

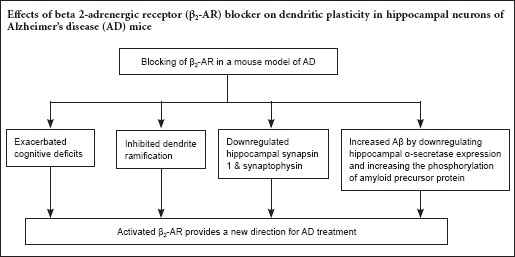
Keywords: nerve regeneration, neurodegeneration, beta-2 adrenergic receptor, Alzheimer's disease, amyloid-β, ICI 118551, cognitive function, dendrite ramification, synapsin 1, synaptophysin, α-secretase, amyloid precursor protein, neural regeneration
Abstract
Dendrite ramification affects synaptic strength and plays a crucial role in memory. Previous studies revealed a correlation between beta 2-adrenergic receptor dysfunction and Alzheimer's disease (AD), although the mechanism involved is still poorly understood. The current study investigated the potential effect of the selective β2-adrenergic receptor antagonist, ICI 118551 (ICI), on Aβ deposits and AD-related cognitive impairment. Morris water maze test results demonstrated that the performance of AD-transgenic (TG) mice treated with ICI (AD-TG/ICI) was significantly poorer compared with NaCl-treated AD-TG mice (AD-TG/NaCl), suggesting that β2-adrenergic receptor blockage by ICI might reduce the learning and memory abilities of mice. Golgi staining and immunohistochemical staining revealed that blockage of the β2-adrenergic receptor by ICI treatment decreased the number of dendritic branches, and ICI treatment in AD-TG mice decreased the expression of hippocampal synaptophysin and synapsin 1. Western blot assay results showed that the blockage of β2-adrenergic receptor increased amyloid-β accumulation by downregulating hippocampal α-secretase activity and increasing the phosphorylation of amyloid precursor protein. These findings suggest that blocking the β2-adrenergic receptor inhibits dendrite ramification of hippocampal neurons in a mouse model of AD.
Introduction
Alzheimer's disease (AD) is a common form of dementia that typically occurs during old age (Schott et al., 2006; Doody et al., 2014; Dubois et al., 2014; Nelson et al., 2014; Aron and Yankner, 2016; Canter et al., 2016; Reiman, 2016; Roy et al., 2016; Shrestha and Klann, 2016; Tycko, 2016). The disease is characterized by a progressive degenerative change in brain regions that leads to serious damage to cognitive functions, including memory, decision-making, language and orientation (Li et al., 2013). Amyloid beta (Aβ) deposits in brain regions are universally present among AD patients and are considered the fundamental cause of the disease (Tanzi, 2005; Gao et al., 2016). However, currently there is no effective cure for this devastating disease (Choi et al., 2016; The Lancet, 2016).
Early symptoms of AD include difficulty in remembering recent events, and as short-term memory loss progresses, patients begin to have problems with languages, disorientation, change of mood, along with other behavior problems that affect personal and social life (Artero et al., 2003; Cummings et al., 2016; Fray et al., 2016; Kida et al., 2016; Lauterbach, 2016; Makovac et al., 2016; Nagata et al., 2016). Histologically, disease usually presents with two major characteristics: neurofibrillary tangles and amyloid plaques. The plaques are composed of Aβ, a peptide cleaved from the amyloid precursor protein (APP) (Querfurth and LaFerla, 2010). The neurofibrillary tangle is formed by hyperphosphorylation of a microtubule-associated protein, tau (Ballatore et al., 2007; Kim et al., 2015; Koliatsos and Xu, 2015). Clinical manifestations suggest that tau and Aβ accumulation play an essential role in the pathology of the disease. However, these histological changes are only observed when AD patients have succumbed to the disease (Querfurth and LaFerla, 2010).
In recent years, a number of studies have demonstrated a link between AD and β2-adrenergic receptor (β2-AR) functions (Ni et al., 2006; Yu et al., 2008). For example, an epidemiological study reported a correlation between low AD risk and the use of non-selective β-ARs antagonists among people with high blood pressure (Khachaturian et al., 2006). A genetic study also found a correlation between β2-AR gene polymorphisms and the high incidence of late-onset AD (Li et al., 2013). Interestingly, an animal study showed that chronic β2-AR agonist treatment resulted in greater numbers of Aβ deposits among mice contained a mixed background of C57Bl/6 and 129 and when the β2-AR was blocked, Aβ induced by acute stress was significantly decreased (Li et al., 2013). In addition, β2-AR is well known for its involvement in memory and normal learning. Studies have shown that the activation of β2-AR promotes the long-term potentiation of synapses. Indeed, studies demonstrated a modulatory effect of β2-AR in memory building and consolidation. β2-AR activation also alleviates the negative effects of amyloid-β on long-term potentiation (Li et al., 2013). All this evidence suggests a relationship between β2-AR and AD. However, the role of β2-AR and the mechanism underlying the correlation of β2-AR and AD in AD pathogenesis are still largely unknown.
In this study, an AD mouse model was used to investigate the potential effect of a selective β2-AR antagonist on Aβ deposits and the AD-related cognitive impairment. The mechanism underlying the relationship between β2-AR and AD was also investigated.
Materials and Methods
Animals
Twenty 6-month old female amyloid precursor protein/presenilin 1 double transgenic (APPswe/PS1dE9) mice (AD-TG) (weight 22–27 g) and 20 female wild type (non-TG) mice (weight 22–27 g) were purchased from Jackson Laboratory (Bar Harbor, ME, USA).
