

Abstract
Objective:
To describe long-term outcomes of children with early-infantile Krabbe disease who underwent hematopoietic stem cell transplantation (HSCT) in the first 7 weeks of life.
Methods:
In this prospective longitudinal study, evaluations performed at baseline and follow-up included brain imaging, neurodiagnostic tests, and neurobehavioral evaluations.
Results:
Of the 18 patients in this study (11 girls, 7 boys; mean follow-up 9.5 years, range 4–15), 5 died (3 of peritransplant complications, 1 of a surgical complication unrelated to Krabbe disease, 1 of disease progression). One of the surviving patients has normal cognitive function and 10 continue to develop cognitive skills at a slightly slower rate than normal. All surviving patients continue to gain receptive language skills, with 7 falling within the normal range. Ten patients receive speech therapy, and 2 of these patients require augmentative communication devices. Gross motor development varies widely, but 3 patients can walk independently, and 7 walk with assistive devices. Spasticity ranges from mild to severe, and 12 patients wear orthotics. Fine motor skills are generally preserved. Brain myelination and atrophy stabilized in 8 patients, improved in 4 patients, and worsened in 1 patient. Nerve conduction velocities initially improved but continue to be abnormal in most patients.
Conclusions:
The surviving patients function at a much higher level than untreated children or symptomatic children who underwent HSCT. These results show that early HSCT changes the natural history of this disease by improving both lifespan and functional abilities.
Classification of evidence:
This study provides Class IV evidence that for children with early-infantile Krabbe disease, early HSCT improves lifespan and functional abilities.
Krabbe disease (globoid cell leukodystrophy) is an autosomal recessive neurodegenerative disease caused by a deficiency of galactocerebrosidase (GALC).1 GALC deficiency results in an excess of the substrate psychosine, which is toxic to myelin-generating cells.1 Krabbe disease is characterized by progressive central and peripheral demyelination and can present as early-infantile, late-infantile, juvenile, or adult disease. Early-infantile disease is the most commonly identified form, with an estimated incidence of 1 in 100,000–394,000 births in the United States.1,2
Early-infantile Krabbe disease predominantly affects the corticospinal tracts and typically presents before 6 months of age with irritability, dystonia, and feeding difficulties. The diagnosis is made by measuring GALC activity in leukocytes or skin fibroblasts and mutation analysis.3 However, more than 140 disease-causing GALC mutations have been reported.1 In addition, genetic background can influence phenotype, and common polymorphisms influence enzyme activity; thus, GALC activity does not always correlate with disease severity.1
Umbilical cord blood transplantation (UCBT) has been shown to significantly improve neurologic outcome in asymptomatic neonates. However, when transplantation is performed in symptomatic patients with early-infantile disease, the neurologic insult remains severe.4 Although almost all treated newborns have normal cognitive function in the short term, motor outcome ranges from mild to severe disability.4 The purpose of this article is to summarize long-term neurodevelopmental outcomes of 18 children with early-infantile Krabbe disease who were transplanted in the first 7 weeks of life.
METHODS
Study population and study design.
The study cohort included all babies with Krabbe disease referred for evaluation at the Program for the Study of Neurodevelopment in Rare Disorders from January 2000 to September 2011 who had received or were being considered for hematopoietic stem cell transplantation (HSCT). These babies were detected by newborn screening or diagnosed prenatally or neonatally because of family history. Inclusion criteria were (1) diagnosis of early-infantile Krabbe disease established by family history or deficiency of GALC activity in peripheral blood leukocytes and (2) HSCT within 7 weeks after birth. Genotype information was collected when available; however, mutational analysis was not a standard part of the diagnostic workup for Krabbe disease at the time most of these patients received diagnoses.
Baseline and follow-up examinations consisted of brain imaging; measurement of CSF protein, peripheral nerve conduction velocity (NCV), and visual and brainstem auditory evoked responses; and assessment of motor, language, cognitive, and adaptive skills using standardized neurobehavioral protocols, as previously reported.4 We reviewed patient records to evaluate growth, independent behavior, and educational needs of patients older than 2 years at their last evaluation. The results of baseline studies and short-term developmental outcomes of 11 of these patients were reported previously.4
Standard protocol approvals, registrations, and patient consents.
