

Abstract
Objective:
To define the natural history of X-linked myotubular myopathy (MTM).
Methods:
We performed a cross-sectional study that included an online survey (n = 35) and a prospective, 1-year longitudinal investigation using a phone survey (n = 33).
Results:
We ascertained data from 50 male patients with MTM and performed longitudinal assessments on 33 affected individuals. Consistent with existing knowledge, we found that MTM is a disorder associated with extensive morbidities, including wheelchair (86.7% nonambulant) and ventilator (75% requiring >16 hours of support) dependence. However, unlike previous reports and despite the high burden of disease, mortality was lower than anticipated (approximate rate 10%/y). Seventy-six percent of patients with MTM enrolled (mean age 10 years 11 months) were alive at the end of the study. Nearly all deaths in the study were associated with respiratory failure. In addition, the disease course was more stable than expected, with few adverse events reported during the prospective survey. Few non–muscle-related morbidities were identified, although an unexpectedly high incidence of learning disability (43%) was noted. Conversely, MTM was associated with substantial burdens on patient and caregiver daily living, reflected by missed days of school and lost workdays.
Conclusions:
MTM is one of the most severe neuromuscular disorders, with affected individuals requiring extensive mechanical interventions for survival. However, among study participants, the disease course was more stable than predicted, with more individuals surviving infancy and early childhood. These data reflect the disease burden of MTM but offer hope in terms of future therapeutic intervention.
X-linked myotubular myopathy (MTM) is a neuromuscular disorder caused by mutations in the myotubularin (MTM1) gene.1 It is a rare condition (estimated at 1:50,000 male births) associated with substantial morbidities and early mortality.2 At present, there are no treatments or disease-modifying therapies. However, several therapeutic strategies, including gene therapy,3 enzyme replacement therapy,4 dynamin-2 modulation,5 and PIK3C2B inhibition,6 are showing promise in preclinical studies.
A key barrier to clinical translation for MTM is the lack of natural history data. Two previous studies examining clinical characteristics were performed in 1999 and 2002,7,8 and each identified considerable disease morbidities and extremely high mortality. The literature also includes several case reports9–11 and a congenital myopathy cohort study (including 8 MTM cases),11 each corroborating the severe weakness and early mortality of patients with MTM. There remains, however, a critical lack of knowledge of the MTM disease process, limiting the ability to develop outcome measures and hindering current clinical care, particularly as it relates to anticipatory guidance and disease prognostication. At present, MTM care is based on generalized practice guidelines established for all congenital myopathies.12
To address the need for natural history data, we performed a cross-sectional plus prospective, longitudinal study of MTM. We found that patients with MTM have severe morbidities, including wheelchair, ventilator, and feeding tube dependence, but markedly longer survival than previously reported. We discovered that the disease course is relatively stable, although with sizeable impact on daily activities for patients and caregivers. In total, we identified important parameters that will guide clinical care and future clinical trials.
METHODS
Study participants, data collection, protocol approvals, registrations, and patient consent.
Our cohort included male patients with confirmed MTM1 mutation or muscle biopsy consistent with MTM plus family history consistent with X-linked inheritance. Patients were recruited at MTM family conferences and through the Cure CMD registry. Medical records were obtained with a signed Authorization to Request Medical Records. Written consent was obtained, either from an English-speaking parent/guardian or from an English-speaking decisionally competent affected male patient >18 years of age. The cross-sectional and longitudinal (NCT01840657) studies were reviewed and approved by the University of Michigan Institutional Review Board (HUM00032624 and HUM00066029). Ethics approval was obtained before study initiation. All participants were assigned deidentified numbers.
Cross-sectional data were collected via parent-reported surveys with the Qualtrics system (Provo, UT) (n = 35). Surveys were administered at enrollment and at a follow-up date when available. Data for the 1-year prospective study were collected from 2 sources: medical records and monthly telephone interviews recorded on a paper survey (n = 33). Additional details are provided in the e-Methods (at Neurology.org).
RESULTS
Cohort characteristics.
