

Alzheimer's disease (AD) is a complex neurological disorder characterized by a progressive dementia. The amyloid hypothesis states that pathogenesis is driven by the accumulation of amyloid β (Aβ) peptides within the brain (Hardy and Higgins, 1992), which have multiple effects including activation of glial cells and synapse degeneration. For many years therapeutic strategies were based upon the discovery of compounds that reduced the production of Aβ in vitro and in vivo. However, the amyloid hypothesis is not universally accepted; critics pointed out poor correlations between concentrations of Aβ in the brain and clinical disease and that, although numerous compounds reduced the production of Aβ in animal models, few had any clinical benefit in AD patients. The failure of Aβ-lowering therapies in translational research has resulted in important modifications of the amyloid hypothesis.
Aβ self-associates and is found in multiple forms ranging from monomers and small soluble oligomers to much larger aggregates seen as fibrils or plaques. It is apparent that not all forms of Aβ have equal biological significance; neuronal toxicity is dependent upon the nature of Aβ, whether that is the length of peptide, the state of aggregation, homogeneity of aggregates or specific Aβ conformations. The key to understanding the amyloid hypothesis is the realization that there exist toxic forms of Aβ, while other forms are less toxic, biologically inert and may even play a role in normal synapse function (Yang et al., 2017). Thus, while many studies reported the effects of compounds on total Aβ, this approach does not distinguish between the toxic and non-toxic forms of Aβ. In our recent paper we argued that the biologically active forms of Aβ could be measured by their effects upon neuronal cultures (Nolan et al., 2017). The pathogenesis of AD is intimately linked with the loss of synapses and many studies have demonstrated close correlations between the loss of synaptic proteins such as synaptophysin and the degree of dementia in AD (Sze et al., 1997). Our paper measured the loss of synaptic proteins including synaptophysin and cysteine string protein from neurons as an in vitro model to detect toxic forms of Aβ.
The production of Aβ was investigated in 7PA2 cells (Chinese hamster ovary cells stably transfected with human amyloid precursor protein (APP)751). These cell release soluble forms of Aβ that are similar to those found in the brains of AD patients and which are considered to be key mediators of synapse damage in AD (Cleary et al., 2005). A key observation was that while conditioned media (CM) from control 7PA2 cells was added to cultured neurons it caused extensive synapse damage, the CM from 7PA2 cells treated with specific sialylated glycosylphosphatidylinositols (GPIs) did not. The surprising observation that CM from treated and control 7PA2 cells contained similar amounts of Aβ emphasized the idea that simply measuring Aβ concentrations alone is insufficient in informing us about the biological effects of those Aβ peptides.
Our observations suggested that treatment affected the forms of Aβ produced, the loss of toxicity in CM from treated cells was associated with 2 phenomena. Firstly, GPI treatment reduced the concentrations of Aβ oligomers in CM; an observation that is consistent with numerous observations that it is the small soluble Aβ oligomers that cause synapse damage (Yang et al., 2017). However, since CM from treated cells still contained Aβ oligomers we postulated the presence of neuroprotective factors also released by treated cells. Notably, GPI treatment increased the concentrations of Aβ monomers, previously reported to have a neuroprotective function (Giuffrida et al., 2009), released into the CM. These results are consistent with the hypothesis that it is the ratio of Aβ monomers to Aβ oligomers that is a critical factor in determining synapse damage.
Opinion is divided on the subject of how Aβ oligomers in the CM form; one theory is that Aβ oligomers are formed intracellularly and released by cells while another suggests that they are formed by the oligomerization of Aβ monomers in the CM. In these studies the Aβ monomers in CM from 7PA2 cells were stable and there was no evidence that they formed Aβ oligomers in a cell-free system. There was a highly significant inverse correlation between the concentrations of Aβ oligomers and Aβ monomers in CM from cells treated with 0.125 to 10 nM sialylated GPIs (Figure 1). To explain this correlation we first excluded the possibility that GPIs had a direct effect on Aβ oligomers by showing that in a cell-free system the GPIs did not cause the dissociation of Aβ oligomers into Aβ monomers. We concluded that the GPIs affected the production of Aβ oligomers and Aβ monomers. Currently we can only speculate how sialylated GPIs might alter the production of Aβ. GPI-anchored proteins triggered the formation, structure and function of lipid rafts (Suzuki et al., 2012). APP and many of the enzymes involved in the generation of Aβ are found in lipid rafts. Treatment with GPIs reduced the % of APP that was found within lipid rafts, an observation consistent with reports that GPIs sequester cholesterol and consequently affect lipid raft composition. The finding that there was a significant positive correlation between the concentrations of Aβ oligomers and the amounts of APP within lipid rafts suggests that membrane targeting of APP is a key factor in production of Aβ oligomers. Since proteins in lipid rafts traffic via different intracellular pathways to those in the normal cell membrane, then APP in lipid rafts may be targeted to different cell compartments (and consequently interacts with a different range of enzymes) than APP found in the normal cell membrane.
Figure 1.
