

Parkinson's disease (PD) is a progressive neurodegenerative disease, which is generally considered a multifactorial disorder that arises owing to a combination of genes and environmental factors. While most cases are idiopathic, in about 10% of the patients a genetic cause can be detected, ascribable to mutations in more than a dozen genes. PD is characterized clinically by tremor, rigidity, reduced motor activity (bradykinesia), and postural instability and pathologically by loss of dopaminergic (DA) neurons in the substantia nigra pars compacta, loss of DA innervation in the striatum, and the presence of α-synuclein positive aggregates in the form of Lewy bodies. The symptomatic treatment of PD with levodopa, which aims at replacing dopamine, remains the gold standard, and no neuroprotective or disease-modifying therapy is available. During treatment, the disease continues to progress, and long-term use of levodopa has important limitations including motor complications termed dyskinesias. Therefore, a pharmacological therapy able to prevent or halt the neurodegenerative process is urgently required.
PARK2 (the gene encoding Parkin) mutations are the most common known cause of early-onset PD, accounting for up to 77% of the familial cases with an age of onset < 30 years (Lücking et al., 2000), and Parkin dysfunction represents a risk factor for sporadic PD. The Parkin protein functions as an E3 ubiquitin ligase, transferring activated ubiquitin to lysine residues of protein substrates. It shows an amino-terminal ubiquitin-like domain, which is involved in substrate recognition, proteasome association, and the regulation of Parkin expression levels and activity, followed by an atypical RING domain, named RING0 or the unique Parkin domain. The carboxy-terminal domain consists of the RING-between-RING domain, comprising RING1 and RING2, and one in-between-ring domain and is responsible for the interaction with the ubiquitination machinery. At steady state, Parkin exists in an inactive state, repressed by several mechanisms of autoinhibition. By studying the function of PARK2 mutations, a direct role of mitochondrial dysfunction in the onset of PD has emerged. Parkin, in concert with PTEN-induced putative kinase 1 (PINK1), another recessively linked PD gene, has been implicated in the degradation of dysfunctional, depolarized mitochondria, a process known as mitophagy. Parkin translocates in a PINK1-dependent manner from the cytosol to dysfunctional mitochondria and ubiquitinates mitochondrial outer membrane proteins to initiate selective autophagy. In addition, Parkin plays a crucial role in the degradative pathways mediated by the ubiquitin-proteasome system, which is required for the clearance of Parkin substrates, like PARIS, which acts as a transcriptional repressor of peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), a transcriptional coactivator and master regulator of mitochondrial biogenesis. Moreover, Parkin mediates nondegradative ubiquitination involved in cell death processes and is implicated in the regulation of inflammatory signaling (Winklhofer, 2014).
Since the full repertoire of Parkin-binding proteins is purely defined, and to shed further light on the diverse spectrum of Parkin functions, we have characterized the profile of binding partners of Parkin, particularly at the mitochondrial level, by tandem affinity purification/mass spectrometry interaction screens (Zanon et al., 2013). We identified a total of 203 candidate Parkin-binding proteins involved in cell death processes, protein folding and response to unfolded protein, the fission/fusion machinery, and the mitophagy pathway. In a subsequent study, we have investigated the functional relevance of the Parkin interaction with one of the newly identified binding partners Stomatin-like protein 2 (SLP-2) in human neurons and Drosophila melanogaster (Zanon et al., 2017).
SLP-2 is a member of the stomatin family (comprising stomatin, SLP-1, SLP-2, SLP-3, and podocin), characterized by the presence of a conserved stomatin domain, which is further related to the SPFH (stomatin, prohibitin, flotillin, HflC/K) protein superfamily (Lapatsina et al., 2012). SPFH proteins are scaffolds, which assemble into ring-like structures and are present in lipid raft microdomains of diverse cellular membranes. The SLP-2 protein is distinguished within the stomatin protein family by its localization at the inner mitochondrial membrane, and an N-terminal mitochondrial leading sequence replaces the hydrophobic membrane anchor of the other family members (Lapatsina et al., 2012). There is increasing evidence for a fine-scale organization also of the inner mitochondrial membrane into functional microdomains, and proteins of the SPFH family assist their formation by specific protein-protein and protein-lipid interactions. SLP-2 was shown to bind to cardiolipin and to prohibitins (PHB-1 and PHB-2) (Christie et al., 2011) and forms cardiolipin and prohibitin-enriched microdomains in the inner mitochondrial membrane, which facilitate the assembly of respiratory chain complexes and their function in T-cells (Christie et al., 2012). Cardiolipin synthesis is increased in SLP-2 overexpressing cells, which translates into increased mitochondrial membrane formation and biogenesis (Christie et al., 2011), and PHB-2 has recently been identified as an inner mitochondrial membrane receptor required for Parkin-induced mitophagy by binding the autophagosomal membrane-associated protein LC3 upon mitochondrial depolarization and rupture of the outer membrane (Wei et al., 2017). In addition, SLP-2 was described as part of a new mitochondrial protein complex in the inner mitochondrial membrane composed of the rhomboid protease PARL and the i-AAA protease YME1L named SPY, which facilitates the cleavage of PINK1 by PARL and limits the activity of the mitochondrial protease OMA1, resulting in the protection of OPA1 and stress-induced hyperfusion under stress conditions (Wai et al., 2016). It was suggested that SLP-2, acting as a membrane scaffold, defines the lipid environment for proteolysis and modulates substrate accessibility (Wai et al., 2016). Furthermore, SLP-2 is known to form a complex with mitofusin-2 (MFN2), which is a mitochondrial outer membrane fusion protein and a Parkin ubiquitination substrate, and it was identified in a proteomic screen performed in brain synaptosomes as a binding partner of monomeric α-synuclein (Betzer et al., 2015), a presynaptic neuronal protein that is genetically and neuropathologically linked to PD. Ultimately, the expression level of SLP-2 affects the composition of detergent-resistant membranes in macrophages and the signaling underlying the innate immune response (Chowdhury et al., 2015) (Figure 1).
