT3] | Abcam | ab44941 |
| Anti-Integrin beta 1 antibody [EP1041Y] (ab52971) | Abcam | ab52971 |
| Anti-ADAM10 antibody (ab1997) | Abcam | ab1997 |
| Anti-TGF beta induced factor 2 antibody [EPR5655(2)] | Abcam | ab155948 |
| Anti-FUT8 antibody (Clone No.: 2F7H2 | Proteintech | 66118 |
| Monoclonal Anti-α-Tubulin | Sigma | T9026 |
| Streptavidin, Alexa Fluor® 647 conjugate | Life Technologies | S32357 |
| Pierce™ High Sensitivity Streptavidin-HRP | Thermo Fisher | 21130 |
| Lectins; see Table S1 | E.Y. or Vector Lab | N/A |
| Biological Samples | ||
| Melanoma FFPE samples; see Table S4 | IMCG, NYUMC | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fluorescein labeled Ulex Europaeus Agglutinin I (UEA I) | Vector lab | FL1061 |
| Fluorescein labeled Lens Culinaris Agglutinin (LCA) | Vector lab | FL1041 |
| Biotinylated Lens Culinaris Agglutinin (LCA) | Vector lab | B-1045 |
| Agarose Streptavidin | Vector lab | SA-5010 |
| Matrigel Basement Membrane Matrix | BD Biosciences | CB-40234 |
| Cy5 NHS ester | Amersham | PA15101 |
| Cy3 NHS Ester | Amersham | PA13101 |
| Lipofectamine 2000 | Thermo Fisher | 11668027 |
| Plasmin | Sigma-Aldrich | P1867 |
| α2-Antiplasmin | Molecular innovations | HA2AP |
| Critical Commercial Assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7571 |
| LightSwitch Luciferase Assay Kit | SwitchGear Genomics | 32031 |
| miRNeasy mini kit | Qiagen | 217004 |
| Immunoprecipitation Kit (Protein G) | Roche | 11719386001 |
| Deposited Data | ||
| Lectin microarray data | This paper | https://www.synapse.org/#!Synapse:syn9748084/files/ |
| Mass spectrometry data | This paper | https://github.com/FenyoLab/Fucosylated_Melanoma/ |
| Experimental Models: Cell Lines | ||
| Human: 113/6-4L (4L) | (Cruz-Munoz et al., 2008) | Authenticated by STR analysis by ATCC |
| Human: MeWo | ATCC | Authenticated by STR analysis by ATCC |
| Human: SkMel147 | Dr. Alan Houghton (MSKCC, New York) | Authenticated by STR analysis by ATCC |
| Human: SkMel85 | Dr. Alan Houghton (MSKCC, New York) | Authenticated by STR analysis by ATCC |
| Human: WM3248 | Wistar Institute | N/A |
| Human: WM3211 | Wistar Institute | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | The Jackson Laboratory | Stock # 005557 |
| Oligonucleotides | ||
| Primers; see Table S3 | This paper | N/A |
| Plasmids | ||
| pLKO.1 | Sigma | N/A |
| TRIPZ Inducible Lentiviral Human FUT8 shRNA | Dharmacon | RHS4740-EG2530 |
| plasmid pReceiver-Lv205 for L1CAM | Genecopeia | EX-Z2881-Lv205 |
| FUT8 overexpression plasmid pCMV6-Myc-DDK | Origene | RC212345 |
| p-Lenti-C-Myc-DDK | Origene | PS100069 |
| pLightSwitch Prom (pLS) | Switchgear | S790005 |
| Software and Algorithms | ||
| LivingImage software | Xenogen Corp., Alameda, CA | http://www.perkinelmer.com/product/spectrum-200-living-image-v4series-1-128113 |
| NDPIS software | Hamamatsu | http://www.hamamatsu.com/us/en/U12388-01.html#1328463611394 |
| ImageStudioLite | LICOR | https://www.licor.com/bio/products/software/image_studio_lite/ |
| ImageJ | NIH | https://fiji.sc |
| PRISM | Graphpad | https://www.graphpad.com |
| Matinspector software | Genomatix | https://www.genomatix.de/online_help/help_matinspector/matinspector_help.html |
| CHEA | Icahn School of Medicine at Mount Sinai | http://amp.pharm.mssm.edu/Enrichr/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Eva Hernando (eva.hernando-monge@nyumc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and cell culture
Cell line 113/6-4L (4L) was cultured as previously described (Cruz-Munoz et al., 2008). SkMel147 and SkMel85 were acquired from Dr. Alan Houghton (MSKCC, New York). The human WM3211 and WM3248 cell lines were acquired from the Wistar Institute. HEK293T was purchased from American Type Culture Collection (ATCC). 4L, SkMel147, SkMel85 and MeWo are metastatic melanoma cell lines, whereas WM3211 and WM3248 were derived from primary melanomas. MeWo is male origin and 4L, SkMel147, WM3248 are female origin cell lines. 4L and SkMel147 cells were cultured in DMEM (Invitrogen) and MeWo was cultured in EMEM (Invitrogen) containing 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin. WM3211 and WM3248 were cultured in medium containing 80% (v/v) MCDB153, 1.2 g/L NaHCO3, 20% (v/v) Leibovitz’s L-15 (Invitrogen), 2% heat inactivated (v/v) FBS, 5 g/mL bovine insulin, 1.68 mM CaCl2 and 1% (v/v) penicillin/streptomycin at 37°C and 5% CO2. Cell lines were maintained in a 5% CO2 incubator at 37°C. Cell lines were routinely tested to exclude Mycoplasma contamination. For in vitro assays, α2-antiplasmin (5.0 μg/ml; Molecular Innovations, ref: HA2AP), and plasmin (3.0 μg/ml: Sigma, ref: P1867) were added to the medium and incubated for 18–24 hr.
Clinical Specimens
Human melanoma specimens (primary, metastatic) were collected at the time of surgery (Table S4). Approval to collect specimens was granted by New York University Institutional Review Board protocol number i10362, “Development of an NYU Interdisciplinary Melanoma Cooperative Group: A clinicopathological database.” Informed consent was obtained from all subjects included.