The studies were approved by the institutional review boards at the University of Pittsburgh (PRO11050036) and University of North Carolina, Chapel Hill (UNC-CH 08-0237). Informed consent was obtained for all patients evaluated at the University of Pittsburgh. Patients evaluated at UNC-CH either gave informed consent or were included under a waiver granted by UNC-CH.
Neurologic measures.
Flash visual evoked potentials were considered abnormal if the P100 wave was absent. Brainstem auditory evoked responses were considered abnormal if wave I–V interpeak latencies were prolonged or any of the obligate waveforms (I, III, or V) were absent. NCVs were considered abnormal if they showed prolongation of distal and F-wave latencies, low amplitude, or no evoked response. CSF protein was collected by lumbar puncture and measured according to institutional guidelines.
Statistical methods.
The probability of survival was calculated by Kaplan-Meier analysis5 using the cutoff date of October 1, 2015. Descriptive statistics were calculated for clinical and neurodevelopmental outcomes. Age at transplantation was examined as a predictor of long-term neurocognitive and clinical outcomes. For neurocognitive outcomes (cognition, speech/language, motor function, and adaptive behavior), a linear mixed model6 was used to estimate growth trajectories based on the patient's age at testing and age at transplantation. In each of these models, the age-equivalent score was the outcome, and age at transplantation and age at testing were predictors. Random effects were estimated for the slope in the linear model. Fixed effects estimates were used to test for the effect of age at transplantation on the neurocognitive trajectories. This study provides Class IV evidence that for children with early-infantile Krabbe disease early HSCT improves lifespan and functional abilities.
RESULTS
Patient characteristics.