Figure 1.
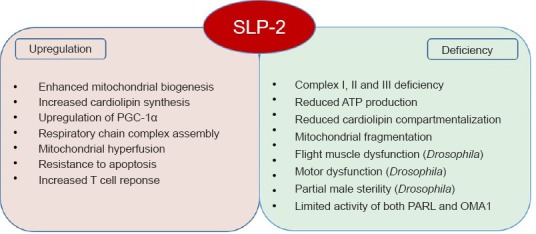
Known functions of the membrane scaffold Stomatin-like protein 2 (SLP-2).
Summary of cellular and physical phenotypes known to be regulated by SLP-2 in cellular and Drosophila models. PGC-1α: Peroxisome proliferator-activated receptor gamma coactivator-1 alpha; ATP: adenosine triphosphate.
In our recent work, we have shown that SLP-2- and Parkin-depleted cells exhibit similar defects in mitochondrial function regarding reduced complex I activity, adenosine triphosphate (ATP) production as well as a fragmented mitochondrial network morphology in neuroblastoma SH-SY5Y cells and induced pluripotent stem cell (iPSC)-derived neurons harboring PD-causing PARK2 mutations (Zanon et al., 2017). In addition, knockdown of SLP-2 in Drosophila mirrors several phenotypes associated with the loss of Parkin function. Reduced levels of SLP-2 resulted in a clear disruption of the mitochondrial network structure accompanied by a significant reduction in ATP levels in energy-demanding flight muscles. Accordingly, flies with reduced SLP-2 expression exhibited reduced flight ability and wing posture phenotypes indicative of muscle dysfunction, as it has been observed with the loss of PD genes like parkin. Further, global knockdown of SLP-2 resulted in partial male sterility, which is again consistent with the male fertility phenotypes observed with reduction of Parkin activity. Notably, tissue-specific reduction in neuronal SLP-2 led to a moderate loss of DA neurons in the PPL1 cluster, which is accompanied by compromised motor function as measured using a climbing assay. Pan-neuronal knockdown of SLP-2 also resulted in significant reduction in ATP levels, suggesting a general requirement of SLP-2 in maintaining mitochondrial bioenergetics, flight muscle function, and neuronal survival. Reduction in both Parkin and SLP-2 expression led to a further worsening of Parkin-deficiency phenotypes in Drosophila suggesting that normal SLP-2 activity in Paekin-deficient flies results in partial mitochondrial protection and thus the double knockdown produces a more severe phenotype as compared to Parkin knockdown flies suggesting a genetic interaction of Parkin and SLP-2 in vivo. Importantly, we have shown that SLP-2 overexpression rescues the mitochondrial dysfunction identified in neuronal cell models of Parkin-deficiency and in Drosophila, where also the loss of disease-relevant DA neurons and the associated motor dysfunction could be restored (Zanon et al., 2017). These data showed that Parkin and SLP-2 interact functionally and work in a common pathway to promote mitochondrial integrity and bioenergetic function in neurons, which might be the critical factor underlying the SLP-2-mediated rescue of Parkin phenotypes, in particular DA neuron survival. Thus, SLP-2 provides an interesting new target for the development of rescue strategies by specifically targeting the mitochondrial dysfunction in neurons implicated in the pathogenesis of PD.
Our data on the rescue of functional defects due to Parkin-deficiency by SLP-2 overexpression places SLP-2 in a pathway downstream or in parallel to Parkin. Unlike parkin knockout mice that are viable, complete removal of SLP-2 in mice is lethal (Christie et al., 2012) suggesting additional functions for SLP-2 that are critical during development and indicating that SLP-2 is required for certain functions downstream and in parallel to Parkin.