Animal studies
All mice experiments were performed in compliance with a referenced protocol (120405-03) approved by the NYU Institutional Animal Care and Use Committee (IACUC). 4–6 weeks old NOD/Shi-scid/IL-2Rgamma null (NSG) mice (male and female) were purchased from Jackson Laboratory and maintained under standard pathogen free conditions. Experimental sample size was based on our previous experience using the same strains. In intracardiac inducible metastasis model, mice were randomized and subdivided into two groups for +/−DOX feed such that there were no differences in average radiance between the two groups.
METHOD DETAILS
Lectin microarray printing, hybridization and analysis
Lectins were purchased from E.Y. Laboratories or Vector Laboratories with the following exceptions: recombinant CVN, SVN and griffithsin were gifts from B. O’Keefe (NCI, Frederick, MD); TJA-I and TJA-II were from NorthStar Bioproducts. Printing, hybridization and data analysis were performed as previously described (Pilobello et al., 2013). Our data were normally distributed as determined by the Lilliefors test in MATLAB. Lectins were excluded from analysis if they did not meet our minimal threshold for activity (Batista et al., 2011). See table S1 for lectin print list.
FFPE primary and metastatic melanoma sample deparaffinization, protein extraction and labeling
Unstained cut sections mounted on slides were macrodissected to remove contaminating normal cells, and 2 × 20 μm sections of each FFPE tissue were scraped into a 1.5-ml microcentrifuge tube. Samples were deparaffinized with xylene and ethanol, rehydrated and solubilized in Cy buffer (0.1 M NaHCO3, pH=9.3) containing 0.5% NP40 (Nonidet P-40) and labeled as previously described (Hsu et al., 2011; Pilobello et al., 2013). Reference sample was a mixture of 6 primary and 6 metastatic samples. Detailed protocols are described in the supplementary section.
Mapping lectin specificity onto glycosylation pathways
Biosynthetic pathways for relevant glycan epitopes were mapped by using the Kyoto Encyclopedia of Genes and Genomes (Kanehisa and Goto, 2000) and the work of Nairn et al. (Nairn et al., 2012).
FFPE primary and metastatic melanoma sample deparaffinization, protein extraction and labeling
Formalin-fixed paraffin-embedded (FFPE) tissues from patients with primary (n=17) or metastastic (n=17) melanoma (n=17 paired cases, 34 total) were analyzed on our lectin microarrays. In brief, Melanoma samples were fixed in 10% neutral buffered formalin. For analysis of tumor samples, hematoxylin and eosin-stained slides were reviewed to ensure enough viable tumor. Unstained cut sections mounted on slides were macrodissected to remove contaminating normal cells, and 2 × 20 μm sections of each FFPE tissue were scraped into a 1.5-ml microcentrifuge tube. One mL xylene and 200 μl ethanol were added to microcentrifuge tube, incubated for 15 min at room temperature, centrifuged at 10,000 × g for 2 min and supernatant was removed. The deparaffinized tissue pellets were then rehydrated with a graded series of ethanol. One mL of 95% ethanol was added to each tissue pellet, incubated for 10 min at room temperature and centrifuged at 10,000 × g for 2 min. This was followed by rehydration with 70% ethanol, spun down to remove excess supernatant and allowed samples to dry at room temperature. The rehydrated tumor tissue and cell sections were re-suspended in 200 μl 10 mM sodium citrate buffer (pH 6.0) and incubated at 95°C for 60 min. The supernatant was removed and tissue was washed with PBS. Pellet was solubilized with 250 μl (100 μl for smaller pellets) Cy buffer (0.1 M NaHCO3, pH=9.3) containing 0.5% NP40 (Nonidet P-40). Sample was sonicated gently (70% power, total time 1 min [10 s on, 10 s off]) and incubated on ice for 60 min. Sample was centrifuged at 12,000 x g for 5 min at 4°C. Supernatant was collected for analysis. Total protein concentration was quantified by BCA assay. Samples were then labeled with NHS-Cy3 or -Cy5 and analyzed on our lectin microarrays in dual color using standard protocols (Hsu et al., 2011; Pilobello et al., 2013). Reference sample was a mixture of 6 primary and 6 metastatic samples. Normalized data was subjected to hierarchical clustering using the Pearson correlation coefficient (R) as the distance metric and average linkage analysis to generate heatmaps.
Reverse transfection of siRNA
Transfection conditions were optimized for each cell line using fluorescein labeled oligos (Invitrogen, Block-IT). Liposomal transfection complexes with siRNA pools (Dharmacon, Smart Pools, 50 nM, PLAT siRNA was purchased from Origene.) were generated with Lipofectamine 2000 (Invitrogen), following manufacturer’s recommendations. Media was changed after 6 hr incubation with liposomal complexes. 48 hr after initiation of transfection, cells were used for RNA extraction or invasion assay seeding.
RNA Extraction and Real-Time qPCR
Total RNA was extracted from samples (miRNeasy mini kit; Qiagen) and quantified by using Nanodrop 8000 and stored at −80 °C. 500 ng of RNA were reverse transcribed using Taqman RT reagents (Applied Biosystems) with random hexamers following manufacturer’s recommendations. Transcripts were quantified by real-time qPCR using Power SYBR Green PCR MasterMix (Applied Biosystems) in StepOne System (ABI). Primers were designed by using PrimerSelect. Some qPCR primers were purchased from Origene (Table S3). Cycle threshold values were normalized to those of the housekeeping genes GAPDH or HPRT1. The average for three biological replicates was plotted as relative transcript abundance.
Plasmids
pLKO.1 plasmids targeting human FUT8 and a non-targeting control were purchased from Sigma. DOX inducible pTRIPZ plasmids targeting human FUT8 and scramble control were purchased from Dharmacon. See Supplemental Table S3 for sequences. L1CAM overexpression plasmid pReceiver-Lv205 and control-Lv205 were purchased from Genecopoiea. FUT8 overexpression plasmid pCMV6-Myc-DDK was purchased from Origene. FUT8 was subcloned into p-Lenti-C-Myc-DDK destination vector. Empty promoter vector pLightSwitch Prom (pLS) was purchased from Switchgear. FUT8 promoter was subcloned in pLS reporter vector. See Table S3 for FUT8 promoter sequence.