The animal study was approved by the Committee of the Ethics of Animal Experiments of Yancheng Vocational Institute of Health Sciences, China. The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1986).
APPswe/PS1dE9 transgenic mice have been widely used as a model of AD (Lin et al., 2016). Mice were raised in accordance with a standard protocol, with a 12-hour light/dark cycle (light on from 7:00 a.m. and light off from 7:00 p.m), at 23 ± 1°C and 52 ± 5% humidity with free access to tap water and rodent diet for 5 days prior to experimental interventions and behavioral tests.
Drug treatment
AD-TG (n = 16) and wild type control (non-TG) mice (n = 16) were sub-grouped as follows: non-TG/NaCl and AD-TG/NaCl saline control groups (n = 8 per group) receiving injections of 0.9% NaCl (1 mg/kg per day, intraperitoneally) for 2 months; and non-TG/ICI and AD-TG/ICI groups receiving β2-AR antagonist treatment, ICI 118551 (Sigma-Aldrich, St Louis, MO, USA) (1 mg/kg per day, intraperitoneally) for 2 months (n = 8 per group). ICI 118551 was all dissolved in saline (0.9% NaCl) before use.
Morris water maze test
To evaluate the memory and spatial learning of all animals, the Morris water maze test was performed in the morning as described in a previous study (Zhang et al., 2012). After AD-TG and non-TG mice were subjected to saline or ICI 118551 for 2 months, the Morris water maze test was performed. A computerized tracking system (DigBeh-MM; Shanghai Jiliang Software Technology Co., Ltd., Shanghai, China) was utilized to monitor the trajectory of all mice. During the test, for the training trials, a hidden platform with a diameter of 5 cm was maintained in the same quadrant, 1.5 cm below the surface of water.
In every trial, all mice had up to 1 minute to locate the hidden platform and escape onto it. If a mouse did not locate the hidden platform within 1 minute, the experimenter would manually guide the mouse to the platform and keep it there for 10 seconds. The trial was carried out four times daily for 6 days. The escape latency refers to the time that a mouse spent in finding the platform, and this formed the spatial learning score.
Following the last training trial, the probe trial was carried out to test the spatial memory by allowing all animals to swim freely for 1 minute in the maze with the platform removed. After finding the platform, the mice were allowed to stay on it for 20 seconds and were then placed in their home cage for 30 seconds until the start of the next acquisition trial. If a mouse did not find the platform within 90 seconds, it was guided onto the platform and allowed to remain on it for 20 seconds. After training, the mice were placed back in their home cage until retention testing 24 hours later. A 60-second probe trial was performed 5 minutes after the injection of propofol or saline. During the probe test, the platform was not present, and the times crossing the platform and the time spent in the target quadrant (3rd quadrant) was recorded. The escape latency before reaching the hidden platform location, the swimming distance in the 3rd quadrant and counts of crossing the 3rd quadrant within 1 minute during the probe trial were recorded.
Golgi staining and immunohistochemistry
After AD-TG and non-TG mice were subjected to saline or ICI 118551 for 2 months, brain tissues were harvested. For Golgi staining, half of the brain was fixed with 400 mL Golgi fixative (0.9% NaCl, 5% formaldehyde, 5% potassium dichromate, and 5% chloral hydrate) in the dark. The brain was removed and used for Golgi staining as previously described (Li et al., 2013). In brief, the brain was post-fixed for 3 days in the same Golgi fixative, and impregnated with 1.0% aqueous silver nitrate solution for 3 days. Coronal brain sections of hippocampal tissue were cut at 35 μm. The images were observed by microscopy (Olympus, Tokyo, Japan).
For immunohistochemistry staining, the brains were subjected to 4% formaldehyde in PBS for 2 days and bathed in PBS with sodium azide (0.02%). The fixed brains were then sliced into 50-μm thick sections. The sections were rinsed with Tris buffered saline (TBS), and incubated with 3% H2O2 for 30 minutes, then subjected to a 15-minute incubation of TBS-A and a 30-minute incubation of TBS-B. The sections were then probed with specific primary antibody rabbit monoclonal anti-Aβ (1:200; Abcam, Cambridge, MA, USA) at 4°C overnight. Sections were rinsed with TBS before a 2-hour incubation of goat anti-rabbit secondary antibody at room temperature. Finally, the reaction product was visualized with diaminobenzidine in 0.1 M phosphate-buffered saline for 30 seconds. Immunohistochemical staining results were observed under a light microscope (Olympus), and quantitative assessment of relative changes in immunohistochemical staining was performed by Image Pro Plus 6.0 software (Media Cybernetics, Atlanta, GA, USA). The average number of dendritic branches of pyramidal neurons in the hippocampus was analyzed by Golgi staining.
Enzyme linked immunosorbent assay
Amyloid beta 40 (Aβ40) and amyloid beta 42 (Aβ42) enzyme linked immunosorbent assay plates were purchased from Invitrogen (Waltham, MA, USA) and experiments were conducted according to the manufacturer's instructions.