Our cohort included 50 individuals with MTM: 35 in the cross-sectional study and 33 in the longitudinal study; 18 individuals participated in both studies, including 4 sibling pairs. Ages ranged from 1 day to 42 years 4 months, with a median age of 8 years 4 months and average age of 10 years 11 months (figure 1A and table e-1).
Figure 1. Survival and burden of disease in an MTM cohort.

(A) Survival of participants over the course of the study. For those who died, age and cause of death are summarized in the table to the right of the graph. Twelve of 50 patients in our cohort were deceased, and the average age at death was 6 years 10 months. Note that 3 died during the longitudinal study, including 1 individual who died 2 months after survey completion. (B) Burden of disease (respiratory, feeding, and ambulation support requirements). Breakdown of technology support required, including wheelchair dependence, feeding tube, and ventilation. Forty-eight of 50 patients with MTM had some technology dependence, and 37 of 48 were wheelchair, ventilator, and tube feed dependent. (C) Change in respiratory status during the 1-year longitudinal survey (listed as percent of the total cohort, n = 33). The majority of individuals had either no change or small fluctuations in support. Small fluctuations were defined as 1 or 2 increases or decreases in time on ventilation that lasted <3 months. Frequent changes were defined as ≥3 periods (≤3 months) of increased or decreased ventilator setting requirements. Changes in time needing ventilation sustained for at least 4 months are indicated as increased requirements or sustained improvement. (D) Breakdown of the extent of feeding support, respiratory support, and highest motor function at the time of survey completion. G-tube = gastrostomy tube; MTM = myotubular myopathy.
Survival.
Data were ascertained for 12 deceased patients with MTM (figure 1A). Age of death ranged from 1 day to 25 years 4 months, with an average age at death of 6 years 10 months and mean survival of 12 years 2 months. Ten of 12 patients died of respiratory complications, with nonrespiratory deaths caused by cardiac arrest (likely precipitated by pseudomonas infection) and fatal hemorrhage at an indwelling catheter site. There were 3 deaths among the 33 individuals who participated in the 1-year prospective study.
Overall disease burden.
To evaluate MTM comorbidities, we looked at 3 parameters: wheelchair, feeding tube, and ventilator dependence (figure 1B). Forty-eight of 50 individuals required at least 1 of these interventions, and 37 required all 3, reflecting the high degree of technology dependence in MTM.
Motor functional status.
Forty-five of 50 participants were >2 years of age and thus considered of ambulatory age. Of this group, only 6 (13.3%) were ambulant. Seven of 33 in the longitudinal study had previously walked but lost the ability before survey initiation. For nonambulant individuals, current maximum motor function was as follows: 1 patients could stand without support for >10 seconds and take steps with assistance; 24 patients could sit without support for >10 seconds; and 16 individuals could not sit or stand independently (figure 1D). There were impairments in other motor skills as well: 9 of 33 patients could feed themselves, 12 of 33 could comb their hair, 14 of 47 could brush their teeth, 5 of 33 could button clothing, and 29 of 33 could pick up small objects. Twelve of 32 patients lost 1 or more of these abilities over time (figure e-1).
Twelve individuals participated in a physical examination, with 5 having 2 examinations separated by 12 months. Muscle weakness was diffuse in all, including striking facial musculature involvement and complete or near-complete ophthalmoparesis. In the 9 nonambulant individuals, extremity weakness was severe. Three had no antigravity strength, while 4 achieved antigravity strength only in distal muscles. The remaining 2 had movements against gravity and with some resistance. Overall, examination features were consistent with previous reports,2,9,11 although we additionally observed neck flexor weakness and the combination of joint hypermobility plus joint contractures in most individuals (figure e-2).
Quantitative testing included timed 10-m walk (3 of 12 individuals were able perform), the Hammersmith scale for children with spinal muscular atrophy,13 and repetitive nerve stimulation (table e-2). For the Hammersmith scale, the mean score was 11 and the median was 3, with scores largely segregated into 2 groups: <10 of 40 (9 of 12 patients) and >35 of 40 (3 of 12 patients). Repetitive nerve stimulation studies were technically limited because of submaximal stimuli in each case; however, no individual demonstrated electrodecrement. No appreciable differences in examination or quantitative testing were noted in the 5 individuals who underwent repeat evaluation.