Viral Production
4×106 HEK293T cells were seeded per 10 cm tissue culture dish and incubated overnight at 37°C and 5% CO2 for 16–20 hr. After seeding, HEK293T were co-transfected with lentiviral expression constructs (12 μg), viral packaging plasmid (psPAX2, 8 μg), and viral envelope plasmid (pMD2.G, 4 μg) using Lipofectamine 2000 (Invitrogen) following manufacturer’s recommendations. Viral supernatant was collected and 0.45 μm filtered at 48 hr post-transfection and stored at 4°C for short-term use (1–5 days) or −20°C for long-term storage (5–30 days).
Viral Transduction
Target cells were seeded and incubated overnight prior to infection. Medium was replaced with 1:2 diluted viral supernatant supplemented with 8 μg/mL polybrene, and incubated for 6–8 hr, followed by replacement with growth medium. Control and shRNA cells were selected using puromycin (2 μg/mL) or neomycin (500 μg/mL) prior to use in experiments.
In vitro invasion assay
Cell invasion was measured using 24-well Fluoroblok inserts (8 μm Becton Dickinson). Optimization was performed for each cell line to identify assay time length and Matrigel concentration. Briefly, 4L, WM3211, WM3248, SkMel147 or MeWo cells (40,000 cells per insert) were suspended in serum-free medium over a Matrigel coating (Becton Dickinson), and medium supplemented with 10% serum was used as a chemoattractant. Cells that invaded after 12–36 hr were stained in 4 μg/ml Calcein AM dye (ThermoFisher) for 1 hr and counted in 5 different fields using a fluorescent microscope. For each independent experiment, six replicates per condition were run. The average of cell counts from six inserts per condition was used for plotting results. Cell counts for each well were normalized to the mean counts (of replicate wells) for the corresponding condition in the cell input plate to control for cell proliferation effects that may have occurred between initiation of transfection and assay seeding.
In vitro proliferation assay
Cells were seeded at 5000 cells/well in 96-well plates. Cell proliferation was measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega) following manufacturer’s instructions.
Mouse Experiments
In vivo metastasis assay: We used a xenograft model of metastasis. 4L cells were first transduced with mCherry/luciferase reporter lentivirus and then with shNTC or FUT8 shA or FUT8 shB lentiviruses. Cells were resuspended in sterile PBS at a concentration of 2 × 106 cells per 150 μl, aliquoted into Eppendorf tubes (150 μl) and maintained on ice until injection. Immediately before injection, cell aliquots were mixed with 150 μl Matrigel (Becton Dickinson). Cell/Matrigel (1:1) suspensions were injected subcutaneously in the right flank of NOD/Shi-scid/IL-2Rgamma null (NSG) 8-weeks-old male mice (n=10 for each group). When primary tumors were palpable (7 days), length (l) and width (w) measurements were made with calipers 2 times weekly until resected. Tumor volume was calculated using the formula π/6 × l × (w2). Primary tumors were resected once tumor volume reached 500 mm3. IVIS was performed weekly or twice in a week to measure bioluminescence produced by metastatic cells in the living mice. Briefly, the substrate luciferin was injected into the intraperitoneal cavity at a dose of 150 mg/kg body weight (30 mg/ml luciferin), approximately 15 min before imaging. Mice were anesthetized with isoflurane/oxygen and placed on the imaging stage. Ventral and dorsal images were collected for automatic exposure (10 s to 2 min) using the IVIS (Xenogen Corp., Alameda, CA). Analysis was performed using LivingImage software (Xenogen) by measurement of photon flux (measured in photons/s/cm2/steradian) with a region of interest (ROI) drawn around the bioluminescence signal to be measured. Data were plotted using GraphPad PRISM and significance was determined by unpaired t test. After sacrificing mice, ventral and dorsal macroscopic images of metastasis-bearing lungs and other organs were taken with a fluorescent dissecting microscope equipped with a color camera. Fluorescent intensity of macrometastases was quantified by Image J software. Intracardiac inducible metastasis model: To check the effect of FUT8 modulation of existing metastasis we used a model based on intracardiac instillation of tumor cells (Morsi et al., 2013). 4L cells were first transduced with GFP/luciferase reporter lentivirus and then with DOX inducible shSCR or FUT8 shC lentiviruses. Cells were resuspended in sterile PBS at a concentration of 5 × 104 cells per 150 μl, aliquoted into Eppendorf tubes (150 μl) and maintained on ice until injection. Female NOD/Shi-scid/IL-2Rgamma null (NSG) 8-weeks-old mice (n=10 for Luc-shSCR, n=20 for Luc-FUT8 shC) were anesthetized by exposure to isoflurane. On day 0, anesthetized mice were injected with 5 × 104 4L cells transduced with Luc-shSCR or Luc-FUT8 shC lentiviruses suspended in 150 μl sterile PBS into the left ventricle of the heart using Visualsonics Vevo 770 Ultrasound Imaging System. A successful intracardiac injection was indicated on day 1 by images showing systemic bioluminescence distributed throughout the animal. Only mice with evidence of a satisfactory injection were kept in the experiment. Once metastasis was established mice within both groups (pTRIPZ/shSCR or pTRIPZ/FUT8 shC) were subdivided into two groups for +/−DOX feed. Mice were fed DOX (200 mg/kg weight) containing food. Assessment of metastasis was monitored in vivo twice a week by imaging and data was analyzed as mentioned earlier.
Lectin fluorescence microscopy
Lectin fluorescence was performed on 4 μm formalin fixed, paraffin embedded tissue sections using LcH-biotin and UEA-I-FITC lectins (Vector lab). Dual immunofluorescent staining was run with Ventana Discovery Ultra autostainer on the protocol platform of RUO DISCOVERY Universal (v0.00.0201) staining module. The targeted signals were detected using series of lectins in order of FITC tagged UEA-I (4 μg/slide) followed by Biotin-LcH (5 μg/slide). Biotin-LcH was detected by Streptavidin, Alexa Fluor® 647 conjugate [(S32357) (Life Technologies) (4 μg/slide)]. Both lectins and secondary antibodies were incubated for 1 hr each. DAPI staining was used to determine the nuclei morphology. Fluorescent slides were stored in −20°C until imaging. Stained slides were imaged by Hamamatsu fluorescent slide scanner and images were extracted using NDPIS software. Lectin fluorescence microscopy for cultured cells was performed as previously described (Agrawal et al., 2014).