Eighteen patients met the inclusion criteria and were evaluated by our program between December 1999 and October 2015. All of the patients (11 girls, 7 boys) were Caucasian, and 3 were Hispanic. Most originated from the Eastern and Midwestern United States and Eastern Canada, representing at least 8 states and 1 province. These patients underwent HSCT between October 1999 and October 2011. Mean follow-up was 9.5 years (range 4–15). Fifteen patients were identified prenatally/neonatally through family history, and 3 were identified by state newborn screening programs. Fifteen children were evaluated by our clinic before receiving HSCT, and 3 were referred and evaluated 3 months–3.3 years after receiving HSCT.
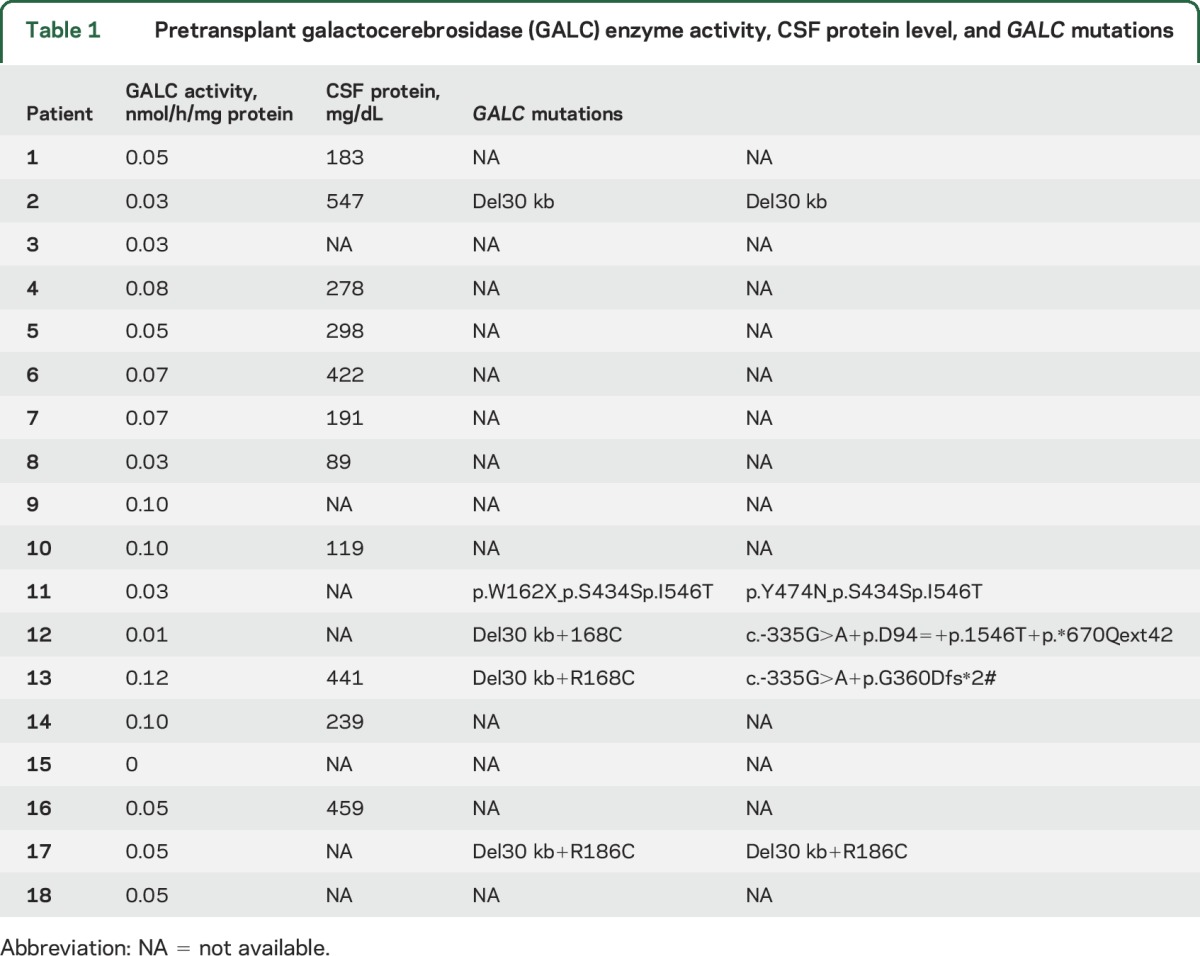
All pretransplant GALC levels were <0.12 nmol/h/mg protein (range 0.0–0.12, mean 0.06, SD 0.03), and pretransplant CSF protein levels ranged from 89 to 547 mg/dL (table 1). Pretransplant GALC genotype information was available for 3 patients identified through newborn screening and 2 of the 15 patients identified through family history. Four of these patients carried the common 30-kb deletion (c.1161+6532_polyA+9kbdel), which is considered a severe mutation. Performing mutational analysis at this time in the remaining surviving patients would be technically challenging, since the genotypes of their blood cells currently reflect those of their hematopoietic stem cell donors.
Table 1.
Pretransplant galactocerebrosidase (GALC) enzyme activity, CSF protein level, and GALC mutations
Survival and engraftment.
Sixteen patients received unrelated UCBT, and 2 patients received bone marrow transplantation. All patients continue to maintain their graft. Thirteen patients are currently surviving. Three patients died of complications during the peritransplant period, 1 patient died of a surgical complication unrelated to Krabbe disease 6 years posttransplant, and 1 patient died of disease progression 15 years posttransplant (table 2). Median overall survival posttransplant was 10.71 years (range 0.09–16.2 years) (figure 1). Among the 15 patients who survived the peritransplant period, median survival posttransplant was 11.3 years (range 5.4–16.2). All patients had normal GALC enzyme activity posttransplant.
Table 2.
