

Abstract
Tp53, a stress response gene, is involved in diverse cell death pathways and its activation is implicated in the pathogenesis of Parkinson's disease. However, whether the neuronal Tp53 protein plays a direct role in regulating dopaminergic (DA) neuronal cell death or neuronal terminal damage in different neurotoxicant models is unknown. In our recent studies, in contrast to the global inhibition of Tp53 function by pharmacological inhibitors and in traditional Tp53 knock-out mice, we examined the effects of DA-specific Tp53 gene deletion after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and methamphetamine exposure. Our data suggests that the Tp53 gene might be involved in both neuronal apoptosis and neuronal terminal damage caused by different neurotoxicants. Additional results from other studies also suggest that as a master regulator of many pathways that regulate apoptosis and synaptic terminal damage, it is possible that Tp53 may function as a signaling hub to integrate different signaling pathways to mediate distinctive target pathways. Tp53 protein as a signaling hub might be able to evaluate the microenvironment of neurons, assess the forms and severities of injury incurred, and determine whether apoptotic cell death or neuronal terminal degeneration occurs. Identification of the precise mechanisms activated in distinct neuronal damage caused by different forms and severities of injuries might allow for development of specific Tp53 inhibitors or ways to modulate distinct downstream target pathways involved.
Keywords: Parkinson's disease; Tp53; 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; neurotoxicity; apoptosis; methamphetamine
Neuronal Damage Caused by Methamphetamine (MA)
MA is a potent psycho-stimulant with an extremely high potential for dependency and abuse. Long term usage of MA has been associated with neurological functional changes in both animals and humans. Several hypotheses regarding the mechanism underlying MA-induced neurotoxicity have been proposed. In particular, it is thought that endogenous dopamine (DA) in the striatum may play an important role in mediating MA-induced neuronal damage. MA induces redistribution of DA from the vesicular storage pool to the cytoplasm, where DA can oxidize to produce quinones and additional reactive oxygen species. This mechanism may account for its selective neurotoxicity in DA systems. MA selectively injures the neurites of DA neurons, promotes a severe loss of dopaminergic axonal arborization, and profoundly decreases striatal DA levels. Chronic MA exposure might lead to a wider spread of neurotoxicity in the brain. In an early morphological study, Miyakawa et al. (1969) administered MA to guinea pigs at a rate of 1 mg/kg per day for up to 1 year. A coalescence of axonal membranes with those of the terminals and dendrites as well as an increase in the number of coated vesicles were reported throughout the telencephalon and diencephalon. These observations might be related to the neurological and psychiatric symptoms observed in humans who chronically abuse MA. Swollen nerve fibers in the striatum following repeated MA exposure were later reported by other studies as well (Kita et al., 2003). These aforementioned studies clearly demonstrate that MA exposure could cause nerve terminal degeneration.
Apoptotic Pathway Activated by MA Exposure
A relationship between the apoptotic pathway and MA-induced neurotoxicity has been suggested through reports of appropriate evidence for activation of apoptotic pathways during the toxic response to MA. For example, Cadet and coworkers have studied the degenerative processes in the central nervous system via apoptotic pathways caused by neurotoxic doses of MA. They reported that MA caused dose-dependent apoptosis and loss of cellular viability in immortalized neural cells, whereas neural cells overexpressing bcl2 were protected against these deleterious effects (Cadet et al., 2005). Immunocytochemistry analysis revealed a marked increase in cytochrome c release from mitochondria in the rat brain after MA exposure, which is correlated with caspase-9, caspase-6, and caspase-3 activation. These results suggest that cellular death genes in the apoptotic pathway may play an important role in terminal degeneration caused by MA application.
