

Abstract
M3 muscarinic receptor (M3R) expression is increased in colon cancer; M3R activation stimulates colon cancer cell invasion via cross-talk with epidermal growth factor receptors (EGFR), post-EGFR activation of mitogen-activated protein kinase (MAPK) ERK1/2, and induction of matrix metalloproteinase-1 (MMP1) expression. MMP1 expression is strongly associated with tumor metastasis and adverse outcomes. Here, we asked whether other MAPKs regulate M3R agonist-induced MMP1 expression. In addition to activating ERK1/2, we found that treating colon cancer cells with acetylcholine (ACh) stimulated robust time- and dose-dependent phosphorylation of p38 MAPK. Unlike ERK1/2 activation, ACh-induced p38 phosphorylation was EGFR-independent and blocked by inhibiting protein kinase C-α (PKC-α). Inhibiting activation of PKC-α, EGFR, ERK1/2, or p38-α/β alone attenuated but did not abolish ACh-induced MMP1 expression, a finding that predicted potentiating interactions between these pathways. Indeed, ACh-induced MMP1 expression was abolished by incubating cells with either an EGFR or MEK/ERK1/2 inhibitor combined with a p38-α/β inhibitor. Activating PKC-α and EGFR directly with the combination of phorbol 12-myristate 13-acetate (PMA) and EGF potentiated MMP1 gene and protein expression, and cell invasion. PMA- and ACh-induced MMP1 expression were strongly diminished by inhibiting Src and abolished by concurrently inhibiting both p38-α/β and Src, indicating that Src mediates the cross-talk between PKC-α and EGFR signaling. Using siRNA knockdown, we identified p38-α as the relevant p38 isoform. Collectively, these studies uncover novel functional interactions between post-muscarinic receptor signaling pathways that augment MMP1 expression and drive colon cancer cell invasion; targeting these potentiating interactions has therapeutic potential.
Keywords: Matrix metalloproteinases, colorectal cancer, p38 MAPK, cell invasion, cell signaling, muscarinic receptors
INTRODUCTION
In the United States, colon cancer is the third leading cause of cancer-related mortality and the most common cause of death from gastrointestinal disease [1]. Each year approximately 150,000 people are diagnosed with colon cancer and nearly 50,000 are anticipated to perish from the disease [2]. While early resection of primary cancers greatly improves prognosis, persons who succumb die primarily as a consequence of unresectable metastases [3]. Despite advances in elucidating the somatic gene mutations that cause progressive intestinal mucosal dysplasia and cancer, the mechanisms governing colon cancer cell invasion and dissemination are less certain [4, 5]. While novel mutations are not observed in metastases, growth factors promote colon cancer cell invasion into adjacent epithelium and blood vessels, and extravasation in other organs; these are key precursors to the establishment of metastatic foci [4, 6].
Whereas the molecular events driving colon cancer progression remain obscure, a growing body of evidence suggests that in addition to serving as growth factors, muscarinic receptor agonists modulate the expression and activation of key molecules that promote cancer cell dissemination [7]. Of the five muscarinic receptor subtypes, M1R, M3R, and M5R are conditional oncogenes when expressed in cells capable of proliferation [8]. M3R are expressed widely in the gut and both M3R and CHRM3, the gene encoding its expression, are over-expressed in colon cancer [7, 9–11]. We previously showed that M3R activation governs key hallmarks of colon cancer progression including cell proliferation, survival, migration, and invasion [12–17]. In mouse models, M3R activation augments, and M3R deficiency attenuates, intestinal neoplasia [18–20].
Many steps precede tissue invasion and metastasis in cancer, including loss of cell adhesion, extracellular matrix degradation, and induction of cell migration [21]. Members of the matrix metalloproteinase (MMP) family of enzymes play key roles in degrading the extracellular matrix and promoting invasion [22–24]. Within this family, expression of MMP1, a secreted enzyme that breaks down interstitial collagens, correlates strongly with advanced colon cancer, metastatic dissemination, and an adverse outcome [25, 26]. Thus our discovery that M3R activation selectively induces MMP1 expression by approximately 50-fold identified a mechanism likely to be important for colon cancer progression [26]. Our finding that blocking MMP1 activity abolishes M3R agonist-induced colon cancer cell invasion lends further credibility to this hypothesis [27]. The focus of the current work was to understand more completely how M3R activation promotes MMP1 expression.
Our previous work suggested that transactivation of EGFR and downstream activation of MEK followed by extracellular signal-related kinase1/2 (ERK1/2) [also known as p44/42 mitogen activated kinase (MAPK)] represented a critical signal transduction pathway mediating post-M3R MMP gene induction [26]. However, whereas blocking this pathway suppressed M3R agonist-induced MMP1 expression efficaciously, it did so incompletely – there remained residual acetylcholine-induced MMP1 expression. This led us to consider the possibility that other post-M3R signaling pathways play a role in regulating MMP1 expression. Mammals possess two other MAPK’s, c-Jun-N-terminal kinase (JNK) and p38 MAPK (p38), which are also focal points for phosphorylation cascades that transduce extra-cellular stimuli into cellular responses and play key roles in promoting cancer growth and dissemination [28]. We considered these MAPK’s worthy of consideration, particularly p38 which mediates growth factor-induced cell migration and MMP1 expression in other cancers [25, 29–31].
As reported here, this line of investigation led us to identify novel potentiating interactions between post-M3R signaling pathways, involving protein kinase C (PKC)-α and p38-α activation that are likely to provide malignant cells with a means of rescuing M3R agonist-induced MMP1 gene induction when only EGFR/ERK signaling is blocked. We describe a gain of pro-invasive characteristics of human colon cancer cells following activation of this M3R/MMP1 axis and uncover intermediary factors and pathways that modulate these interactions between signaling pathways.
MATERIALS AND METHODS
Reagents and antibodies
The following antibodies were used: total and phospho-ERK1/2, total p38, phospho-p38, total JNK and phospho-JNK, and β-actin from Cell Signaling (Danvers, MA); p38-α and p38-β from Thermo Fisher (Waltham, MA). Chemicals were purchased as follows: ACh and phorbol 12-myristate 13-acetate (PMA) from Sigma-Aldrich; EGF from Cell Signaling. See Supplemental Table 1 for list and sources of the inhibitors used in this study.
Cell lines and cell culture
HT-29, H508, and Caco-2 human colon cancer cell lines were authenticated and purchased from American Type Culture Collection (ATCC). H508 cells were grown in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum (FBS) plus antibiotics. HT-29 cells were grown in McCoy’s 5A medium (Life Technologies) supplemented with 10% FBS plus antibiotics. Caco-2 cells were grown in E-MEM (Quality Biological) supplemented with 10% FBS plus antibiotics. Cells were grown at 37°C, with 5% CO2 in a humidified incubator and passaged weekly at subconfluence after trypsinization.
Immunoblot analysis
After the indicated treatments, cells were lysed in a solution containing 20 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 1 mM NaF, 1 mM NaVO3, 1 mM EDTA, 1% Triton X-100, 1 μg/ml pepstatin, and 1 μg/ml leupeptin. Cell lysates were centrifuged at 15,000 × g at 4°C for 10 min. Supernatants were collected and protein concentration was determined by the BCA method (Thermo Scientific). Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes that were probed against total (Cell Signaling catalog #4695) and phospho-ERK1/2 for sites p-T202 and p-Y204 (Cell Signaling catalog #4376), total (Cell Signaling catalog #9212) and phospho-p38 for sites p-T180 and p-Y182 (Cell Signaling catalog #9211), total (Cell Signaling catalog #9252) and phospho-JNK for sites p-T183 and p-Y185 (Cell Signaling catalog #9251), p38-α (Thermo Fisher #PA1-41321) and p38-β (Thermo Fisher #PA5-14050). To confirm equal protein loading, blots were stripped and re-probed with antibody to β-actin. Image quantification was performed by scanning densitometry using NIH Image J 1.50i software. In most experiments, conditions were normalized and expressed relative to the positive control.
RNA extraction and real-time PCR
Total cellular RNA was isolated with Trizol reagent (Invitrogen) according to the manufacturer’s instructions. First strand cDNAs were synthesized using the Superscript III First Strand Synthesis System (Invitrogen). Real-time PCR was performed with ABI StepOnePlus real-time PCR System (Applied Biosystems) using Veriquest Sybr Green qPCR Master (Affymetrix) with specific primers for MMP1 or GAPDH. PCR conditions were 2 min at 50°C, 10 min at 95°C, and then 40 cycles at 95°C for 15 s and 60°C for 60 s. PCR specificity was confirmed by melting-curve analysis. The standard cycle threshold (2− ΔΔCt CT) method was used to calculate relative levels of mRNA expression. Individual expression values were normalized by comparison to GAPDH. Sense and antisense PCR primers for RT-PCR were as follows: MMP1, 5′- AACTGCCAAATGGGCTTG -3′ and 5′- CCGTGTAGCACATTCTGTCC -3′. GAPDH, 5- CTCCTCACAGTTGCCATGTA -3′ and 5′- GTTGAGCACAGGGTACTTTATTG -3′.
MMP1 ELISA
After cells were treated with the indicated agents, aliquots of the supernatants were collected and assessed for MMP1 release using the Human MMP-1 ELISA Kit (Sigma-Aldrich) according to the manufacturer’s instructions. Inhibitors were pre-incubated for 45 min at 37°C prior to 4-h stimulation with indicated agents. All results were normalized to the unstimulated control.
Cell transfection
Lipofectamine® RNAiMAX Reagent (Life Technologies) was used to transfect silencing RNA. Pre-designed MISSION siRNAs specific to human p38-α (esiRNA1, Sigma-Aldrich) were used to knock down p38-α expression. MISSION siRNAs consist of a heterogeneous mixture of siRNAs that are endoribonuclease-prepared siRNA pools targeting the same mRNA sequence. Cells were transfected with siRNAs for 72 h prior to treatment with designated agents.
Cell invasion assay
Invasion assays were performed using Biocoat Matrigel Invasion Chambers (Corning) following the manufacturer’s instructions. Briefly, HT-29 and Caco-2 cells were trypsinized and resuspended in serum-free medium and placed in the upper chamber of the Transwell inserts at a density of 5 × 104 cell/ml. Chemoattractant (fetal bovine serum) was placed in the lower chamber, and the cells were treated with the indicated agents. Cells were incubated for 48 h in a 37°C, 5% CO2 humidified incubator. Non-invading cells were dislodged with a cotton-tipped swab. Invading cells were fixed and stained with 0.1% crystal violet (Sigma). Imaging of the invaded cells was acquired from an inverted light microscope, and analysis was performed with NIS-Elements imaging software (Nikon).
Statistical analysis
Data are presented as mean ± SE of at least three separate experiments, and analyzed by two-tailed unpaired Student’s t-test. P < 0.05 was considered statistically significant.
RESULTS
Treating human colon cancer cells with a M3R agonist stimulates p38 MAPK phosphorylation
First, we explored the possibility that in addition to ERK1/2 either JNK or p38 might play a role in M3R agonist-induced induction of MMP1 expression in colon cancer [28]. To do so, we employed two well-characterized human colon cancer cell lines which primarily and robustly express the M3R muscarinic receptor subtype; HT-29 and H508 cells, respectively, are derived from human male and female well-differentiated colon adenocarcinomas [9, 32]. Our published dose-response experiments revealed that changes in colon cancer cell signaling and function are first detected with 1 μM acetylcholine (ACh) and maximal with 100–300 μM ACh [26].
Treating HT-29 cells with maximal concentrations of ACh for 5 min stimulated 10- to 20-fold increases in p38 and ERK1/2 phosphorylation, but did not alter JNK phosphorylation (Figure 1A, B). No changes in total levels of p38, ERK1/2, or JNK were detected (Figure 1A). Pre-incubating cells with atropine (5 µM) consistently abolished ACh-induced p38 and ERK1/2 phosphorylation, confirming these actions were MR-dependent (Figure 1A, B). Similar results were observed with H508 cells; ACh stimulated 15-fold increased p38 phosphorylation with no change in total p38 (Figure 1C, D). p38 phosphorylation was rapid and reversible; detected within 5 min, maximal by 10 min, and nearly returned to basal levels within 1 h (Figure 1C, D). In accord with our previous ACh dose-response findings [26], p38 phosphorylation was detectable after 10-min incubation with 1 µM and maximal with 100–300 µM ACh (Figure 1E, F).
Figure 1. M3R activation stimulates p38 MAPK and ERK1/2 phosphorylation.
