

Abstract
Stereolithography (SLA) holds great promise in fabrication of cell-laden hydrogels with biomimetic complexity for use in tissue engineering and pharmaceutics. However, the availability of biodegradable photocrosslinkable hydrogel polymers for SLA is very limited. In this study, a water-soluble methacrylated poly(ethylene glycol-co-depsipeptide) was synthesized to yield a biodegradable photocrosslinkable macromer for SLA. Structural analysis confirmed the inclusion of biodegradable peptide and ester groups and photocrosslinkable methacrylate groups into the polymer backbone. The new macromer combined with RGDS peptide was used for SLA fabrication of hydrogels in absence and presence of cells. With the increasing light exposure time in SLA, mechanical stiffness of the hydrogels increased from 3 ± 1 kPa to 38 ± 13 kPa. Total mass loss of the samples within 7 days in PBS was 13%–21% and within 24 days 35%–66%. Due to degradation, the mechanical stiffness decreased by one order magnitude within 7-day incubation in PBS. Encapsulated endothelial cells proliferated in the hydrogels during 10-day in vitro cell culturing study. The macromer was further used in SLA to fabricate bifurcating tubular structures as preliminary vessel grafts. The new biodegradable, photocrosslinkable polymer is a significant addition to the very limited material selection currently available for SLA-based fabrication of cell-laden tissue engineering constructs.
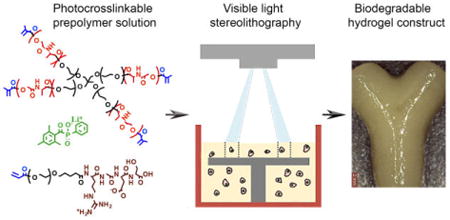
Graphical abstract
3D defined cell-laden hydrogel constructs were fabricated using stereolithography and a new biodegradable photocrosslinkable poly(ethylene glycol-co-depsipeptide) prepolymer.
1. Introduction
Stereolithography (SLA) is increasingly used for fabrication of biomimetic complex scaffolds and hydrogels for tissue engineering (TE) and pharmaceutics. It enables high-resolution and relatively fast scaffold fabrication in absence of harsh chemicals and high fabrication temperature.1-3 Of various SLA techniques, digital light processing (DLP)-based SLAs are particularly suitable for three-dimensional (3D) fabrication of cell-laden hydrogels due to their shorter production times compared with traditional laser-based SLA techniques1 and the use of visible light instead of UV light, reducing the potential risk of harming DNA of encapsulated cells.4 The recent progress of biomaterials and 3D fabrication technology makes SLA increasingly attractive for fabrication of hydrogel constructs with complex designs and enhanced functionality that cannot be achieved via more traditional fabrication methods.
To fabricate cell-laden hydrogel constructs by SLA, a biocompatible, biodegradable hydrogel polymer with appropriate photocrosslinking kinetics and cell attachment properties is crucially needed. However, the availability of such polymers for SLA is currently very limited. Previously, naturally derived, biodegradable methacrylated gelatin has been used for SLA-based fabrication of cell-free hydrogels,5,6 and synthetic (meth)acrylated poly(ethylene glycol) (PEG) has been applied in several cell-laden, non-degradable hydrogels.7-11 Even though naturally derived polymers demonstrate biodegradability and good cell adhesion properties, they do not typically allow for controlled tuning of their properties without simultaneously interfering their bioactivity.12 Instead, synthetic PEG allows for systematic modification of its physical and mechanical properties to satisfy the requirements of a variety of target applications.12,13 However, PEG is known to be resistant to protein adsorption and cell attachment14 and is inherently non-degradable, which restricts the migration and proliferation of encapsulated cells inside the crosslinked hydrogel.9 The lack of controlled degradation of PEG has been previously addressed by incorporating enzymatically cleavable peptides12,15 or hydrolytically labile ester bonds3,16 into its backbone. However, the photocrosslinking properties and fabrication parameters of these biodegradable polymers have not been optimized for use in SLA-based fabrication of cell-laden hydrogels. To successfully 3D fabricate cell-laden hydrogels, it is essential to understand how parameters of both the material and the SLA process affect the resulting hydrogel properties. In a photocrosslinked hydrogel, properties like swelling capacity, material degradation, and mechanical stiffness depend directly on the crosslinking density of the gel, which in turn is known to depend on the parameters like polymer concentration and molecular weight as well as photoinitiator concentration and crosslinking energy.13,17,18 The understanding of correlation between fabrication parameters and hydrogel properties will allow for better control over the characteristics of cell-laden hydrogel and will help to regulate and promote the intended functions of encapsulated cells in the hydrogel matrix.15,19,20
In our previous study,21 we synthesized a biodegradable photocrosslinkable poly(ε-caprolactone-co-depsipeptide) for use in DLP-based SLA to fabricate porous hydrophobic TE scaffolds. Previously, depsipeptides have been used to synthesize hydrophilic PEG-based copolymers for use as non-photocrosslinkable, temperature responsive gels and drug delivery micelles.22,23 In our current work, we used l-alanine-derived depsipeptide to synthesize a new biodegradable, photocrosslinkable poly(ethylene glycol-co-depsipeptide) (PEG-co-PDP) macromer for SLA-based fabrication of cell-laden hydrogel constructs. We hypothesized that the new photocrosslinkable macromer, which combines both naturally derived and synthetic building blocks, will allow for 3D fabrication of biodegradable tissue engineering grafts that support cell proliferation within the hydrogel matrix. Subsequently, we studied the effect of light exposure time in SLA on the hydrogel characteristics and hypothesized that by controlling the light exposure time, we can tune the hydrogel properties without need for changing the parameters of the hydrogel solution during the fabrication process. We evaluated the suitability of our new polymer for fabrication of cell-laden hydrogels by first characterizing its chemical structure and gelling properties as well as its hydrolytic mass loss and mechanical properties, and next we studied its cell encapsulation capacity with human umbilical vein endothelial cells (HUVECs). Finally, we demonstrated the use of our new polymer for SLA-based fabrication of tubular structures as preliminary vessel graft models.