Whereas terminal damage in both the striatum and the substantia nigra pars recitulata has consistently been reported in many previous studies, whether or not MA induces DA neuronal apoptosis or neuronal loss in vivo remains controversial. It has been reported that transient decreases of tyrosine hydroxylase (TH) expression in both the striatum and substantia nigra (SN) is followed by a spontaneous recovery that then results in an apparent lack of dopaminergic neuronal loss within the SN in rodents (Luo et al., 2010). Since the Tp53 gene is a master regulator of apoptosis and neuronal terminal damage, we therefore examined whether Tp53 affects the neurotoxicity of MA and whether regulation of apoptosis or neuronal terminal damage through Tp53 is involved in MA neurotoxicity in dopaminergic neurons in vivo (Lu et al., 2017).
Tp53 and Neurotoxicity Induced by MA
Apoptosis-inducing transcription factor Tp53 is a pleiotropic protein involved in a very large number of biological processes, including cell cycle regulation, cell differentiation, and apoptosis. It is implicated in MA neurotoxicity based on the findings of attenuated MA-induced dopaminergic cell damage, especially in dopaminergic terminals, in Tp53-knockout (KO) mice (Hirata and Cadet, 1997). In a previous report, repeated MA injections increased Tp53-DNA binding activity in the striatum, which was markedly attenuated in Cu, Zn-superoxide dismutase transgenic mice, but not affected by treatment with N-methyl-D-aspartate or D1-receptor antagonists. These authors indicate that Tp53 activation might be part of the mechanism that causes the long-term deleterious and neurotoxic effects of MA on the cerebral dopaminergic system. In adult Tp53 KO mice, traditional Tp53 gene deletion has been described as leading to learning deficits and behavioral alterations. Therefore, to precisely evaluate Tp53 function in different neural systems and to evaluate Tp53's role under different toxicological insults, it is critical to utilize a cell type-specific Tp53 conditional knockout that we have recently generated and characterized. Utilizing this DA-specific Tp53 KO mouse model, we evaluated the role of Tp53 in dopaminergic neurotoxicity in a MA binge model. Notably, although Tp53 pathway-related genes were upregulated by MA binge exposure, we did not observe loss of TH-positive neurons at 10 days following MA binge, consistent with previous studies. Despite the absence of DA neuronal loss in the MA binge model, we observed attenuated neurotoxicity in DA-specific Tp53 KO mice in terms of neuronal terminal damage and behavioral outcomes. This suggests that rather than inducing DA neuronal apoptosis and cell death, Tp53 may instead have a role in regulating the neuronal terminal damage evident in MA binge models. In support of this, previous studies have demonstrated that Tp53 is present in synaptic terminals, has the ability to regulate synaptosome survival, and plays a role in synaptic plasticity and function (Gilman et al., 2003). Recently, it has been reported that Tp53 and Bax are involved in mediating either neuronal terminal degeneration or cell body apoptosis (Cusack et al., 2013) that is selectively regulated through distinct pathways. This can be considered essential to support the extensive neuronal apoptosis and axonal pruning that are each separately required when establishing specific neuronal circuits during development, as well as to support the selective pruning of axons that is continuously necessary to permit plasticity in the adult nervous system.