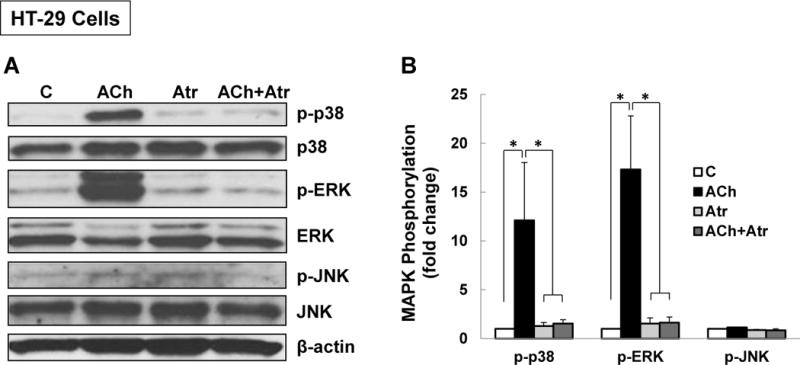
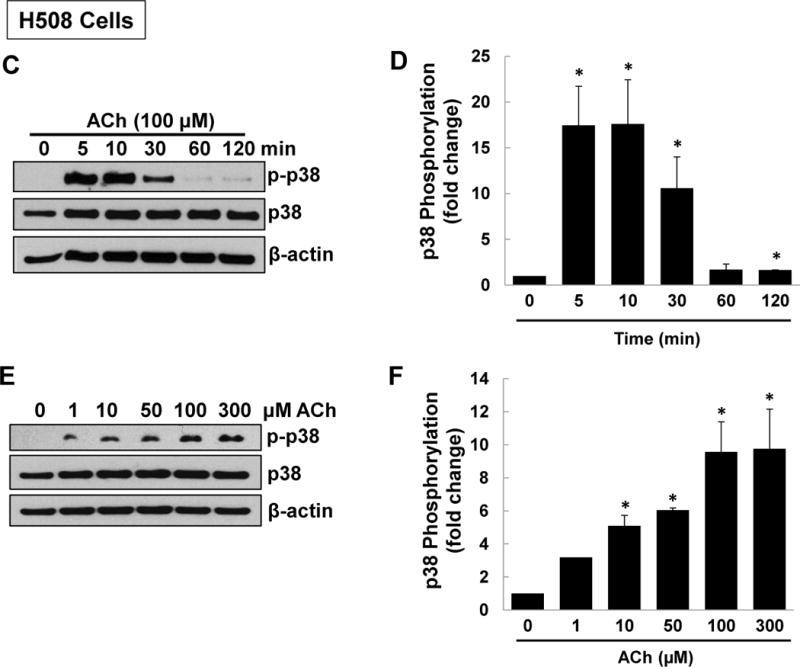
(A) Acetylcholine (ACh) stimulates p38 and ERK1/2 phosphorylation. HT-29 colon cancer cells were incubated for 5 min with 200 μM ACh and cell extracts were immunoblotted with antibodies against phosphorylated and total p38, ERK1/2, and JNK. (B) Densitometry of immunoblots from four separate experiments show that ACh stimulated robust phosphorylation of p38 and ERK1/2, but did not alter levels of total or phosphorylated JNK, total ERK1/2 or total p38. The actions of ACh were blocked by pre-incubating cells with 5 μM atropine (Atr) for 45 min. C, untreated control; *P < 0.05. (C) ACh stimulates time-dependent p38 phosphorylation. H508 colon cancer cells were incubated with 100 μM ACh for the indicated times. Cell extracts were immunoblotted with antibodies against phosphorylated and total p38. (D) Densitometry of immunoblots from three separate experiments show that ACh stimulated maximal p38 phosphorylation within 5 to 10 min which was nearly back to basal levels within 1 h. *P < 0.05 compared to Time 0. (E) ACh stimulates dose-dependent p38 phosphorylation. H508 colon cancer cells were incubated with the indicated concentrations of ACh for 10 min. Cell extracts were immunoblotted with antibodies against phosphorylated and total p38. (F) Densitometry of immunoblots from three separate experiments show ACh-induced p38 phosphorylation was detectable with 1 μM ACh and maximal with 100 to 300 μM ACh. *P < 0.05 compared to vehicle alone (no ACh). In all immunoblots, β-actin was used as a loading control.
Regulation of M3R agonist-induced p38 activation
Next, to uncover the intermediary signaling molecules that regulate p38 activation in ACh-stimulated colon cancer cells we used a series of previously validated selective chemical inhibitors [12–14, 16–18]; we confirmed effects by using two separate inhibitors chosen by their selectivity for blocking the kinase of interest [33] (Supplementary Table 1). Control experiments with atropine and other selective inhibitors used in these and subsequent experiments did not alter basal p38 phosphorylation (Supplemental Figure 1A). As a positive control, pre-incubating HT-29 cells with 5 μM atropine before treatment with 100 μM ACh for 5 min consistently inhibited both p38 and ERK1/2 activation (Figure 2A, B). Incubating cells with atropine alone did not alter levels of total or phospho-p38 or phospho-ERK1/2 (Figures 1, 2A, B). In contrast, neither MEK (10 μM PD98059, 10 μM U0126) nor EGFR (5 μM PD168393, 5 μM PD153035) inhibitors altered ACh-induced p38 phosphorylation (Figure 2A–D). Immunoblot controls verified MEK and EGFR inhibitors robustly blocked ACh-induced ERK1/2 phosphorylation (Figure 2A–D).
Figure 2. ACh-induced p38 phosphorylation is not altered by inhibiting MEK/ERK1/2, EGFR, or PI3K/AKT activation.
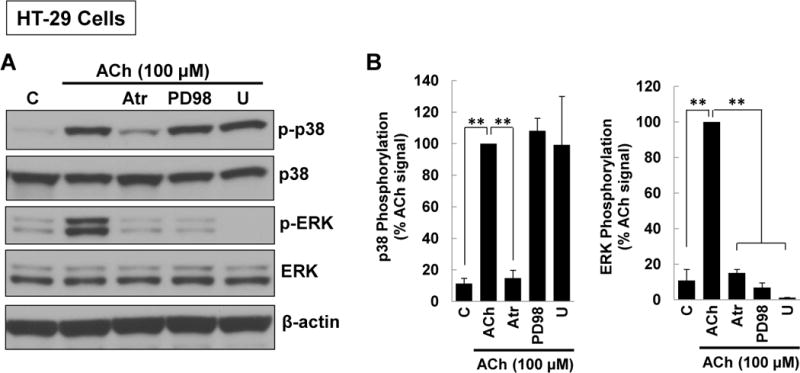
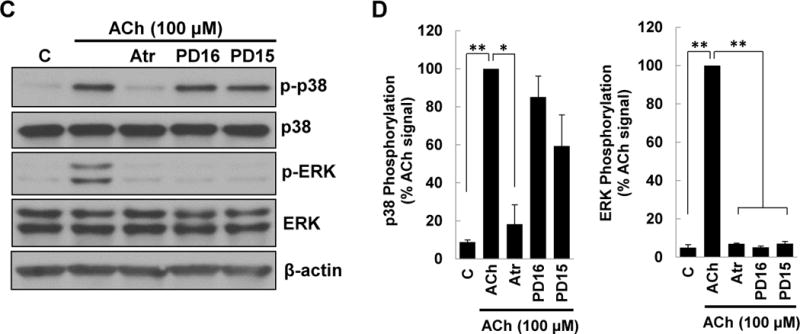
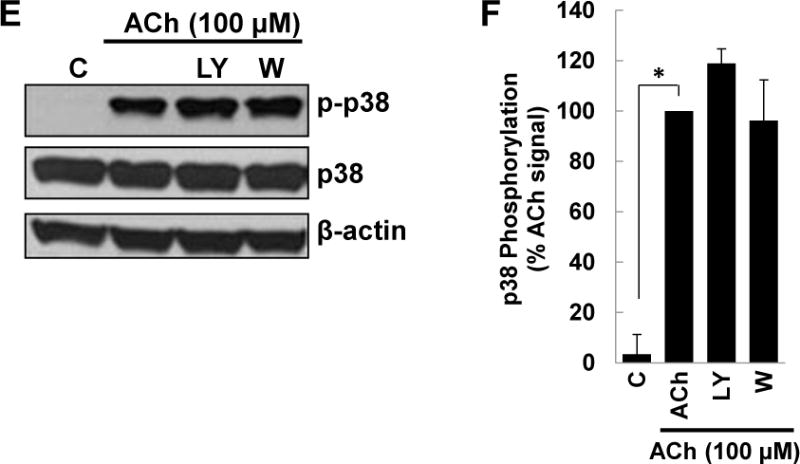
(A) HT-29 cells were pre-incubated for 45 min with M3R or MEK/ERK1/2 inhibitors before adding ACh (100 μM) for an additional 5-min incubation. Cell extracts were immunoblotted with antibodies against phosphorylated and total p38 and ERK1/2. (B) Densitometry of immunoblots from three separate experiments shows ACh-induced p38 phosphorylation was blocked by pre-incubating cells with 5 μM atropine (Atr) but not by MEK (ERK1/2) inhibitors [10 μM PD98059 (PD98), 10 μM U0126 (U)]. As positive controls, atropine and MEK (ERK1/2) inhibitors consistently blocked ACh-induced ERK1/2 phosphorylation. **P < 0.01 compared to 100 μM ACh alone. (C) HT-29 cells were pre-incubated for 45 min with inhibitors of M3R or EGFR inhibitors [5 μM PD168393 (PD16), 5 μM PD153035 (PD15)] before adding ACh (100 μM) for an additional 5-min incubation. Cell extracts were immunoblotted with antibodies against phosphorylated and total p38 and ERK1/2. (D) Densitometry of immunoblots from three separate experiments shows ACh-induced p38 phosphorylation was blocked by pre-incubating cells with atropine but not by EGFR inhibitors. As positive controls, atropine and EGFR inhibitors consistently blocked ACh-induced ERK1/2 phosphorylation. *P < 0.05, **P < 0.01 compared to 100 μM ACh alone. (E) HT-29 cells were pre-incubated for 45 min with PI3K/AKT signaling inhibitors [10 μM LY 294002 (LY), 5 nM Wortmannin (W)] before adding 100 μM ACh for an additional 5-min incubation. Cell extracts were immunoblotted with antibodies against phosphorylated and total p38. (F) Densitometry of immunoblots from three separate experiments shows ACh-induced p38 phosphorylation was not blocked by pre-incubating cells with PI3K/AKT inhibitors. *P < 0.05 compared to 100 μM ACh alone.
Previously, we showed that ACh-induced transactivation of EGFR activates downstream PI3K/AKT signaling [16, 18]. As anticipated by the failure of EGFR inhibitors to block ACh-induced p38 phosphorylation (Figure 2C, D), neither of two PI3K inhibitors tested (10 μM LY294002, 5 nM Wortmannin) altered p38 phosphorylation (Figure 2E, F). These results indicated unambiguously that ACh-induced p38 activation is not regulated by EGFR, ERK1/2, or PI3K/AKT activation.
Protein kinase C (PKC) is an important post-M3R signaling molecule [34, 35]. To determine whether PKC played a role in mediating post-M3R p38 activation in colon cancer cells we used two selective PKC-α/β1 inhibitors, Gӧ6976 and Gӧ6983. Notably, pre-incubating HT-29 cells with either 5 μM Gӧ6976 or 5 μM Gӧ6983 consistently attenuated ACh-induced p38 phosphorylation but did not alter levels of total p38 (Figure 3A, B). These findings were replicated in H508 colon cancer cells (Figure 3A, B). In these experiments, atropine and a MEK inhibitor (U0126) were used, respectively, as positive and negative controls. Others have reported that HT-29 colon cancer cells express little to no PKC-β [36–38]. Hence, our findings implicated PKC-α as the key intermediary between M3R and p38 activation in colon cancer.
Figure 3. ACh-induced p38 phosphorylation is mediated by activation of PKC.