Posttransplant patient characteristics, galactocerebrosidase (GALC) enzyme activity, CSF protein level, and treatment outcomes
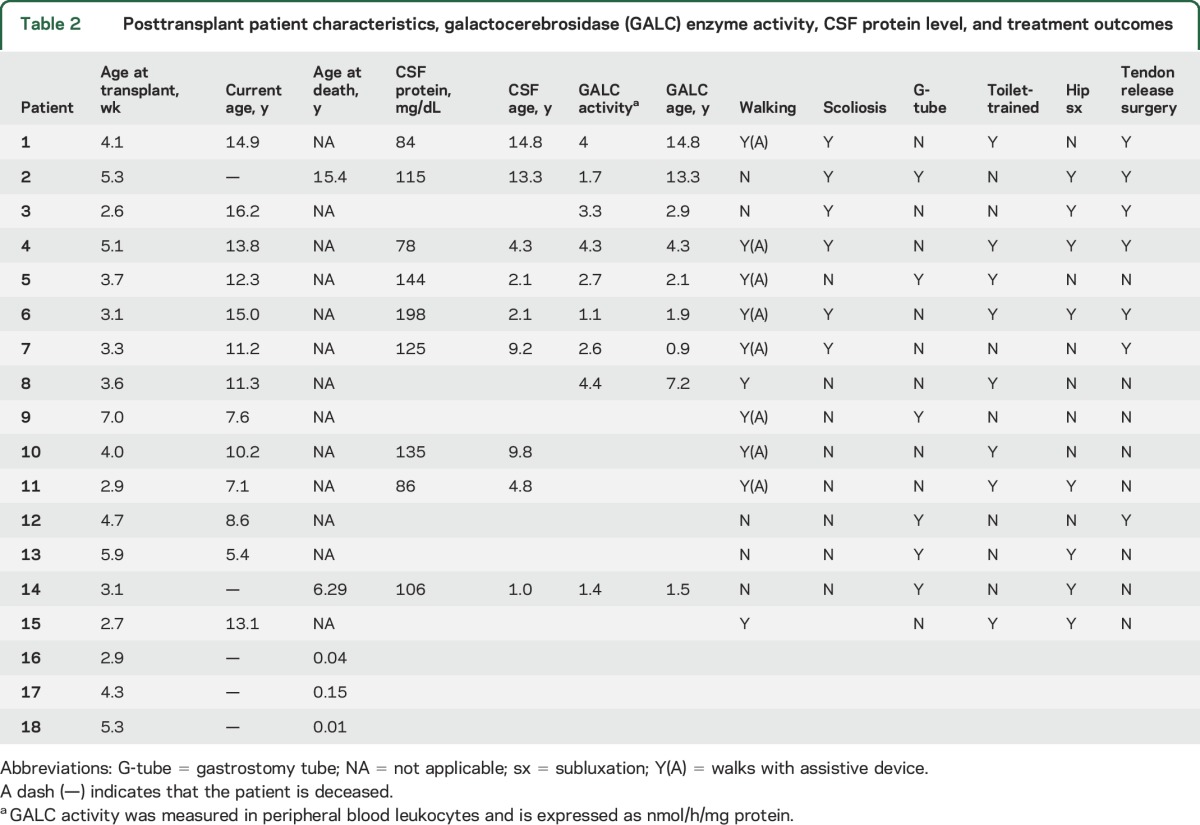
Figure 1. Survival of children with early-infantile Krabbe disease after hematopoietic stem cell transplantation.

Kaplan-Meier curve of survival for 18 children who underwent hematopoietic stem cell transplantation for infantile Krabbe disease before 7 weeks of age.
Neurodevelopmental outcomes.
Follow-up data were available for at least 2 years posttransplantation for all 15 patients who survived the peritransplant period (median follow-up evaluations 8, range 1–21).
Growth and nutrition.
Twelve of the 15 children with available data were <5th percentile for weight (figure 2, A and B); however, 8 of these children maintain a normal body mass index, remaining between the 5th and 97th percentiles. Graphs showing height and head circumference growth are provided in figure e-1, A–D, at Neurology.org. All babies had feeding difficulties during the neonatal period, ranging from weak suck to uncoordinated suck and swallow, or slow feeding. Six of the 15 children had gastrostomy tubes at their last evaluation, but 12 were able to eat by mouth. Three used the gastrostomy tube only for nutritional supplementation and because they ate very slowly.
Figure 2. Growth after hematopoietic stem cell transplantation for children with early-infantile Krabbe disease.

Growth charts show weight gain for the (A) 6 boys and (B) 10 girls with follow-up data after hematopoietic stem cell transplantation before 7 weeks of age. The inset in the upper left corner of each graph magnifies and highlights growth during the first 24 months of age. The red lines represent individual patients. The gray and blue lines represent standard growth curves (blue = 50th percentile, gray lines = 3rd, 5th, 10th, 25th, 75th, 90th, 95th, and 97th percentiles). Additional graphs are included in the supplemental material.