2. Experimental
2.1 Materials
l-alanine and chloroacetyl chloride (Acros Organics), tin(II) 2-ethylhexanoate (Sn(Oct)2) and methacrylic anhydride (Sigma-Aldrich), 2,4,6-trimethylbenzoyl chloride (Chem-Impex Int'l Inc.), dimethyl phenylphosphonite (Alfa Aesar), lithium bromide (Strem Chemicals), and 2-butanone (Acros Organics) were used as received. Star-shaped four-arm PEG (10,000 g/mol, Creative PEGworks) was used after drying under vacuum at 100 °C for 3 h. Acryl-PEG-SVA (3,400 g/mol, Laysan Bio, Inc.) and RGDS peptide (Bachem) were used as received. Immortalized HUVECs expressing green fluorescent protein (GFP) were a generous gift from the late Dr. J. Folkman, Children's Hospital, Boston. Fetal bovine serum (FBS), antibiotic solution, and trypsin-EDTA solution were purchased from Invitrogen Co. EBM™ endothelial basal medium containing and EGM™ Single Quots™ kit were purchased from Lonza, Inc. AlamarBlue® cell viability assay was from Bio-Rad Laboratories.
2.2 Photoinitiator synthesis
Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) photoinitiator was synthesized as previously described by Fairbanks et al.24 Briefly, 2,4,6-trimethylbenzoyl chloride was added dropwise to an equimolar amount of dimethyl phenylphosphonite at RT under nitrogen atmosphere. After mixing for 18 h, a fourfold excess of lithium bromide dissolved in 2-butanone was added to the mixture, and the reaction was continued for 10 min at 50 °C. After heating, the mixture was kept at RT for 4 h; the resulting precipitate was filtered and washed once with 2-butanone, and then twice with diethyl ether, and finally dried under vacuum.
2.3 Synthesis of photocrosslinkable macromers and adhesion peptide
Biodegradable, water-soluble PEG-co-PDP oligomer was synthesized by ring-opening polymerization (ROP) of l-alanine-derived depsipeptide using 4-arm PEG as a macroinitiator as shown in Scheme 1A. Before ROP, 3-methylmorpholine-2,5-dione (MMD) monomer was synthesized as described in our previous study.21 Briefly, l-alanine was reacted with an equimolar amount of chloroacetyl chloride in diethyl ether/water mixture at -5 °C by simultaneously adding 4 M NaOH solution into the flask to keep the pH at 11. After adding dropwise the chloroacetyl chloride and NaOH for 20 min, the aqueous layer was acidified to pH 1 with 4 M HCl and was extracted twice with ethyl acetate. The combined organic layers were washed twice with saturated NaCl solution and dried with MgSO4 and concentrated under vacuum. The resulting chloroacetyl alanine was then dissolved in dry N,N-dimethylformamide (DMF) and mixed dropwise with triethylamine. After 8 h at 90 °C under nitrogen and overnight at room temperature, the reaction solution was filtered and most of the solvent was removed under reduced pressure. The resulting cyclic MMD in a small amount of DMF was then purified with chloroform and diethyl ether and dried under vacuum. To polymerize the hydroxyl-ended PEG-co-PDP oligomer with a designated molecular weight of 11,000 g/mol, MMD monomer was added with dried PEG macroinitiator and Sn(Oct)2 catalyst to a flask, which was then evacuated and flushed several times with nitrogen. ROP was continued for 6 days at 130 °C, after which the resulting oligomer was purified by dialysis and freeze-drying. To obtain a photocrosslinkable macromer, the oligomer was further reacted with an excess of methacrylic anhydride at 60 °C for 24 h, after which it was precipitated in cold diethyl ether and dried under vacuum. The macromer was then purified by dialysis and freeze-drying. As a control polymer, PEG macromer was synthesized by methacrylating four-arm PEG macroinitiator with methacrylic anhydride at 60 °C for 24 h. In addition, to enable covalent linking of RGDS adhesion peptide to the PEG-co-PDP hydrogel polymer, RGDS was coupled with an acryl-PEG linker as shown in Scheme 1B. Acryl-PEG-SVA and RGDS were dissolved in 50 mM NaHCO3 solution (pH 8.3) and mixed for 4 h at RT. The resulting acryl-PEG-RGDS was then purified by dialysis and freeze-drying.