Tp53 as a Signaling Hub
As a master regulator of many pathways that regulates apoptosis and synaptic terminal damages, it is possible that Tp53 may function as a signaling hub to integrate different signaling pathways to mediate distinctive target pathways (Table 1). The topic of cell death in PD has been summarized in excellent reviews (Burke, 2007; Perier et al., 2012; Venderova and Park, 2012). Tp53 immunoreactivity is increased in brains of PD patients and is accompanied by an increased phosphorylation of p38 MAPK, which phosphorylates and stabilizes Tp53. It has been reported that neuroprotective effects of delta opioid (D-Ala 2, D-Leu 5) enkephalin (DADLE) against MA neurotoxicity was accompanied by attenuation of mRNA expression of a tumor necrosis factor Tp53 (Borlongan et al., 2004). Imam et al. (2005) found an induction of Tp53 was observed in Nurr1+/– mice, while MA significantly increased Tp53 levels in Nurr1+/– mice as compared with wild-type. This might be one of the potential mechanisms for the previously reported data that demonstrated that reduced expression of Nurr1 increases the vulnerability of mesencephalic dopamine neurons to dopaminergic toxins, as reported by us and others (Luo et al., 2010). In a previous study, we also demonstrated that inhibition of Drp1-mediated mitochondrial fission by the P110 peptide mitigated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced loss of dopaminergic neurons, inhibited MPTP-induced reduction in striatal dopaminergic neuronal terminal density, and attenuated the behavioral deficits induced by MPTP. Our data showed that this neuroprotection provided by the P110 treatment might be a result of inhibition of the Drp1-dependent Tp53-mediated apoptotic pathway (Filichia et al., 2016). Consistent with the potential role of Tp53 and BAX in neuronal cell body apoptosis, in a previous study using the same dopaminergic neuron-specific Tp53 conditional knockout mouse, we have demonstrated that dopaminergic neuron-specific deletion of Tp53 gene attenuates DA neuronal death in the MPTP mouse model of Parkinson's disease and attenuates MPTP-induced Bax upregulation (Qi et al., 2016). Previously, traditional Tp53 KO mice demonstrated a global role of Tp53 in MPTP-induced apoptosis in DA neurons (Trimmer et al., 1996; Perier et al., 2007). Moreover, inhibition of Tp53 function by pharmacological inhibitors or a dominant negative form of Tp53 provided protection to DA neurons in MPTP animal models or 6-OHDA in vitro cell models (Duan et al., 2002; Biswas et al., 2005; Bernstein et al., 2011). In addition to neurotoxicant-induced DA damage, Tp53 might be involved in DA damage caused by other factors. Duplan et al. (2016) have identified α-synuclein, a key protein that accumulates in PD-related Lewy bodies, as a new transcriptional target of Tp53 and delineated a cellular mechanism feeding the accumulation of toxic aggregated α-synuclein in PD. Pharmacological and genetic upregulation of Tp53 expression leads to a strong increase of α-synuclein protein, promoter activity, and mRNA levels. Moreover, during development and differentiation of DA neurons, it has also been reported that suppression of Tp53, in conjunction with cell cycle arrest at G1 and maintenance of an appropriate extracellular environment, markedly increases the efficiency in the transdifferentiation of human fibroblasts to induced dopaminergic (iDA) neurons by Ascl1, Nurr1, Lmx1a and miR124 (Jiang et al., 2015). All the aforementioned studies indicate that Tp53 plays a key role in dopaminergic neurotoxicity in different neurodegenerative models as it has been widely demonstrated now that both genetic KO and pharmacological inhibition of Tp53 shows a protective effect to dopaminergic neuronal damage. It has also been shown that post-translational modification of Tp53 protein by PARP-1 stabilizes Tp53 and alters its transaction of downstream genes in MPTP models, suggesting additional means for Tp53 to incorporate signals from various pathways (Mandir et al., 2002).