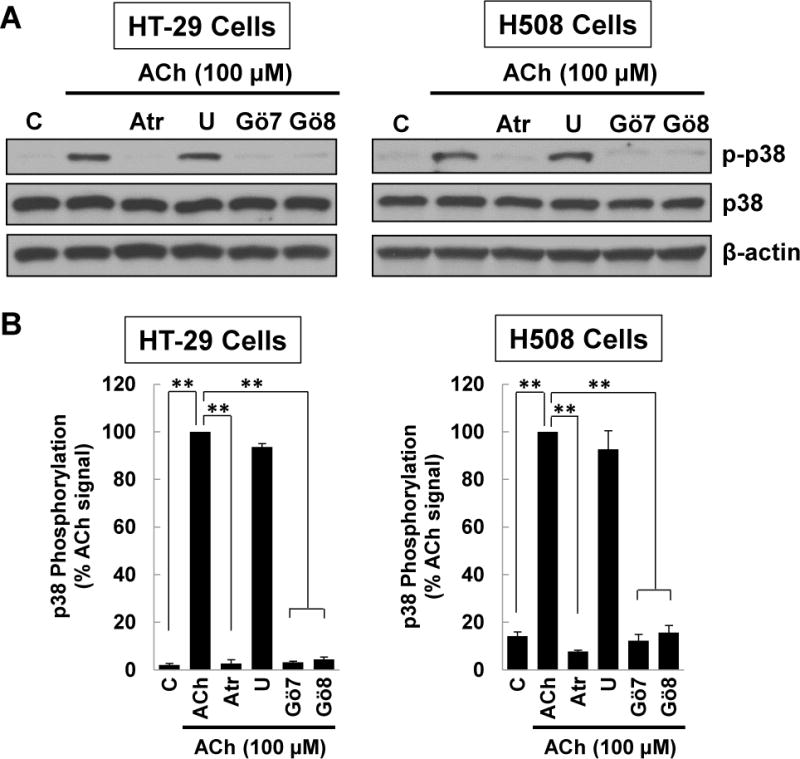
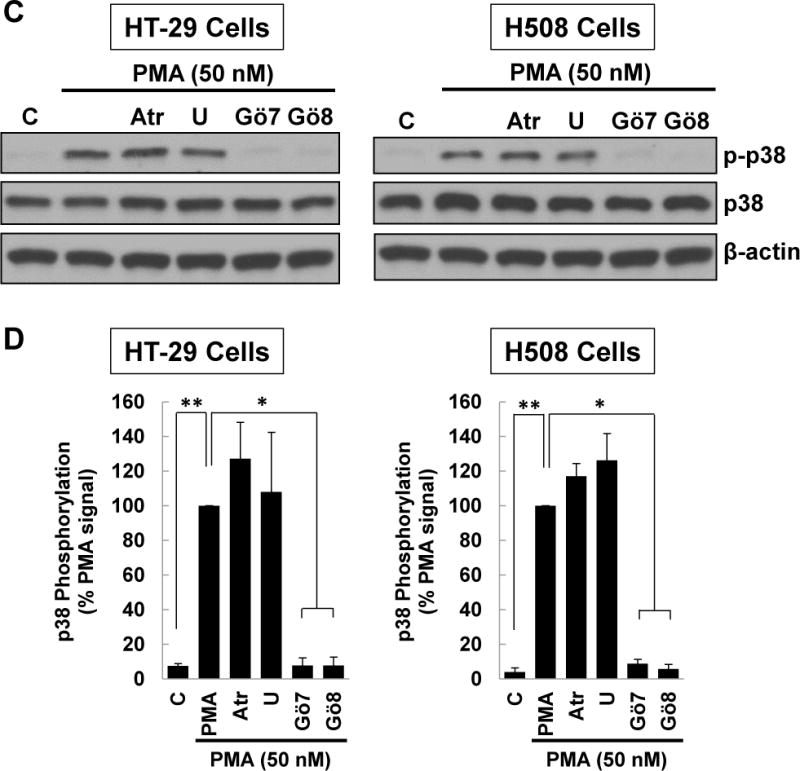
(A) HT-29 and H508 cells were pre-incubated for 45 min with atropine, a MEK inhibitor [10 μM U0126 (U)] or PKC-α/β1 inhibitors [5 μM Gӧ6976 (Gӧ7), 5 μM Gӧ6983 (Gӧ8)] before adding 100 μM ACh for an additional 5-min incubation. Cell extracts were immunoblotted with antibodies against phosphorylated and total p38. (B) Densitometry of immunoblots from three separate experiments shows pre-incubating HT-29 and H508 cells with atropine or PKC-α/β1 inhibitors consistently attenuated ACh-induced p38 phosphorylation, but pre-incubation with atropine or the MEK inhibitor had no effect. **P < 0.01 compared to 100 μM ACh alone. (C) HT-29 and H508 cells were pre-incubated for 45 min with atropine, a MEK inhibitor [10 μM U0126 (U)], or PKC-α/β1 inhibitors [5 μM Gӧ6976 (Gӧ7), 5 μM Gӧ6983 (Gӧ8)] before adding 50 mM PMA for an additional 5-min incubation. Cell extracts were immunoblotted with antibodies against phosphorylated and total p38. (D) Densitometry of immunoblots from three separate experiments shows pre-incubating HT-29 and H508 cells with PKC-α/β1 inhibitors consistently attenuated PMA (50 nM)-induced p38 phosphorylation, but pre-incubation with atropine or the MEK inhibitor had no effect. *P < 0.05, **P < 0.01 compared to 50 nM PMA alone. β-actin was a loading control. C, untreated control.
To test the hypothesis that post-M3R PKC activation mediates downstream p38 phosphorylation, we examined the effects of activating PKC directly with a phorbol ester. We incubated HT-29 and H508 cells with phorbol 12-myristate 13-acetate (PMA) to activate PKC directly and employed selective inhibitors to show that these actions were neither MR- nor EGFR-dependent. As shown in Figure 3C and D, neither control (atropine or MEK inhibitor U0126) altered PMA-induced p38 phosphorylation. In contrast, in both cell lines, PKC-α/β1 inhibitors, Gӧ6976 and Gӧ6983, consistently inhibited PMA-induced p38 phosphorylation (Figure 3C, D). These findings confirmed that PKC-α regulates p38 activation in colon cancer cells and provided further evidence supporting the role of PKC-α as a key intermediary molecule between M3R and p38 activation.
Intrigued by the novel observation that M3R activation resulted in PKC-α-mediated activation of p38, we sought a functional role for this finding. We considered whether this signaling pathway could contribute to a more invasive phenotype. Previously, we reported that M3R signaling promoted colon cancer cell invasion by increasing expression and activation of MMP1 [26, 27]. Whereas stimulating colon cancer cells with ACh induced MMP1 expression by activating EGFR/ERK1/2 signaling [26], it was conceivable that other post-M3R signaling pathways contributed to this action.
Interactions between post-M3R signaling pathways contribute to ACh-induced MMP1 expression
As shown in Figure 4, we found incubating HT-29 cells with 100 μM ACh stimulated a nearly 40-fold increase in MMP1 mRNA expression, an action completely blocked by pre-incubating cells with atropine (Fig. 5A; 40.8 ± 0.7-fold increase in MMP1 mRNA with ACh alone compared to 1.3 ± 0.2-fold increase with ACh plus atropine; P < 0.05). Pre-incubating HT-29 cells with inhibitors of PKC-α/β1, p38-α/β, EGFR, and MEK strongly attenuated but did not completely abolish ACh-induced MMP1 expression (Figure 4). Notably, pre-incubating cells with the combination of a p38-α/β inhibitor plus either an EGFR or MEK inhibitor completely blocked ACh-induced MMP1 expression; i.e., the levels of ACh-induced MMP1 mRNA following pre-incubation with these combinations of inhibitors were significantly lower than those observed with these inhibitors acting alone and not different from control basal values (Figure 4). Pre-incubating cells with the triple combination of an EGFR, PKC-α/β1, and p38-α/β inhibitor caused no further reduction in MMP1 mRNA levels (Figure 4). Similar findings were observed when we repeated this experiment using H508 instead of HT-29 cells (Supplemental Fig. 2). Control experiments showed atropine and other selective inhibitors used in these and subsequent experiments did not stimulate MMP1 expression (Supplemental Figure 1B).
Figure 4. Potentiating interactions between post-M3R signaling pathways govern ACh-induced MMP1 gene induction.
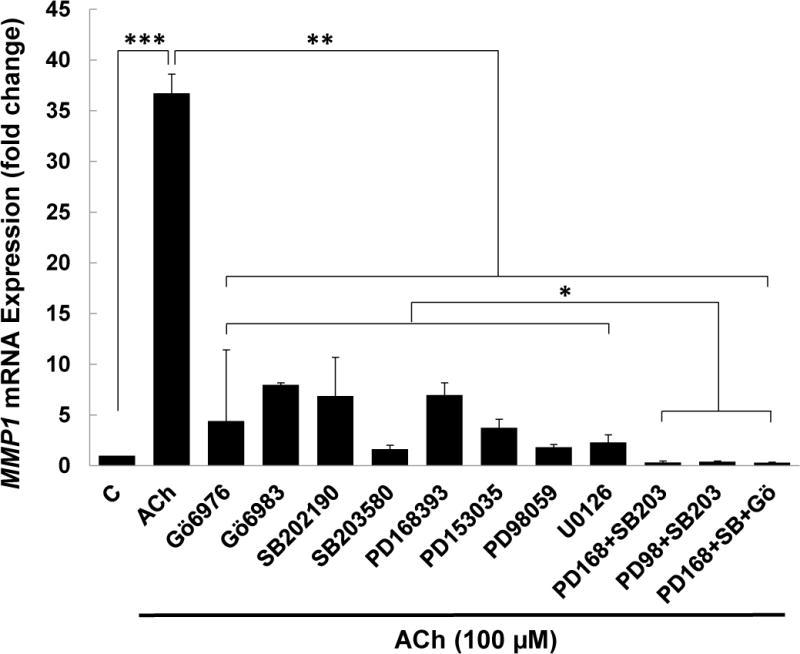
HT-29 cells were pre-incubated for 45 min with PKC-α/β1, p38-α/β, EGFR, or MEK inhibitors, and then incubated for an additional 4 h with no additional test agents (C) or with ACh (100 μM). MMP1 mRNA levels were measured by qPCR. ACh-induced MMP1 expression was attenuated by inhibitors of PKC-α/β1 (5 μM Gӧ6976, 5 μM Gӧ6983), p38-α/β (10 μM SB202190, 10 μM SB203580), EGFR (5 μM PD168393, 5 μM PD153035), and MEK (10 μM PD98059, 10 μM U0126). Pre-incubating cells with a combination of p38-α/β plus EGFR inhibitors or p38-α/β plus MEK inhibitors abolished ACh-induced MMP1 gene expression, as did pre-incubation with a combination of p38-α/β, EGFR and PKC-α/β1 inhibitors. qPCR data were normalized to GAPDH and are means of four separate experiments. C, untreated control; *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 5. Simultaneous activation of PKC and EGFR signaling potentiates MMP1 gene expression.