Respiratory status.
At the time of survey completion, 48 of 50 individuals required some element of respiratory support, and 38 of 50 had tracheostomies in active use. Thirty-nine individuals used support ≥8 hours per day, with 33 requiring >16 hours of ventilation.
We investigated respiratory variability over the prospective 1-year analysis (n = 33) (figure 1C). In general, patients remained stable, with 8 having no change in ventilation requirements and 16 experiencing only minor fluctuations, related primarily to increased ventilator dependence during illness or in the immediate postoperative period (figure e-3). Three individuals had modest, sustained functional improvements lasting >3 months, while 6 required increased support.
Other medical problems and surgeries.
A full review of medical comorbidities is presented at http://phenostacks.org/AmburgeyMTM (figures e-2 and e-4) and summarized in table 1. Scoliosis was the most common comorbidity, occurring in 72% of individuals (mean age at presentation 31.5 months). In 13 cases, scoliosis worsened over time, and 10 patients reported rapid change (mean age for change 10 years 1 month) (figure e-1). Twelve individuals used spinal bracing, and 9 underwent scoliosis surgery. Of patients who developed scoliosis before 5 years of age, 8 of 22 required scoliosis surgery, as opposed to 1 of 7 with later onset.
Table 1.
Nonmuscular comorbidities in X-linked myotubular myopathy
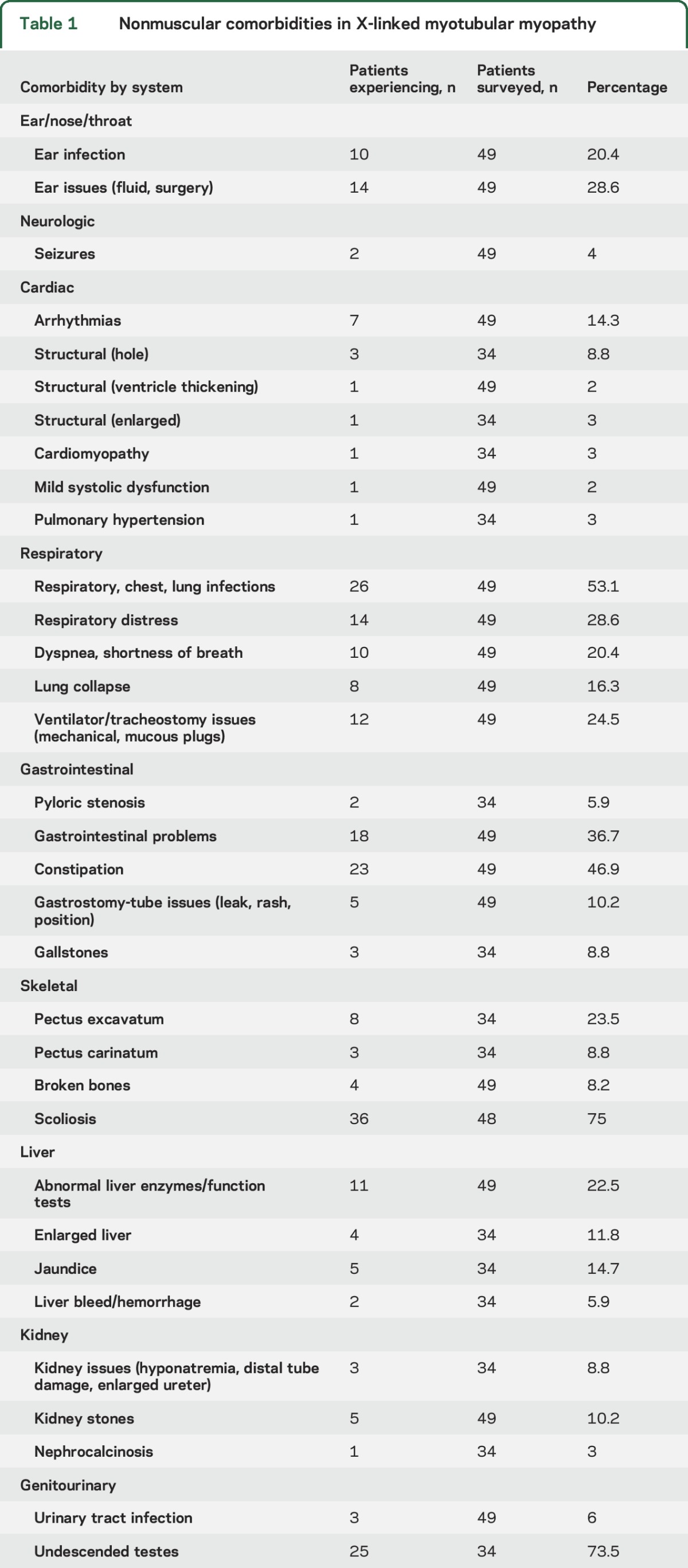
Other commonly encountered problems were acid reflux, constipation, and bone fractures. Hearing difficulties were surprisingly reported in 15 of 33 and vision difficulties in 16 of 33. Chronic/recurrent otitis media occurred frequently, as did respiratory infections. Cardiac involvement was rare, with only 1 report of cardiomyopathy, 2 reports of hypertension, and 6 reports of tachycardia. Three individuals were reported to have septal defects at birth, which is slightly higher than the general population risk (estimated at 1:2000). Liver involvement, including the feared complication of hepatic peliosis, was uncommon. We recorded 2 instances of liver hemorrhage (table 1 and figure e-1); neither was confirmed as related to peliosis (although it was suspected), and neither was fatal.