Sensory function.
Ten patients wear corrective lenses for strabismus, astigmatism, or myopia. One patient has severe cerebral visual impairment. Visual evoked potentials were available for 11 patients at 1.7–15.4 years posttransplantation (median 6.9). Normal visual evoked potentials were demonstrated by 7 children at 2–14.7 years posttransplant.
Six of the 13 patients with available data had abnormal brainstem auditory evoked responses but normal behavioral audiometry, a phenomenon that has been reported in auditory neuropathy and brainstem dysfunction.7 Although responses were normal, the rate of response to audiometric stimuli was slower than that of typical children, which should be taken into account during testing for accurate results. None of the patients had conductive hearing loss.
Cognitive development.
Evaluating cognitive function in infantile Krabbe disease is challenging because motor difficulties affect test performance. Figure 3A, which presents the individual developmental trajectory for each patient, shows that one patient has developed normally, with scores around the 50th percentile. Ten surviving children continue to develop skills at a rate that is slightly slower than that of normally developing children, and 2 children show a plateauing of skills below the developmental age of 2 years. All patients except for the patient developing normally receive special education services.
Figure 3. Development after hematopoietic stem cell transplantation for early-infantile Krabbe disease before 7 weeks of age.
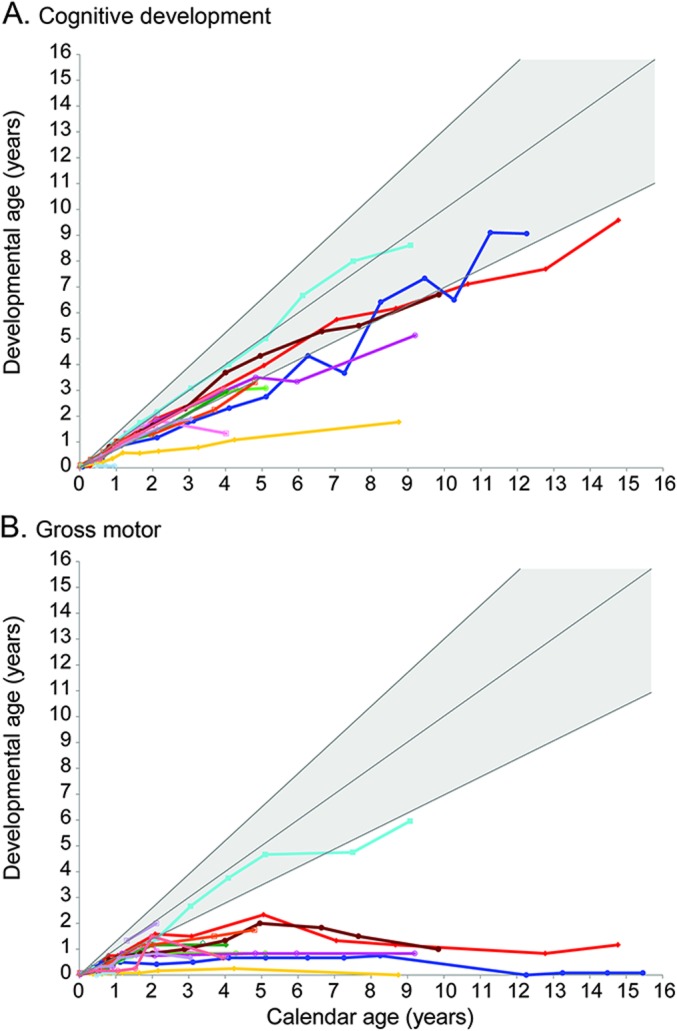
For the 15 children with follow-up data, we used age-equivalent scores (i.e., developmental age) for (A) cognitive development and (B) gross motor function to allow comparisons across tests and monitor development over time. The colored lines represent individual patients. The shaded gray area represents typical development. Additional graphs showing adaptive behavior, receptive language, expressive language, and fine motor function are included in the supplemental material.