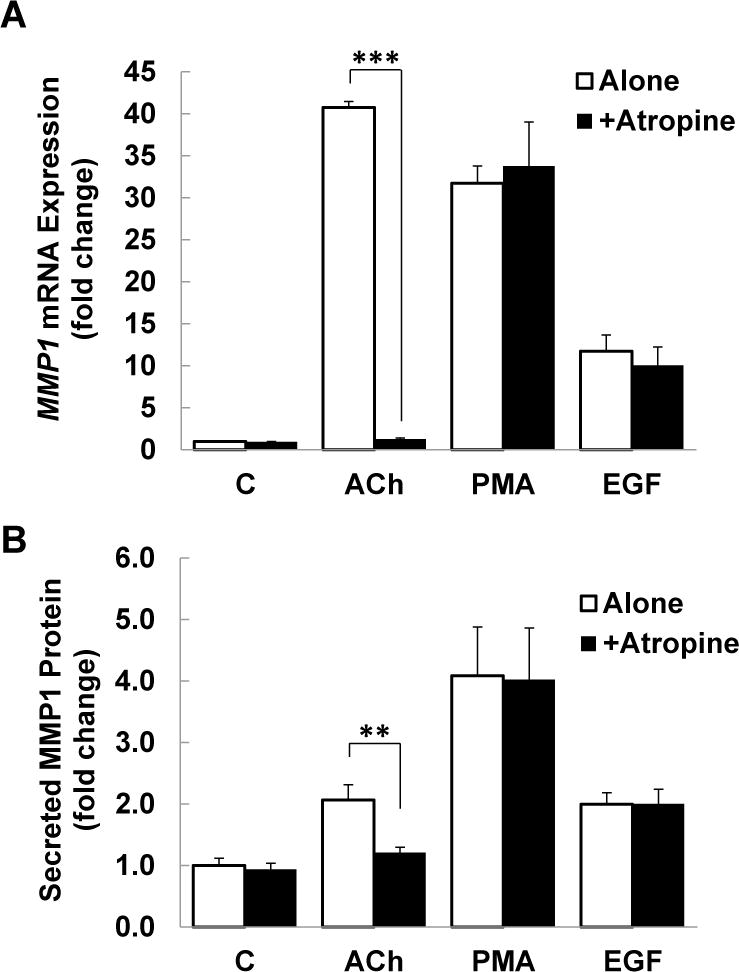
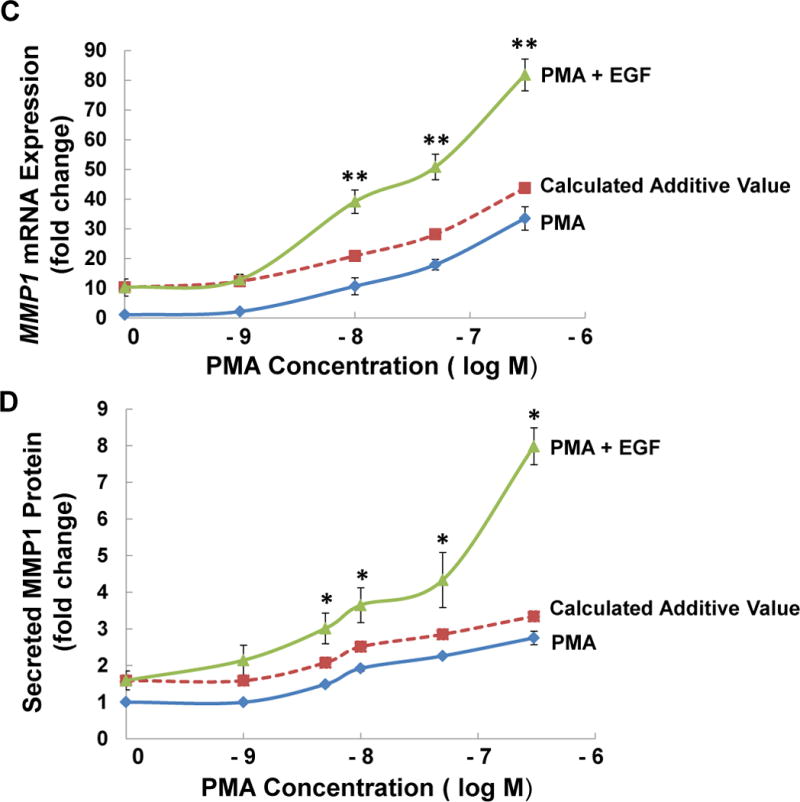
(A) HT-29 cells were pre-incubated with 5 μM atropine (Atr) for 45 min and then incubated for an additional 4 h with no test agents (C), or with ACh (100 μM), a PKC activator (50 nM PMA), or an EGFR activator (10 ng/ml EGF). MMP1 mRNA levels were measured by qPCR. Treating HT-29 cells with PMA or EGF increased levels of MMP1 mRNA. Whereas the effects of ACh were blocked by pre-incubating cells with atropine, PMA- and EGF-induced changes in MMP1 expression were unaffected. C, untreated control; ***P < 0.001. (B) Similar findings were observed when HT-29 cells were treated under the same conditions and MMP1 protein expression was measured using ELISA. C, untreated control; **P < 0.01. (C) HT-29 cells were treated with increasing concentrations of PMA, as indicated, for 4 h and MMP1 gene expression was measured by qPCR. Increasing concentrations of PMA progressively increased MMP1 gene expression. Adding 10 ng/ml of EGF to each concentration of PMA significantly potentiated MMP1 gene expression. The dashed line represents the calculated additive values for 10 ng/ml of EGF plus the indicated concentration of PMA. (D) Similar findings were observed when HT-29 cells were treated under the same conditions and secreted MMP1 protein was measured using ELISA. qPCR data were normalized to GAPDH. Data represent mean ± SE of at least three separate experiments. *P < 0.05, **P < 0.01 compared to calculated additive value.
We hypothesized that the effects of inhibitor combinations on ACh-induced MMP1 expression were most likely explained by potentiating interactions between post-M3R signaling pathways. In particular, a potentiating interaction between post-M3R PKC/p38 and EGFR/ERK signaling would explain the observations that blocking any component of these two signaling pathways attenuated ACh-induced MMP1 expression and simultaneously blocking both pathways abolished it. To test this hypothesis we designed experiments to activate PKC/p38 and EGFR/ERK signaling directly and determined the impact on MMP1 expression.
Simultaneous activation of PKC/p38 and EGFR/ERK signaling potentiates MMP1 gene and protein expression
Based on our findings that blocking activation of both PKC/p38 and EGFR/ERK signaling downstream of M3R modulated ACh-induced MMP1 expression, we hypothesized that incubating HT-29 cells with a combination of PMA, a phorbol ester that directly activates PKC, and EGF, an EGFR agonist, would potentiate MMP1 expression. First, we confirmed that the effects of PMA and EGF did not require activation of M3R; pre-incubating HT-29 cells with atropine alone did not alter MMP1 expression whereas pre-treating cells with atropine blocked ACh-induced MMP1 expression (Figure 5A). Treating HT-29 cells with PMA (50 nM) or EGF (10 ng/ml) increased levels of MMP1 mRNA and, as predicted, these changes were not blocked by pre-incubating cells with atropine (Figure 5A).
To confirm that inducing MMP1 mRNA expression resulted in similar effect on protein expression, we used ELISA to measure MMP1 protein levels in the culture media after stimulating HT-29 cells with test agents. As shown in Figure 5B, incubating cells with 100 μM ACh stimulated a greater than two-fold increase in the levels of secreted MMP1 (P < 0.01) that was completely blocked by pre-treating cells with 5 μM atropine. PMA and EGF stimulated approximately four- and two-fold increases in MMP1 release, respectively, effects that were not altered by pre-incubating cells with atropine (Figure 5B). Collectively, these findings indicate that activating M3R, PKC, and EGFR directly induces MMP1 gene and protein expression.
We next examined the effects of simultaneous direct activation of PKC and EGFR signaling. Based on the robust increase in MMP1 mRNA expression observed with independent activation of PKC and EGFR and the evidence for potentiating interactions suggested by the data in Figure 4, we hypothesized that simultaneously activating these two post-M3R signaling pathways would potentiate both MMP1 gene and protein expression. Indeed, as expected, incubating HT-29 cells with 10 ng/ml EGF plus concentrations of PMA greater than 1 nM significantly augmented MMP1 mRNA levels compared to the calculated additive value for those concentrations of PMA and EGF acting alone (Figure 5C). Moreover, the degree of potentiation appeared to increase in concert with increases in the concentration of PMA (Figure 5C). Similar findings were observed when we examined EGF- and PMA-induced changes in MMP1 protein release from HT-29 cells (Figure 5D). Augmented MMP1 release was first detected in cells incubated with 5 nM PMA plus 10 ng EGF/ml, and the degree of potentiation increased as the concentration of PMA was progressively increased (Figure 5D). These results indicate that direct co-stimulation of PKC and EGFR signaling potentiates increases in MMP1 mRNA and protein levels, a finding consistent with our hypothesis that potentiating interactions between these pathways mediate the effects of M3R activation and explain the findings shown in Figure 4.
PKC regulates p38 and EGR/ERK activation and potentiation of post-M3R MMP1 expression
To investigate further the potentiating interactions that augment MMP1 mRNA and protein expression we explored post-PKC signaling pathways. Prior studies reporting that PKC cross-talk with EGFR may be mediated by Src [39, 40] led us to consider the likelihood that Src plays an important intermediary role in post-M3R induction of MMP1 expression in colon cancer. To address this we used chemical inhibitors to parse the relative contributions of EGFR, MEK/ERK1/2, p38, and Src signaling to MMP1 mRNA expression in PMA-activated cells. As shown in Figure 6A, as we anticipated pre-incubating HT-29 cells with PKC-α/β1 inhibitors (5 μM Gӧ6976 and 5 μM Gӧ6983) abolished PMA-induced increases in MMP1 gene expression. Pre-incubating cells with inhibitors of p38-α/β (10 μM SB202190, 10 μM SB203580), EGFR (5 μM PD168393, 5 μM PD153035), MEK (10 μM PD98059, 10 μM U0126), and Src (10 μM PP2) alone significantly attenuated PMA-induced increases in levels of MMP1 mRNA (Figure 6A). These findings were consistent with two discrete actions of PKC, downstream activation of p38 and Src-mediated cross-talk with EGFR/MEK/ERK signaling. As predicted, PMA-induced MMP1 gene expression was abolished when we tested this hypothesis by simultaneously blocking p38-α/β plus either EGFR or MEK/ERK1/2 signaling (Figure 6A).
Figure 6. Potentiating interactions between post-PKC signaling pathways govern MMP1 gene induction.
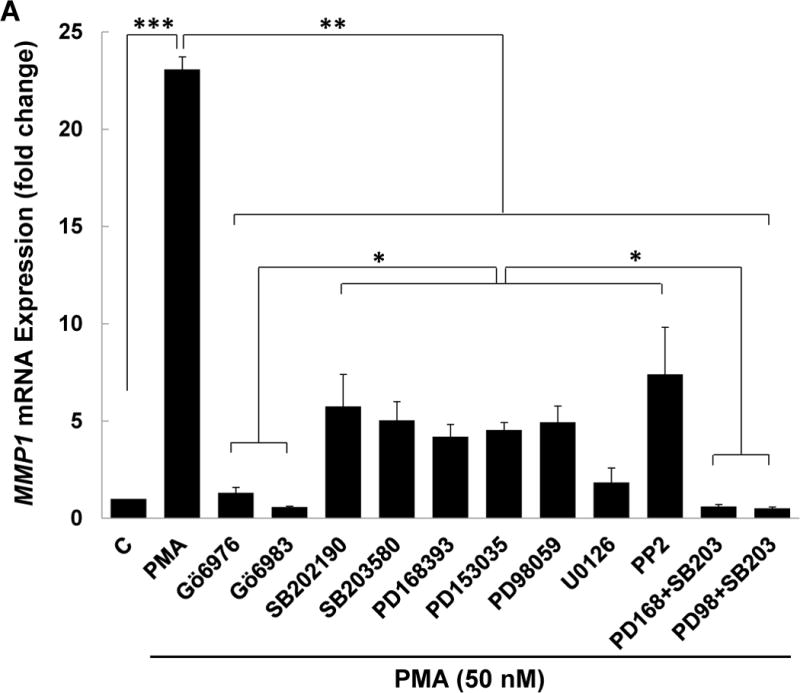
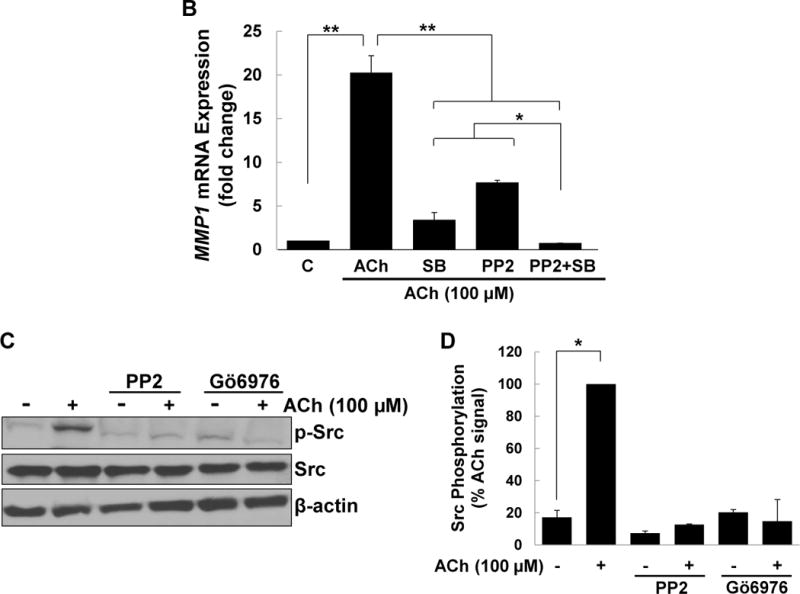
(A) HT-29 cells were pre-incubated for 45 min alone or with PKC-α/β1 inhibitors (5 μM Gӧ6976, 5 μM Gӧ6983), p38-α/β inhibitors (10 μM SB202190, 10 μM SB203580), EGFR inhibitors (5 μM PD168393, and PD153035), MEK inhibitors (10 μM PD98059, 10 μM U0126), or a Src inhibitor (10 μM PP2), alone or in combination, before adding 50 nM PMA for an additional 4-h incubation. MMP1 mRNA levels were measured by qPCR. PMA-induced MMP1 gene expression was abolished by pre-incubating cells with PKC-α/β1 inhibitors and attenuated by pre-incubating cells with inhibitors of p38-α/β, EGFR, MEK, and Src. Simultaneously blocking p38-α/β and EGFR, p38-α/β and MEK, or p38-α/β and Src abolished PMA-induced MMP1 gene expression. (B) HT-29 cells were pre-incubated for 45 min alone or with a p38-α/β (10 μM SB203580) or Src (10 μM PP2) inhibitor, alone or in combination, followed by an additional 4-h incubation with 100 μM ACh. MMP1 mRNA levels were measured by qPCR. ACh-induced MMP1 expression was attenuated by pre-incubation with p38-α/β and Src inhibitors and abolished by these inhibitors in combination. qPCR data were normalized to GAPDH and are the mean ± SE of three separate experiments. C, untreated control; *P < 0.05, **P < 0.01. (C) H508 cells were pre-incubated for 45 min with inhibitors of Src (10 μM PP2) and PKC-α/β1 (5 μM Gö6976) before adding ACh (100 μM) for an additional 5-min incubation. Cell extracts were immunoblotted with antibodies against phosphorylated and total Src. (D) Densitometry of immunoblots from two separate experiments shows ACh-induced Src phosphorylation was blocked by pre-incubating cells with Src and PKC-α/β1 inhibitors. *P < 0.05 compared to 100 μM ACh alone.