Twelve of 28 individuals were documented to have learning disability. In those who had formal neuropsychological testing (n = 10), 8 had abnormal studies, and 6 had impairments in all learning areas. Only 1 patient carried a formal diagnosis of intellectual disability. In terms of other neurobehavioral diagnoses, only 1 of 26 had attention-deficit/hyperactivity disorder. Speech abnormalities paralleled learning disabilities. The mean age for first words was 17 months and for first sentences was 29 months. Hypophonic voice was common (16 of 33), and 12 of 33 had speech articulation difficulties.
Prenatal and birth history.
Consistent with previous reports, a high degree of prenatal and perinatal disease was noted.7 Twelve of 32 mothers reported decreased fetal movements, and 19 of 33 pregnancies were complicated by polyhydramnios. Prematurity (<36 weeks) was noted in 12 of 34 births. Forty-three of 48 patients required respiratory support at birth. Hypotonia was uniformly noted. Congenital contractures were relatively common (observed in 10 of 28), although only 1 dislocation (hip) was noted. Diagnosis was achieved in all but 1 patient during the neonatal period, accomplished by muscle biopsy in 24 of 33 and by gene testing in the remainder.
In the 2002 genotype–phenotype study,7 the authors used a perinatal evaluation score (PES; determined by 6 perinatal factors) as a means of grading severity at birth and correlating severity with future outcomes. We calculated PES for our cohort (figure 2A). We observed high neonatal severity (low PES) in nearly all cases, as well as a general correlation between PES and eventual respiratory requirement, with individuals scoring in the 0% to 25% category requiring >16 hours of support. The PES poorly correlated with survival, with a similar distribution of death across all score ranges. Of note, the most mildly affected patients in our cohort had on average lower PES (more severe) than the previous study (0.5 vs 0.8 in the previous study).
Figure 2. Perinatal evaluation score (PES) and neonatal respiratory support in relation to patient outcome.