Scheme 1.

A) Synthesis of cyclic MMD monomer and further ROP and methacrylation of PEG-co-PDP copolymer. B) Synthesis of acryl-PEG-RGDS adhesion peptide.
2.4 Fabrication of cell-free hydrogels by stereolithography
To prepare a photocrosslinkable hydrogel solution for use in SLA, photocrosslinkable PEG-co-PDP macromer (10% w/v) and LAP photoinitiator (0.25% w/v) were dissolved in EBM-2 medium. The viscosity of the filtered solution was increased by adding 7% w/v of Percoll® medium to the polymer solution.10 For degradation and mechanical studies, cell-free PEG-co-PDP hydrogels (d = 8.5 mm, h = 0.6 mm) were fabricated with an in-house built visible light projection SLA (3,200 lumens) in a layer-by-layer manner with a layer thickness of 200 μm. The layer exposure time was 100 s, 120 s, or 160 s. For SLA fabrication, the digital 3D model of the hydrogels was designed with SolidWorks CAD software and was converted into printing instructions with free Slic3r software (Slic3r.org). The light exposure time was controlled using free Pronterface software (pronterface.com).
2.5 Physicochemical characterization of the polymers and the cell-free hydrogels
1H NMR spectra were recorded on a Varian Inova 300 MHz NMR spectrometer. Samples were dissolved in deuterated chloroform (CDCl3) or in deuterium oxide (D2O), and the spectrum was acquired in a 5-mm NMR tube at RT. The presence of peptide and ester bonds in the PEG-co-PDP copolymer was analyzed using a Bruker Vertex 70 FTIR spectrometer with a diamond ATR. Thermal properties of the oligomers and macromers were determined using a Q2000 differential scanning calorimeter (DSC, TA Instruments). Samples were first heated from 25 °C to 100 °C at a rate of 20 °C/min, after which the temperature was decreased to -100 °C at 10 °C/min. After 2 min at -100 °C, the sample was heated from -100 °C to 100 °C at 10 °C/min. Melting point (Tm) and melting enthalpy (ΔH) were determined from the second heating scan. Gel content (G) of the photocrosslinked hydrogels was measured by extracting the pre-weighed hydrogel in distilled water for 10 h and subsequently drying the sample to obtain its dry weight. G was then calculated by dividing the dry weight of the sample after extraction (md) by its dry weight before extraction (m0) according to Equation (1):25
| (1) |
To measure the initial swelling degree (Q) of hydrogels, the samples were immersed in distilled water for 24 h to reach the swelling equilibrium, after which the weighed samples were dried and weighed again. By using the wet weight (msw) and dry weight (md) of the samples and the density values of 1.0 g/ml for water (ρs) and 1.2 g/ml for photocrosslinked PEG-based polymer (ρp),26 the swelling degree was calculated using Equation (2):25
| (2) |
In vitro mass loss of hydrogels was monitored by measuring the dry weight of the samples after incubation in PBS (pH 7.4, with 0.02% sodium azide) at 37 °C. At day 7, 14, and 24, triplicate samples were removed from the PBS, weighed, and dried first at ambient pressure for 2 d and then in a freeze-dryer for 3 d. After drying, the remaining mass was divided by the initial mass to get the remaining mass percentage. The swelling degree during the degradation study was calculated using Equation (2) as described before. Viscoelastic behavior of 3D fabricated hydrogels was analyzed with an Ares G2 rheometer (TA Instruments) using an 8-mm parallel plate geometry. The viscoelastic regime of the samples was first determined with an amplitude sweep from 0.1 to 50% strain, after which the angular frequency sweep was run from 0.1 to 10 rad/s at 0.5% strain and 25 °C. AFM force-distance measurements were done with an Agilent 5500 AFM in a liquid cell with PBS as the liquid phase. Tip sensitivity calibration was performed on glass surface after the end of each set of experiments with the respective tip. The tip used for the experiments (NanoAndMore, k = 0.08 N/m, R = 950 nm) had a SiO2 sphere on its apex. The stiffness of tips from this batch was confirmed by Sader method and was found to be <20% different from the nominal reported value. FDS curve data analysis was done using commercially available SPIP software from Image Metrology A/S. The baseline correction from the laser reflection and the determination of Young's modulus using Hertz sphere approximation was performed in batch processing regime, with the results checked visually for mistakes.
