

Abstract
Objective:
To analyze gastrointestinal (GI) manifestations, their progression over time, and medications being used to treat GI symptoms in a large cohort of patients with myotonic dystrophy types 1 (DM1) and 2 (DM2).
Methods:
We analyzed patient-reported data and medical records in a national registry cohort at baseline and 5 years.
Results:
At baseline, the majority of patients reported trouble swallowing in DM1 (55%; n = 499 of 913) and constipation in DM2 (53%; n = 96 of 180). Cholecystectomy occurred in 16.5% of patients with DM1 and 12.8% of patients with DM2, on average before 45 years of age. The use of medications indicated for gastroesophageal reflux disease was reported by 22.5% of DM1 and 18.9% of patients with DM2. Greater risk of a GI manifestation was associated with higher body mass index and longer disease duration in DM1 and female sex in DM2. At the 5-year follow-up, the most common new manifestations were trouble swallowing in patients with DM1 and constipation in patients with DM2.
Conclusions:
GI manifestations were common in both DM1 and DM2, with a relatively high frequency of gallbladder removal in DM1 and DM2 occurring at a younger age compared to normative data in the literature. Studies are needed to determine the pathomechanism of how sex, weight gain, and duration of disease contribute to GI manifestations and how these manifestations affect quality of life and clinical care for patients with DM1 and DM2.
The myotonic dystrophies (DMs) are dominantly inherited disorders with multisystem manifestations.1 DM type 1 (DM1) results from a trinucleotide repeat expansion (CTG) in the myotonic dystrophy protein kinase (DMPK) gene.2–4 DM type 2 (DM2) has similar multisystem manifestations but is clinically and pathologically distinct.1 DM2 is caused by an expansion of CCTG repeats in the CCHC-type zinc finger, nucleic acid binding protein (CNBP) gene.5,6
Gastrointestinal (GI) manifestations have been reported to occur along the entire digestive tract in patients with DM.7–10 These investigations were case reports and clinical studies of <30 patients with DM1.7–10 Only 1 study has analyzed GI symptoms in patients with DM2.11 These researchers reported a high frequency of dysphagia, abdominal pain, and constipation in 29 patients with DM2, which were similar to the percentages reported in 29 patients with DM1 and greater in frequency than reported for healthy controls enrolled in the study.11 Forty-five percent of these patients with DM2 reported GI symptoms as their most disabling complaint.11 To the best of our knowledge, no studies have analyzed the progression of GI manifestations in DM1 and DM2, and none have reported the types of medications used to manage these problems.
In this study, we analyze GI manifestations and their progression in large cohorts of patients with DM1 (n = 913) and DM2 (n = 180) enrolled in the NIH-sponsored National Registry of Myotonic Dystrophy and Facioscapulohumeral Muscular Dystrophy Patients and Family Members.12
METHODS
Enrollment process of the National Registry and cohort selection.
The registry has enrolled patients since September 2000 (ClinicalTrials.gov identifier NCT00082108).12 The registry is located at the University of Rochester (Rochester, NY).
Applications to enroll in the registry consist of a patient-reported questionnaire that collects disease-specific data, a Release of Medical Information Form, and Patient Consent Forms.12 Eligibility is determined when registry staff and leadership establish the diagnostic classification of each patient on the basis of a review of their clinical examinations, EMG and muscle biopsy reports, and results of genetic testing.12 Members of the Scientific Advisory Committee developed the diagnostic classifications for registry members for genetically and clinically confirmed patients.13 Annual update forms are sent to members yearly to track changes in patient-reported outcomes.
In this study, deidentified data were analyzed from patients enrolled in the registry who were at least 18 years old, US citizens, and classified with genetically proven or clinically defined DM1 or DM2.12 Patients analyzed in this study were enrolled in the National Registry from 2002 to 2016. Patients with congenital DM of any age were excluded from analyses.
Standard protocol approvals, registrations, and patient consents.
The Human Subjects Review Board of the University of Rochester Medical Center approved the protocol and all forms associated with the registry. All members of the registry provided written informed consent at enrollment.
Characteristics of the registry participants.
Demographic and clinical information in the registry included age, sex, age at symptom onset, body mass index (BMI), years since symptom onset, and CTG (DMPK gene) or CCTG size (CNBP gene). The lengths of the repeats were derived from medical record review. Patient-reported GI manifestations were categorized on the questionnaire at baseline as trouble with swallowing, acid reflux or heartburn, gallbladder trouble (including gallbladder surgery), stomach ulcer, liver trouble, constipation, and other. The questions related to GI manifestations are asked on the form as follows: “Have you ever had or do you have any of these conditions?”
Information on current GI medication use was also collected, including name of the medication, duration of use, and daily dosage. We reviewed the medical records of the patients who reported liver problems to extract data on their specific conditions.
Progression of GI symptoms was documented from the annual update forms. For the longitudinal dataset, we analyzed eligible members of the registry who had completed an updated form 5 years after baseline. The question on the form reads, “Have you ever had or do you have any of these conditions?” We describe the percentage of patients who did not have the symptom at baseline and then reported it by year 5.
Statistical analyses.
We used t tests and χ2 tests to compare baseline demographic and clinical characteristics between patients with DM1 and those with DM2.