Western blot assay
Proteins (20 μg) from each sample of hippocampus were loaded on a 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. After electrophoresis, the proteins were transblotted to a polyvinylidene difluoride membrane (Millipore) blocked with 5% non-fat milk solution with Tris buffered saline Tween (TBST) for 60 minutes. The protein blots were then incubated with the following specific primary antibodies: rabbit monoclonal anti-Synapsin-1 (1:500), rabbit polyclonal anti-synaptophysin (1:1,000), rabbit anti-monoclonal 6E10 (1:500), rabbit polyclonal anti-PS1(1:1,000), rabbit monoclonal anti-APP (1:1,000) (all from Abcam, Cambridge, MA, USA); rabbit polyclonal anti-p-APP, rabbit monoclonal anti-secreted amyloid precursor protein-α (sAPPα) and rabbit monoclonal anti-secreted amyloid precursor protein-β (sAPPβ), and rabbit monoclonal anti-β-actin (all from Cell Signaling, Beverly, MA, USA) at 4°C overnight. The membranes were then washed with TBST and subjected to a 2-hour incubation with the following secondary antibody: donkey anti-rabbit IgG (H&L) antibody (1:5,000; Rockland Immunochemicals, Inc., Pottstown, PA, USA) at room temperature for 2 hours. Finally, the protein blots were visualized using the ECL Plus Western Blotting Substrate (TIANGEN, Beijing, China). Optical density values were analyzed with PDQuest 7.2.0 software (Bio-Rad, Richmond, CA, USA). β-Actin was used as an internal reference.
Statistical analysis
Continuous variables are expressed as the mean ± SD. All statistical analyses were performed using SPSS 19.0 software (IBM, Armonk, NY, USA). Significant differences between groups were assessed by one-way analysis of variance followed by the least significant difference test. A two-sided significant value of P < 0.05 was considered statistically significant.
Results
Blocking β2-AR exacerbated memory deficits in AD-TG mice
To determine the effect of blocking β2-AR on spatial memory in AD-TG mice, the Morris water maze test was conducted. A previous study demonstrated that AD-TG mice showed Aβ accumulation, synaptic deficits and memory impairments at 8 months (Li et al., 2013). Therefore, AD-TG mice were injected with an β2-AR antagonist (ICI 118551) at 8 months of age and their learning and memory behaviors were assessed at 10 months of age (Chai et al., 2017). For the Morris water maze training, mice received 4 rounds of training each day, for 5 days until the probe trial. The latency for finding the hidden platform was significantly increased in AD-TG mice compared with the wild type control (non-TG) mice (P < 0.01) (Figure 1A). The performances of AD-TG mice treated with ICI (AD-TG/ICI) in the Morris water maze were significantly poorer at 3 and 4 days, compared with NaCl-treated AD-TG mice (AD-TG/NaCl) (Figure 1A) (P < 0.01). In addition, AD-TG/ICI mice had poorer Morris water maze performance at 4 and 5 days, compared with the non-TG group. However, ICI had no significant impact on the learning and memory behavior of non-TG mice (Figure 1A; P < 0.01).
Figure 1.
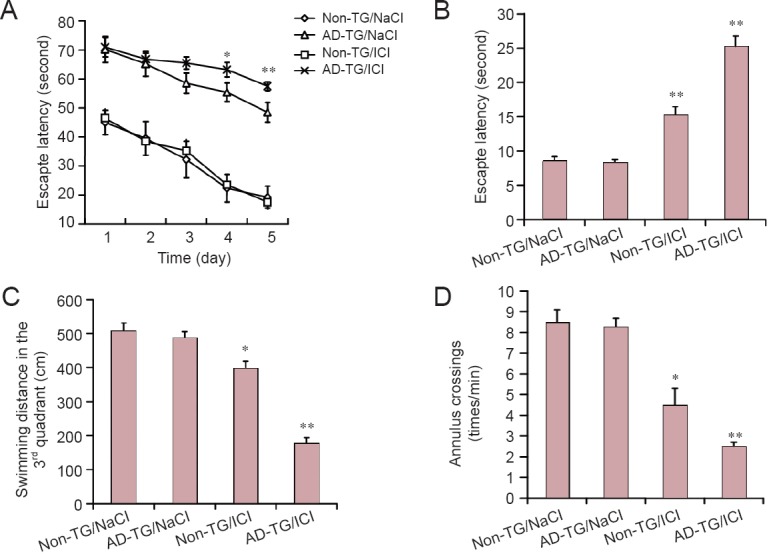
Blocking beta 2-adrenergic receptor (β2-AR) exacerbated memory deficits in amyloid precursor protein/presenilin 1 double transgenic (AD-TG) mice.
(A) The escape latency before reaching the hidden platform for 5 days of training trial. (B–D) The escape latency before reaching the hidden platform (B), the swimming distance in the 3rd quadrant (C) and counts of crossing the 3rd quadrant within 1 minute during the probe trial (D). Data are expressed as the mean ± SD (n = 8, one-way analysis of variance followed by the least significant difference test). *P < 0.05, **P < 0.01, vs. non-TG/NaCl group. Wild type control (non-TG) and AD-TG mice were sub-grouped: saline control group, injections of 0.9% NaCl, termed non-TG/NaCl and AD-TG/NaCl; β2-AR antagonist treatment, mice were administered ICI 118551 (1 mg/kg per day, intraperitoneally), termed AD-TG/ICI.
To evaluate the spatial memory of mice, probe trials were performed after 5 days of training trial. The reference memory of AD-TG/ICI mice was significantly poorer (with significantly longer latency before reaching the location of the platform), compared with the other groups (Figure 1B; P < 0.01). In addition, the AD-TG/ICI group had a slower swim speed in the probe test compared with the AD-TG group (Figure 1C). Moreover, fewer counts of location crossing during the trial were reported for AD-TG/ICI mice compared with untreated AD-TG mice (Figure 1D; P < 0.01). These findings suggest that ICI has a significant negative effect on the cognitive function of AD-TG mice.