To confirm the role of Src, we examined the actions of blocking this intermediary on ACh-induced MMP1 gene expression. As shown in Figure 6B, pre-incubating cells with either a p38-α/β or Src inhibitor robustly attenuated ACh-induced MMP1 gene expression. Notably, pre-incubating cells with the combination of the p38-α/β and Src inhibitors abolished ACh-induced MMP1 expression (Figure 6B), similar to the effects of combining the Src and p38-α/β inhibitors on PMA-induced MMP1 expression (Figure 6A). Moreover, immunoblotting revealed that ACh treatment stimulated Src phosphorylation, an action blocked by pre-incubating cells with the Src inhibitor PP2 or the PKC-α/β1 inhibitor Gö6976; these interventions did not alter levels of total Src (Figure 6C, D). These findings strongly support the hypothesis that in human colon cancer cells Src serves as a key intermediary between PKC-α and EGFR/MEK/ERK signaling.
Collectively, these observations identify PKC-α as a central regulator of post-M3R signaling. Once activated, PKC-α stimulates Src-mediated activation of EGFR/MEK/ERK1/2 signaling that induces MMP1 gene expression. By a separate, distinct mechanism PKC-α activates p38-α/β signaling that also induces MMP1 gene expression. Simultaneous activation of EGFR/MEK/ERK1/2 and p38-α/β signaling downstream of PKC-α, as occurs following M3R activation, potentiates MMP1 gene expression.
Identification of p38-α as the p38 isoform relevant to potentiating interactions in colon cancer cells
Since the inhibitors we used block both α and β isoforms of p38, we employed siRNA knockdown of p38-α to define more clearly the role of this isoform in mediating M3R agonist-induced increases in MMP1 expression. As shown in Figure 7A, following HT-29 cell transfection with siRNA we were able to knock down p38-α expression by more than 90%; transfecting cells with non-targeting ‘mock’ siRNA did not substantially alter levels of total p38 or p38-α. Immunoblotting for p38-β resulted in a weak signal that was not substantially altered by siRNA knockdown of p38-α (Figure 7A). These findings identify p38-α as the predominant isoform in this cell line, a supposition confirmed by markedly reduced expression of total p38 following siRNA knockdown of p38-α (Figure 7A).
Figure 7. Effects of p38-α knockdown on ACh- and PMA-induced MMP1 expression.
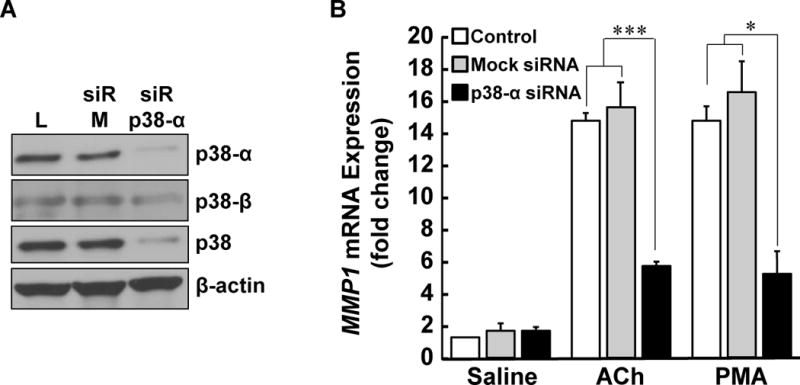
(A) HT-29 cells were transfected with Lipofectamine (L), 50 pmoles non-targeting mock siRNA (siR-M), or 50 pmoles siRNA targeting p38-α (siR-p38-α). p38-α, p38-β, total p38, and β-actin expression were measured by immunoblotting with specific antibodies. (B) HT-29 cells transfected with Lipofectamine (control), non-targeting mock siRNA, and siRNA targeting p38-α were incubated with saline, 100 μM ACh, or 50 nM PMA for 4 h at 37°C. MMP1 mRNA levels were measured by qPCR. qPCR data were normalized to GAPDH and are means of four separate experiments. *P < 0.05; ***P < 0.001.
As shown in Figure 7B, transfecting HT-29 cells with mock or p38-α siRNA did not alter basal levels of MMP1 mRNA. In contrast, transfecting cells with siRNA targeting p38-α significantly reduced both ACh- and PMA-induced expression of MMP1 mRNA, an effect not observed following transfection with non-targeting mock siRNA (Figure 7B). These findings confirm the p38-α isoform mediates downstream actions of M3R and PKC activation on MMP1 expression.
Potentiating interactions between EGFR/ERK and PKC/p38 signaling augment colon cancer cell invasion
Lastly, we explored the functional consequences of the potentiating interactions we uncovered. Invasion is an important cellular attribute required for colon cancer cell dissemination to other organs such as the liver. Our previous work supported a role for M3R signaling in stimulating colon cancer cell invasion; blocking M3R activation with atropine or the actions of MMP1 with a neutralizing antibody abolished muscarinic agonist-induced colon cancer cell invasion [17, 27]. Here, we compared the impact of selectively blocking the post-M3R signal transduction pathways studied above on ACh-induced HT-29 cell invasion. We measured cell invasion using a Boyden chamber/Matrigel assay and quantified changes with imaging software after 48-h incubation with test agents.
As shown in Figure 8A, we first confirmed that treatment with 100 μM ACh robustly stimulated HT-29 cell invasion and that this was completely blocked by pre-incubating cells with atropine. Then we confirmed that, as we observed for changes in MMP1 mRNA (Figure 5C) and MMP1 protein (Figure 5D), the combination of PMA and EGF potentiated cell invasion (Figure 8B); that is, whereas PMA and EGF induced ~5- and 2-fold increases in cell invasion individually, when added together these PKC and EGFR agonists stimulated a 10-fold increase in cell invasion. The combination of PMA and EGF stimulated cell invasion to a greater degree than the additive value predicted from the results of these agents acting alone.
Figure 8. Interactions between PKC and EGFR signaling potentiate colon cancer cell invasion.
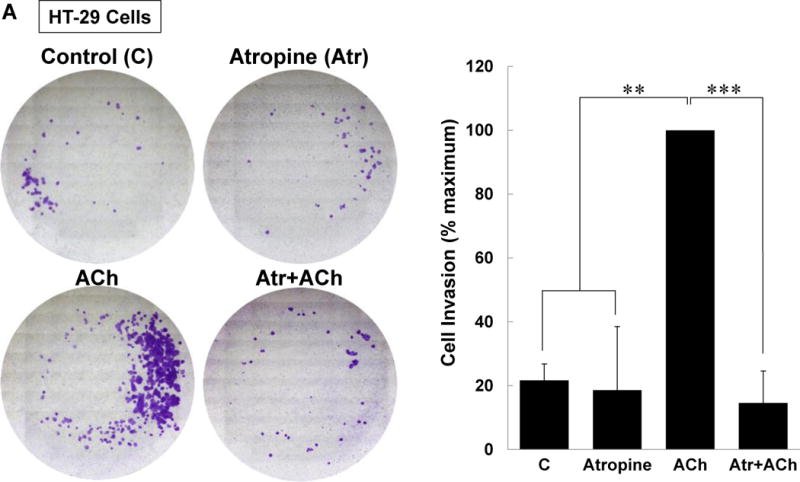
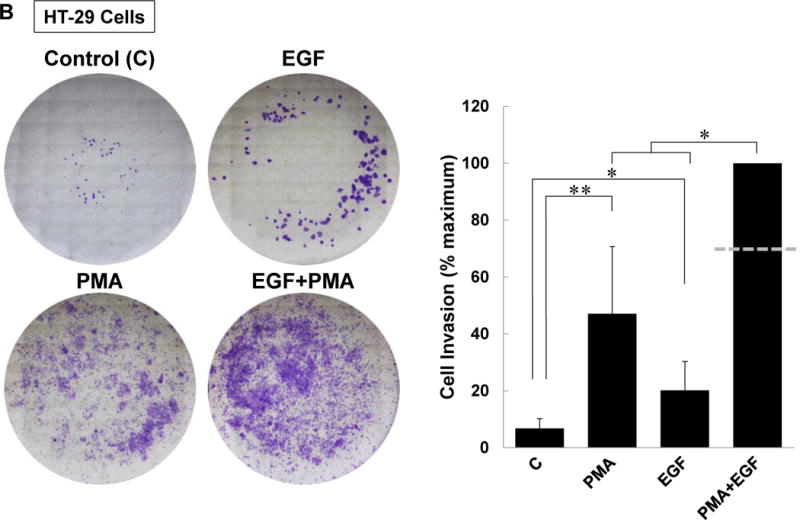
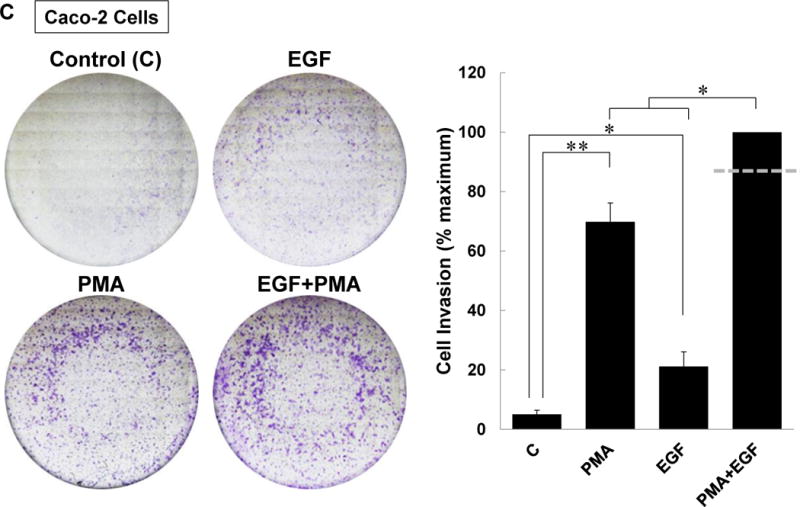
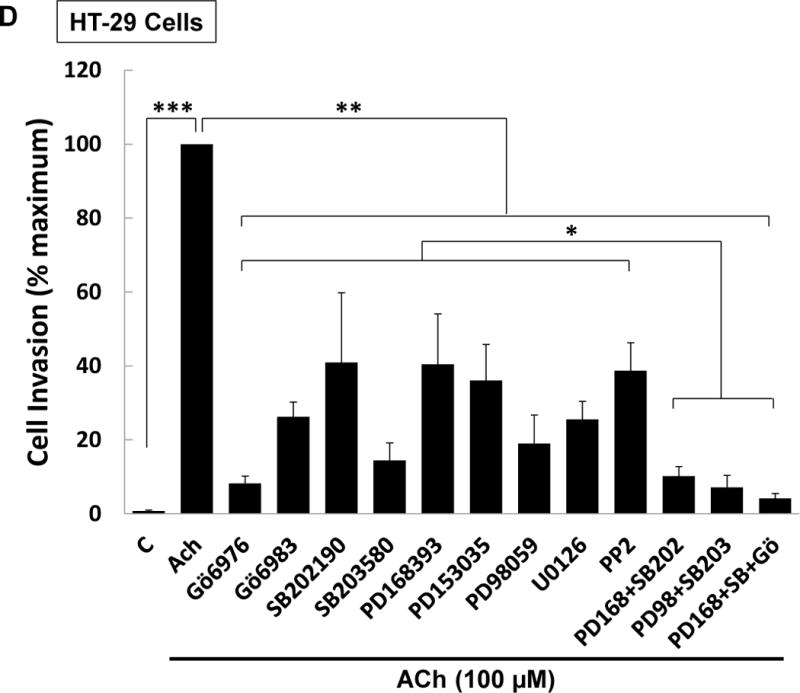
(A) ACh-induced colon cancer cell invasion is blocked by atropine. HT-29 cells were pre-incubated with 5 μM atropine (Atr) for 45 min and then incubated for an additional 48 h with no test agents, or with 100 μM ACh. Atropine alone had no effect but blocked ACh-induced cell invasion. (B) Directly activating PKC plus EGFR potentiated colon cancer cell invasion. HT-29 cells were incubated with PMA (50 nM) and EGF (10 ng/ml), alone or in combination, for 48 h. Simultaneous activation of PKC and EGFR significantly potentiates cell invasion; the dashed line represents the calculated additive value for PMA plus EGF (P < 0.05 for the observed vs. the calculated addition of PMA plus EGF). (C) Caco-2 cells were incubated with PMA (50 nM) and EGF (10 ng/ml), alone or in combination, for 48 h. Simultaneous activation of PKC and EGFR signaling potentiated cell invasion; the dashed line represents the calculated additive value for PMA plus EGF (P < 0.05 for the observed vs. the calculated addition of PMA plus EGF). (D) ACh-induced colon cancer cell invasion is blocked by inhibitors of PKC-α/β1 (5 μM Gӧ6976, 5 μM Gӧ6983), p38-α/β (10 μM SB202190, 10 μM SB203580), EGFR (5 μM PD168393, 5 μM PD153035), MEK (10 μM PD98059, 10 μM U0126), and Src (10 μM PP2). Pre-incubating cells with a combination of p38-α/β plus EGFR inhibitors or p38-α/β plus MEK inhibitors nearly abolished ACh-induced cell invasion, as did pre-incubation with a combination of EGFR, p38-α/β, and PKC-α/β1 inhibitors. In this set of experiments, cell invasion was measured using BD Biocoat Invasion Chambers with Matrigel inserts. Representative images of crystal violet-stained HT-29 cells that invaded through Matrigel inserts are shown for Fig. 8A–C. Data shown in bar graphs represent mean ± SE of at least three separate experiments. C, untreated control; *P < 0.05; **P < 0.01; ***P < 0.001.