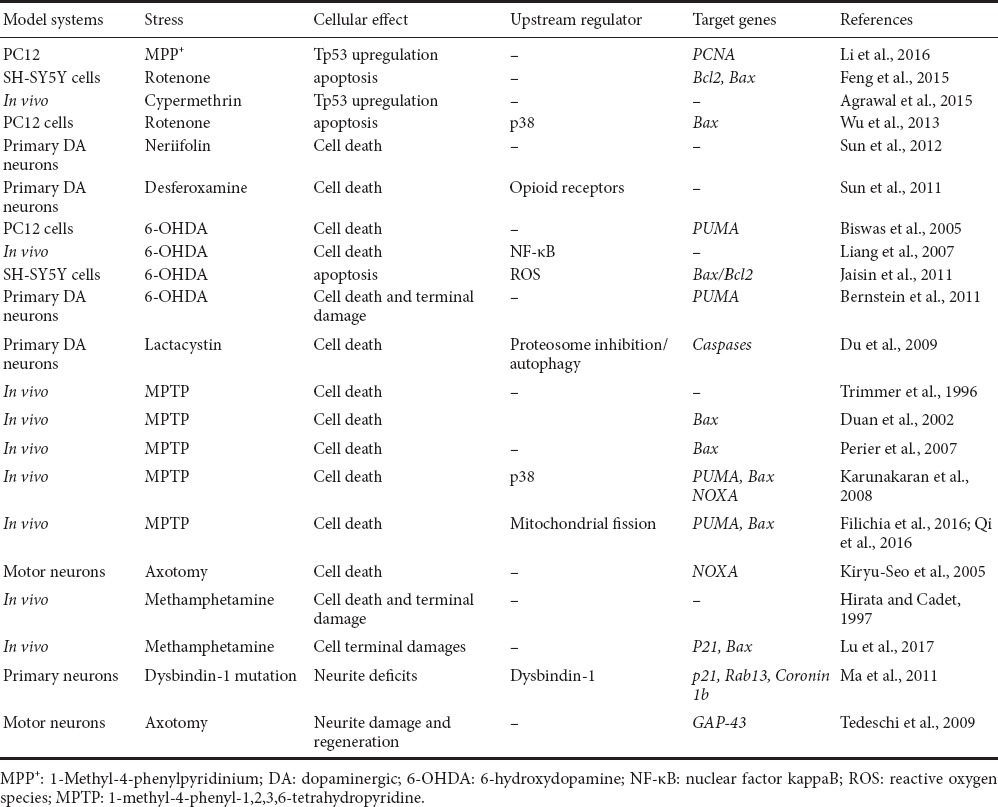
Table 1.
Summary of Tp53 involvement in neuronal degeneration
Conclusion and Summary
Traditionally, Tp53 is a major neuronal pro-apoptotic factor that is the center of gravity of multiple physiological and pathological cascades, some of which are implicated in several key neurodegenerative disorder-linked proteins. The role of Tp53 in the pathophysiology of central nervous system injuries remains complex and warrants further elucidation, particularly in light of recent findings of the involvement of Tp53 function in mediating either neuronal terminal degeneration or cell body apoptosis (Cusack et al., 2013) that is selectively regulated through distinct pathways (Figure 1). Our studies in both MPTP and MA binge models using DA-specific Tp53 KO mice support this hypothesis as DA-specific Tp53 deletion showed protection in the DA system both in the presence (MPTP model) and absence (MA binge model) of DA neuronal death. In addition, our data showed that although MPTP and MA binge exposure both induced Tp53 gene expression, distinct downstream genes are upregulated in these two different models (PUMA in the MPTP model and Bax/p21 in the MA binge model), which might contribute to the different types of neurotoxicity in the DA system. It is possible that the Tp53 gene serves as an information and signaling hub which allows it to evaluate the microenvironment of neurons, assess the forms and severities of injury incurred, and determine whether apoptotic cell death or neuronal terminal degeneration occurs. Moreover, it is now known that Tp53 can induce biological responses through both its transcriptional and mitochondrial activity. Recently, mitochondrial Tp53 inhibitor pifithrin-mu has been reported to attenuate MPTP neurotoxicity (Shin et al., 2016). Whether these two distinct molecular mechanisms of Tp53 activity contribute to distinct types of neuronal damage (apoptotic and non-apoptotic) still warrants further investigation. Identification of the precise mechanisms activated in distinct neuronal damage caused by different forms and severities of injuries might allow for development of specific Tp53 inhibitors or ways to modulate distinct downstream target pathways involved.
Figure 1.