(A) Breakdown of PES and patient outcome. PES was defined by McEntagart et al.7 Individuals with scores for at least 4 of the 6 PES factors were included. Most individuals in our cohort were in the severe or moderate categories for PES (PES <3 of 6 or <50%). There was no obvious correlation between PES and need for future respiratory support and survival. PES was determined by 6 factors: delivery (normal = 1, abnormal = 0), fetal movement (normal = 1, weak or absent = 0), polyhydramnios (no = 1, yes = 0), tendon reflexes at birth (present = 1, absent = 0), joint contractures or club feet (no = 1, yes = 0), and intubation at birth (no = 1, yes = 0). All 34 had scores (0 or 1) for at least 4 of 6 factors. (B) Flow diagram of respiratory requirement at birth and patient outcome. Modeled after the flow diagram in McEntagart et al.7 Sufficient perinatal data were available for 35 individuals with MTM in the study. Note that essentially all individuals who required respiratory support at birth went on to need it after the neonatal period. While a small sample size, 2 of 3 who did not require breathing support at birth were ventilator free at study completion (ages 2 years 2 months, 32 years 10 months, and 42 years 4 months). XLMTM = X-linked myotubular myopathy.
We also looked at duration of neonatal breathing support as a predictor of disease outcomes. Twenty-nine of 32 patients required respiratory support at birth. All 3 individuals not requiring neonatal respiratory support were alive at the time of survey (figure 2B), as opposed to 9 of 29 dead in the group requiring it. There was no obvious correlation between the length of respiratory support and outcomes; equal percentages of individuals currently needing ventilator support were in the group requiring <1 week and the group needing >30 days of neonatal support.
Genotype–phenotype correlation.
MTM1 genetic testing reports were available for 35 participants, representing 27 unique mutations and 5 recurrent mutations.14 Mutations were located throughout the gene with no obvious hotspots and no clear clustering to known functional domains (figure 3).15 We examined clinical phenotype in comparison to mutation subtype (figure e-5). Sixteen of 17 individuals with nonsense mutations were in the severely impaired category (nonambulant plus mechanical respiratory support). The 1 individual with milder presentation achieved the ability to walk and did not require ventilator support; however, he died at the age of 2.5 years. Three of 10 individuals with missense mutations were ambulatory, and 4 of 10 required only noninvasive respiratory support. There was more variability among the 8 individuals with splice-site mutations, with 75% requiring a ventilator, 13% requiring noninvasive support, and 13% requiring no breathing support. Of note, scoliosis and feeding tube dependence were common across all genotypes, and survival did not obviously correlate with mutation type. Overall, our mutational analyses matched previous genotype–phenotype studies.7,16
Figure 3. Genotype–phenotype correlations.

Genetic testing results were available for 35 individuals. Twenty-seven unique mutations were identified. For genotype–phenotype assessment, mutations were categorized as nonsense (n = 17), missense (n = 10), or splice site (n = 8). Of note, 1 recurrent missense mutation (R69C in the PH-GRAM domain) has been shown to alter splicing (Pierson et al.14). (A) Mutation type and location. Mutations in MTM1 were found throughout the gene, and there was no obvious clustering in sections of the MTM1 gene coding for the known functional domains. Protein domain locations were defined by Laporte et al.15 (B) Mutation type and location in relationship to respiratory requirement. Because nearly all individuals required ventilatory support, there was no clear correlation between ventilation and mutation type, although a higher proportion of individuals with missense mutations did not need invasive support (4 of 10 missense on noninvasive support vs 0 of 17 nonsense and 1 of 8 splice site). (C) Mutation location and motor function. Other than 1 individual with a nonsense mutation, only individuals with missense mutations achieved the ability to ambulate independently (3 of 10).
Prospective evaluation of disease characteristics.
We obtained prospective data for a 1-year time period (n = 33, age range 1 year 4 months–26 years, mean age 11 years). Three individuals died during the course of the study (figure 1A). Outside of these obviously severe events, the disease course was relatively stable (figure 1C).