Logistic regression analyses were performed to determine whether select demographic/clinical variables were associated with GI manifestations at baseline separately in patients with DM1 and DM2. The variables analyzed include sex, age, BMI, symptom duration, smoking, and CTG or CCTG repeat size. The results of these analyses are reported as odds ratios (ORs), their associated 95% confidence intervals (CIs), and p values.
A significance level of 0.05 was used for all hypothesis tests.
RESULTS
Characteristics of patients with adult-onset DM1 and DM2.
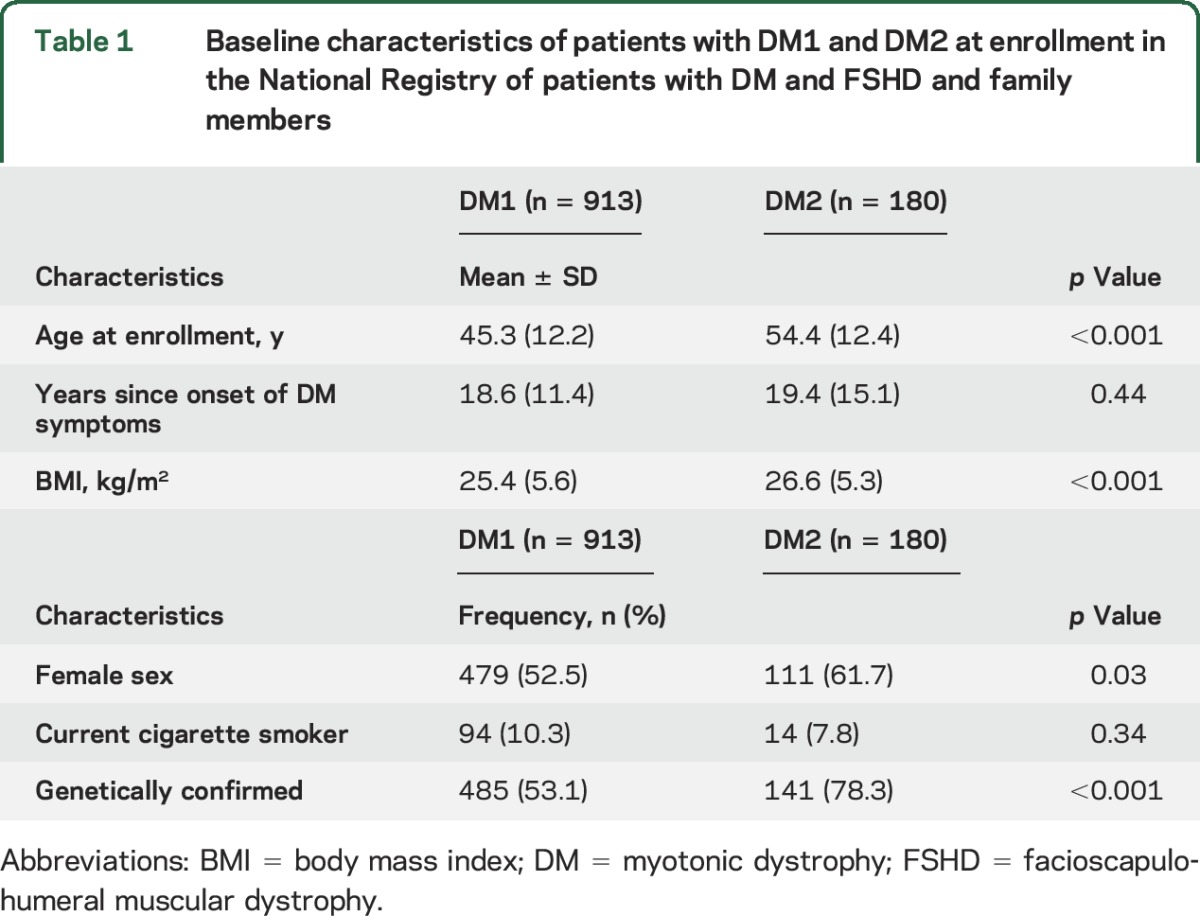
A total of 913 DM1 (52.5% female) and 180 patients with DM2 (61.7% female) were enrolled in the registry at the time of this study (table 1). Mean ± SD age at enrollment was older in DM2 (54.4 ± 12.4 years) compared to DM1 (45.3 ± 12.2 years) (p < 0.001). Mean duration of disease symptoms was similar: 18.5 ± 11.4 years in DM1 and 19.4 ± 15.1 years in DM2 (p = 0.44). Average CTG size of the DMPK nucleotide repeat in circulating blood in the DM1 cohort was 415 ± 304 repeats (n = 413, range 50–1,734), and the average DM2 allele size was 12,337 ± 3,883 base pairs (n = 84, range 373–15,600). Approximately one third of all patients with DM1 and DM2 were overweight (BMI >25 kg/m2), and ≈15% of patients with DM1 and 20% of patients with DM2 were obese as defined by BMI >30 kg/m2.
Table 1.
Baseline characteristics of patients with DM1 and DM2 at enrollment in the National Registry of patients with DM and FSHD and family members
GI manifestations at baseline.