2.6 Cell cultures and 3D fabrication of cell-laden hydrogels
HUVECs were cultured in endothelial basal medium (EBM-2) mixed with a Single Quots (EGM-2, excluding hydrocortisone) endothelial growth supplement, 10% FBS, and 1% antibiotic solution at standard conditions (5% CO2, 37 °C, 95% humidity). To study material cytocompatibility, two cell-free hydrogel groups were fabricated by SLA using the hydrogel solutions consisting of neat PEG-co-PDP macromer or PEG-co-PDP macromer mixed with 2 mM acryl-PEG-RGDS. HUVECs (1×103 cells) were seeded on top of the hydrogels (d = 8.5 mm, h = 0.6 mm). For cell encapsulation studies, cell-laden hydrogel discs were prepared by SLA using the filtered PEG-co-PDP/RGDS solution mixed with HUVECs (5×106 cells/ml) and the same fabrication parameters as for the cell-free hydrogels (Fig. 1). Before hydrogel fabrication, the container for hydrogel solution was sterilized with 70% ethanol followed by thorough rinsing with PBS, and the air in the SLA system was continuously filtered with a HEPA filter. To further demonstrate the use of PEG-co-PDP polymer for 3D fabrication of vascular tissue engineering grafts, cell-laden hydrogel rings and bifurcating hydrogel vessels were fabricated by SLA as described for the disc samples using 200 μm as a layer thickness and 120 s as a light exposure time.
Fig. 1.
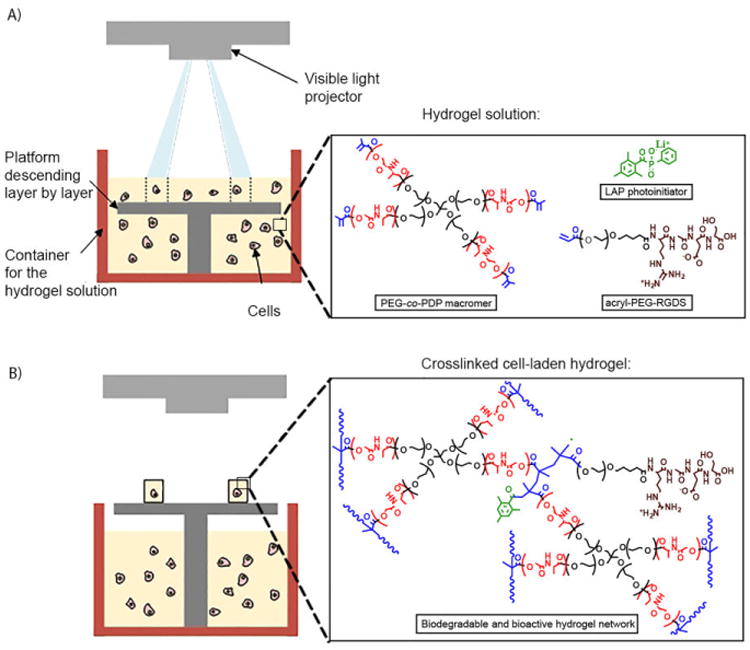
A schematic presentation of SLA-based fabrication of cell-laden hydrogels: A) Light is projected to photocrosslinkable hydrogel solution to form the designed pattern, and the hydrogel is crosslinked in a layer-by-layer manner; B) After crosslinking the last layer, the platform is moved up and the resulting hydrogel is removed from the SLA.
2.7 Cell proliferation
Metabolic activity of seeded or encapsulated HUVECs was measured with a colorimetric AlamarBlue® cell viability assay. At predetermined time points, the hydrogel samples were transferred to a new well-plate, and 500 μL of fresh medium and 50 μL of AlamarBlue® solution was added into the wells. After incubation for 4 h, the medium was aliquoted to three parallel 100 μL samples, and the optical density was read using a SpectraMax® M2e microplate reader (Molecular Devices) at 555 nm/585 nm excision/emission.
2.8 Statistical analysis
For the statistical analysis, samples were used in triplicates, and the statistical significance was determined with the IBM SPSS Statistics software using a one-way ANOVA followed by Tukey's post hoc test (p < 0.05).
3. Results
3.1 The macromer synthesis
The water-soluble photocrosslinkable PEG-co-PDP macromer for use in 3D fabrication of cell-laden hydrogels was synthesized by ROP of l-alanine-derived depsipeptide and a 4-arm PEG macroinitiator followed by the methacrylation. The reactions were monitored by 1H NMR characterization as shown in Fig. 2. The cyclic MMD monomer that was synthesized from l-alanine and chloroacetyl chloride gave the 1H NMR peaks 1-3 in Fig. 2A. The MMD was then copolymerized with PEG, resulting in a water-soluble, hydroxyl-terminated oligomer. In the 1H NMR spectrum (Fig. 2B), the monomer peaks transferred to the corresponding oligomer peaks 1-3, while new peak b′ appeared and was attributed to the last CH2 protons of the PEG arms before depsipeptide units. 2D COSY 1H NMR revealed a strong coupling between peaks 2 and 3 of the oligomer. As calculated from the 1H NMR spectrum, the polymerization degree of the MMD monomer was around 85%, when the molecular weight of the oligomer was approximately 10,850 g/mol. The yield of oligomer was 98%. The oligomer was further functionalized with methacrylate end-groups to obtain the macromer with photocrosslinkable double bonds. The successful methacrylation was observed as a disappearance of peak 1 in Fig. 2B attributed to the last CH2 protons before the hydroxyl groups of the oligomer and as an appearance of the peaks i, j, and k in Fig. 2C attributed to the methacrylate groups of the macromer. No trace of the oligomeric peak 1 was left in the macromer, showing the complete methacrylation. The dialyzed and freeze-dried macromer was a light-brown powder showing great water-solubility at room temperature. The yield of purified macromer was 97%. Some residual water remained in the polymer despite the extended freeze-drying, giving a broad peak near 2 ppm in the 1H NMR spectrum.