Immunohistochemistry
Unconjugated mouse α-human α-1,6 fucosyltransferase (FUT8), clone 2F7H2 (ProteinTech Group, catalog number 66118). Antibody optimization was performed on formalin-fixed, paraffin embedded cell blocks prepared from 4L cell line transduced with shNTC or FUT8shA or FUT8 shB lentiviruses and then validated on human tissue microarrays containing multiple normal and cancerous tissues. We regarded cells clearly expressing granular (Golgi) cytoplasmic staining as positive consistent with immunohistochemical results published on the Human Protein Atlas (http://www.proteinatlas.org/). All paraffin embedded material was sectioned at 4 μm. Chromogenic immunohistochemistry was performed on a Ventana Medical Systems Discovery XT instrument with online deparaffinization and using Ventana’s reagents and detection kits unless otherwise noted. Antigen retrieval was done in Ventana Cell Conditioner 1 (Tris-Borate-EDTA) for 52 min. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 4 min. Non-specific protein interactions were blocked using Dulbecco’s phosphate buffered saline (PBS; Life Technologies) with 1% non-fat dry milk, 1% BSA and 0.05% Tween-20 added. FUT8 was diluted 1:200 in Dulbecco’s PBS and incubated for 6 hr at room temperature. Primary antibody was detected with α-mouse, horseradish peroxidase conjugated multimer incubated for 8 min. The complex was visualized with alpha-naphthol pyronin incubated for 8 min. Slide were washed in distilled water, counterstained with hematoxylin, dehydrated and mounted with permanent media. Negative controls were incubated with Dulbecco’s PBS instead of primary antibody and did not show any signal. A blinded pathologist who has experience in melanoma evaluated IHC staining. Perinuclear granular cytoplasmic staining was considered as FUT8 positive. IHC score was calculated using staining intensity x percentage of positive cells and three-tiered system also provided (negative=0, weak=1–50, moderate=51–100 and strong=101–200).
Histopathological analysis
The primary tumors and the lungs were harvested and fixed in 10% formalin for 48 hr followed by washing with PBS. Then the fixed samples were transferred in 70% ethanol. The fixed samples were then embedded in paraffin and three non-sequential serial sections per animal were obtained. The sections were stained with hematoxylin/eosin and analyzed for the presence of metastases by light microscopy. The total number of metastases per lung section was counted by a blinded pathologist and averaged among the animals.
Chromatin immunoprecipitation (ChIP) and ChIP-qPCR
For ChIP, 4L cells were used for each immunoprecipitation (Di Micco et al., 2014). Five micrograms of TGIF2 antibody (Abcam, ab155948) were used per immunoprecipitation. Next, ChIP qPCR was performed to validate pull down sample. Primers designed to expand regions of the genome not anticipated to be pulled down in the ChIP (GAPDH, HPRT1 or FUT8 exon 5/8) served as negative controls. See Supplementary Table S3 for ChIP-qRT-PCR primer sequences.
Luciferase Reporter Assay
The FUT8 promoter was cloned from human genomic cDNA using Q5 Hotstart Polymerase (New England Biolabs) and inserted into the pLightSwitch-Prom reporter vector (SwitchGear Genomics) using the forward primer 5′-TTTCAGTTGGAAGGAGGTAGGG-3′, and reverse primer 5′-CCGCTCGGACTCGGA-3′. Plasmids were purified using Endo-Free Plasmid Maxi Kit (Qiagen). HEK 293/T17 cells (1.26 × 104) were co-transfected with plasmid (250 ng) and siRNA (60 nM) using Lipofectamine 2000 and plated in a well of a 96-well plate (100 μL total volume). After 24 hr, the assay was developed with the LightSwitch Assay Reagent (SwitchGear Genomics), and luminescence read on a Biotek microplate reader. For each assay, we calculated the average fold change as a ratio (siRNA TGIF2)/(siRNA negative control) for six replicate wells per condition. Data presented represents the average of three independent assays.
Western Blotting
Cells were lysed in cold RIPA buffer supplemented with protease inhibitors. Equal amounts of protein were resolved by 4–12% Bis-Tris/PAGE, transferred onto PVDF membranes, and blocked in buffer [5% (wt/vol) milk, TBST (TBS, pH 7.4, 0.05% Tween-20), 1 hr, room temperature]. Primary antibodies were diluted in blocking buffer, as follows: FUT8 (1:1,000; Proteintech, 66118), L1CAM, also referred as L1CAM Ab1 (1:5,000; Abcam, ab20148), L1CAM Ab2 (1:2000; Abcam, ab208155), L1CAM Ab3 (1:2000, Abcam, ab24345), GAPDH (1:5,000; Abcam, ab8245); NRP2 (1:5,000; Santa Cruz, sc13117), LAMB1 (1:1000; Abcam, ab44941), Integrin beta 1 (1:2000; Abcam, ab52971), ADAM10 (1:1000; Abcam, ab1997), α-Tubulin (1:5000; Sigma, T9026). Secondary antibodies were α-mouse or α-rabbit-HRP (1:5,000; Bio-Rad). Blots were developed by using Clarity™ Western ECL Blotting Substrate (BioRad) and imaged in LICOR Odyssey Fc imaging system.
LcH chromatography
For lectin enrichment assays, FUT8 siRNA or non-targeting control siRNA transfected cells were lysed with lysis buffer containing 1% Nonidet P-40. 1000 μg of lysate was mixed with 100 μl of biotinylated LcH lectin and volume made up to 1000 μl with PBS containing 1 mM MnCl2, 1 mM MgCl2, and 1 mM CaCl2 and incubated with rotation at 4°C overnight. Then, 60 μl of a 1:1 suspension of agarose-coupled streptavidin was added, and incubation was continued for 4 hr. The beads were washed five times with PBST (Tween, 0.05%) buffer and subsequently extracted with SDS-PAGE sample buffer at 95°C for 5 min. The samples were separated by 4–12% Bis-Tris/PAGE and subjected to immunoblotting with α-L1CAM, α-NRP2, α-LAMB1, α-Integrin B1 and α-ADAM10 antibodies.