At baseline, 79% (n = 721 of 913) of patients with DM1 and 77% (n = 139 of 180) of patients with DM2 reported a history of ≥1 GI manifestations (figure e-1 at Neurology.org). Only 1.3% of patients with DM1 and none of the patients with DM2 reported a GI problem as the initial manifestation of their disease. The most common GI manifestation in DM1 was trouble swallowing (55%, n = 499 of 913), whereas the most common GI symptom in DM2 was constipation (53%, n = 96 of 180; figure 1). Combining the GI manifestations classified as other in patients with DM1 and DM2, we identified 1.7% (n = 19 of 1,093) of DM patients with a history of irritable bowel syndrome (IBS), 1.3% (n = 14 of 1,093) with diarrhea, and <3 patients each with lactose intolerance, obstruction, fecal incontinence, abdominal pain, megacolon, upset stomach, and spasms/delayed gastric emptying.
Figure 1. Percentages of patients with DM1 and DM2 who reported GI manifestations at enrollment in the registry.

Percentages are from totals of the baseline cohort of 913 patients with DM1 and 180 with DM2. DM = myotonic dystrophy; GI = gastrointestinal. *Significant difference between the DM1 and DM2 baseline cohorts (p < 0.001).
Similar frequencies of liver problems occurred in 5.8% of patients with DM1 (n = 53 of 913) and 8.3% of patients with DM2 (n = 15 of 180) (p = 0.20). We reviewed the medical records for all 68 patients with liver problems. The majority of records from these patients were obtained from neurologists (70.6%, n = 48 of 68). Abnormal liver problems were noted in the medical records of 34 patients. Fifty percent of these patients (n = 16 of 34) had elevated liver enzyme levels, 20.6% (n = 7 of 34) had evidence of fatty liver disease, and 16.1% (n = 5 of 34) had hepatitis C. Two patients (5.9%, n = 2/34) had liver tumors that were not otherwise specified, and 4 patients were noted for having hepatitis B, hepatitis not otherwise specified, autoimmune hepatitis, or a “prediabetic” liver conditions (2.9%, n = 1 of 34).
Of the patients with DM1 with liver problems (n = 53), 41.5% (n = 22 of 53) also had gallbladder problems. In the patients with DM2 with liver problems (n = 15), the co-occurrence of gallbladder problems was less frequent (20%, n = 3 of 15). Cholecystectomy had been performed in 16.5% (n = 151 of 913) of patients with DM1. The mean age at the time of gallbladder removal was 38.1 ± 10.9 years. More women (20.0%, n = 96 of 479) than men (12.7%, n = 55 of 434) with DM1 reported having a cholecystectomy. For patients with DM2, 12.8% (n = 23 of 180) reported a cholecystectomy at a mean age of 43.7 ± 13.3 years. Cholecystectomy was reported by more women (16.2%; n = 18 of 111) than men (7.2%; 5 of 69) with DM2.
Variables associated with GI manifestations.
Female sex was significantly associated with a history of constipation at baseline in both DM1 (OR 2.14, 95% CI 1.61–2.85, p < 0.001) and DM2 (OR 3.12, 95% CI 1.67–5.84, p < 0.001) (table e-1). In addition, female sex was associated with a history of gallbladder problems at baseline in patients with DM1 (OR 1.59, 95% CI 1.14–2.23, p < 0.01) and with a history of acid reflux at baseline in patients with DM2 (OR 2.35, 95% CI 1.26–4.39, p < 0.01) (table e-1).
Higher BMI was associated with histories of acid reflux, gallbladder trouble, and liver trouble in patients with DM1 (table e-1). Patients with DM1 with longer symptom duration were at greater risk of having histories of trouble swallowing, constipation, gallbladder trouble, and liver trouble at baseline (table e-1). Older patients with DM1 were at increased risk of a history of gallbladder trouble and liver trouble at baseline (table e-1).
No other variables were associated with a history of GI manifestations in patients with DM1 or DM2 (table e-1).
Medications used to manage GI manifestations.
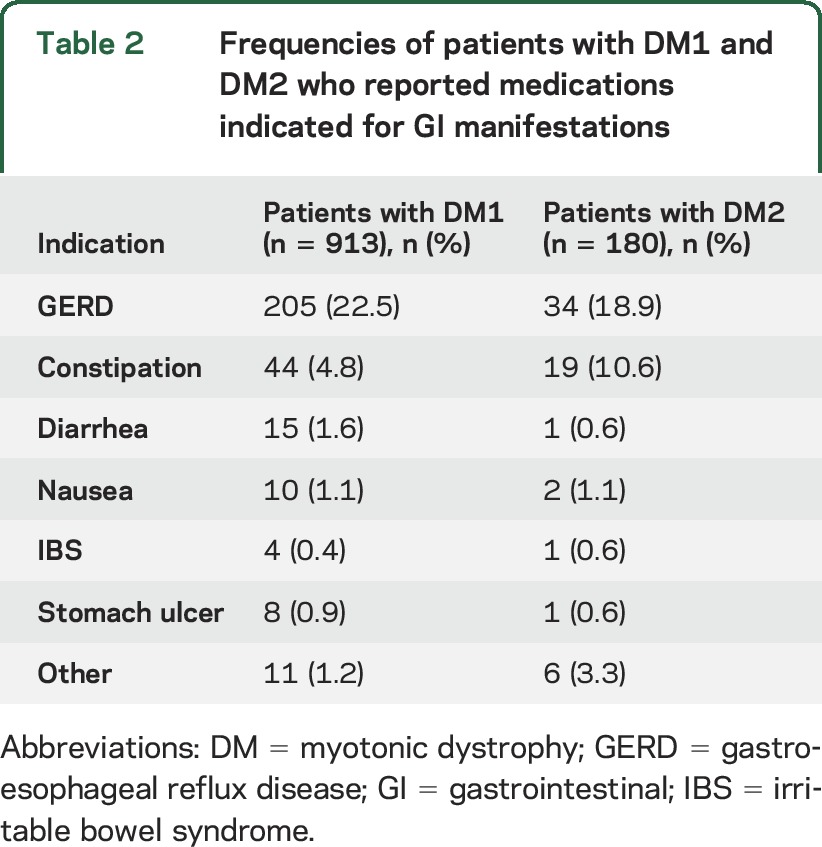
We identified 59 medications being used to treat GI manifestations in the DM1 cohort and 28 GI-related medications in the DM2 cohort. The medications are classified by indication in table 2. The most common indication reported was gastroesophageal reflux disease (GERD) in both the DM1 (22.5%, n = 205 of 913) and DM2 (18.9%, n = 34 of 180, table 2) cohorts. The frequencies and percentages of patients with DM1 and DM2 using 1 or more of 15 anti-GERD medications are shown in table e-2.