We focused on 3 areas that reflected disease burden for patients and families (figure 4 and table e-3). The first was hospital visits. There were 38 emergency room visits or an average of 1.15 annual visits per patient. Fourteen of 33 had 1 or 2 visits, 1 of 33 had 3 visits, and 1 of 33 had 5 visits, while 16 of 33 had none. The most common reasons for emergency room visits were infection (10 of 38), fever (8 of 38), and equipment issues (7 of 38). Of note, from our retrospective data (n = 30), the average number of emergency room visits per year during the first 2 years of life was 2.53 per patient. There were 13 emergent hospitalizations, for an average of 0.39 per individual. Infection (7 of 13) and fever (2 of 13) were the most common reasons for hospitalization. In addition, there were 27 elective procedures (0.82 per patient). Myringotomy tube placement was the most common surgery (7 of 27), followed by spinal surgery (4 of 27). Nine of 33 patients avoided hospital encounters; of the 24 patients requiring at least 1 hospital visit, 6 experienced almost half of all events (36 of 78).
Figure 4. Hospitalizations and surgeries during a 1-year prospective study of myotubular myopathy.

We tracked hospital visits (ER and admissions) and surgeries during our 1-year prospective analysis. Twenty-four of 33 individuals in the study required at least 1 of these services (pie chart, top left). There were 38 ER visits (pie chart, top right, listed by chief complaint), with fever and infection the most common reasons. Infections included pneumonia, pseudomonas, enterovirus, and rhinovirus. aRespiratory issues included respiratory distress, lung collapse, and high CO2 levels. bEquipment issues included ventilator malfunction, mucous plugs, and leaky gastrostomy tubes. cOther reasons for ER visits included drug allergy, power chair accident, postsurgical hip pain, and inability to urinate. There were 13 emergent hospitalizations (i.e., not scheduled or elective admissions), with infection the overwhelmingly most common reason for admission (pie chart, bottom right). There were 27 elective surgical procedures reported, with ear tube placement (i.e., myringotomy tube placement) the most common surgery. ER = emergency room.
Next, we examined number of school days missed (n = 21 school-aged individuals, with 14 attending traditional school and 7 home school). The total number of missed days was 476, an average of 22.7 per eligible patient. This was not distributed uniformly; 4 patients missed >50 days (range 53–91, all secondary to illness or postsurgical recovery). Forty-eight percent of patients with MTM missed >12 days of school compared to the US average of 6% overall and 14.9% for children with special care needs (www.qualitymeasures.ahrq.gov/summaries/summary/47477).
The third parameter measured was number of workdays missed. Four hundred thirteen days of work were missed, averaging 1 d/mo per family (12.52 d/y). This contrasts with the US average, which is 4 d/y lost to childcare. The most common reasons for missing work were lack of nursing coverage and medical appointments.
DISCUSSION
This comprehensive MTM natural history study enables new assessment of clinical care parameters, improved prognostication, and evaluation of potential clinical trial outcome measures.
Perhaps our most important observation is that survival in MTM is increased over previous reports. We report deaths in 12 of 50 participants, with an average age of 6 years 10 months and 83% of deaths occurring before age 9. This contrasts with previous studies describing up to 50% of patients dying in the first year of life and 90% dying by age 10.2,7 This was not due to lack of severity in our cohort because 88% were nonambulant and required >8 hours of ventilatory support. One caveat is that our study potentially underascertained cases associated with neonatal death (and MTM infants in general) because parents in this setting may be less likely to participate in a study such as ours. Two other considerations are that our cohort is composed of patients in North America (where care decisions for critically ill neonates can be different from those in other continents) and that neuromuscular respiratory care has advanced compared to 2002. True determination of survival in MTM requires a prospective study in which all patients are enrolled and followed up from diagnosis.
Our data predict an approximate annual death rate in patients with MTM of 10%. This agrees with the general examination of our full cohort, in which we observed a linear death rate in the first decade of life, although with a relative increase in infants. In the second decade and beyond, however, the death rate appears lower, although our numbers are comparatively small for these age ranges.
The improved survival in our MTM cohort does not reflect a reduction in overall disease severity. Only 2 individuals had no technology requirements, and most (75%) needed invasive care in essentially all domains. This high disease burden was echoed by a sizeable effect on school and work life; students with MTM missed nearly 1 month of school on average, and families missed at least 1 day of work per month. Of note, despite the fact that most patients with MTM share a similar extent of disability, the effect on school/work was not uniformly distributed, with a few individuals missing the majority of school days. Additional investigation is needed to understand why these differences were observed, although most frequently the long school absences were associated with medical complications.