Immunoprecipitation
L1CAM and NRP2 immunoprecipitation was performed using protein G Immunoprecipitation (Roche) according to the manufacturer’s protocol. Briefly, 4L, WM3248 or MeWo cells were transfected with 50 nM FUT8 siRNA or non-targeting control siRNA per 10 cm plate. 72 hr post-transfection, cells were lysed (lysis buffer: 50 mM Tris, 150 mM NaCl, 2 mM EDTA, protease inhibitor cocktail (Complete Mini, Roche), 0.5% Triton X-100) and incubated 1 hr on ice. Cell lysate was centrifuged 30 min at 4°C, and an aliquot of the supernatant was kept aside on ice (“input”). 1000 μg of cell lysate was incubated with 5 μg of primary antibody per IP at 4°C with mild shaking for overnight. The following antibodies were used for IP: L1CAM/L1CAM ab1 (Abcam, ab20148) and NRP2 (Santa Cruz, sc13117). Next day protein G-agarose beads were loaded for 4 hr at room temperature and washed according to the manufacturer’s protocol. Beads were resuspended in SDS sample buffer, boiled 5 min, and loaded onto a 4–12% acrylamide gel. Treatment with PNGase F (NEB) was performed as described by manufacturer. For lectin blotting, samples were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked in blocking buffer [5% (wt/vol) BSA, TBST (TBS, pH 7.4, 0.05% Tween-20), 1 hr, room temperature], washed three times with TBST, and incubated with biotinylated LcH lectin (5 μg/ml) in TBST at 4°C overnight. The blots were incubated with HRP-conjugated streptavidin (0.2 μg/ml) at room temperature for 1 hr and then developed with chemiluminescence detection.
Mass spectrometry
The lectin affinity enriched samples were suspended in 30 μl of PBS and reduced with 20 mM DTT at 57°C for 1 hr and alkylated with 50 mM iodoacetamide at room temperature in the dark for 45 min. Immediately after alkylation the samples were digested using 100 ng of sequencing grade-modified trypsin (Promega). The digestion proceeded overnight at room temperature with gentle agitation.
Peptide extraction was performed, as described (Cristea et al., 2005), by adding a slurry of R2 20 μm Poros beads (Life Technologies Corporation) in 30 μl of 5% formic acid and 0.2% trifluoroacetic acid (TFA) to each sample. Samples were incubated with agitation at 4°C for 4 hr. The beads were loaded onto equilibrated C18 Ziptips (Millipore) using a microcentrifuge for 30 s at 6000 rpm. The sample vials were rinsed three times with 0.1% TFA and each rinse was added to the corresponding ziptip followed by microcentrifugation. Extracted poros beads were further rinsed with 0.5% acetic acid. Peptides were eluted by addition of 40% acetonitrile in 0.5% acetic acid followed by the addition of 80% acetonitrile in 0.5% acetic acid. The organic solvent was removed using a SpeedVac concentrator and the samples were reconstituted in 0.5% acetic acid.
An aliquot of each sample was loaded onto an EASY spray 50 cm C18 analytical HPLC column with <2 μm bead size equilibrated in solvent A (2% acetonitrile, 0.5% acetic acid) using the auto sampler of an EASY-nLC 1000 HPLC (ThermoFisher). The peptides were gradient eluted directly into a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) using a 1 hr gradient from 2% to 31% solvent B (90% acetonitrile, 0.5% acetic acid), followed by 10 min from 31% to 40% solvent B and 10 min from 40% to 100% solvent B. The Q Exactive Orbitrap mass spectrometer acquired high resolution full MS spectra with a resolution of 70,000, an AGC target of 1e6, with a maximum ion time of 120 ms, and a scan range of 400 to 1500 m/z. Following each full MS twenty data dependent MS/MS spectra were acquired using the following instrument parameters: resolution of 17,500, AGC target of 5e4, maximum ion time of 120 ms, one microscan, 2 m/z isolation window, fixed first mass of 150 m/z, normalized collision energy (NCE) of 27 and dynamic exclusion of 30 s. The raw data was searched against the Uniprot Human database (downloaded on May 12, 2016) using SEQUEST within Proteome Discoverer 1.4 (ThermoFisher) allowing carbamidomethylation of cysteine as a fixed modification and oxidation of methionine, deamidation of glutamine and asparagine as variable modifications. The results were filtered for <1% False Discovery Rate (FDR) searched against a decoy database and proteins with less than two unique peptides were excluded. The raw data of the 4L cell line sample was also searched using the Byonic search algorithm to identify glycosylated peptides (Bern et al., 2012). The search was performed against a focused protein database created from the proteins identified in the 4L sample enabling glycopeptide identifications using the known human N-glycan database incorporated in the Byonic software suite. The Byonic output was filtered using a peptide score of > 300 and identified glycopeptides manually verified.
PSM filtering and Enrichment Analysis
Following liquid chromatography-mass spectrometry (LC-MS) analysis of samples, a custom python script (provided) was used to parse the Sequest output to generate gene name information to form a useable matrix for enrichment analysis using the R project for statistical computing (provided). Since PSM values represent a qualitative measure of protein enrichment but depend on the sample matrix (as noted before), we determined that location from the median would give us insight into protein enrichment for downstream analysis. The Right Skewed distributions contained 471 overlapping proteins (complete cases) from a total of 1100 identified. Since sample medians are heavily influenced by lower PSM values in Right Skewed distributions, univariate analysis was performed from a matrix of the overlapping proteins by looking for common protein enrichment from the median within each. Downstream ontological analysis was performed following accumulation of the enriched overlapping proteins between the samples from the individual lists (Huang da et al., 2009). These lists were filtered for contaminant proteins such as association with ribosomal function using a custom python script (provided) that parsed the description of the protein for removal before GO analysis. These lists were also filtered for other cytoplasmic and non-glycosylated proteins such as tubulins and heat shock proteins.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed with GraphPad Prism (GraphPad Software, Inc.) Data are presented as the mean ± SD. Significance was determined using an unpaired/paired t test. Statistical details of experiments can be found in the figure legends also. In mouse experiments, n represents number of mice utilized in each treatment group.