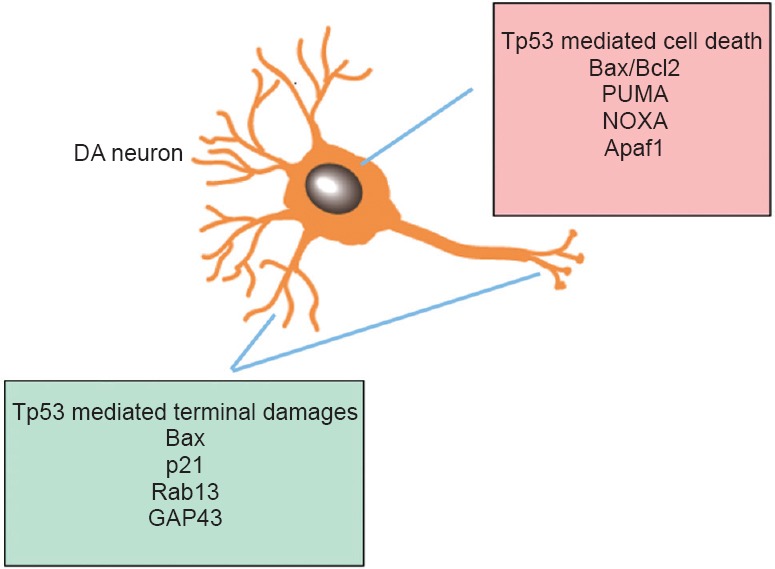
Bimodal mechanism of Tp53 in dopaminergic (DA) neuronal death or neuronal terminal damage.
Differential gene targets for cell death vs. terminal damages are summarized. References for gene targets are included in Table 1.
Footnotes
Funding: This paper is supported by NINDS R01NS094152 and R01 NS091213.
Conflicts of interest: None declared.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review reports:
Reviewer 1: Michal Hetman, University of Louisville, USA.
Comments to authors: This is a mini-review on the role of Tp53 in neurotoxin-associated degeneration of dopaminergic neurons. This is an important topic and, with the Tp53 field being very large, a good quality, synthetic review of that subject would be useful.
Reviewer 2: Takao Yasuhara, Okayama University, Japan.
Comments to authors: This is a nice mini-review. In this manuscript, the authors would like to show the importance of p53 gene in DA neuronal damage by neurotoxin. This is an elegant mini-review to show the readers apoptotic/non-apoptotic mechanisms of p53 and further direction of this research field.
References
- Agrawal S, Singh A, Tripathi P, Mishra M, Singh PK, Singh MP. Cypermethrin-induced nigrostriatal dopaminergic neurodegeneration alters the mitochondrial function: a proteomics study. Mol Neurobiol. 2015;51:448–465. doi: 10.1007/s12035-014-8696-7. [DOI] [PubMed] [Google Scholar]
- Bernstein AI, Garrison SP, Zambetti GP, O’Malley KL. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol Neurodegener. 2011;6:2. doi: 10.1186/1750-1326-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SC, Ryu E, Park C, Malagelada C, Greene LA. Puma and p53 play required roles in death evoked in a cellular model of Parkinson disease. Neurochem Res. 2005;30:839–845. doi: 10.1007/s11064-005-6877-5. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Wang Y, Su TP. Delta opioid peptide (D-Ala 2, D-Leu 5) enkephalin: linking hibernation and neuroprotection. Front Biosci. 2004;9:3392–3398. doi: 10.2741/1490. [DOI] [PubMed] [Google Scholar]
- Burke RE. Programmed cell death in Parkinson's disease. Handb Clin Neurol. 2007;83:591–605. doi: 10.1016/S0072-9752(07)83029-6. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Jayanthi S, Deng X. Methamphetamine-induced neuronal apoptosis involves the activation of multiple death pathways. Review. Neurotox Res. 2005;8:199–206. doi: 10.1007/BF03033973. [DOI] [PubMed] [Google Scholar]
- Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun. 2013;4:1876. doi: 10.1038/ncomms2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Yang D, Li L, Luo G, Li T, Fan X, Wang Q, Zhang X, Wang Y, Le W. An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy. 2009;5:663–675. doi: 10.4161/auto.5.5.8377. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol. 2002;52:597–606. doi: 10.1002/ana.10350. [DOI] [PubMed] [Google Scholar]
- Duplan E, Giordano C, Checler F, Alves da Costa C. Direct alpha-synuclein promoter transactivation by the tumor suppressor p53. Mol Neurodegener. 2016;11:13. doi: 10.1186/s13024-016-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Liu T, Dong SY, Guo YJ, Jankovic J, Xu H, Wu YC. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J Neurochem. 2015;134:668–676. doi: 10.1111/jnc.13172. [DOI] [PubMed] [Google Scholar]