Table 2.
Frequencies of patients with DM1 and DM2 who reported medications indicated for GI manifestations
Progression of GI manifestations.
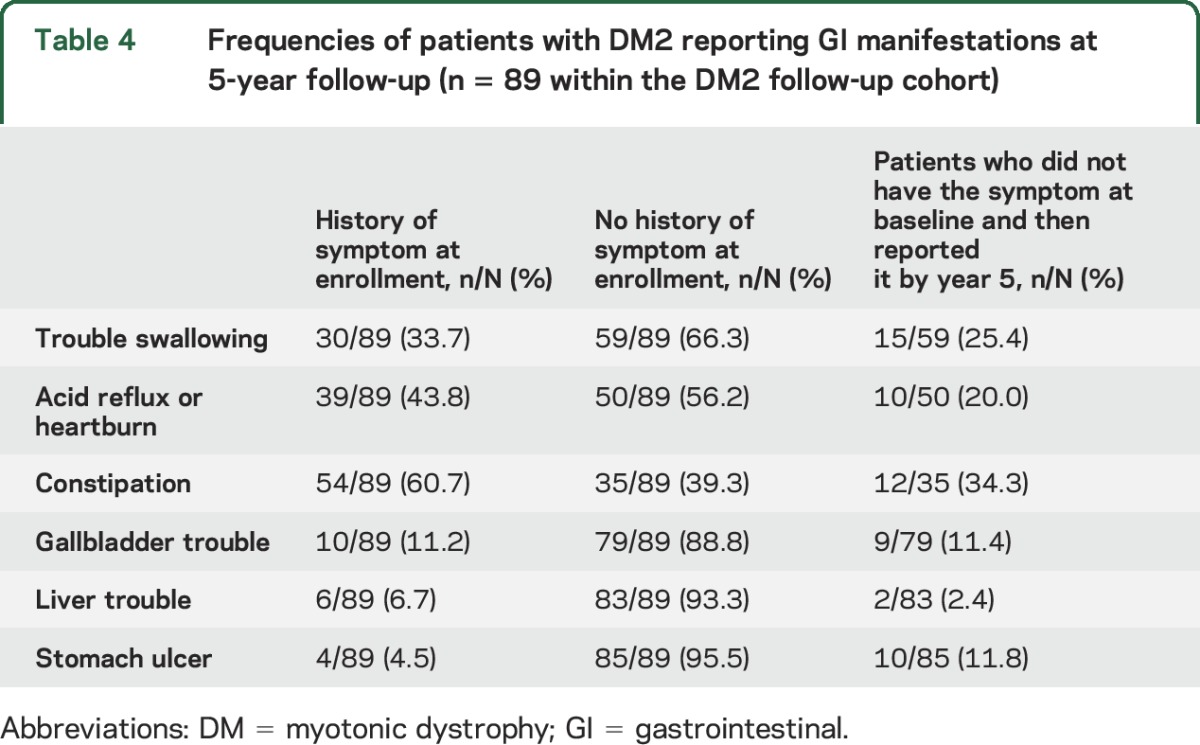
Forty-six percent of patients with DM1 (n = 418 of 913) and 49% of patients with DM2 (n = 89 of 180) completed an annual update form at year 5. The occurrence of new GI manifestations over 5 years is shown in tables 3 and 4 for patients with DM1 and DM2, respectively. The most frequent, new reports of GI manifestations at year 5 were trouble swallowing and stomach ulcers in DM1. For example, 220 of the 418 patients (52.6%) with DM1 in the follow-up cohort reported having a history of swallowing difficulty at baseline; of the 198 patients who did not have this history, 54 (27.3%) reported having it at their 5-year follow-up (table 3). The most frequent changes in the DM2 follow-up cohort were reports of new manifestations of constipation and trouble swallowing by year 5 (table 4).
Table 3.