DATA AND SOFTWARE AVAILABILITY
Data resources
Lectin microarray data of melanoma FFPE primary and metastatic samples is available at https://www.synapse.org/#!Synapse:syn9748084/files/
Custom scripts of mass spectrometry data analysis are available at https://github.com/FenyoLab/Fucosylated_Melanoma/
Following melanoma GEO datasets were used for gene expression analysis GSE8401 (Xu et al., 2008), GSE15605 (Raskin et al., 2013), GSE46517 (Kabbarah et al., 2010), GSE7553 (Riker et al., 2008). mRNA expression (normalized RNAseq results) of Skin Cutaneous Melanoma (SKCM) cases were obtained from the Cancer Genome Atlas (TCGA) Data Portal (http://tcga-data.nci.nih.gov). DNA methylation data from Illumina 450K human methylation arrays available for melanoma patients was collected from TCGA HM450K level 3. Median β value for each sample was calculated for the specified CpG dinucleotides included in the Illumina 450K Human Methylation array. β value methylation score ranges from 0 (lack of methylation) to 1 (complete methylation).
Software
We used MatInspector software (Cartharius et al., 2005) and ChIP Enrichment Analysis (ChEA) database (Lachmann et al., 2010) to predict transcription factors bind to FUT8 promoter.
Supplementary Material
Lectin print conditions.
HIGHLIGHTS.
Primary and metastatic melanoma display distinct core and α-1,2 fucosylation
FUT8 promotes melanoma metastasis
FUT8 is transcriptionally controlled by TGIF2
FUT8-mediated core fucosylation alters L1CAM proteolytic cleavage and cell invasion
SIGNIFICANCE.
Our work, which is a systematic attempt to identify key glycosylation changes associated with metastasis in melanoma patient samples and map them onto enzymes, revealed the fucosyltransferase FUT8 as an anti-tumor target for the prevention and treatment of metastases. Using multiple state-of the-art complementary approaches, we characterized the role of FUT8 in melanoma metastasis, cementing core fucosylation as a critical agent in adaptation of cancer cells to a new host environment, such as the metastatic site, and identifying specific target proteins whose aberrant core fucosylation confers aggressive behavior to melanoma cells.
Acknowledgments
We thank NYU School of Medicine Histopathology and IHC Core Laboratories for tissue processing and histology, and the NYUMC Proteomics Resource Center, NYUMC for mass spectrometry data. These shared resources are partially supported by the Cancer Center Support Grant, P30CA016087, at the Laura and Isaac Perlmutter Cancer Center. We thank Dr. Tin Htwe Thin (Pathology core, Icahn School of Medicine at Mount Sinai) for lectin fluorescence staining. We thank Dr. Rana S. Moubarak and Alejandro Ulloa for critical reading of the manuscript. This work was funded by the Department of Defense PRCRP Discovery Award CA110602, a NYULMC (Louise and Joshua Samuelson)-Melanoma Research Alliance established investigator award and the NIH-NCI Cancer Center Support Grant P30CA016087. P.A. has been supported by a National Cancer Center fellowship. The authors declare no conflict of interest.
Footnotes
AUTHOR CONTRIBUTIONS
Experimental design: P.A., L.K.M. and E.H.; data mining and glycan pathway analysis: P.A. and L.K.M.; Western blots and qRT-PCRs: P.A. and D.A.; Lectin microarray, Lectin fluorescence: P.A., M.C.; Generation of the expression vectors: P.A.; In vivo mouse experiments: P.A., B.F., E.S. and F.D.; Invasion and proliferation assays: P.A.; Immunohistochemistry: P.A., M.C and F.D.; ChIP and Luciferase reporter assay: P.A., C.V. and M.M.; Clinical samples acquisition and pathological evaluation: I.O., F.D. and M.C.; Proteomic analysis: P.A., S.J., S.N., D.F. and B.U.; DNA methylation data analysis: V.D.; Manuscript writing: P.A., L.K.M. and E.H. All authors reviewed and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott KL, Nairn AV, Hall EM, Horton MB, McDonald JF, Moremen KW, Dinulescu DM, Pierce M. Focused glycomic analysis of the N-linked glycan biosynthetic pathway in ovarian cancer. Proteomics. 2008;8:3210–3220. doi: 10.1002/pmic.200800157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal P, Kurcon T, Pilobello KT, Rakus JF, Koppolu S, Liu Z, Batista BS, Eng WS, Hsu KL, Liang Y, Mahal LK. Mapping posttranscriptional regulation of the human glycome uncovers microRNA defining the glycocode. Proc Natl Acad Sci U S A. 2014;111:4338–4343. doi: 10.1073/pnas.1321524111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agre P, Bertozzi C, Bissell M, Campbell KP, Cummings RD, Desai UR, Estes M, Flotte T, Fogleman G, Gage F, et al. Training the next generation of biomedical investigators in glycosciences. The Journal of clinical investigation. 2016;126:405–408. doi: 10.1172/JCI85905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt P, Doberstein K, Fogel M. L1CAM in human cancer. International journal of cancer. 2016;138:1565–1576. doi: 10.1002/ijc.29658. [DOI] [PubMed] [Google Scholar]
- Batista BS, Eng WS, Pilobello KT, Hendricks-Munoz K, Mahal LK. Identification of a Conserved Glycan Signature for Microvesicles. J Proteome Res. 2011;10:4624–4633. doi: 10.1021/pr200434y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M, Kil YJ, Becker C. Byonic: advanced peptide and protein identification software. Current protocols in bioinformatics/editoral board, Andreas D Baxevanis [et al] 2012;Chapter 13(Unit13):20. doi: 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi C, Soffientini U, Piacente F, Tonetti MG. Effects of MicroRNAs on Fucosyltransferase 8 (FUT8) Expression in Hepatocarcinoma Cells. PLoS One. 2013;8:e76540. doi: 10.1371/journal.pone.0076540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird-Lieberman EL, Neves AA, Lao-Sirieix P, O’Donovan M, Novelli M, Lovat LB, Eng WS, Mahal LK, Brindle KM, Fitzgerald RC. Molecular imaging using fluorescent lectins permits rapid endoscopic identification of dysplasia in Barrett’s esophagus. Nature medicine. 2012;18:315–321. doi: 10.1038/nm.2616. [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, Yang PC, Hsiao M, Hsu TL, Wong CH. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci U S A. 2013;110:630–635. doi: 10.1073/pnas.1220425110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X, Cheng X, Euthine V, Hu P, Guo JY, et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell. 2015;28:569–581. doi: 10.1016/j.ccell.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Gao S, Song X, Dong W, Zhou H, Zhao L, Jia L. Comprehensive N-glycan profiles of hepatocellular carcinoma reveal association of fucosylation with tumor progression and regulation of FUT8 by microRNAs. Oncotarget. 2016 doi: 10.18632/oncotarget.11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell surface protein glycosylation in cancer. Proteomics. 2014;14:525–546. doi: 10.1002/pmic.201300387. [DOI] [PubMed] [Google Scholar]