We sought to validate the results shown in Figure 8B with another colon cancer cell line but H508 cells are too large to pass through 8-micron pores in Boyden chambers. To address this technical limitation, we used smaller Caco-2 human colon cancer cells. Supplemental Figure 3 confirms that the combination of PMA plus EGF stimulates potentiation of MMP1 expression in Caco-2 cells. As shown in Figure 8C, similar to our observations in HT-29 cells, both PMA and EGF stimulated significant increases in Caco-2 cell invasion that were augmented by treating cells with the combination of these agents. Altogether, these findings confirm that potentiating interactions between post-M3R signaling pathways augment colon cancer cell invasion.
Finally, we examined the actions of pre-incubating HT-29 cells with selective inhibitors of post-M3R signaling on ACh-induced cell invasion. None of the inhibitors used in the invasion assays altered basal cell invasion (Supplemental Figure 1C). As shown in Figure 8D, the results of this experiment were consistent with potentiating interactions between post-M3R signaling pathways. Moreover, the findings strongly suggested that these potentiating interactions are central to ACh-induced cell invasion. That is, whereas inhibiting only PKC-α, p38-α, EGFR, or MEK/ERK1/2 activation attenuated ACh-induced cell invasion, combining inhibitors to block EGFR plus p38-α, MEK plus p38-α, or Src plus p38-α nearly abolished cell invasion (Fig. 8D). Thus, achieving maximal ACh-induced colon cancer cell invasion appears to depend largely on potentiating interactions between post-M3R signaling pathways.
DISCUSSION
No specific genetic mutations convert a primary colon tumor into an invasive metastatic cancer [41]. Rather, it appears that cancer cells that already possess the ability to invade adjacent normal tissue have metastatic potential and the more invasive the phenotype the more likely tumor cells will metastasize to other organs. Matrix metalloproteinases, which encompass a family of zinc-dependent endopeptidases that play critical roles in a wide variety of physiological and pathological processes, are key drivers of cell invasion [42]. Whereas low levels of MMP1 are important for tissue remodeling, in cancer increased levels of MMP1 facilitate cell invasion and dissemination; hence, it is not surprising that the level of MMP1 expression correlates with advanced colon cancer stage, metastasis, and adverse outcome [43–45].
Our group showed that M3R agonists like acetylcholine that promote colon cancer cell proliferation, survival, migration, and invasion, are also highly efficacious regulators of MMP1 expression [26]. In the colon, these actions may be mediated by neurocrine, paracrine, or autocrine release of acetylcholine [46, 47] or alternatively by the actions of luminal bile acids that may also interact functionally with muscarinic receptors on neoplastic epithelial cells and induce MMP1 gene expression [27, 48]. In a mouse model, we found that bethanechol, another M3R agonist, robustly augmented colon neoplasia and expression of Mmp13, the murine homologue of MMP1 [19]. Blocking either M3R or MMP1 activation in human colon cancer cells abolished M3R agonist-induced colon cancer cell invasion [27], confirming that MMP1 plays a critical role in mediating these effects of M3R activation. Overall, we acquired a strong body of in vitro and in vivo evidence supporting a functional M3R/MMP1 axis that promotes colon cancer cell invasion.
The present communication addresses the regulation of this M3R/MMP1 axis – we found that M3R agonist-induced transcriptional activation of MMP1 is regulated by a complex set of potentiating interactions between post-M3R signaling pathways. To validate our findings, we used at least two selective inhibitors to block key molecules in at least two colon cancer cell lines, used selective activators of these signaling molecules to verify their roles, and confirmed a key finding using siRNA knockdown. In so doing we identified PKC-α as a focal point of the signaling cascade downstream of M3R activation. ACh-induced MMP1 expression was abolished by blocking activation of PKC-α directly, or by blocking downstream activation of p38-α and either EGFR or ERK1/2.
As anticipated from these findings, direct co-activation of PKC and EGFR robustly potentiated both MMP1 gene and protein expression, and augmented cell invasion. Consistent with reports that PKC cross-talk with EGFR may be mediated by Src, we found that ACh-induced MMP1 expression and cell invasion were attenuated by inhibiting Src and totally blocked by concurrently inhibiting p38-α plus Src; similar findings were observed for ACh-induced MMP1 expression. As expected from these findings, we also discovered that ACh-induced Src phosphorylation (activation) is blocked by inhibiting activation of PKC-α. Collectively, these studies uncover novel functional interactions between post-M3R signaling pathways involving PKC-α-mediated p38-α activation and Src-mediated cross-talk between PKC-α and EGFR/ERK1/2 signaling which augment expression of MMP1, and drive cell invasion (Figure 9).
Figure 9. Potentiating interactions between post-M3R signaling pathways govern MMP1 gene transcription and colon cancer cell invasion.
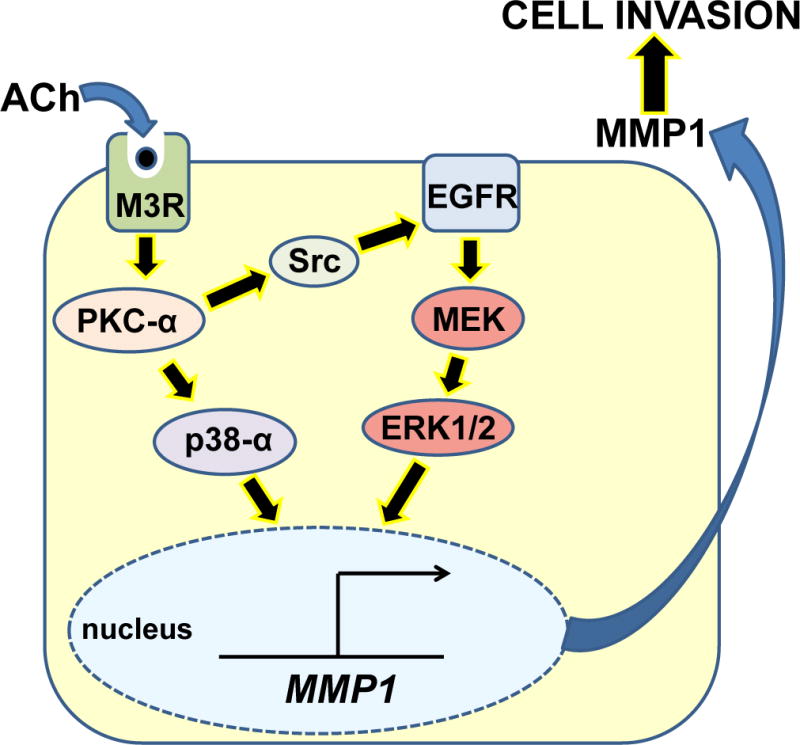
The interaction of a muscarinic receptor agonist (acetylcholine; ACh) with M3 muscarinic receptors triggers downstream PKC-α activation which, in turn, activates Src and p38-α. Src interaction with EGFR stimulates signaling through the MEK/ERK1/2 pathway. Activated p38-α and ERK1/2 coordinately induce MMP1 gene transcription and enhance MMP1 protein expression. Besides degrading interstitial collagen and facilitating cell invasion, the resulting augmented release of MMP1 into the colon cancer cell microenvironment may catalyze release of EGFR ligands and further enhance ERFR activation. These interactions between post-M3R signaling pathways potentiate MMP1 expression and release, and culminate in augmented cancer cell invasion, thereby providing potential rescue mechanisms in the event that only one post-M3R signal transduction pathway is blocked.
Members of the PKC family regulate basic cellular processes including proliferation differentiation, survival, and migration [49]. In particular, activation of PKC-α has been reported to promote colon cancer progression [50–52] and resistance to chemotherapy [53]. Nonetheless, although PKC isozyme expression is altered in many types of cancer, the role of PKC in the initiation and progression of cancers remains uncertain [54]. Indeed, it appears that fundamental actions of PKC are isoenzyme- and context-dependent; in some cancer cells PKC isozymes act as tumor promoters whereas in others they are tumor suppressors [55]. In concert with our findings, in cell lines derived from cancers of the breast, liver, brain, and skin, PKC-α stimulates cell invasion [55]. Our data show that indirect activation of PKC-α as part of a post-M3R signaling cascade or direct activation of PKC using the phorbol ester PMA stimulates colon cancer cell invasion. Moreover, we find that this effect is mediated by downstream activation of separate circuits involving both EGFR/ERK1/2 and p38-α signaling, the former mediated by Src activation (Figure 5). These findings highlight the key concept reviewed by Hannahan and Weinberg [5] – the mechanistic underpinnings of different stages in cancer progression (e.g. cell proliferation vs. cell invasion) may be very different and a particular molecule or pathway may be pro-neoplastic at one stage and anti-neoplastic at another. There are now several examples of the context-dependent, dual nature of key signaling molecules. A recent report suggests that reactive oxygen species (ROS) may promote cancer cell proliferation but inhibit cell invasion and dissemination [56]. TGF-β may be either pro-tumorigenic or tumor suppressive depending on Sox4 co-expression [57]. Likewise, PKC-α may play seemingly opposing roles in mediating cell proliferation and invasion.
Four p38 isozymes, designated α, β, γ, and δ, have been identified. The principal p38 inhibitor used in our studies is p38-α/β-selective and our siRNA knockdown data (Figure 7) provides strong evidence that the p38-α isoform is the downstream effector of PKC-induced MMP1 gene expression in colon cancer cells. Although not previously linked to muscarinic receptor agonist-induced induction of matrix metalloproteinase genes in colon cancer, p38-α is reported to play a role in interleukin-1-induced MMP1 gene transcription in fibroblasts and endothelial cells [58], plays a critical role in squamous epithelial cell invasion [59], and mediates an H-Ras-induced invasive phenotype in breast epithelial cells [60]. Moreover, PKC-α is reported to alter p38 activity in microglia [61], macrophages [62], vascular smooth muscle cells [63], and mast cells [64]. These findings in other cell types provide additional support for our findings that upstream activation of PKC-α regulates activation of p38-α in colon cancer cells.