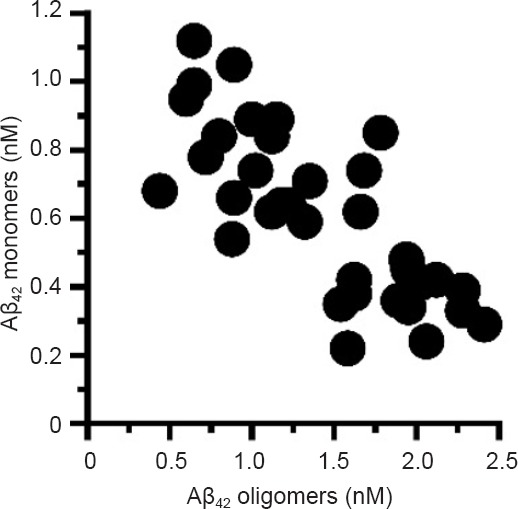
Glycosylphosphatidylinositol (GPI) treatment altered the release of amyloid β (Aβ) oligomers and Aβ monomers.
Treatment with GPIs caused dose-dependent reductions in the concentrations of Aβ oligomers and increases in Aβ monomers in conditioned media. There was a significant inverse correlation between the concentrations of Aβ oligomers and Aβ monomers in conditioned media from 7PA2 cells treated with sialylated GPIs (0.125 to 10 nM), Pearson's coefficient = –0.68, P < 0.01.
How is this work relevant to neural regeneration? Synapse degeneration occurs in the early stages of AD and in vitro is reversible when toxic Aβ is removed (Bate, 2015). Drugs that pass the blood-brain barrier and reduce toxic Aβ oligomers could allow the regeneration of synapses by neurons. Like all in vitro work there are a number of important caveats that should be considered. It should be stressed that our paper examined the production of small soluble Aβ monomers and oligomers and that other factors involved in synapse damage were not considered. In addition, neurons from different parts of the brain may have different sensitivities to the neurons used in our assays.
Last year saw the publication of a clinical trial demonstrating that a mAb that bound to Aβ (aducanumab) altered the progression of AD (Sevigny et al., 2016). This report raised important questions as to why many of the preceding trials with other Aβ-binding mAbs, that had reduced Aβ within the brain, had failed to show clinical benefit. One explanation may be the forms of Aβ that these mAbs react with; it was noteworthy that aducanumab did not bind to Aβ monomers. If the current paradigm that Aβ oligomers are highly synaptotoxic, while Aβ monomers are not and may even be neuroprotective is correct, then a mAb that binds and removes only Aβ oligomers would have more impact than a mAb that binds to both Aβ monomers and Aβ oligomers. It remains a possibility that compounds that reduce Aβ may increase clinical symptoms by removing neuroprotective Aβ monomers. Conversely, treatments that may not affect total Aβ concentrations may have clinical relevance if they selectively reduce or remove Aβ oligomers.
In summary, our study demonstrated two key findings. Firstly, it is possible to switch production of Aβ away from toxic Aβ oligomers to generate neuroprotective Aβ monomers. Secondly, the release of toxic Aβ from 7PA2 cells is controlled by a pathway sensitive to the presence of sialylated GPIs. While the mechanism(s) of this switch remain elusive, the discovery of brain-penetrant compounds that affect this aspect of Aβ production is an interesting new therapeutic approach.
Footnotes
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review reports:
Reviewer 1: Panteleimon Giannakopoulos, Université de Genève, Switzerland.
Reviewer 2: Antonio G. García, Universidad Autonoma De Madrid, Spain.
Comments to author: This is a perspective mini-review on an interesting topic, whether toxic Aβ oligomers can be switched to neuroprotective Aβ monomers, a potential strategy to treat AD. The Ms. rises two interesting topics: 1) different forms of Aβ are produced in the brain of patients with AD; 2) clinical trials with antibodies against Aβ have so far failed because the antibodies targeting Aβ did not consider these heterogeneous forms. Authors comment a recent successful clinical trial with aducanumab, a mAb that does not bind to neuroprotective Aβ monomers.
References
- Bate C. Enhanced neuronal degradation of amyloid-beta oligomers allows synapse regeneration. Neural Regen Res. 2015;10:700–701. doi: 10.4103/1673-5374.156955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Nolan W, McHale-Owen H, Bate C. Sialylated glycosylphosphatidylinositols suppress the production of toxic amyloid-beta oligomers. Biochem J. 2017;474:3045–3058. doi: 10.1042/BCJ20170239. [DOI] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- Suzuki KG, Kasai RS, Hirosawa KM, Nemoto YL, Ishibashi M, Miwa Y, Fujiwara TK, Kusumi A. Transient GPI-anchored protein homodimers are units for raft organization and function. Nat Chem Biol. 2012;8:774–783. doi: 10.1038/nchembio.1028. [DOI] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. Large soluble oligomers of amyloid β-protein from alzheimer brain are far less neuroactivethan the smaller oligomers to which they dissociate. J Neurosci. 2017;37:152–163. doi: 10.1523/JNEUROSCI.1698-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]