Several other forms of monogenic PD, like mutations in PINK1, DJ-1, LRRK2 (encoding the leucine-rich repeat kinase 2), and α-synuclein, relate to mitochondrial dysfunction with often overlapping phenotypes like reduced complex I activity and reactive oxygen species accumulation. Maintenance of a healthy population of mitochondria requires a balance between mitophagic elimination of dysfunctional organelles after their fission and mitochondrial biogenesis. The involvement of SLP-2 in mitochondrial biogenesis (Christie et al., 2011) and the identification of PHB-2, a member of the SPFH protein superfamily and interacting partner of SLP-2 at the inner mitochondrial membrane, as a mitophagy receptor (Wei et al., 2017), might provide the rationale for testing the rescue potential of SLP-2 induction not only for Parkin-deficiency but also for mitochondrial dysfunction caused by mutations in other PD genes. Ultimately, it will be important to test this approach of boosting mitochondrial function and neuronal survival in genetic and toxin-induced animal models of PD.
An interesting aspect of SLP-2 function is that its expression is upregulated during T cell activation while down-regulation of SLP-2 correlates with decreased T cell responses in vivo and in vitro (Christie et al., 2011). This is of particular interest as Parkin is implicated in the regulation of inflammatory signaling. In particular, Parkin deficiency was shown to increase the vulnerability of DA neurons to inflammation-related degeneration, which might be explained in part by recent data showing that Parkin, together with PINK1, is able to inhibit mitochondrial self antigen presentation at the cellular surface to trigger immune responses upon inflammatory stress (Matheoud et al., 2016). Therefore, it is possible that the Parkin-SLP-2 interaction will turn out to be involved in the regulation of inflammatory pathways and innate immunity, processes that may play an important role in PD pathogenesis.
We are currently working on further characterizing the function of SLP-2 in neurons in the context of mitochondrial function and neuronal survival in PD and hope to further develop this interesting protein as a drug target for interfering with or preventing the disease process. Several lines of evidence are being pursued including in vivo overexpression using viral vectors or targeting processes in parallel or downstream of SLP-2 that may be more amenable to therapeutic manipulation.
This work was supported by the Ministry of Health and Department of Educational Assistance, University and Research of the Autonomous Province of Bolzano.
We apologize to all authors whose work could not be referenced here due to paper length restrictions.
Footnotes
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Aarnoud van der Spoel, Dalhousie University, Canada.
References
- Betzer C, Movius AJ, Shi M, Gai WP, Zhang J, Jensen PH. Identification of synaptosomal proteins binding to monomeric and oligomeric alpha-synuclein. PLoS One. 2015;10:e0116473. doi: 10.1371/journal.pone.0116473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury SM, Zhu X, Aloor JJ, Azzam KM, Gabor KA, Ge W, Addo KA, Tomer KB, Parks JS, Fessler MB. Proteomic analysis of ABCA1-null macrophages reveals a role for stomatin-like protein-2 inraft composition and toll-like receptor signaling. Mol Cell Proteomics. 2015;14:1859–1870. doi: 10.1074/mcp.M114.045179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie DA, Mitsopoulos P, Blagih J, Dunn SD, St-Pierre J, Jones RG, Hatch GM, Madrenas J. Stomatin-like protein 2 deficiency in T cells is associated with altered mitochondrial respiration and defective CD4+ T cell responses. J Immunol. 2012;189:4349–4360. doi: 10.4049/jimmunol.1103829. [DOI] [PubMed] [Google Scholar]
- Christie DA, Lemke CD, Elias IM, Chau LA, Kirchhof MG, Li B, Ball EH, Dunn SD, Hatch GM, Madrenas J. Stomatin-like protein 2 binds cardiolipin and regulates mitochondrial biogenesis and function. Mol Cell Biol. 2011;31:3845–3856. doi: 10.1128/MCB.05393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW, Agid Y, Brice A French Parkinson's Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson's Disease. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- Lapatsina L, Brand J, Poole K, Daumke O, Lewin GR. Stomatin-domain proteins. Eur J Cell Biol. 2012;91:240–245. doi: 10.1016/j.ejcb.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau LE, Burelle Y, Gagnon E, McBride HM, Desjardins M. Parkinson's disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016;166:314–327. doi: 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- Wai T, Saita S, Nolte H, Müller S, König T, Richter-Dennerlein R, Sprenger HG, Madrenas J, Mühlmeister M, Brandt U, Krüger M, Langer T. The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 2016;17:1844–1856. doi: 10.15252/embr.201642698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R, Jr, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238.e10. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 2014;24:332–341. doi: 10.1016/j.tcb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Zanon A, Rakovic A, Blankenburg H, Doncheva NT, Schwienbacher C, Serafin A, Alexa A, Weichenberger CX, Albrecht M, Klein C, Hicks AA, Pramstaller PP, Domingues FS, Pichler I. Profiling of Parkin-binding partners using tandem affinity purification. PLoS One. 2013;8:e78648. doi: 10.1371/journal.pone.0078648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanon A, Kalvakuri S, Rakovic A, Foco L, Guida M, Schwienbacher C, Serafin A, Rudolph F, Trilck M, Grunewald A, Stanslowsky N, Wegner F, Giorgio V, Lavdas AA, Bodmer R, Pramstaller PP, Klein C, Hicks AA, Pichler I, Seibler P. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum Mol Genet. 2017;26:2412–2425. doi: 10.1093/hmg/ddx132. [DOI] [PMC free article] [PubMed] [Google Scholar]