Frequencies of patients with DM1 reporting GI manifestations at 5-year follow-up (n = 418 within the DM1 follow-up cohort)
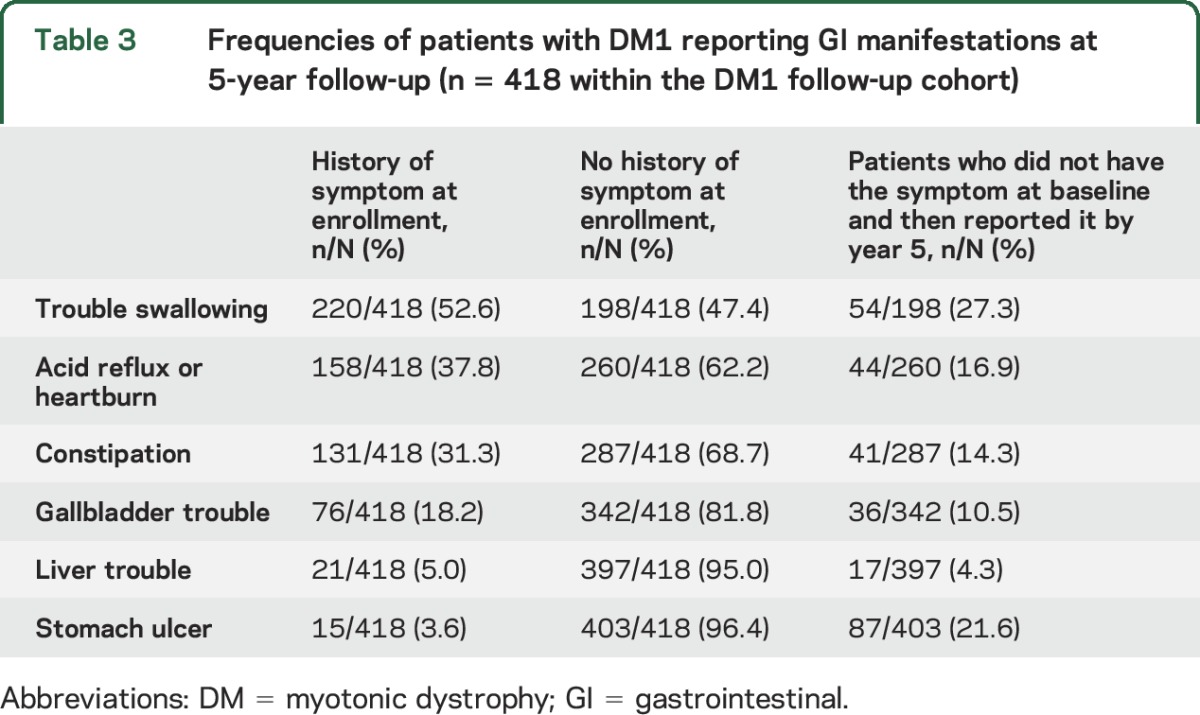
Table 4.
Frequencies of patients with DM2 reporting GI manifestations at 5-year follow-up (n = 89 within the DM2 follow-up cohort)
DISCUSSION
Our data support the hypothesis that GI manifestations are common in DM1 and DM2.11 During the 5 years of follow-up, the most common changes were new reports of stomach ulcers and trouble swallowing in patients with DM1 and the development of trouble swallowing and constipation in patients with DM2.
Dysphagia often exacerbates risks of pneumonia, decreases nutritional intake, and reduces quality of life.14,15 Unfortunately, literature reviews indicate little consensus on the pathophysiology, assessment, and management of dysphagia in neuromuscular disorders.14,15 Diagnostic approaches include a detailed medical history, clinical examination, and multidisciplinary tests such as manometry, videofluoroscopy, and fiberoptic endoscopic evaluation of swallowing.14,15 Additional studies are needed to assess the reliability of these tests and their ability to detect changes over time. Examples of management of dysphagia include dietary changes, exercise, and rehabilitation strategies that focus on quality-of-life issues and safety.14,15
The emergence of constipation also complicates disease management and reduces quality of life in patients. Prior research suggests that patients with DM1 often report stomach pain, bloating, and fluctuations between diarrhea and constipation that are similar to IBS.1,7 Other small intestine and colon concerns in DM include bacterial overgrowth, pseudo-obstruction, and bile acid malabsorption.1,7 One study showed positive results of antibiotic treatment of bacterial overgrowth in patients with DM1.16 In an uncontrolled clinical trial, 3 patients with DM1 with severe IBS showed marked improvement after 24 weeks of recombinant human insulin-like growth factor (IGF)-1 complexed with IGF binding protein-3.17 More studies are needed to determine whether similar IGF-1–like compounds or other growth factors may improve IBS-like symptoms in DM. Proposed pathomechanisms of both diarrhea and constipation in DM include endocrine disturbances, smooth and striated muscle dysfunction, myotonia, inflammation, and alterations in motility.7
Similar mechanisms may help explain the high frequency of gallbladder problems and cholecystectomy reported in the patients with DM1 and DM2. Our data corroborate previous findings of studies that assessed smaller samples of patients with DM1 only.18–21 Moreover, our study shows that members of the registry had their gallbladder removed ≈10 years earlier on average than individuals in the general population based on available normative data.22 Additional studies are needed to confirm our findings and to determine the risks of cholecystectomy at a comparatively younger age in patients with DM1 and DM2, including potential bile duct injury, hemorrhage, pulmonary insufficiency, atelectasis, and other DM-related anesthesia complications. Studies are also needed to assess the pathophysiology of the gallbladder problems in patients with DM1 and DM2.