Blocking β2-AR inhibited dendrite ramification in the mouse hippocampus
Synaptic plasticity is essential for learning and memory and can be evaluated by the observation of dendrite morphology (Li et al., 2013). To investigate the mechanisms involved in exacerbated memory deficits in AD-TG mice by blocking β2-AR, the average number of dendritic branches of pyramidal neurons in the hippocampus was analyzed by Golgi staining. The average counts of dendritic branches were significantly decreased in AD-TG mice compared with non-TG mice (Figure 2A and B; P < 0.01). Noticeably, the blocking of β2-AR with ICI treatment further decreased the number of dendritic branches (Figure 2A and B; P < 0.05).
Figure 2.

Blocking of beta 2-adrenergic receptor (β2-AR) inhibits the number of dendritic branches in the hippocampus of amyloid precursor protein/presenilin 1 double transgenic (AD-TG) mice.
(A) Probe trials were performed after 5 days of training. Representative Golgi stained hippocampal pyramidal neurons in groups of wild type control (non-TG/Nacl), amyloid precursor protein/presenilin 1 double transgenic (AD-TG/NaCl), and AD-TG/NaCl mice given ICI 118551 (AD-TG/ICI) under light microscopy. The dendritic branches (indicated by arrows) were decreased in AD-TG/NaCl mice compared with non-TG mice. Blocking β2-AR with ICI treatment further decreased the number of dendritic branches. Scale bars: 100 μm. (B) The number of dendritic branches was quantified from randomly selected neurons. The average counts of dendritic branches were significantly decreased in AD-TG/NaCl mice compared with non-TG/NaCl mice, and blocking β2-AR with ICI treatment further decreased the number of dendritic branches. *P < 0.05, **P < 0.01, vs. non-TG/NaCl. Data are expressed as the mean ± SD (n = 8, one-way analysis of variance followed by the least significant difference test). Wild type control (non-TG) and AD-TG mice were sub-grouped: saline control group, injections of 0.9% NaCl, termed non-TG/NaCl and AD-TG/NaCl; β2-AR antagonist treatment group, mice were administered ICI 118551 (1 mg/kg per day, intraperitoneally), termed AD-TG/ICI.
To further explore the molecular mechanisms underlying β2-AR blocking exacerbated memory deficits, the expression of several synaptic proteins was examined by western blot assay. Results showed that ICI treatment in AD-TG mice further decreased the expression of synaptophysin and synapsin 1 (P < 0.01, P < 0.05; Figure 3A and B).
Figure 3.

Effect of ICI 118551 on the expression of synaptophysin and synapsin 1 in the hippocampus of amyloid precursor protein/presenilin 1 double transgenic (AD-TG) mice.
(A) Protein samples of hippocampus were harvested after 5 days of training. The protein levels of synaptophysin and synapsin I in the hippocampus of wild type control (non-TG/NaCl), amyloid precursor protein/presenilin 1 double transgenic (AD-TG/NaCl), and AD-TG/NaCl mice given ICI 118551 (AD-TG/ICI) were determined by western blot assay. (B) Synaptophysin and synapsin I signal intensity was quantified by ImageJ software and normalized by β-actin expression. *P < 0.05, **P < 0.01, vs. non-TG/NaCl. Data are expressed as the mean ± SD (n = 8, one-way analysis of variance followed by the least significant difference test). Wild type control (non-TG) and AD-TG mice were sub-grouped: saline control group, injections of 0.9% NaCl, termed non-TG/NaCl and AD-TG/NaCl; β2-AR antagonist treatment group, mice were administered ICI 118551 (1 mg/kg per day, intraperitoneally), termed non-TG/ICI and AD-TG/ICI. Experiments were conducted in triplicate.
Blocking of β2-AR increased Aβ accumulation in AD-TG mice
The mice were treated with ICI at 8 months of age and the alteration of Aβ was measured at 10 months of age. Results showed that 2 months of ICI treatment on AD-TG mouse could increase Aβ accumulation in hippocampus (Figure 4A and B; P < 0.01). The blocking of β2-AR with ICI increased Aβ40-42 production (Figure 4C and D; P < 0.01, P < 0.05).
Figure 4.

Blocking beta 2-adrenergic receptor (β2-AR) increases amyloid beta (Aβ) accumulation in amyloid precursor protein/presenilin 1 double transgenic (AD-TG) mice.
(A) Immunohistochemical staining of the hippocampus was performed 1 day after 5 days of training. The representative images show the levels of plaque load in the hippocampus of wild type control (non-TG), AD-TG/NaCl, and AD-TG/NaCl mice given ICI 118551 (AD-TG/ICI) immunostained with Aβ antibody under a light microscope. Results showed that ICI treatment of AD-TG mice increased Aβ accumulation in the hippocampus (indicated by arrow). Scale bars: 100 μm. (B) Quantification of Aβ staining was performed by Image Pro Plus 6.0 software. (C and D) The levels of amyloid beta 40 (Aβ40) and amyloid beta 42 (Aβ42) in hippocampi were detected by enzyme linked immunosorbent assay. *P < 0.05, **P < 0.01, vs. non-TG/NaCl. Data are expressed as the mean ± SD (n = 8, one-way analysis of variance followed by the least significant difference test). Wild type control (non-TG) and AD-TG mice were sub-grouped: saline control group, injections of 0.9% NaCl, termed non-TG/NaCl and AD-TG/NaCl; β2-AR antagonist treatment group, mice were administered ICI 118551 (1 mg/kg per day, intraperitoneally), termed AD-TG/ICI.