Fig. 2.
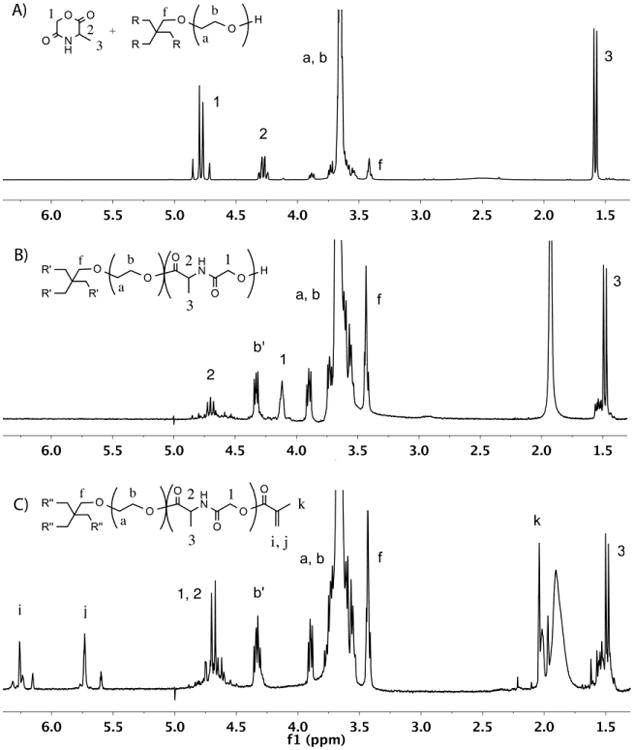
1H NMR spectra of A) MMD monomer and PEG before ROP, B) PEG-co-PDP oligomer after ROP, and C) photocrosslinkable PEG-co-PDP macromer after methacrylation, all in CDCl3.
RGDS conjugation with acryl-PEG-SVA was confirmed with 1H NMR. After 4 h of mixing, the proton peak g of acryl-PEG-SVA end-group in Fig. 3A disappeared and new peaks assigned to the RGDS peptide appeared in Fig. 3B. After dialysis and freeze-drying, the acryl-PEG-RGDS was a water-soluble white powder with a molecular weight of 3,700 g/mol.
Fig. 3.
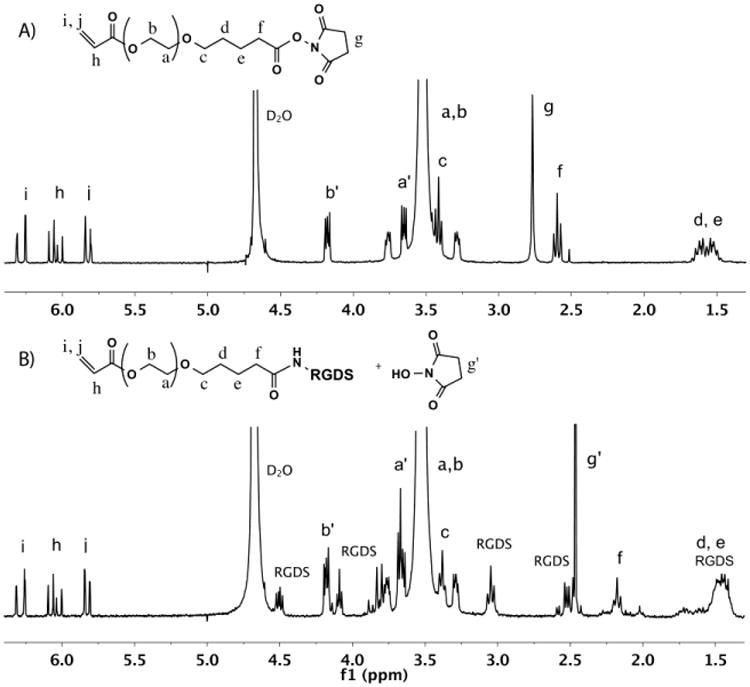
1H NMR spectra of A) acryl-PEG-SVA and B) acryl-PEG-RGDS in D2O.