A second important observation is that the disease course appears relatively stable, as reflected by the low rate of emergency room and hospital visits. This was surprising to us, given that most individuals with MTM have extensive respiratory requirements and are considered medically fragile. One potential explanation is the skill (and resources) of parents and caregivers in terms of managing respiratory difficulties, a factor that may be skewed in our cohort (which relied on families who could attend family conferences or participate in monthly phone surveys).
Another important observation is that the incidence of “nonmuscle” comorbidities was lower than predicted by previous reports.8 In particular, we observed only a single instance of suspected hepatic peliosis in our retrospective cohort (with 1 additional incident of liver hemorrhage during the prospective survey) and no other severe medical issues not related to infection or respiratory exacerbation. While there is little doubt that nonmuscle problems occur in MTM, our data suggest that they are relatively uncommon. As with survival, determining the true incidence of issues such as hepatic peliosis requires a long-term prospective longitudinal study. In addition, because these events may be age dependent, fully understanding their prevalence may need evaluation of a larger cohort of older patients.
One surprising morbidity was learning impairment, with 43% of patients reported to have a learning disability. A contributor to this problem is likely impaired speech mechanics and the potentially high risk for perinatal hypoxia. Whether there additionally is a primary cognitive component to the disease is harder to know. Myotubularin is expressed not only in muscle but also in many tissues, including the brain. However, no CNS abnormalities have been reported in MTM animal models, although they have not been rigorously investigated. Future study is necessary to sort out this potentially important issue.
Our other major goal was to identify disease characteristics suitable for application as outcome measures. We achieved this in part, but primarily by identifying characteristics unlikely to be suitable for clinical trials. For example, the Hammersmith scale gave both floor and ceiling effects, with the cohort basically divided between extremes of very low and very high scores. Alternative motor scales such as the Hammersmith Function Motor Scale Expanded (modified for ambulant patients with spinal muscular atrophy) or the Motor Functional Measure 20/32 (used for many neuromuscular diseases and enables separation of skills into domains) could provide better discrimination in MTM and thus be more appropriate for future studies. In addition, events such as emergency room visits and hospitalizations were lower than expected and thus likely not suitable; the same could be said for survival. In contrast, respiratory status appears promising as an outcome measure. Most patients with MTM require some level of intervention that is relatively unchanging over a 1-year period. Our data could thus enable estimation of sample size in treatment studies targeted at reducing ventilator dependence, a clinically meaningful primary outcome.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Elizabeth DeChene for valuable assistance with recruitment and data collection. They also thank the University of Michigan study team for assistance with patient enrollment and data collection and acknowledge Valerion Therapeutics for providing a grant-in-aid and for assisting with data collation and statistical analysis. Mary Britt (University of Michigan), Kimberly Creamer (Memorial Healthcare), Elizabeth DeChene (Children's Hospital of Philadelphia), Elizabeth Howell (University of Michigan), and Teresa Spiegelberg (University of Michigan) assisted with data collection. Janet Young (Cure CMD volunteer) provided clerical assistance. Daniel Wigdor (University of Toronto) and Azam Khan (Autodesk Research) are Michael Glueck’s supervisors. Christopher Fitzpatrick (Valerion) assisted with statistical analysis, and Hal Landy (Valerion) assisted with manuscript editing.
GLOSSARY
- MTM
myotubular myopathy
- MTM1
myotubularin
- PES
perinatal evaluation score
Footnotes
Supplemental data at Neurology.org
Editorial, page 1316
AUTHOR CONTRIBUTIONS
Kimberly Amburgey: study concept and design, acquisition, analysis, and interpretation of data, writing, and critical revision of manuscript for intellectual content. Etsuko Tsuchiya: analysis and interpretation of data, writing and critical revision of manuscript for intellectual content. Sabine de Chastonay: adverse event study concept and design, acquisition, execution. Michael Glueck: analysis and interpretation of data, visualization of data. Rachel Alverez: adverse event study concept and design, acquisition, execution. Cam-Tu Nguyen: acquisition, analysis, and interpretation of data. Anne Rutkowski and Joseph Hornyak: study concept and design. Alan H. Beggs: study design and critical revision of manuscript for intellectual content. James J. Dowling, study concept and design, study supervision, acquisition and interpretation of data, writing, and critical revision of manuscript for intellectual content.