To explore the mechanisms underlying the upregulation of Aβ production by ICI, the expression of PS1, APP and p-APP was evaluated by western blot assay. A significant increase of APP phosphorylation and PS1 was observed in AD-TG mice compared with non-TG control mice. Notably, ICI treatment further increased the protein expression of p-APP and PS1 (Figure 5A and B; P < 0.01). Furthermore, changes in the sAPPα, sAPPβ, and 6E10 (Figure 5A and B) proteins were detected, which are involved in Aβ synthesis and degradation in AD-TG mice (Nussbaum and Ellis, 2003). Compared with non-TG mice, the protein levels of sAPPα and sAPPβ were significantly decreased in AD-TG mice (P < 0.01). Blocking β2-AR significantly reduced sAPPα and sAPPβ levels compared with untreated AD-TG mice (P < 0.01).
Figure 5.

Effect of ICI 118551 (ICI) treatment on the expression of amyloid beta (Aβ) accumulation related protein in amyloid precursor protein/presenilin 1 double transgenic (AD-TG) mice.
(A) The protein levels of presenilin 1 (PS1), amyloid precursor protein (APP), phosphorylation of APP (p-APP), secreted amyloid precursor protein-α (sAPPα), secreted amyloid precursor protein-β (sAPPβ) and amyloid beta (6E10) in the hippocampus were measured by western blot assay. (B) Protein signal intensity was quantified by ImageJ software and normalized by β-actin expression. *P < 0.05, **P < 0.01, vs. non-TG/NaCl. Data are expressed as the mean ± SD (n = 8, one-way analysis of variance followed by the least significant difference test). Wild type control (non-TG) and AD-TG mice were sub-grouped: saline control group, injections of 0.9% NaCl, termed non-TG/NaCl and AD-TG/NaCl; β2-AR antagonist treatment group, mice were administered ICI 118551 (1 mg/kg per day, intraperitoneally), termed non-TG/ICI and AD-TG/ICI. Experiments were conducted in triplicate.
Discussion
Because the global population age is increasing, it has been suggested that the prevalence of AD might be increased 4-fold by the middle of this century (Alzheimer's Association, 2013; Kunz et al., 2015; Wang et al., 2015). Aging might also lead to many other conditions, such as cardiovascular diseases (Yang and Ming, 2012; Carvalho-de-Souza et al., 2013; Qi and Xie, 2013; Corella and Ordovas, 2014; Shah et al., 2014; Baralla et al., 2015; Chang et al., 2016; Harshman and Shea, 2016; Sahoo et al., 2016; Sallam and Laher, 2016; Silva-Palacios et al., 2016). Because cardiovascular diseases such as hypertension are commonly comorbid with AD, it is important to clarify the effect of certain treatments on both conditions (Li et al., 2013). In this study, we investigated β-blockers, and showed that blocking β2-AR by ICI in AD-TG mice substantially inhibited dendrite ramification in hippocampal neurons and downregulated the postsynaptic density proteins synapsin 1 and synaptophysin. Moreover, blocking β2-AR increased Aβ accumulation by downregulating α-secretase activity and increasing the phosphorylation of APP. Our findings suggest that β2-AR might be a novel target for AD treatment.
Postsynaptic dendritic spine remodeling plays a fundamental role in the formation of memory (Becker et al., 2017; Kulasiri et al., 2017; Schiff et al., 2017). Morphological diversity represents functional properties of dendritic spine (Jiang et al., 2015). Recently, Li and colleagues reported that the chronic oral administration of a β-adrenergic agonist mitigated Aβ-related hippocampal impairments in wild type mice (Li et al., 2013). In the present study, blocking β2-AR decreased dendritic complexity and decreased spine density. This suggests that β2-AR antagonists might lead to memory impairment, consistent with the above evidence.
Morphological changes in dendrites are multifactorial, including the influence of neuronal activity (Whitford et al., 2002), and cadherin and β-catenin (Abe et al., 2004). A protective effect of non-selective β-ARs antagonists against AD was previously reported (Khachaturian et al., 2006). Moreover, genetic studies of AD revealed that β2-AR gene polymorphisms are associated with the risk of late-onset AD (Insel, 2011). Presynaptic neurotransmitter release involves many vesicle-related proteins such as synapsin I and synaptophysin. These proteins are biomarkers for synaptic plasticity of neural networks (Pieribone et al., 1995). In this study, the role of β2-AR in AD was investigated. Blocking β2-AR exacerbated the decreased expression of synaptophysin and synapsin I. These data might be related to the functional and structural mechanisms underlying the β2-AR-mediated amelioration of memory deficits in AD-TG mice.