Copolymerization of depsipeptide units into PEG was monitored by FTIR. Fig. 4 shows the spectra of hydroxyl-ended PEG before copolymerization and the resulting PEG-co-PDP oligomer. The FTIR analysis revealed that the ROP of PEG with MMD resulted in new peaks at 1750 cm-1 and 1670 cm-1 attributed to the C=O ester and C=O amide stretches, respectively, and a new peak at 1540 cm-1 attributed to the N-H bend of the depsipeptide units. The FTIR data together with the 1H NMR data thereby confirmed that the designated poly(ether ester amide) copolymer was successfully synthesized.
Fig. 4.
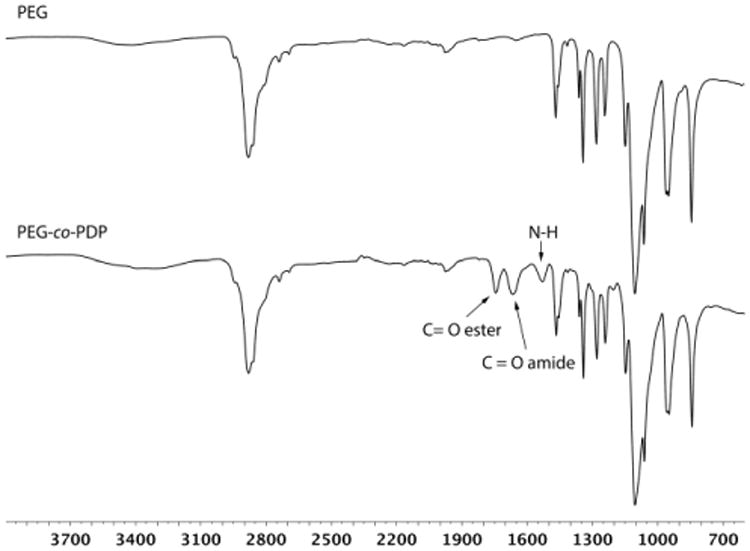
FTIR spectra of PEG homopolymer and PEG-co-PDP copolymer showing the new ester and amide bonds of the copolymer.
The effect of the new depsipeptide units and the subsequent methacrylation on thermal properties of PEG was characterized with the DSC analysis. The DSC curves revealed a clear melting peak for all the oligomers and macromers. Table 1 shows that the copolymerization of depsipeptides into PEG decreased Tm of the oligomer by around 10 °C and its ΔH to the half of the initial value. The methacrylation decreased the Tm and ΔH values both for the PEG control polymer and for the PEG-co-PDP copolymer.
Table 1.
Melting point and melting enthalpy of the oligomers and macromers.
| Sample | Oligomer | Macromer | ||
|---|---|---|---|---|
| Tm (°C) | ΔH (J/g) | Tm (°C) | ΔH (J/g) | |
| PEG | 55.82 | 160.3 | 43.98 | 100.9 |
| PEG-co-PDP | 45.43 | 81.38 | 42.59 | 76.60 |
3.2 Cytocompatibility of photocrosslinked hydrogels
To study in vitro cytocompatibility of methacrylated PEG-co-PDP polymer and to study the effect of RGDS adhesion peptide on cell proliferation, PEG-co-PDP and PEG-co-PDP/RGDS hydrogels were photocrosslinked and HUVECs were seeded on the hydrogels. The metabolic activity of HUVECs, which is an indicator of the number of living cells, was monitored with the colorimetric AlamarBlue® assay. Fig. 5 shows that on both hydrogel groups, metabolic activity of cells increased from day 4 to day 14. RGDS peptide increased significantly the cell activity on PEG-co-PDP/RGDS hydrogels compared to neat PEG-co-PDP hydrogels.
Fig. 5.
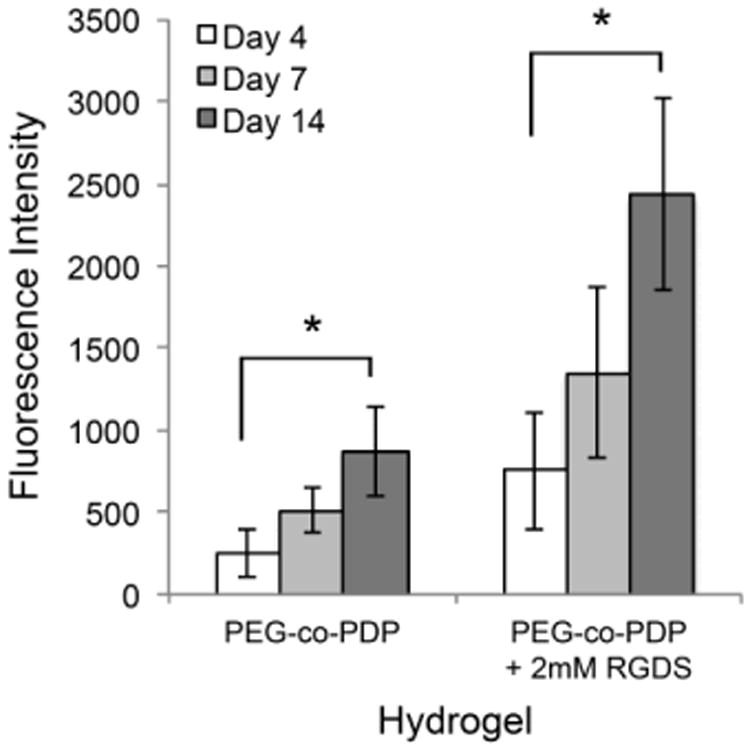
Metabolic activity of HUVECs seeded on PEG-co-PDP and PEG-co-PDP/RGDS hydrogels as measured with a colorimetric AlamarBlue® cell viability assay.