- Filichia E, Hoffer B, Qi X, Luo Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson's disease model induced by MPTP. Sci Rep. 2016;6:32656. doi: 10.1038/srep32656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman CP, Chan SL, Guo Z, Zhu X, Greig N, Mattson MP. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Med. 2003;3:159–172. doi: 10.1385/NMM:3:3:159. [DOI] [PubMed] [Google Scholar]
- Hirata H, Cadet JL. p53-knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J Neurochem. 1997;69:780–790. doi: 10.1046/j.1471-4159.1997.69020780.x. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Jankovic J, Ali SF, Skinner JT, Xie W, Conneely OM, Le WD. Nitric oxide mediates increased susceptibility to dopaminergic damage in Nurr1 heterozygous mice. FASEB J. 2005;19:1441–1450. doi: 10.1096/fj.04-3362com. [DOI] [PubMed] [Google Scholar]
- Jaisin Y, Thampithak A, Meesarapee B, Ratanachamnong P, Suksamrarn A, Phivthong-Ngam L, Phumala-Morales N, Chongthammakun S, Govitrapong P, Sanvarinda Y. Curcumin I protects the dopaminergic cell line SH-SY5Y from 6-hydroxydopamine-induced neurotoxicity through attenuation of p53-mediated apoptosis. Neurosci Lett. 2011;489:192–196. doi: 10.1016/j.neulet.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Jiang H, Xu Z, Zhong P, Ren Y, Liang G, Schilling HA, Hu Z, Zhang Y, Wang X, Chen S, Yan Z, Feng J. Cell cycle and p53 gate the direct conversion of human fibroblasts to dopaminergic neurons. Nat Commun. 2015;6:10100. doi: 10.1038/ncomms10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran S, Saeed U, Mishra M, Valli RK, Joshi SD, Meka DP, Seth P, Ravindranath V. Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. J Neurosci. 2008;28:12500–12509. doi: 10.1523/JNEUROSCI.4511-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Hirayama T, Kato R, Kiyama H. Noxa is a critical mediator of p53-dependent motor neuron death after nerve injury in adult mouse. J Neurosci. 2005;25:1442–1447. doi: 10.1523/JNEUROSCI.4041-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T, Wagner GC, Nakashima T. Current research on methamphetamine-induced neurotoxicity: animal models of monoamine disruption. J Pharmacol Sci. 2003;92:178–195. doi: 10.1254/jphs.92.178. [DOI] [PubMed] [Google Scholar]
- Li DW, Li GR, Zhang BL, Feng JJ, Zhao H. Damage to dopaminergic neurons is mediated by proliferating cell nuclear antigen through the p53 pathway under conditions of oxidative stress in a cell model of Parkinson's disease. Int J Mol Med. 2016;37:429–435. doi: 10.3892/ijmm.2015.2430. [DOI] [PubMed] [Google Scholar]
- Liang ZQ, Li YL, Zhao XL, Han R, Wang XX, Wang Y, Chase TN, Bennett MC, Qin ZH. NF-kappaB contributes to 6-hydroxydopamine-induced apoptosis of nigral dopaminergic neurons through p53. Brain Res. 2007;1145:190–203. doi: 10.1016/j.brainres.2007.01.130. [DOI] [PubMed] [Google Scholar]
- Lu T, Kim PP, Greig NH, Luo Y. Dopaminergic Neuron-Specific Deletion of p53 Gene Attenuates Methamphetamine Neurotoxicity. Neurotox Res. 2017;32:218–230. doi: 10.1007/s12640-017-9723-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Wang Y, Kuang SY, Chiang YH, Hoffer B. Decreased level of Nurr1 in heterozygous young adult mice leads to exacerbated acute and long-term toxicity after repeated methamphetamine exposure. PLoS One. 2010;5:e15193. doi: 10.1371/journal.pone.0015193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Fei E, Fu C, Ren H, Wang G. Dysbindin-1, a schizophrenia-related protein, facilitates neurite outgrowth by promoting the transcriptional activity of p53. Mol Psychiatry. 2011;16:1105–1116. [Google Scholar]