Environmental factors such as stress-activated receptors including β2-AR, are considered risk factors for AD (Fratiglioni et al., 2004). Ni et al. (2006) reported that a β2-AR agonist stimulated γ-secretase activity and accelerated Aβ plaque formation in vivo and in vitro (Ni et al., 2006). Their experiments mimicked abnormal β2-AR activation, which might result from a response to stress and might contribute to Aβ accumulation in the pathology of AD. Moreover, Aβ may induce β2-AR internalization and degradation, leading to the impairment of adrenergic and glutamatergic activities (Wang et al., 2011). Indeed, a decrease in β2-AR levels was observed in different areas of AD brains, and the β2-AR response was also decreased in fibroblasts isolated from AD patients (Huang and Gibson, 1993). Aβ is generated by the consecutive cleavage of APP by two proteases, i.e., β-secretase and γ-secretase (presenilin, PS1/PS2) (Nishitomi et al., 2006). In our study, although the levels of Aβ40 and Aβ42 were increased in ICI-treated AD-TG mice, the total levels of APP and PS1 were not significantly changed compared with NaCl-treated AD-TG mice. Thus, alterations of APP in ICI-treated AD-TG mice were rare. These findings, along with other previous animal studies, indicated that chronic β2-AR agonist treatment might increase Aβ deposits in transgenic mice, while β-blocker treatment reduced stress-related acute Aβ production (Yu et al., 2010). We also showed that blocking β2-AR with ICI significantly increased the phosphorylation of APP and increased Aβ production in AD-TG mice whilst downregulating α-secretase activity. These results might shed light on the mechanism underlying the correlation between β2-AR and AD.
In summary, blocking β2-AR exacerbated cognitive deficits and decreased dendritic branches accompanied by the downregulation of synaptic protein levels. Furthermore, blocking β2-AR increased Aβ accumulation by downregulating α-secretase activity and increasing the phosphorylation of APP in AD-TG mice. These results suggest that targeting β2-AR pharmacologically might provide a new direction for AD treatment.
Footnotes
Funding: This work was financially supported by the Key Laboratory of Brain Disease Bioinformation of Jiangsu Province of China, No. Jsbl1202.
Conflicts of interest: None declared.
Research ethics: The animal study was approved by the Committee on the Ethics of Animal Experiments of Yancheng Vocational Insititute of Health Sciences. The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1986).
Data sharing statement: The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Copyedited by Wang J, Li CH, Qiu Y, Song LP, Zhao M
References
- Abe K, Chisaka O, Van Roy F, Takeichi M. Stability of dendritic spines and synaptic contacts is controlled by alpha N-catenin. Nat Neurosci. 2004;7:357–363. doi: 10.1038/nn1212. [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association. 2013 Alzheimer's disease facts and figures. Alzheimers Dement. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Aron L, Yankner BA. Neurodegenerative disorders: Neural synchronization in Alzheimer's disease. Nature. 2016;540:207–208. doi: 10.1038/540207a. [DOI] [PubMed] [Google Scholar]
- Artero S, Tierney MC, Touchon J, Ritchie K. Prediction of transition from cognitive impairment to senile dementia: a prospective, longitudinal study. Acta Psychiatr Scand. 2003;107:390–393. doi: 10.1034/j.1600-0447.2003.00081.x. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Baralla A, Sotgiu E, Deiana M, Pasella S, Pinna S, Mannu A, Canu E, Sotgiu G, Ganau A, Zinellu A, Sotgia S, Carru C, Deiana L. Plasma clusterin and lipid profile: a link with aging and cardiovascular diseases in a population with a consistent number of centenarians. PLoS One. 2015;10:e0128029. doi: 10.1371/journal.pone.0128029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker B, Steffens M, Zhao Z, Kendrick KM, Neumann C, Weber B, Schultz J, Mehta MA, Ettinger U, Hurlemann R. General and emotion-specific neural effects of ketamine during emotional memory formation. NeuroImage. 2017;150:308–317. doi: 10.1016/j.neuroimage.2017.02.049. [DOI] [PubMed] [Google Scholar]
- Canter RG, Penney J, Tsai LH. The road to restoring neural circuits for the treatment of Alzheimer's disease. Nature. 2016;539:187–196. doi: 10.1038/nature20412. [DOI] [PubMed] [Google Scholar]
- Carvalho-de-Souza JL, Varanda WA, Tostes RC, Chignalia AZ. BK channels in cardiovascular diseases and aging. Aging Dis. 2013;4:38–49. [PMC free article] [PubMed] [Google Scholar]