One research team analyzed gallbladder tissue in a 30-year-old patient with DM1 who underwent elective cholecystectomy for chronic gallbladder disease.23 The researchers found alterations of muscleblind-like proteins in the smooth muscle of the gallbladder.23 This finding is typical of the skeletal and cardiac muscle molecular pathology in DM1. The presence of bound muscleblind-like mutant DM mRNA inclusions in the smooth muscle of the gallbladder supports the hypothesis that RNA toxicity, perhaps related to its role in causing a decreased production of chloride channel protein, may lead to altered function of gallbladder smooth muscle. Such alterations may decrease normal contractility of the gallbladder and contribute to the formation of gallstones.23 Studies are also needed to assess the pathomechanisms of gallbladder dysfunction in DM2 and to compare the pathophysiology to DM1.
Another concern is that ≈40% of our patients with DM1 and DM2 reported acid reflux. GERD occurs in 18.1% to 27.8% in general US cohorts.24 Untreated GERD often reduces health-related quality of life, impairs sleep, alters eating habits, and reduces productivity at work.25 Pharmacologic treatment of GERD consists of acid suppression drugs, prokinetics, histamine receptor type 2 antagonists, and proton pump inhibitors.26 Proton pump inhibitors are the most effective treatments for GERD and account for ≈77% of the market for GI medications.27 Long-term use of proton pump inhibitors poses several adverse side effects to balance against their benefits.26 Examples of risks include hypomagnesemia, pneumonia, Clostridium difficile infections, fractures, acute and chronic kidney disease, and dementia.26,28
To the best of our knowledge, no clinical trials have evaluated the efficacy and safety of various formulations of proton pump inhibitors in patients with DM1 and DM2. No clear clinical guidelines exist on length of treatment needed in DM. There are issues specific to their use in DM1 and DM2 such as how other disease manifestations may interact with such medications and potential socioeconomic and compliance factors that may hinder their use.29 Further studies are necessary to evaluate alternative treatment approaches such as nutrition, exercise, and weight-loss counseling alone or in conjunction with GERD medications.
Women with DM1 were at greater risk than men for constipation and gallbladder problems. Our data support findings from a recent study in a large sample of French patients with DM1.30 In that study, the researchers reported that a higher percentage of female participants had digestive tract dysfunction, defined as either constipation or diarrhea.30 A higher percentage of women with DM1 also reported more cataracts, dysphagia, incontinence, thyroid dysfunction, and obesity compared to men.30 The authors highlight previous research linking differences between men and women to variabilities in gene expression and metabolism of skeletal muscle, increased oxidative stress (e.g., in cardiovascular disease), and hormonal effects on myotonia.30 A literature review focuses on varying mitochondrial dysfunctions between men and women, in particular, highlighting sex differences in patients with amyotrophic lateral sclerosis, Parkinson disease, and Alzheimer disease.31 Other researchers report a greater frequency of certain types of cancers in female compared to male patients with DM1.32,33 One of these studies suggests that cancer risk, especially in women, is related to changes in downstream regulation of specific cancer genes instead of a direct effect of the DMPK mutation.32
In the present study, no associations were found between repeat sizes in the DMPK or CNBP mutant genes in circulating leukocytes and the GI manifestations in our cohorts. A prior study of 15 patients with DM1 showed a correlation between CTG repeat sizes and severity of swallowing measured by videofluoroscopy.34 Other investigators have observed no associations between the severity of skeletal muscle damage and GI disturbances.35,36 Data from the registry support previous studies that show an association between disease duration and GI manifestations in DM1.36,37 More research is needed to better assess biomarkers and other predictors of GI manifestations in DM.
The strengths of our study include cohorts of patients with a broad range of disease severity and age at onset; diverse demographics, including sex, age, and a broad geographic representation within the United States; stringent review of clinical and genetic medical records to verify disease classification; and annually updated information up to a maximum of 13 years.12
There are also limitations to our study. One limitation is that these GI manifestations are patient reported. These manifestations may actually be underreported because patients often do not attribute their GI manifestations to DM. We often prompt patients in clinic to discuss GI manifestations, especially patients with DM1 who are more severely affected and have cognitive or apathetic personality related to their disease.38 More detailed information from clinical examinations and diagnostic testing is needed to confirm and expand our results. Another limitation is the lack of information on risk factors of the patients who have experienced GI manifestations (e.g., stress, diet, family history). A follow-up questionnaire is needed to expand on our data to assess comorbidities and lifestyle factors that increase the risk of GI disturbances and the exact timing of their onset. The registry is tailored to allow easy recruitment of patients to participate in such future questionnaire-based studies.
Another limitation is that our cohorts may be less severely affected than the overall populations of patients with DM1 and DM2. A future opportunity is to partner with international registries to compare GI manifestations in larger patient cohorts and across broader socioeconomic backgrounds and nationalities. Such collaborations may also facilitate the development of studies to bridge patient-reported data with studies on how GI disturbances may or may not relate to our current understanding of RNA toxicity in DM and better ways to assess future targeted therapies.39
Supplementary Material
ACKNOWLEDGMENT
The authors thank Eileen Eastwood and Kathy Knab for their assistance in enrolling patients in the National Registry and facilitating data entry and management. They also thank the patients and family members of the National Registry for sharing new insights about their disease manifestations and for their participation in research.