- Mandir AS, Simbulan-Rosenthal CM, Poitras MF, Lumpkin JR, Dawson VL, Smulson ME, Dawson TM. A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism. J Neurochem. 2002;83:186–192. doi: 10.1046/j.1471-4159.2002.01144.x. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Sumiyoshi S, Deshimaru M, Murayama E, Tatetsu S. Electron microscopic studies concerning the structural mechanism of the development of mental disturbance in experimental chronic methamphetamine poisoning. Acta Neuropathol. 1969;14:215–225. doi: 10.1007/BF00685301. [DOI] [PubMed] [Google Scholar]
- Perier C, Bové J, Vila M. Mitochondria and programmed cell death in Parkinson's disease: apoptosis and beyond. Antioxid Redox Signal. 2012;16:883–895. doi: 10.1089/ars.2011.4074. [DOI] [PubMed] [Google Scholar]
- Perier C, Bové J, Wu DC, Dehay B, Choi DK, Jackson-Lewis V, Rathke-Hartlieb S, Bouillet P, Strasser A, Schulz JB, Przedborski S, Vila M. Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson's disease. Proc Natl Acad Sci U S A. 2007;104:8161–8166. doi: 10.1073/pnas.0609874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Davis B, Chiang YH, Filichia E, Barnett A, Greig NH, Hoffer B, Luo Y. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson's disease model. J Neurochem. 2016;138:746–757. doi: 10.1111/jnc.13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin EJ, Nam Y, Lee JW, Nguyen PT, Yoo JE, Tran TV, Jeong JH, Jang CG, Oh YJ, Youdim MBH, Lee PH, Nabeshima T, Kim HC. N-Methyl, N-propynyl-2-phenylethylamine (MPPE), a selegiline analog, attenuates MPTP-induced dopaminergic toxicity with guaranteed behavioral safety: involvement of inhibitions ofmitochondrial oxidative burdens and p53 gene-elicited pro-apoptotic change. Mol Neurobiol. 2016;53:6251–6269. doi: 10.1007/s12035-015-9527-1. [DOI] [PubMed] [Google Scholar]
- Sun Y, Dong Z, Khodabakhsh H, Chatterjee S, Guo S. Zebrafish chemical screening reveals the impairment of dopaminergic neuronal survival by cardiac glycosides. PLoS One. 2012;7:e35645. doi: 10.1371/journal.pone.0035645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YM, Hoang T, Neubauer JA, Walters AS. Opioids protect against substantia nigra cell degeneration under conditions of iron deprivation: a mechanism of possible relevance to the Restless Legs Syndrome (RLS) and Parkinson's disease. J Neurol Sci. 2011;304:93–101. doi: 10.1016/j.jns.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Tedeschi A, Nguyen T, Puttagunta R, Gaub P, Di Giovanni S. A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 2009;16:543–554. doi: 10.1038/cdd.2008.175. [DOI] [PubMed] [Google Scholar]
- Trimmer PA, Smith TS, Jung AB, Bennett JP., Jr Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration. 1996;5:233–239. doi: 10.1006/neur.1996.0031. [DOI] [PubMed] [Google Scholar]
- Venderova K, Park DS. Programmed cell death in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009365. doi: 10.1101/cshperspect.a009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Wang Z, Gu JH, Ge JB, Liang ZQ, Qin ZH. p38(MAPK)/p53-mediated Bax induction contributes to neurons degeneration in rotenone-induced cellular and rat models of Parkinson's disease. Neurochem Int. 2013;63:133–140. doi: 10.1016/j.neuint.2013.05.006. [DOI] [PubMed] [Google Scholar]