- Chai GS, Wang YY, Zhu D, Yasheng A, Zhao P. Activation of beta2-adrenergic receptor promotes dendrite ramification and spine generation in APP/PS1 mice. Neurosci Lett. 2017;636:158–164. doi: 10.1016/j.neulet.2016.11.022. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chang YC, Hu WL, Hung YC. Oxidative stress and salvia miltiorrhiza in aging-associated cardiovascular diseases. Oxid Med Cell Longev. 2016;2016:4797102. doi: 10.1155/2016/4797102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Quinti L, Tanzi RE, Kim DY. 3D culture models of Alzheimer's disease: a road map to a “cure-in-a-dish”. Mol Neurodegener. 2016;11:75. doi: 10.1186/s13024-016-0139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corella D, Ordovas JM. Aging and cardiovascular diseases: the role of gene-diet interactions. Ageing Res Rev. 2014;18:53–73. doi: 10.1016/j.arr.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Cummings J, Lai TJ, Hemrungrojn S, Mohandas E, Yun Kim S, Nair G, Dash A. Role of donepezil in the management of neuropsychiatric symptoms in Alzheimer's disease and dementia with lewy bodies. CNS Neurosci Ther. 2016;22:159–166. doi: 10.1111/cns.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R Alzheimer's Disease Cooperative Study Steering C, Solanezumab Study G. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, DeKosky ST, Gauthier S, Selkoe D, Bateman R, Cappa S, Crutch S, Engelborghs S, Frisoni GB, Fox NC, Galasko D, Habert MO, Jicha GA, Nordberg A, Pasquier F, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- Fratiglioni L, Paillard-Borg S, Winblad B. An active and socially integrated lifestyle in late life might protect against dementia. Lancet Neurol. 2004;3:343–353. doi: 10.1016/S1474-4422(04)00767-7. [DOI] [PubMed] [Google Scholar]
- Fray S, Ali NB, Rassas AA, Kechaou M, Oudiaa N, Cherif A, Echebbi S, Messaoud T, Belal S. Early psychiatrics symptoms in familial Alzheimer's disease with presenilin 1 mutation (I83T) J Neural Transm (Vienna) 2016;123:451–453. doi: 10.1007/s00702-015-1498-x. [DOI] [PubMed] [Google Scholar]
- Gao ML, Zhang YD, Li N, Qiao J, Yu M. Bone marrow mesenchymal stem cells transplanted into a rat model of Alzheimer's disease: improvement in the learning and memory ability. Zhongguo Zuzhi Gongcheng Yanjiu. 2016;20:2059–2065. [Google Scholar]
- Harshman SG, Shea MK. The role of vitamin k in chronic aging diseases: inflammation, cardiovascular disease, and osteoarthritis. Curr Nutr Rep. 2016;5:90–98. doi: 10.1007/s13668-016-0162-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HM, Gibson GE. Altered beta-adrenergic receptor-stimulated cAMP formation in cultured skin fibroblasts from Alzheimer donors. J Biol Chem. 1993;268:14616–14621. [PubMed] [Google Scholar]
- Insel PA. beta(2)-Adrenergic receptor polymorphisms and signaling: do variants influence the “memory” of receptor activation? Sci Signal. 2011;4:e37. doi: 10.1126/scisignal.2002352. [DOI] [PubMed] [Google Scholar]
- Jiang X, Chai GS, Wang ZH, Hu Y, Li XG, Ma ZW, Wang Q, Wang JZ, Liu GP. Spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice. Sci Rep. 2015;5:9488. doi: 10.1038/srep09488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, Tschanz JT, Mayer LS, Welsh-Bohmer KA, Breitner JC. Antihypertensive medication use and incident Alzheimer disease: the cache county study. Arch Neurol. 2006;63:686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- Kida J, Nemoto K, Ikejima C, Bun S, Kakuma T, Mizukami K, Asada T. impact of depressive symptoms on conversion from mild cognitive impairment subtypes to Alzheimer's disease: a community-based longitudinal study. J Alzheimer's Dis. 2016;51:405–415. doi: 10.3233/JAD-150603. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kim SS, Lee YD. Fas-associated factor 1 promotes in neurofibrillary tangle-mediated cell death of basal forebrain cholinergic neurons in P301L transgenic mice. Neuroreport. 2015;26:767–772. doi: 10.1097/WNR.0000000000000423. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Xu L. The problem of neurodegeneration in cumulative sports concussions: emphasis on neurofibrillary tangle formation. In: Kobeissy F. H, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): 2015. [PubMed] [Google Scholar]
- Kulasiri D, Liang J, He Y, Samarasinghe S. Global sensitivity analysis of a model related to memory formation in synapses: model reduction based on epistemic parameter uncertainties and related issues. J Theor Biol. 2017;419:116–136. doi: 10.1016/j.jtbi.2017.02.003. [DOI] [PubMed] [Google Scholar]
- Kunz L, Schroder TN, Lee H, Montag C, Lachmann B, Sariyska R, Reuter M, Stirnberg R, Stocker T, Messing-Floeter PC, Fell J, Doeller CF, Axmacher N. Reduced grid-cell-like representations in adults at genetic risk for Alzheimer's disease. Science. 2015;350:430–433. doi: 10.1126/science.aac8128. [DOI] [PubMed] [Google Scholar]
- Lauterbach EC. Six psychotropics for pre-symptomatic & early Alzheimer's (MCI), Parkinson's, and Huntington's disease modification. Neural Regen Res. 2016;11:1712–1726. doi: 10.4103/1673-5374.194708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Zhang D, Yang T, Koeglsperger T, Fu H, Selkoe DJ. Environmental novelty activates beta2-adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron. 2013;77:929–941. doi: 10.1016/j.neuron.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Cheng YS, Liao TY, Lin C, Chen ZT, Twu WI, Chang CW, Tan DT, Liu RS, Tu PH, Chen RP. Intranasal administration of a polyethylenimine-conjugated scavenger peptide reduces amyloid-beta accumulation in a mouse model of alzheimer's disease. J Alzheimer's Dis. 2016;53:1053–1067. doi: 10.3233/JAD-151024. [DOI] [PubMed] [Google Scholar]
- Makovac E, Serra L, Spano B, Giulietti G, Torso M, Cercignani M, Caltagirone C, Bozzali M. Different patterns of correlation between grey and white matter integrity account for behavioral and psychological symptoms in alzheimer's disease. J Alzheimer's Dis. 2016;50:591–604. doi: 10.3233/JAD-150612. [DOI] [PubMed] [Google Scholar]