GLOSSARY
- BMI
body mass index
- CI
confidence interval
- CNBP
CCHC-type zinc finger, nucleic acid binding protein
- DM
myotonic dystrophy
- DMPK
myotonic dystrophy protein kinase
- GERD
gastroesophageal reflux disease
- GI
gastrointestinal
- IBS
irritable bowel syndrome
- IGF
insulin-like growth factor
- OR
odds ratio
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: National Registry Scientific Advisory Committee/Investigators, Tetsuo Ashizawa, Richard J. Barohn, Paula R. Clemens, P. Michael Conneally, John W. Day, Denise A. Figlewicz, Jacqueline M. Jackson, John T. Kissel, Shannon Lord, Katherine D. Mathews, Donald B. Sanders, and Stephen J. Tapscott
AUTHOR CONTRIBUTIONS
James Hilbert designed the study, facilitated data collection, interpreted the data, and wrote and revised the manuscript. Richard Barohn, Paula Clemens, Rabi Tawil, and Charles Thornton helped lead the development of the National Registry, facilitated the design of the study, and revised the manuscript. Amy Parkhill analyzed the data and revised the manuscript. Elizabeth Luebbe facilitated data collection, helped design the study, and revised the manuscript. Michael McDermott helped lead the development of the National Registry, conducted statistical analyses, interpreted the data, and revised the manuscript. William Martens managed the data, conducted statistical analyses, interpreted the data, and revised the manuscript. Richard Moxley helped lead the development of the National Registry, facilitated the design of the study, wrote and revised the manuscript, and oversaw the study.
STUDY FUNDING
The National Registry is supported through the National Institute of Neurologic Disorders and Stroke (NIH Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center grant U54-NS048843), the Saunders Family Fund, and the Abrams Family Fund for Myotonic Dystrophy Research. From 2000 to 2010, the National Registry was supported through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (contracts N01-AR-5-2274 and NO1-AR-0-2250).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Harper PS, Engelen BG, Eymard B, Wilcox DE. Myotonic Dystrophy: Present Management, Future Therapy. New York: Oxford University Press; 2004. [Google Scholar]
- 2.Fu YH, Pizzuti A, Fenwick RG Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255:1256–1258. [DOI] [PubMed] [Google Scholar]
- 3.Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science 1992;255:1253–1255. [DOI] [PubMed] [Google Scholar]
- 4.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell 1992;68:799–808. [DOI] [PubMed] [Google Scholar]
- 5.Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001;293:864–867. [DOI] [PubMed] [Google Scholar]
- 6.Ranum LP, Rasmussen PF, Benzow KA, Koob MD, Day JW. Genetic mapping of a second myotonic dystrophy locus. Nat Genet 1998;19:196–198. [DOI] [PubMed] [Google Scholar]
- 7.Bellini M, Biagi S, Stasi C, et al. Gastrointestinal manifestations in myotonic muscular dystrophy. World J Gastroenterol 2006;12:1821–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avez-Couturier J, Michaud L, Cuisset JM, et al. Encopresis revealing myotonic dystrophy in 2 children [in French]. Arch Pediatr 2009;16:430–434. [DOI] [PubMed] [Google Scholar]
- 9.Ronnblom A, Forsberg H, Danielsson A. Gastrointestinal symptoms in myotonic dystrophy. Scand J Gastroenterol 1996;31:654–657. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y, Kato T, Nishida H, et al. Is there a difference in gastric emptying between myotonic dystrophy type 1 patients with and without gastrointestinal symptoms? J Neurol 2013;260:1611–1616. [DOI] [PubMed] [Google Scholar]
- 11.Tieleman AA, van Viet J, Jansen JBMJ, van der Kooi AJ, Borm GF, van Engelen BGM. Gastrointestinal involvement is frequent in myotonic dystrophy type 2. Neuromuscul Disord 2008;18:646–649. [DOI] [PubMed] [Google Scholar]
- 12.Hilbert JE, Kissel JT, Luebbe EA, et al. If you build a rare disease registry, will they enroll and will they use it? Methods and data from the national registry of myotonic dystrophy (DM) and facioscapulohumeral muscular dystrophy (FSHD). Contemp Clin Trials 2012;33:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Myotonic Dystrophy Physician Checklist Form; NIH National Registry [online]. Available at: https://www.urmc.rochester.edu/MediaLibraries/URMCMedia/neurology/documents/Physician-Checklist-DM-7-25-11.pdf. Accessed April 26, 2017. [Google Scholar]
- 14.Jones K, Pitceathly RD, Rose MR, et al. Interventions for dysphagia in long-term, progressive muscle disease. Cochrane Database Syst Rev 2016;2:CD004303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pilz W, Baijens LW, Kremer B. Oropharyngeal dysphagia in myotonic dystrophy type 1: a systematic review. Dysphagia 2014;29:319–331. [DOI] [PubMed] [Google Scholar]