Adaptive behavior.
Only 2 of 14 patients exhibited an age-appropriate adaptive behavior at the last evaluation (figure e-2B). Six of the remaining children feed themselves, 2 dress independently, and 1 assists with dressing. Of the 8 children who are toilet trained, 3 can walk independently to the toilet, but 5 require assistance (table 2). Only 2 patients reported sleeping problems (i.e., sleep apnea).
Speech and language.
Receptive language is the least affected developmental domain. Seven of the 13 surviving children show language development in the normal range, and the remaining 6 patients continue to gain skills (figure e-2C). Expressive language scores are similar (figure e-2D) but are more variable due to abnormalities in muscle tone in the tongue and a very high-arched palate in 12 children. Ten patients receive speech therapy for articulation difficulties (mild dyspraxia to severe apraxia); however, use of augmentative communication devices was reported for only 2 patients.
Motor development and musculoskeletal problems.
Most patients continue to have a wide range of motor disability, from slightly abnormal gait to severely impaired ambulation (figure 3B). Three patients are able to walk independently, 7 walk with assistive devices, and 5 are unable to walk (table 2). All of the 13 children with follow-up motor examinations developed spasticity ranging from very mild (not affecting function) to severe, muscle atrophy, and contractures. Twelve patients wear orthotics. As a complication of spasticity, 7 children developed hip subluxation. Six patients developed mild to severe scoliosis, 6 developed kyphosis, 3 developed lordosis, and 2 children have all 3 conditions. Six patients required tendon release surgery, and 4 had fractures due to osteopenia. All patients have received physical therapy at some point. One patient who initially required physical therapy advanced to average abilities within 2 years posttransplant. The rest continue to receive physical therapy to improve range of motion and for equipment adaptations to improve gait. One patient who did not have standardized motor assessments on follow-up shows normal motor function and is able to run and jump.
Fine motor skills are generally preserved (figure e-2A); however, some patients developed truncal weakness and a tendency to internally pronate their arms, which affects distal finger movements and the ability to manipulate objects. Interventions to stabilize the trunk and arms are required in 12 children. Although the children are able to use their hands and fingers, 12 receive occupational therapy focusing on the use of adaptive equipment.
Additional neurodiagnostic testing.
Brain MRI.
Myelination and atrophy were evaluated by brain MRI in 13 patients. In 8 patients, the disease remained stable, with no new signs of demyelination, and in 2 patients with mildly abnormal findings at baseline, brain scans were subsequently read as normal. Two other patients' brain scans improved, and in 1 patient a possible new area of concern in the corticospinal tract was found 8 years after transplantation; this patient also developed mild brain atrophy. The degree of atrophy remained stable in 11 patients and worsened in only 1 patient, who was being treated with steroids for chronic graft-versus-host disease.
Nerve conduction velocities.
NCVs continued to be abnormal in 12 patients. In 9 of the 11 patients with longitudinal follow-up data, NCVs improved during the first year after transplantation but then worsened over the following 1–3 years. In one patient, NCVs worsened after transplantation with no temporary improvement, and in the remaining patient, who had mild peripheral nerve disease, NCVs normalized for 1 year and then became mildly abnormal 6 years later.
CSF protein.
In all patients CSF protein concentration decreased after transplantation but remained elevated 1–8 years posttransplant (mean 127 mg/dL, range 74–173 mg/dL), decreasing with time.
EEG.
Five of the 11 patients tested had normal EEG results. Only 2 had electrographic abnormalities and clinical seizures that required medical treatment. In one case, treatment was tapered, and although electrographic abnormalities continued, the patient did not continue to have clinical seizures.
Relationship between age at transplantation and outcomes.
Mixed models were used to test for associations between age at transplantation and neurocognitive outcomes. Although age at transplantation negatively correlated with measure estimates for all 6 domains, the correlations were significant only for gross motor (β = −0.13; p = 0.027) and expressive language (β = −0.16; p = 0.029) (full results of the regression models are reported in table e-1). An examination of effect sizes showed that domains requiring motor involvement were more closely associated with age at transplantation.