- Nagata T, Shinagawa S, Nakajima S, Plitman E, Mihashi Y, Hayashi S, Mimura M, Nakayama K. Classification of neuropsychiatric symptoms requiring antipsychotic treatment in patients with alzheimer's disease: analysis of the CATIE-AD study. J Alzheimer's Dis. 2016;50:839–845. doi: 10.3233/JAD-150869. [DOI] [PubMed] [Google Scholar]
- Nelson L, Gard P, Tabet N. Hypertension and inflammation in Alzheimer's disease: close partners in disease development and progression. J Alzheimer's Dis. 2014;41:331–343. doi: 10.3233/JAD-140024. [DOI] [PubMed] [Google Scholar]
- Ni Y, Zhao X, Bao G, Zou L, Teng L, Wang Z, Song M, Xiong J, Bai Y, Pei G. Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat Med. 2006;12:1390–1396. doi: 10.1038/nm1485. [DOI] [PubMed] [Google Scholar]
- Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, Becker AG, Hosono M, Sakaguchi I, Minami SS, Nakajima Y, Li HF, Takeyama C, Kihara T, Ota A, Wong PC, Aisen PS, Kato A, Kinoshita N, Matsuoka Y. BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J Neurochem. 2006;99:1555–1563. doi: 10.1111/j.1471-4159.2006.04178.x. [DOI] [PubMed] [Google Scholar]
- Nussbaum RL, Ellis CE. Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Qi YC, Xie XH. Hutchinson-gilford progeria syndrome and its relevance to cardiovascular diseases and normal aging. Biomed Environ Sci. 2013;26:382–389. doi: 10.3967/0895-3988.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Reiman EM. Alzheimer's disease: Attack on amyloid-beta protein. Nature. 2016;537:36–37. doi: 10.1038/537036a. [DOI] [PubMed] [Google Scholar]
- Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer's disease. Nature. 2016;531:508–512. doi: 10.1038/nature17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo S, Meijles DN, Pagano PJ. NADPH oxidases: key modulators in aging and age-related cardiovascular diseases? Clin Sci. 2016;130:317–335. doi: 10.1042/CS20150087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallam N, Laher I. Exercise modulates oxidative stress and inflammation in aging and cardiovascular diseases. Oxid Med Cell Longev. 2016;2016:7239639. doi: 10.1155/2016/7239639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff HC, Johansen JP, Hou M, Bush DE, Smith EK, Klein JE, LeDoux JE, Sears RM. Beta-adrenergic receptors regulate the acquisition and consolidation phases of aversive memory formation through distinct, temporally regulated signaling pathways. Neuropsychopharmacology. 2017;42:895–903. doi: 10.1038/npp.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott JM, Kennedy J, Fox NC. New developments in mild cognitive impairment and Alzheimer's disease. Curr Opin Neurol. 2006;19:552–558. doi: 10.1097/01.wco.0000247611.44106.76. [DOI] [PubMed] [Google Scholar]
- Shah NC, Shah GJ, Li Z, Jiang XC, Altura BT, Altura BM. Short-term magnesium deficiency downregulates telomerase, upregulates neutral sphingomyelinase and induces oxidative DNA damage in cardiovascular tissues: relevance to atherogenesis, cardiovascular diseases and aging. Int J Clin Exp Med. 2014;7:497–514. [PMC free article] [PubMed] [Google Scholar]
- Shrestha P, Klann E. Alzheimer's disease: Lost memories found. Nature. 2016;531:450–451. doi: 10.1038/nature17312. [DOI] [PubMed] [Google Scholar]
- Silva-Palacios A, Konigsberg M, Zazueta C. Nrf2 signaling and redox homeostasis in the aging heart: A potential target to prevent cardiovascular diseases? Ageing Res Rev. 2016;26:81–95. doi: 10.1016/j.arr.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nat Neurosci. 2005;8:977–979. doi: 10.1038/nn0805-977. [DOI] [PubMed] [Google Scholar]
- The Lancet N. Finding a cure for Alzheimer's disease starts with prevention. Lancet Neurol. 2016;15:649. doi: 10.1016/S1474-4422(16)30047-3. [DOI] [PubMed] [Google Scholar]
- Tycko R. Alzheimer's disease: Structure of aggregates revealed. Nature. 2016;537:492–493. doi: 10.1038/nature19470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Yuen EY, Zhou Y, Yan Z, Xiang YK. Amyloid beta peptide-(1-42) induces internalization and degradation of beta2 adrenergic receptors in prefrontal cortical neurons. J Biol Chem. 2011;286:31852–31863. doi: 10.1074/jbc.M111.244335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford KL, Dijkhuizen P, Polleux F, Ghosh A. Molecular control of cortical dendrite development. Annu Rev Neurosci. 2002;25:127–149. doi: 10.1146/annurev.neuro.25.112701.142932. [DOI] [PubMed] [Google Scholar]
- Yang Z, Ming XF. mTOR signalling: the molecular interface connecting metabolic stress, aging and cardiovascular diseases. Obes Rev 13 Suppl. 2012;2:58–68. doi: 10.1111/j.1467-789X.2012.01038.x. [DOI] [PubMed] [Google Scholar]
- Yu JT, Tan L, Ou JR, Zhu JX, Liu K, Song JH, Sun YP. Polymorphisms at the beta2-adrenergic receptor gene influence Alzheimer's disease susceptibility. Brain Res. 2008;1210:216–222. doi: 10.1016/j.brainres.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Yu NN, Wang XX, Yu JT, Wang ND, Lu RC, Miao D, Tian Y, Tan L. Blocking beta2-adrenergic receptor attenuates acute stress-induced amyloid beta peptides production. Brain Res. 2010;1317:305–310. doi: 10.1016/j.brainres.2009.12.087. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bai M, Xi Y, Hao J, Zhang Z, Su C, Lei G, Miao J, Li Z. Multiple inflammatory pathways are involved in the development and progression of cognitive deficits in APPswe/PS1dE9 mice. Neurobiol Aging. 2012;33:2661–2677. doi: 10.1016/j.neurobiolaging.2011.12.023. [DOI] [PubMed] [Google Scholar]