- 16.Tarnopolsky MA, Pearce E, Matteliano A, James C, Armstrong D. Bacterial overgrowth syndrome in myotonic muscular dystrophy is potentially treatable. Muscle Nerve 2010;42:853–855. [DOI] [PubMed] [Google Scholar]
- 17.Heatwole CR, Eichinger KJ, Friedman DI, et al. Open-label trial of recombinant human insulin-like growth factor 1/recombinant human insulin-like growth factor binding protein 3 in myotonic dystrophy type 1. Arch Neurol 2011;68:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pruzanski W, Huvos AG. Smooth muscle involvement in primary muscle disease, I: myotonic dystrophy. Arch Pathol 1967;83:229–233. [PubMed] [Google Scholar]
- 19.Marchi S, Polloni A, Bellini M, et al. Cisapride improves gallbladder kinetics in patients affected with myotonic muscular dystrophy. Int J Clin Pharmacol Res 1993;13:53–58. [PubMed] [Google Scholar]
- 20.Schwindt WD, Bernhardt LC, Peters HA. Cholelithiasis and associated complications of myotonia dystrophica. Postgrad Med 1969;46:80–83. [DOI] [PubMed] [Google Scholar]
- 21.Harvey JC, Sherbourne DH, Siegel CI. Smooth muscle involvement in myotonic dystrophy. Am J Med 1965;39:81–90. [DOI] [PubMed] [Google Scholar]
- 22.Zacks SL, Sandler RS, Rutledge R, Brown RS Jr. A population-based cohort study comparing laparoscopic cholecystectomy and open cholecystectomy. Am J Gastroenterol 2002;97:334–340. [DOI] [PubMed] [Google Scholar]
- 23.Cardani R, Mancinelli E, Saino G, Bonavina L, Meola G. A putative role of ribonuclear inclusions and MBNL1 in the impairment of gallbladder smooth muscle contractility with cholelithiasis in myotonic dystrophy type 1. Neuromuscul Disord 2008;18:641–645. [DOI] [PubMed] [Google Scholar]
- 24.El-Serag HB, Sweet S, Winchester CC, Dent J. Update on the epidemiology of gastro-oesophageal reflux disease: a systematic review. Gut 2014;63:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liker HR, Ducrotte P, Malfertheiner P. Unmet medical needs among patients with gastroesophageal reflux disease: a foundation for improving management in primary care. Dig Dis 2009;27:62–67. [DOI] [PubMed] [Google Scholar]
- 26.Sandhu DS, Fass R. Current trends in the management of gastroesophageal reflux disease. Gut Liver. Epub 2017 Apr 24. [DOI] [PMC free article] [PubMed]
- 27.Shaheen NJ, Hansen RA, Morgan DR, et al. The burden of gastrointestinal and liver diseases, 2006. Am J Gastroenterol 2006;101:2128–2138. [DOI] [PubMed] [Google Scholar]
- 28.Schoenfeld AJ, Grady D. Adverse effects associated with proton pump inhibitors. JAMA Intern Med 2016;176:172–174. [DOI] [PubMed] [Google Scholar]
- 29.Fitzgerald BP, Conn KM, Smith J, et al. Medication adherence in patients with myotonic dystrophy and facioscapulohumeral muscular dystrophy. J Neurol 2016;263:2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dogan C, De Antonio M, Hamroun D, et al. Gender as a modifying factor influencing myotonic dystrophy type 1 phenotype severity and mortality: a nationwide multiple databases cross-sectional observational study. PLoS One 2016;11:e0148264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ventura-Clapier R, Moulin M, Piquereau J, et al. Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond) 2017;131:803–822. [DOI] [PubMed] [Google Scholar]
- 32.Fernandez-Torron R, Garcia-Puga M, Emparanza JI, et al. Cancer risk in DM1 is sex-related and linked to miRNA-200/141 downregulation. Neurology 2016;87:1250–1257. [DOI] [PubMed] [Google Scholar]
- 33.Das M, Moxley RT III, Hilbert JE, et al. Correlates of tumor development in patients with myotonic dystrophy. J Neurol 2012;259:2161–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcon M, Briani C, Ermani M, et al. Positive correlation of CTG expansion and pharyngoesophageal alterations in myotonic dystrophy patients. Ital J Neurol Sci 1998;19:75–80. [DOI] [PubMed] [Google Scholar]
- 35.Modolell I, Mearin F, Baudet JS, Gamez J, Cervera C, Malagelada JR. Pharyngo-esophageal motility disturbances in patients with myotonic dystrophy. Scand J Gastroenterol 1999;34:878–882. [DOI] [PubMed] [Google Scholar]
- 36.Horowitz M, Maddox A, Maddern GJ, Wishart J, Collins PJ, Shearman DJ. Gastric and esophageal emptying in dystrophia myotonica: effect of metoclopramide. Gastroenterology 1987;92:570–577. [DOI] [PubMed] [Google Scholar]
- 37.Bellini M, Alduini P, Costa F, et al. Gastric emptying in myotonic dystrophic patients. Dig Liver Dis 2002;34:484–488. [DOI] [PubMed] [Google Scholar]
- 38.Bugiardini E, Meola G. Consensus on cerebral involvement in myotonic dystrophy: workshop report: May 24–27, 2013, Ferrere (AT), Italy. Neuromuscul Disord 2014;24:445–452. [DOI] [PubMed] [Google Scholar]
- 39.Thornton CA, Wang E, Carrell EM. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev 2017;44:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.