Collectively, our results led us to propose the model shown in Figure 9 wherein signaling interactions downstream of M3R activation promote enhanced transcription of MMP1, increased release of MMP1 into the cell microenvironment, and augmented cell invasion. Activation of PKC-α provokes cooperative signaling that results from Src-dependent activation of the EGFR/ERK1/2 pathway and EGFR-independent activation of p38-α. The robust 30- to 50-fold increase in MMP1 gene expression observed following ACh interaction with M3R can be mimicked by directly co-activating PKC-α and EGFR/ERK1/2. This amplified signaling network results in potentiated levels of MMP1 mRNA and protein, and enhanced colon cancer cell invasion.
Cooperative interactions between transcription factors have long been implicated in gene regulation [65, 66]. Thus, we believe the most likely explanation for the findings described herein is that the coordinated recruitment of downstream transcription factors and cofactors to regulatory elements of the MMP1 gene mediates the potentiating interaction of post-muscarinic receptor signaling pathways. Although beyond the scope of the present work, this speculation will be tested in future experiments.
Finally, although M1R and M3R may both be expressed by gastrointestinal epithelial cells, our work and the cartoon depicted in Figure 9 identifies M3R as the operative muscarinic receptor subtype that regulates the signaling cascades studied herein. This may be considered speculative as acetylcholine is a non-selective M3R agonist. Nonetheless, we believe this conclusion is justified based on the strong body of evidence that M3R regulates these signaling cascades in human colon cancer cells [13, 46, 48], the identification of M3R as the primary M3R subtype expressed in both HT-29 and H508 colon cancer cells [17], and the key finding in animal models that M3R deficiency robustly attenuates the development of colon neoplasia whereas M1R deficiency has no effect [11, 18, 20].
Medicinal approaches to treating cancer using broad-spectrum MMP inhibitors have met with failure and selective therapeutic targeting of MMP1 has not yet been achieved [42]. Whereas EGFR inhibitors have demonstrated efficacy for colon cancer, these effects are commonly transient and emerging drug resistance generally results in tumor resurgence, cancer dissemination, and death. Thus, understanding the mechanisms whereby colon cancer cells develop resistance to EGFR-based therapies is important. Several such mechanisms have been elucidated, including downstream activating mutations in RAS and BRAF, c-MET signaling, and the mutation, hyperactivation or over-expression of EGFR itself. Our findings suggest that convergence of signaling pathways with molecules downstream of EGFR may also play a role. In this instance, activation of PKC-α downstream of M3R stimulates EGFR-independent activation of p38-α and induction of MMP1 gene expression, a series of events that favor cell invasion and dissemination. Our findings suggest that combining an agent that blocks EGFR activity with an agent that blocks either M3R, PKC-α, or p38-α activity may have therapeutic promise for preventing or retarding colon cancer cell metastasis or blocking cancer cell escape when resistance to one therapy (e.g. EGFR inhibitors) emerges. Our findings provide additional support for the notion that combination therapy simultaneously targeting several pathways is needed to incapacitate redundant post-receptor signal transduction mechanisms that regulate MMP1 expression in colon cancer.
Supplementary Material
Supplemental Figure 1. Actions of inhibitors alone. Pre-incubating HT-29 colon cancer cells for 45 min with inhibitors of PKC-α/β1 (5 μM Gӧ6976, 5 μM Gӧ6983), p38-α/β (10 μM SB203580, 10 μM SB202190), EGFR (5 μM PD168393, 5 μM PD153035), and MEK (10 μM U0126, 10 μM PD98059) alone did not alter p38 phosphorylation (A), MMP1 mRNA expression (B), or cell invasion (C). ACh (100 μM) is used as a positive control in these experiments. Measurement of p38 phosphorylation by immunoblotting, MMP1 mRNA expression by qPCR, and cell invasion using Boyden chambers with Matrigel inserts was performed as described in Materials and Methods.
Supplemental Figure 2. Effect of inhibitors on MMP1 mRNA expression in H508 human colon cancer cells. H508 cells were pre-incubated for 45 min with PKC-α/β1, p38-α/β, EGFR, and MEK inhibitors and then incubated for an additional 4 h with no additional test agents (C) or with ACh (100 μM). MMP1 mRNA levels were measured by qPCR. ACh-induced MMP1 expression was attenuated by inhibitors of PKC-α/β1 (5 μM Gӧ6976, 5 μM Gӧ6983), p38-α/β (10 μM SB202190, 10 μM SB203580), EGFR (5 μM PD168393, 5 μM PD153035), and MEK (10 μM PD98059, 10 μM U0126). Pre-incubating cells with a combination of p38-α/β plus EGFR inhibitors or p38-α/β plus MEK inhibitors abolished ACh-induced MMP1 gene expression, as did pre-incubation with a combination of p38-α/β, EGFR and PKC-α/β1 inhibitors. qPCR data were normalized to GAPDH and are means of three separate experiments. C, untreated control; *P < 0.05.
Supplemental Figure 3. Directly activating PKC plus EGFR potentiates MMP1 expression in Caco-2 cells. Caco-2 cells were incubated with PMA (50 nM) and EGF (10 ng/ml), alone or in combination, for 4h. MMP1 mRNA levels were measured by qPCR. Simultaneous activation of PKC and EGFR signaling significantly potentiates MMP1 mRNA levels; the dashed line represents the calculated additive value for PMA plus EGF (P<0.05 for the observed vs. the calculated addition of PMA plus EGF). Results are representative of three separate experiments. qPCR data were normalized to GAPDH and are the means ± SE of three separate experiments. C, untreated control; *P < 0.05, **P < 0.01.
Supplemental Table 1. Inhibitors used in this study.
Acknowledgments
FUNDING
This work was supported in part by Merit Review Awards # BX002129 and BX002777 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This work was also supported by NIDDK-T32-DK067872 and NIDDK-R21-DK093406.
ABBREVIATIONS
- ACh
acetylcholine
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-related kinase
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen activated protein kinase
- MMP1
matrix metalloproteinase-1
- M3R
subtype 3 muscarinic receptors
- p38
p38 MAPK
- PI3K
phosphoinositol-3 kinase
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- qPCR
quantitative real-time PCR
Footnotes
AUTHOR CONTRIBUTIONS
Anan H. Said, Shien Hu, Ameer Abutaleb, Tonya Watkins, Kunrong Cheng, and Panjamurthy Kuppusamy performed experiments and analyzed data. Ahmed Chahdi, Neeraj Saxena, Guofeng Xie, and Jean-Pierre Raufman developed the concepts. Anan Said, Shien Hu, and Jean-Pierre Raufman prepared and edited (pre-submission) the paper. All authors reviewed the results and approved the final version of the manuscript.
References
- 1.Peery AF, Crockett SD, Barritt AS, Dellon ES, Eluri S, Gangarosa LM, Jensen ET, Lund JL, Pasricha S, Runge T, Schmidt M, Shaheen NJ, Sandler RS. Burden of Gastrointestinal, Liver, and Pancreatic Diseases in the United States. Gastroenterology. 2015;149:1731–1741 e1733. doi: 10.1053/j.gastro.2015.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, Boscoe FP, Cronin KA, Lake A, Noone AM, Henley SJ, Eheman CR, Anderson RN, Penberthy L. Annual Report to the Nation on the Status of Cancer, 1975–2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv048. djv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayat MJ, Howlader N, Reichman ME, Edwards BK. Cancer statistics, trends, and multiple primary cancer analyses from the Surveillance, Epidemiology, and End Results (SEER) Program. Oncologist. 2007;12:20–37. doi: 10.1634/theoncologist.12-1-20. [DOI] [PubMed] [Google Scholar]
- 4.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, Traulsen A, Nowak MA, Siegel C, Velculescu VE, Kinzler KW, Vogelstein B, Willis J, Markowitz SD. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruse AC, Li J, Hu J, Kobilka BK, Wess J. Novel insights into M3 muscarinic acetylcholine receptor physiology and structure. J Mol Neurosci. 2014;53:316–323. doi: 10.1007/s12031-013-0127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutkind JS, Novotny EA, Brann MR, Robbins KC. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc Natl Acad Sci U S A. 1991;88:4703–4707. doi: 10.1073/pnas.88.11.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frucht H, Jensen RT, Dexter D, Yang WL, Xiao Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin Cancer Res. 1999;5:2532–2539. [PubMed] [Google Scholar]
- 10.Yang WL, Frucht H. Cholinergic receptor up-regulates COX-2 expression and prostaglandin E(2) production in colon cancer cells. Carcinogenesis. 2000;21:1789–1793. doi: 10.1093/carcin/21.10.1789. [DOI] [PubMed] [Google Scholar]
- 11.Cheng K, Xie G, Khurana S, Heath J, Drachenberg CB, Timmons J, Shah N, Raufman JP. Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol Cancer. 2014;13:77. doi: 10.1186/1476-4598-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raufman JP, Cheng K, Zimniak P. Activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig Dis Sci. 2003;48:1431–1444. doi: 10.1023/a:1024733500950. [DOI] [PubMed] [Google Scholar]
- 13.Cheng K, Chen Y, Zimniak P, Raufman JP, Xiao Y, Frucht H. Functional interaction of lithocholic acid conjugates with M3 muscarinic receptors on a human colon cancer cell line. Biochim Biophys Acta. 2002;1588:48–55. doi: 10.1016/s0925-4439(02)00115-1. [DOI] [PubMed] [Google Scholar]
- 14.Raufman JP, Chen Y, Cheng K, Compadre C, Compadre L, Zimniak P. Selective interaction of bile acids with muscarinic receptors: a case of molecular mimicry. Eur J Pharmacol. 2002;457:77–84. doi: 10.1016/s0014-2999(02)02690-0. [DOI] [PubMed] [Google Scholar]
- 15.Raufman JP, Shant J, Guo CY, Roy S, Cheng K. Deoxycholyltaurine rescues human colon cancer cells from apoptosis by activating EGFR-dependent PI3K/Akt signaling. J Cell Physiol. 2008;215:538–549. doi: 10.1002/jcp.21332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shant J, Cheng K, Marasa BS, Wang JY, Raufman JP. Akt-dependent NF-kappaB activation is required for bile acids to rescue colon cancer cells from stress-induced apoptosis. Exp Cell Res. 2009;315:432–450. doi: 10.1016/j.yexcr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belo A, Cheng K, Chahdi A, Shant J, Xie G, Khurana S, Raufman JP. Muscarinic receptor agonists stimulate human colon cancer cell migration and invasion. Am J Physiol Gastrointest Liver Physiol. 2011;300:G749–760. doi: 10.1152/ajpgi.00306.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raufman JP, Samimi R, Shah N, Khurana S, Shant J, Drachenberg C, Xie G, Wess J, Cheng K. Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res. 2008;68:3573–3578. doi: 10.1158/0008-5472.CAN-07-6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng Z, Heath J, Drachenberg C, Raufman JP, Xie G. Cholinergic muscarinic receptor activation augments murine intestinal epithelial cell proliferation and tumorigenesis. BMC Cancer. 2013;13:204. doi: 10.1186/1471-2407-13-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raufman JP, Shant J, Xie G, Cheng K, Gao XM, Shiu B, Shah N, Drachenberg CB, Heath J, Wess J, Khurana S. Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis. 2011;32:1396–1402. doi: 10.1093/carcin/bgr118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overland AC, Insel PA. Heterotrimeric G proteins directly regulate MMP14/membrane type-1 matrix metalloprotease: a novel mechanism for GPCR-EGFR transactivation. J Biol Chem. 2015;290:9941–9947. doi: 10.1074/jbc.C115.647073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J Cell Biol. 2012;196:375–385. doi: 10.1083/jcb.201105153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Said AH, Raufman JP, Xie G. The role of matrix metalloproteinases in colorectal cancer. Cancers (Basel) 2014;6:366–375. doi: 10.3390/cancers6010366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar JD, Steele I, Moore AR, Murugesan SV, Rakonczay Z, Venglovecz V, Pritchard DM, Dimaline R, Tiszlavicz L, Varro A, Dockray GJ. Gastrin stimulates MMP-1 expression in gastric epithelial cells: putative role in gastric epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2015;309:G78–86. doi: 10.1152/ajpgi.00084.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klekotka PA, Santoro SA, Zutter MM. alpha 2 integrin subunit cytoplasmic domain-dependent cellular migration requires p38 MAPK. J Biol Chem. 2001;276:9503–9511. doi: 10.1074/jbc.M006286200. [DOI] [PubMed] [Google Scholar]