3.3 Fabrication of hydrogels with adjusted light exposure times
The cell-free PEG-co-PDP hydrogels were 3D fabricated with the visible light projection SLA in a layer-by-layer manner at RT. To study the effect of crosslinking time on the swelling capacity, mass loss, and mechanical properties of hydrogels, three different light exposure times, 100 s, 120 s, and 160 s/layer, were used in SLA. Table 2 shows that the gel content of the hydrogels increased from 85% to 88% with the increasing crosslinking time, while the swelling degree decreased from 21 to 16.
Table 2. Gel content and swelling degree of the hydrogels prepared by SLA.
| Light exposure time/layer (s) | Gel content (%) | Swelling degree |
|---|---|---|
| 100 | 85 ± 2 | 21 ± 1 |
| 120 | 86 ± 1 | 18 ± 1 |
| 160 | 88 ± 1 | 16 ± 1 |
3.4 In vitro mass loss and swelling of the 3D hydrogels
In vitro hydrolytic mass loss of photocrosslinked PEG-co-PDP hydrogels was monitored by weighing the remaining dry mass of cell-free hydrogels after predetermined times in PBS. Fig. 6A shows that the total mass loss of hydrogels decreased with increasing crosslinking time. During 24 d in PBS, the 100 s samples lost 66% of their initial dry mass, while the 120 s samples lost 45% and the 160 s samples lost 35% of their initial mass. Fig. 6B shows that the swelling degree of hydrogels increased during the 24-day degradation study from 21 to 58 for the 100 s samples, from 18 to 45 for the 120 s samples, and from 16 to 40 for the 160 s samples.
Fig. 6.
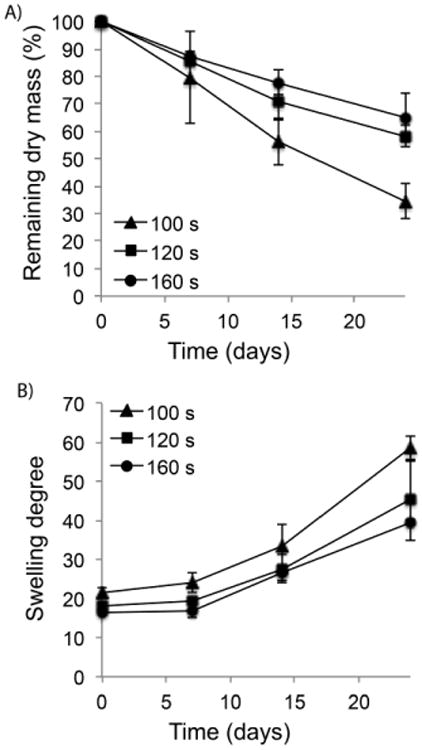
A) In vitro mass loss and B) swelling degree of PEG-co-PDP hydrogels with various crosslinking times during 24-day degradation study in PBS (pH 7.4).
3.5 Mechanical and viscoelastic properties of 3D hydrogels
To evaluate the effect of light exposure time and polymer degradation on the local mechanical stiffness of 3D fabricated hydrogels, AFM was used to measure the Young's modulus of wet samples right after SLA fabrication and after 7 d in PBS. Fig. 7A shows that the stiffness increased with crosslinking time, and the initial Young's modulus was 3.0 ± 1.0 kPa, 9.4 ± 1.2 kPa, and 37.9 ± 13.3 kPa for the 100 s, 120 s, and 160 s samples, respectively. Within 7 d in PBS (Fig. 7C), Young's modulus decreased to 0.24 ± 0.02 kPa, 0.96 ± 0.50 kPa, and 1.52 ± 0.55 kPa, respectively. Rheometric analysis of the hydrogels revealed that the bulk storage modulus of the samples increased with increasing crosslinking time. The storage modulus was initially in the order of magnitude of 1–10 kPa (Fig. 7B), while it decreased to 0.1–1 kPa after 7 d in PBS (Fig. 7D). The one order magnitude decrease within the 7-day hydrolysis period was consistent with both the AFM and rheometric studies.
Fig. 7.