DISCUSSION
This prospective study used longitudinal, standardized assessments of function to describe the outcomes of patients with early-infantile Krabbe disease who received unrelated HSCT within the first 7 weeks of life. Comprehensive longitudinal follow-up of 15 of these babies diagnosed early because of family history or newborn screening allowed us to describe in detail their long-term outcomes. The surviving children function at a much higher level than untreated patients or any of the symptomatic children who underwent HSCT and were previously reported by our group.4 The school-age children continue to learn and improve cognitive skills when given appropriate supports for motor disabilities. The degree of motor function in these children varies from average ability to severe disability: 2 children are developing in an age-appropriate manner and are able to run and jump, whereas other children require power wheelchairs in school and augmentative communication devices to be understood by others. This variability in motor function can be explained by several factors. One, the degree of corticospinal tract involvement at birth differs among patients,8,9 and our results may represent the sequelae of this early damage. Two, peripheral nerve disease initially improves after transplantation but subsequently deteriorates with time,10 causing severe muscular atrophy and scoliosis. Thus, motor disease in treated children varies according to the degree of brain and peripheral nerve involvement at the time of transplantation.
When considering those patients who responded best to treatment, it is evident that treatment with HSCT has allowed them to live relatively normal lives up to the early teen years. Our results show that age at transplantation accounts for some of the variability in long-term outcomes. We have reported pretransplant GALC genotype and enzyme activity of these patients, when available, but their roles in treatment outcome are unclear. Thus further research is needed to better understand the interplay among GALC mutations, enzyme activity, pathophysiology, and mechanisms underlying improvement after HSCT.
Our results can provide further guidance to those evaluating asymptomatic patients identified through newborn screening or family history regarding expected outcomes after transplant. HSCT is an aggressive treatment for a devastating disease, where treatment outcomes have proven to be significantly better than if children are left untreated. Despite failing to cure Krabbe disease, HSCT performed before the onset of severe symptoms delays disease progression and improves both length and quality of life. The clinically and statistically significant associations between age at transplantation and expressive language and gross motor function highlight the importance of transplantation as soon as possible for children with early-infantile disease. To achieve these outcomes, state newborn screening programs must be designed to diagnose and refer for treatment. Ideally, a child with early-infantile disease should begin the transplant procedure before 2 weeks of age.
This study reports only on children with the most severe form of Krabbe disease, expected to become symptomatic before 6 months of age. Therefore, they may represent the worst possible outcomes for the treatment of Krabbe disease. In patients with later-onset forms of Krabbe disease, the effects of transplantation outcomes are expected to be much better since brain myelination slows after 6 months, especially in the corticospinal tracts; therefore, damage to these tracts can be prevented. In addition, peripheral nerve disease is less severe in later-onset forms; therefore, treatment may prevent peripheral nerve disease for many years. Thus, given the significantly improved outcomes for babies with early-infantile Krabbe disease, we expect a better response for those with the late-infantile, juvenile, and adult forms.11–13 However, patients with later-onset disease must also treated as soon as possible and before obvious symptoms develop, because outcomes will be directly related to the degree of neurologic compromise at the time of treatment.
Recent advances in HSCT including reduced-intensity chemotherapy (RIC) may improve long-term outcomes. Only one of the patients in this cohort received RIC before transplant. Additional patients who have received RIC have not been followed posttransplant long enough to report on long-term outcomes; however, peritransplant morbidity and mortality are significantly reduced. This less toxic regimen is safer but not yet available in other centers.14
A great deal of attention has been placed on the rapid referral of newborns identified through newborn screening. Patients at high or moderate risk for infantile Krabbe disease should be referred to specialized centers for evaluation since clinical expertise is needed to recognize the subtle signs of disease, provide optimal counseling, and achieve the best possible outcomes. Counseling should include providing information about the increased risks of transplantation if the procedure is performed in inexperienced centers or if the child is older and more symptomatic. Families should be made aware that HSCT does not cure Krabbe disease but delays for many years the progression of motor disease. They should also be informed about the likelihood of long-term motor and speech difficulties and need for special services.