- Ciborowski P, Finn OJ. Non-glycosylated tandem repeats of MUC1 facilitate attachment of breast tumor cells to normal human lung tissue and immobilized extracellular matrix proteins (ECM) in vitro: potential role in metastasis. Clinical & experimental metastasis. 2002;19:339–345. doi: 10.1023/a:1015590515957. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Williams R, Chait BT, Rout MP. Fluorescent proteins as proteomic probes. Molecular & cellular proteomics: MCP. 2005;4:1933–1941. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- Cruz-Munoz W, Man S, Xu P, Kerbel RS. Development of a preclinical model of spontaneous human melanoma central nervous system metastasis. Cancer research. 2008;68:4500–4505. doi: 10.1158/0008-5472.CAN-08-0041. [DOI] [PubMed] [Google Scholar]
- Cummings RD, Pierce JM. The challenge and promise of glycomics. Chemistry & biology. 2014;21:1–15. doi: 10.1016/j.chembiol.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel M, Crispin M, Scanlan CN, Zitzmann N, Dwek RA. Emerging principles for the therapeutic exploitation of glycosylation. Science. 2014;343:1235681. doi: 10.1126/science.1235681. [DOI] [PubMed] [Google Scholar]
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fontanals-Cirera B, Low V, Ntziachristos P, Yuen SK, Lovell CD, Dolgalev I, Yonekubo Y, Zhang G, Rusinova E, et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell reports. 2014;9:234–247. doi: 10.1016/j.celrep.2014.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube DH, Bertozzi CR. Glycans in cancer and inflammation--potential for therapeutics and diagnostics. Nature reviews Drug discovery. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- Dwek RA, Butters TD, Platt FM, Zitzmann N. Targeting glycosylation as a therapeutic approach. Nature reviews Drug discovery. 2002;1:65–75. doi: 10.1038/nrd708. [DOI] [PubMed] [Google Scholar]
- Gaziel-Sovran A, Segura MF, Di Micco R, Collins MK, Hanniford D, Vega-Saenz de Miera E, Rakus JF, Dankert JF, Shang S, Kerbel RS, et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer Cell. 2011;20:104–118. doi: 10.1016/j.ccr.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giblin AV, Thomas JM. Incidence, mortality and survival in cutaneous melanoma. Journal of plastic, reconstructive & aesthetic surgery: JPRAS. 2007;60:32–40. doi: 10.1016/j.bjps.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Gloster TM, Vocadlo DJ. Developing inhibitors of glycan processing enzymes as tools for enabling glycobiology. Nat Chem Biol. 2012;8:683–694. doi: 10.1038/nchembio.1029. [DOI] [PubMed] [Google Scholar]
- Gorelik E, Xu F, Henion T, Anaraki F, Galili U. Reduction of metastatic properties of BL6 melanoma cells expressing terminal fucose(alpha)1-2-galactose after alpha1,2-fucosyltransferase cDNA transfection. Cancer Res. 1997;57:332–336. [PubMed] [Google Scholar]
- Gu W, Fukuda T, Isaji T, Hashimoto H, Wang Y, Gu J. alpha1,6-Fucosylation regulates neurite formation via the activin/phospho-Smad2 pathway in PC12 cells: the implicated dual effects of Fut8 for TGF-beta/activin-mediated signaling. Faseb J. 2013;27:3947–3958. doi: 10.1096/fj.12-225805. [DOI] [PubMed] [Google Scholar]
- Hsu KL, Pilobello K, Krishnamoorthy L, Mahal LK. Ratiometric lectin microarray analysis of the mammalian cell surface glycome. Methods Mol Biol. 2011;671:117–131. doi: 10.1007/978-1-59745-551-0_6. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kabbarah O, Nogueira C, Feng B, Nazarian RM, Bosenberg M, Wu M, Scott KL, Kwong LN, Xiao Y, Cordon-Cardo C, et al. Integrative genome comparison of primary and metastatic melanomas. PLoS One. 2010;5:e10770. doi: 10.1371/journal.pone.0010770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpanen T, Heckman CA, Keskitalo S, Jeltsch M, Ollila H, Neufeld G, Tamagnone L, Alitalo K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. Faseb J. 2006;20:1462–1472. doi: 10.1096/fj.05-5646com. [DOI] [PubMed] [Google Scholar]
- Kiefel H, Bondong S, Erbe-Hoffmann N, Hazin J, Riedle S, Wolf J, Pfeifer M, Arlt A, Schafer H, Muerkoster SS, Altevogt P. L1CAM-integrin interaction induces constitutive NF-kappaB activation in pancreatic adenocarcinoma cells by enhancing IL-1beta expression. Oncogene. 2010;29:4766–4778. doi: 10.1038/onc.2010.230. [DOI] [PubMed] [Google Scholar]
- Kiefel H, Bondong S, Hazin J, Ridinger J, Schirmer U, Riedle S, Altevogt P. L1CAM: a major driver for tumor cell invasion and motility. Cell adhesion & migration. 2012;6:374–384. doi: 10.4161/cam.20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Mitsui Y, Kakoi N, Yamada K, Hayakawa T, Kakehi K. Common glycoproteins expressing polylactosamine-type glycans on matched patient primary and metastatic melanoma cells show different glycan profiles. J Proteome Res. 2014;13:1021–1033. doi: 10.1021/pr401015b. [DOI] [PubMed] [Google Scholar]
- Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang TC, Xie XJ, He L, Mangala LS, Lopez-Berestein G, et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature. 2014;512:431–435. doi: 10.1038/nature13375. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26:2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidler P, Litynska A, Hoja-Lukowicz D, Labedz M, Przybylo M, Ciolczyk-Wierzbicka D, Pochec E, Trebacz E, Kremser E. Characterization of glycosylation and adherent properties of melanoma cell lines. Cancer Immunol Immunother. 2006;55:112–118. doi: 10.1007/s00262-005-0019-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau E, Feng Y, Claps G, Fukuda MN, Perlina A, Donn D, Jilaveanu L, Kluger H, Freeze HH, Ronai ZA. The transcription factor ATF2 promotes melanoma metastasis by suppressing protein fucosylation. Science signaling. 2015;8:ra124. doi: 10.1126/scisignal.aac6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, Chen CH, Khoo KH, Yu CJ, Yang PC, et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci U S A. 2011;108:11332–11337. doi: 10.1073/pnas.1107385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo JA, Fisher DE. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science. 2014;346:945–949. doi: 10.1126/science.1253735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretzky T, Schulte M, Ludwig A, Rose-John S, Blobel C, Hartmann D, Altevogt P, Saftig P, Reiss K. L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Molecular and cellular biology. 2005;25:9040–9053. doi: 10.1128/MCB.25.20.9040-9053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meany DL, Chan DW. Aberrant glycosylation associated with enzymes as cancer biomarkers. Clinical proteomics. 2011;8:7. doi: 10.1186/1559-0275-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Comunale MA, Rawat S, Casciano JC, Lamontagne J, Herrera H, Ramanathan A, Betesh L, Wang M, Norton P, et al. Intrinsic hepatocyte dedifferentiation is accompanied by upregulation of mesenchymal markers, protein sialylation and core alpha 1,6 linked fucosylation. Scientific reports. 2016;6:27965. doi: 10.1038/srep27965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier F, Busch S, Gast D, Goppert A, Altevogt P, Maczey E, Riedle S, Garbe C, Schittek B. The adhesion molecule L1 (CD171) promotes melanoma progression. International journal of cancer. 2006;119:549–555. doi: 10.1002/ijc.21880. [DOI] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nature reviews Molecular cell biology. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsi A, Gaziel-Sovran A, Cruz-Munoz W, Kerbel RS, Golfinos JG, Hernando E, Wadghiri YZ. Development and characterization of a clinically relevant mouse model of melanoma brain metastasis. Pigment cell & melanoma research. 2013;26:743–745. doi: 10.1111/pcmr.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairn AV, Aoki K, dela Rosa M, Porterfield M, Lim JM, Kulik M, Pierce JM, Wells L, Dalton S, Tiemeyer M, Moremen KW. Regulation of glycan structures in murine embryonic stem cells: combined transcript profiling of glycan-related genes and glycan structural analysis. J Biol Chem. 2012;287:37835–37856. doi: 10.1074/jbc.M112.405233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Pilobello KT, Agrawal P, Rouse R, Mahal LK. Advances in lectin microarray technology: optimized protocols for piezoelectric print conditions. Curr Protoc Chem Biol. 2013;5:1–23. doi: 10.1002/9780470559277.ch120035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilobello KT, Slawek DE, Mahal LK. A ratiometric lectin microarray approach to analysis of the dynamic mammalian glycome. Proc Natl Acad Sci U S A. 2007;104:11534–11539. doi: 10.1073/pnas.0704954104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–555. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- Rakus JF, Mahal LK. New Technologies for Glycomic Analysis: Toward a Systematic Understanding of the Glycome. Annu Rev Anal Chem (Palo Alto Calif) 2011 doi: 10.1146/annurev-anchem-061010-113951. [DOI] [PubMed] [Google Scholar]
- Raskin L, Fullen DR, Giordano TJ, Thomas DG, Frohm ML, Cha KB, Ahn J, Mukherjee B, Johnson TM, Gruber SB. Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. The Journal of investigative dermatology. 2013;133:2585–2592. doi: 10.1038/jid.2013.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riker AI, Enkemann SA, Fodstad O, Liu S, Ren S, Morris C, Xi Y, Howell P, Metge B, Samant RS, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics. 2008;1:13. doi: 10.1186/1755-8794-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackstein R, Merzaban J, Cain D, Dagia N, Spencer J, Lin C, Wohlgemuth R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- Silletti S, Mei F, Sheppard D, Montgomery AM. Plasmin-sensitive dibasic sequences in the third fibronectin-like domain of L1-cell adhesion molecule (CAM) facilitate homomultimerization and concomitant integrin recruitment. J Cell Biol. 2000;149:1485–1502. doi: 10.1083/jcb.149.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. Journal of pharmaceutical sciences. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annual review of pathology. 2015;10:473–510. doi: 10.1146/annurev-pathol-012414-040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara D, Kaji H, Sugihara K, Asano M, Narimatsu H. Large-scale identification of target proteins of a glycosyltransferase isozyme by Lectin-IGOT-LC/MS, an LC/MS-based glycoproteomic approach. Scientific reports. 2012;2:680. doi: 10.1038/srep00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massague J. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Fukuda T, Isaji T, Lu J, Im S, Hang Q, Gu W, Hou S, Ohtsubo K, Gu J. Loss of alpha1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. Faseb J. 2015;29:3217–3227. doi: 10.1096/fj.15-270710. [DOI] [PubMed] [Google Scholar]
- Xu L, Shen SS, Hoshida Y, Subramanian A, Ross K, Brunet JP, Wagner SN, Ramaswamy S, Mesirov JP, Hynes RO. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res. 2008;6:760–769. doi: 10.1158/1541-7786.MCR-07-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Ikeda Y, Takahashi T, Ihara H, Tanaka T, Sasho C, Uozumi N, Yanagidani S, Inoue S, Fujii J, Taniguchi N. Genomic structure and promoter analysis of the human alpha1, 6-fucosyltransferase gene (FUT8) Glycobiology. 2000;10:637–643. doi: 10.1093/glycob/10.6.637. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Chijiwa T, Inoue Y, Abe Y, Suemizu H, Kawai K, Wakui M, Furukawa D, Mukai M, Kuwao S, et al. Overexpression of the miR-34 family suppresses invasive growth of malignant melanoma with the wild-type p53 gene. Experimental and therapeutic medicine. 2012;3:793–796. doi: 10.3892/etm.2012.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. The New England journal of medicine. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lectin print conditions.