STUDY FUNDING
This study was funded by contributions from Valerion Therapuetics and support from the Joshua Frase Foundation and Cure CMD. A.H.B. received grant support from the Muscular Dystrophy Association of USA (MDA) grant MDA383249, NIH grant R01 AR044345 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Where There's a Will There's a Cure, and the Frase Foundation. J.J.D. received support for the study from the University of Michigan and the Hospital for Sick Children, as well as salary support from an MDA development grant and an NIH NIAMS K08 award (K08AR054835).
DISCLOSURE
K. Amburgey, E. Tsuchiya, S. de Chastonay, M. Glueck, R. Alverez, C. Nguyen, A. Rutkowski, and J. Hornyak report no disclosures relevant to the manuscript. A. Beggs is on the scientific advisory board of Audentes Therapeutics, a biotechnology company developing molecular therapies for MTM. J. Dowling is on the scientific advisory boards of the RYR1 Foundation and the MTM/Centronuclear myopathies patient registry. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet 1996;13:175–182. [DOI] [PubMed] [Google Scholar]
- 2.Das S, Dowling J, Pierson CR. X-linked centronuclear myopathy. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993–2017. [Google Scholar]
- 3.Childers MK, Joubert R, Poulard K, et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med 2014;6:220ra210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawlor MW, Armstrong D, Viola MG, et al. Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X-linked myotubular myopathy. Hum Mol Genet 2013;22:1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cowling BS, Chevremont T, Prokic I, et al. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest 2014;124:1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabha N, Volpatti JR, Gonorazky H, et al. PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J Clin Invest 2016;126:3613–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McEntagart M, Parsons G, Buj-Bello A, et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord 2002;12:939–946. [DOI] [PubMed] [Google Scholar]
- 8.Herman GE, Finegold M, Zhao W, de Gouyon B, Metzenberg A. Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr 1999;134:206–214. [DOI] [PubMed] [Google Scholar]
- 9.Abath Neto O, Silva MR, Martins Cde A, et al. A study of a cohort of X-linked myotubular myopathy at the clinical, histologic, and genetic levels. Pediatr Neurol 2016;58:107–112. [DOI] [PubMed] [Google Scholar]
- 10.Lee EH, Yum MS, Park SJ, et al. Two cases of X-linked myotubular myopathy with novel MTM1 mutations. J Clin Neurol 2013;9:57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colombo I, Scoto M, Manzur AY, et al. Congenital myopathies: natural history of a large pediatric cohort. Neurology 2015;84:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang CH, Dowling JJ, North K, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol 2012;27:363–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Main M, Kairon H, Mercuri E, Muntoni F. The Hammersmith Functional Motor Scale for children with spinal muscular atrophy: a scale to test ability and monitor progress in children with limited ambulation. Eur J Paediatr Neurol 2003;7:155–159. [DOI] [PubMed] [Google Scholar]
- 14.Pierson CR, Dulin-Smith AN, Durban AN, et al. Modeling the human MTM1 p.R69C mutation in murine Mtm1 results in exon 4 skipping and a less severe myotubular myopathy phenotype. Hum Mol Genet 2012;21:811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laporte J, Blondeau F, Gansmuller A, Lutz Y, Vonesch JL, Mandel JL. The PtdIns3P phosphatase myotubularin is a cytoplasmic protein that also localizes to Rac1-inducible plasma membrane ruffles. J Cell Sci 2002;115:3105–3117. [DOI] [PubMed] [Google Scholar]
- 16.Fattori F, Maggi L, Bruno C, et al. Centronuclear myopathies: genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. J Neurol 2015;262:1728–1740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.