Continued follow-up studies are needed to monitor the long-term outcomes of treated patients as they age into adolescence and young adulthood. It will also be critical to identify a presymptomatic diagnostic biomarker that will enable clinicians to confidently follow babies at moderate and high risk for Krabbe disease identified through newborn screening programs and those treated with HSCT or any other future therapies.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the families who traveled from other states to receive clinical services at the NDRD Program; the physicians, nurses, and clinical staff at UNC-CH; and transplant physicians, in particular Dr. Joanne Kurtzberg (Duke University), Dr. Paul Szabolcs (University of Pittsburgh), and Dr. Paul Orchard (University of Minnesota).
GLOSSARY
- GALC
galactocerebrosidase
- HSCT
hematopoietic stem cell transplantation
- NCV
nerve conduction velocity
- RIC
reduced-intensity chemotherapy
- UCBT
umbilical cord blood transplantation
- UNC-CH
University of North Carolina, Chapel Hill
Footnotes
Supplemental data at Neurology.org
Editorial, page 1318
AUTHOR CONTRIBUTIONS
M.W. drafted a portion of the manuscript. M.P. analyzed data and drafted a portion of the manuscript. A.D. and S.H. contributed to the acquisition and analysis of data. M.E. was responsible for conception and design of the study, detailed editing, and acquisition of data.
STUDY FUNDING
Partially supported by The Legacy of Angels Foundation and NIH/NINDS 1R01NS061965-01.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wenger DA, Escolar ML, Luzi P, Rafi MA. Krabbe disease (globoid cell leukodystrophy). In: Valle D, Beaudet AL, Vogelstein B, et al., editors. Scriver's the Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). New York: McGraw-Hill; 2013. [Google Scholar]
- 2.Orsini JJ, Kay DM, Saavedra-Matiz CA, et al. Newborn screening for Krabbe disease in New York State: the first eight years' experience. Genet Med 2016;18:239–248. [DOI] [PubMed] [Google Scholar]
- 3.Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab 2000;70:1–9. [DOI] [PubMed] [Google Scholar]
- 4.Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med 2005;352:2069–2081. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Statist Assn 1958;53:457–481. [Google Scholar]
- 6.SAS Institute Inc. SAS/STAT 9.2 User's Guide, the Mixed Procedure. Cary: SAS Institute Inc; 2008. [Google Scholar]
- 7.Kraus N, Ozdamar O, Stein L, Reed N. Absent auditory brain stem response: peripheral hearing loss or brain stem dysfunction? Laryngoscope 1984;94:400–406. [DOI] [PubMed] [Google Scholar]
- 8.Escolar ML, Poe MD, Smith JK, et al. Diffusion tensor imaging detects abnormalities in the corticospinal tracts of neonates with infantile Krabbe disease. AJNR Am J Neuroradiol 2009;30:1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta A, Poe MD, Styner MA, Panigrahy A, Escolar ML. Regional differences in fiber tractography predict neurodevelopmental outcomes in neonates with infantile Krabbe disease. Neuroimage Clin 2014;7:792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siddiqi ZA, Sanders DB, Massey JM. Peripheral neuropathy in Krabbe disease: effect of hematopoietic stem cell transplantation. Neurology 2006;67:268–272. [DOI] [PubMed] [Google Scholar]
- 11.Krivit W, Shapiro EG, Peters C, et al. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N Engl J Med 1998;338:1119–1126. [DOI] [PubMed] [Google Scholar]
- 12.Lim ZY, Ho AY, Abrahams S, et al. Sustained neurological improvement following reduced-intensity conditioning allogeneic haematopoietic stem cell transplantation for late-onset Krabbe disease. Bone Marrow Transpl 2008;41:831–832. [DOI] [PubMed] [Google Scholar]
- 13.Sharp ME, Laule C, Nantel S, et al. Stem cell transplantation for adult-onset Krabbe disease: report of a case. JIMD Rep 2013;10:57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vander Lugt M, Chen X, Chong HJ, et al. Excellent outcomes using reduced-intensity conditioning for patients with inborn errors of immunity, hematopoiesis, and metabolism. Biol Blood Marrow Transpl 2016;22:S100–S101. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.