- 26.Xie G, Cheng K, Shant J, Raufman JP. Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am J Physiol Gastrointest Liver Physiol. 2009;296:G755–763. doi: 10.1152/ajpgi.90519.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raufman JP, Cheng K, Saxena N, Chahdi A, Belo A, Khurana S, Xie G. Muscarinic receptor agonists stimulate matrix metalloproteinase 1-dependent invasion of human colon cancer cells. Biochem Biophys Res Commun. 2011;415:319–324. doi: 10.1016/j.bbrc.2011.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 29.Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002;277:49212–49219. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- 30.Arechederra M, Priego N, Vazquez-Carballo A, Sequera C, Gutierrez-Uzquiza A, Cerezo-Guisado MI, Ortiz-Rivero S, Roncero C, Cuenda A, Guerrero C, Porras A. p38 MAPK down-regulates fibulin 3 expression through methylation of gene regulatory sequences: role in migration and invasion. J Biol Chem. 2015;290:4383–4397. doi: 10.1074/jbc.M114.582239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rousseau S, Dolado I, Beardmore V, Shpiro N, Marquez R, Nebreda AR, Arthur JS, Case LM, Tessier-Lavigne M, Gaestel M, Cuenda A, Cohen P. CXCL12 and C5a trigger cell migration via a PAK1/2-p38alpha MAPK-MAPKAP-K2-HSP27 pathway. Cell Signal. 2006;18:1897–1905. doi: 10.1016/j.cellsig.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Roomans GM. Evidence for muscarinic 3 receptor mediated ion transport in HT29 cells studied by X-ray microanalysis. Cell Struct Funct. 1997;22:379–385. doi: 10.1247/csf.22.379. [DOI] [PubMed] [Google Scholar]
- 33.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slack BE. The m3 muscarinic acetylcholine receptor is coupled to mitogen-activated protein kinase via protein kinase C and epidermal growth factor receptor kinase. Biochem J. 2000;348(Pt 2):381–387. [PMC free article] [PubMed] [Google Scholar]
- 35.Nakajima K, Jain S, Ruiz de Azua I, McMillin SM, Rossi M, Wess J. Minireview: Novel aspects of M3 muscarinic receptor signaling in pancreatic beta-cells. Mol Endocrinol. 2013;27:1208–1216. doi: 10.1210/me.2013-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi PM, Tchou-Wong KM, Weinstein IB. Overexpression of protein kinase C in HT29 colon cancer cells causes growth inhibition and tumor suppression. Mol Cell Biol. 1990;10:4650–4657. doi: 10.1128/mcb.10.9.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peiffer LP, Peters DJ, McGarrity TJ. Differential effects of deoxycholic acid on proliferation of neoplastic and differentiated colonocytes in vitro. Dig Dis Sci. 1997;42:2234–2240. doi: 10.1023/a:1018806431866. [DOI] [PubMed] [Google Scholar]
- 38.Hong DH, Forstner JF, Forstner GG. Protein kinase C-epsilon is the likely mediator of mucin exocytosis in human colonic cell lines. Am J Physiol. 1997;272:G31–37. doi: 10.1152/ajpgi.1997.272.1.G31. [DOI] [PubMed] [Google Scholar]
- 39.Xiao H, Bai XH, Wang Y, Kim H, Mak AS, Liu M. MEK/ERK pathway mediates PKC activation-induced recruitment of PKCzeta and MMP-9 to podosomes. J Cell Physiol. 2013;228:416–427. doi: 10.1002/jcp.24146. [DOI] [PubMed] [Google Scholar]
- 40.Hsieh HL, Sun CC, Wang TS, Yang CM. PKC-delta/c-Src-mediated EGF receptor transactivation regulates thrombin-induced COX-2 expression and PGE(2) production in rat vascular smooth muscle cells. Biochim Biophys Acta. 2008;1783:1563–1575. doi: 10.1016/j.bbamcr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 41.Vogelstein B, Kinzler KW. The Path to Cancer – Three Strikes and You’re Out. N Engl J Med. 2015;373:1895–1898. doi: 10.1056/NEJMp1508811. [DOI] [PubMed] [Google Scholar]
- 42.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murray GI, Duncan ME, O’Neil P, Melvin WT, Fothergill JE. Matrix metalloproteinase-1 is associated with poor prognosis in colorectal cancer. Nat Med. 1996;2:461–462. doi: 10.1038/nm0496–461. [DOI] [PubMed] [Google Scholar]
- 44.Baker EA, Bergin FG, Leaper DJ. Matrix metalloproteinases, their tissue inhibitors and colorectal cancer staging. Br J Surg. 2000;87:1215–1221. doi: 10.1046/j.1365-2168.2000.01531.x. [DOI] [PubMed] [Google Scholar]
- 45.Sunami E, Tsuno N, Osada T, Saito S, Kitayama J, Tomozawa S, Tsuruo T, Shibata Y, Muto T, Nagawa H. MMP-1 is a prognostic marker for hematogenous metastasis of colorectal cancer. Oncologist. 2000;5:108–114. doi: 10.1634/theoncologist.5-2-108. [DOI] [PubMed] [Google Scholar]
- 46.Cheng K, Samimi R, Xie G, Shant J, Drachenberg C, Wade M, Davis RJ, Nomikos G, Raufman JP. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am J Physiol Gastrointest Liver Physiol. 2008;295:G591–597. doi: 10.1152/ajpgi.00055.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah N, Khurana S, Cheng K, Raufman JP. Muscarinic receptors and ligands in cancer. Am J Physiol Cell Physiol. 2009;296:C221–232. doi: 10.1152/ajpcell.00514.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng K, Raufman JP. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharmacol. 2005;70:1035–1047. doi: 10.1016/j.bcp.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 49.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu B, Zhou H, Hu L, Mu Y, Wu Y. Involvement of PKCalpha activation in TF/VIIa/PAR2-induced proliferation, migration, and survival of colon cancer cell SW620. Tumour Biol. 2013;34:837–846. doi: 10.1007/s13277-012-0614-x. [DOI] [PubMed] [Google Scholar]
- 51.Masur K, Lang K, Niggemann B, Zanker KS, Entschladen F. High PKC alpha and low E-cadherin expression contribute to high migratory activity of colon carcinoma cells. Mol Biol Cell. 2001;12:1973–1982. doi: 10.1091/mbc.12.7.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konopatskaya O, Poole AW. Protein kinase Calpha: disease regulator and therapeutic target. Trends Pharmacol Sci. 2010;31:8–14. doi: 10.1016/j.tips.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gravitt KR, Ward NE, Fan D, Skibber JM, Levin B, O’Brian CA. Evidence that protein kinase C-alpha activation is a critical event in phorbol ester-induced multiple drug resistance in human colon cancer cells. Biochem Pharmacol. 1994;48:375–381. doi: 10.1016/0006-2952(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 54.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein kinase C and cancer: what we know and what we do not. Oncogene. 2014;33:5225–5237. doi: 10.1038/onc.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harris IS, Brugge JS. Cancer: The enemy of my enemy is my friend. Nature. 2015;527:170–171. doi: 10.1038/nature15644. [DOI] [PubMed] [Google Scholar]
- 57.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massague J. TGF-beta Tumor Suppression through a Lethal EMT. Cell. 2016;164:1015–1030. doi: 10.1016/j.cell.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, DeWitt DL, Saklatvala J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]
- 59.Johansson N, Ala-aho R, Uitto V, Grenman R, Fusenig NE, Lopez-Otin C, Kahari VM. Expression of collagenase-3 (MMP-13) and collagenase-1 (MMP-1) by transformed keratinocytes is dependent on the activity of p38 mitogen-activated protein kinase. J Cell Sci. 2000;113(Pt 2):227–235. doi: 10.1242/jcs.113.2.227. [DOI] [PubMed] [Google Scholar]
- 60.Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003;63:5454–5461. [PubMed] [Google Scholar]
- 61.Nakajima K, Tohyama Y, Kohsaka S, Kurihara T. Protein kinase C alpha requirement in the activation of p38 mitogen-activated protein kinase, which is linked to the induction of tumor necrosis factor alpha in lipopolysaccharide-stimulated microglia. Neurochem Int. 2004;44:205–214. doi: 10.1016/s0197-0186(03)00163-3. [DOI] [PubMed] [Google Scholar]
- 62.Furundzija V, Fritzsche J, Kaufmann J, Meyborg H, Fleck E, Kappert K, Stawowy P. IGF-1 increases macrophage motility via PKC/p38-dependent alphavbeta3-integrin inside-out signaling. Biochem Biophys Res Commun. 2010;394:786–791. doi: 10.1016/j.bbrc.2010.03.072. [DOI] [PubMed] [Google Scholar]
- 63.Min DS, Shin EY, Kim EG. The p38 mitogen-activated protein kinase is involved in stress-induced phospholipase D activation in vascular smooth muscle cells. Exp Mol Med. 2002;34:38–46. doi: 10.1038/emm.2002.6. [DOI] [PubMed] [Google Scholar]
- 64.Seo JY, Kim DY, Lee YS, Ro JY. Cytokine production through PKC/p38 signaling pathways, not through JAK/STAT1 pathway, in mast cells stimulated with IFNgamma. Cytokine. 2009;46:51–60. doi: 10.1016/j.cyto.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 65.Lin YS, Carey M, Ptashne M, Green MR. How different eukaryotic transcriptional activators can cooperate promiscuously. Nature. 1990;345:359–361. doi: 10.1038/345359a0. [DOI] [PubMed] [Google Scholar]
- 66.Sogaard-Andersen L, Valentin-Hansen P. Protein-protein interactions in gene regulation: the cAMP-CRP complex sets the specificity of a second DNA-binding protein, the CytR repressor. Cell. 1993;75:557–566. doi: 10.1016/0092-8674(93)90389-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Actions of inhibitors alone. Pre-incubating HT-29 colon cancer cells for 45 min with inhibitors of PKC-α/β1 (5 μM Gӧ6976, 5 μM Gӧ6983), p38-α/β (10 μM SB203580, 10 μM SB202190), EGFR (5 μM PD168393, 5 μM PD153035), and MEK (10 μM U0126, 10 μM PD98059) alone did not alter p38 phosphorylation (A), MMP1 mRNA expression (B), or cell invasion (C). ACh (100 μM) is used as a positive control in these experiments. Measurement of p38 phosphorylation by immunoblotting, MMP1 mRNA expression by qPCR, and cell invasion using Boyden chambers with Matrigel inserts was performed as described in Materials and Methods.
Supplemental Figure 2. Effect of inhibitors on MMP1 mRNA expression in H508 human colon cancer cells. H508 cells were pre-incubated for 45 min with PKC-α/β1, p38-α/β, EGFR, and MEK inhibitors and then incubated for an additional 4 h with no additional test agents (C) or with ACh (100 μM). MMP1 mRNA levels were measured by qPCR. ACh-induced MMP1 expression was attenuated by inhibitors of PKC-α/β1 (5 μM Gӧ6976, 5 μM Gӧ6983), p38-α/β (10 μM SB202190, 10 μM SB203580), EGFR (5 μM PD168393, 5 μM PD153035), and MEK (10 μM PD98059, 10 μM U0126). Pre-incubating cells with a combination of p38-α/β plus EGFR inhibitors or p38-α/β plus MEK inhibitors abolished ACh-induced MMP1 gene expression, as did pre-incubation with a combination of p38-α/β, EGFR and PKC-α/β1 inhibitors. qPCR data were normalized to GAPDH and are means of three separate experiments. C, untreated control; *P < 0.05.
Supplemental Figure 3. Directly activating PKC plus EGFR potentiates MMP1 expression in Caco-2 cells. Caco-2 cells were incubated with PMA (50 nM) and EGF (10 ng/ml), alone or in combination, for 4h. MMP1 mRNA levels were measured by qPCR. Simultaneous activation of PKC and EGFR signaling significantly potentiates MMP1 mRNA levels; the dashed line represents the calculated additive value for PMA plus EGF (P<0.05 for the observed vs. the calculated addition of PMA plus EGF). Results are representative of three separate experiments. qPCR data were normalized to GAPDH and are the means ± SE of three separate experiments. C, untreated control; *P < 0.05, **P < 0.01.
Supplemental Table 1. Inhibitors used in this study.