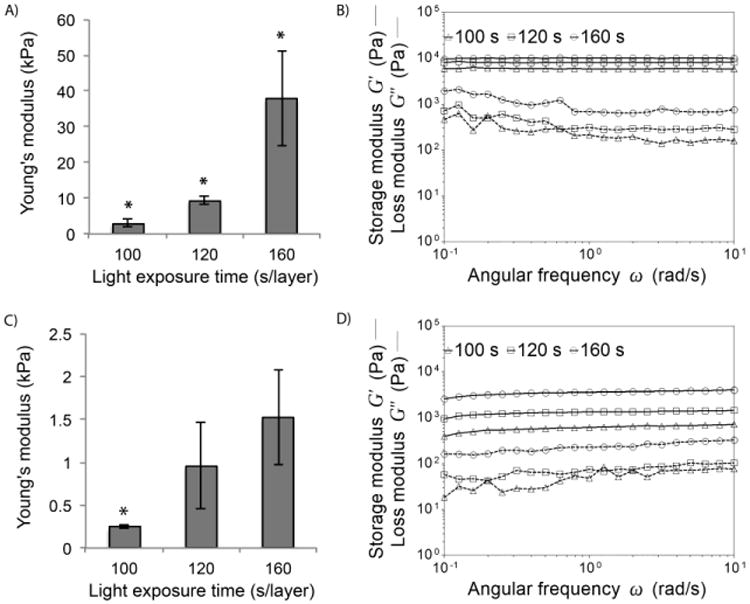
Local Young's modulus measured with AFM and bulk storage and loss modulus of PEG-co-PDP hydrogels measured with a rheometric analysis A-B) right after SLA fabrication and C-D) after 7 d in PBS.
3.6 Proliferation of encapsulated HUVECs in cell-laden hydrogels and vessel graft fabrication
Cell-laden hydrogels were fabricated by photocrosslinking the mixture of a filtered PEG-co-PDP/RGDS hydrogel solution and HUVEC suspension using a visible light projection SLA. The effect of crosslinking time on cell proliferation was tested by fabricating cell-laden hydrogels using the same light exposure times as for the cell-free hydrogels and by monitoring the metabolic activity of encapsulated cells with a colorimetric AlamarBlue® assay. Fig. 8 shows that cells significantly increased their metabolic activity in all sample groups from day 1 to day 10. At each designated time point, there was no significant difference in cell activity among the sample groups.
Fig. 8.
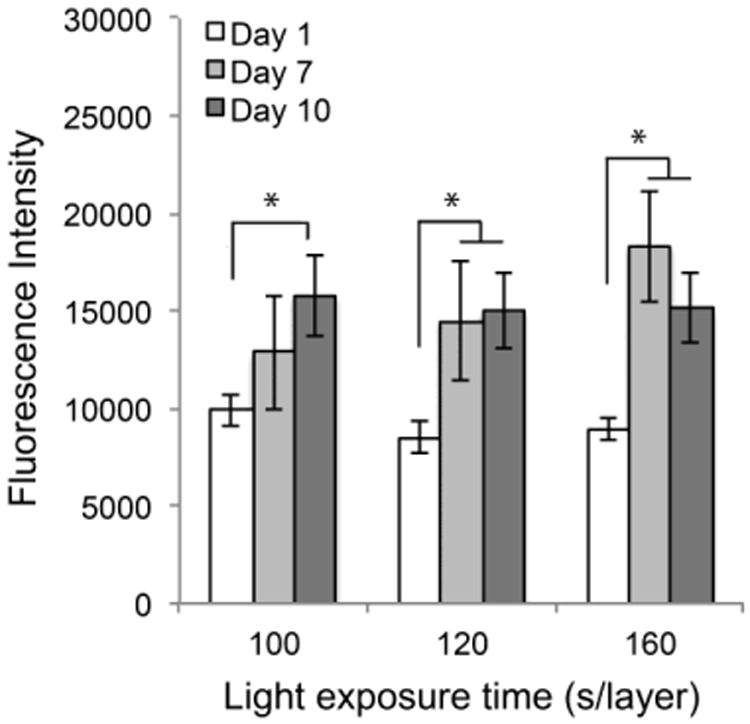
Metabolic activity of encapsulated cells in PEG-co-PDP/RGDS hydrogels within 10 d of cell culturing as measured with a colorimetric AlamarBlue® cell viability assay.
To demonstrate the capacity of the new PEG-co-PDP macromer in SLA-based fabrication of tubular hydrogel constructs for vascular tissue engineering, ring-like cell-laden hydrogels and bifurcating vascular tubes were designed and 3D fabricated with SLA. The fluorescence images in Fig. 9A and 9B show that the cells were homogenously distributed in the hydrogel ring after 1 d of cell culturing. The uniform and round shape of the hydrogel ring is shown in Fig. 9C. Fig. 9D shows that the 3D fabricated bifurcating hydrogel constructs closely resembled their digital models and had a uniform and smooth wall structure as required for functional vascular tissue engineering grafts.
Fig. 9.
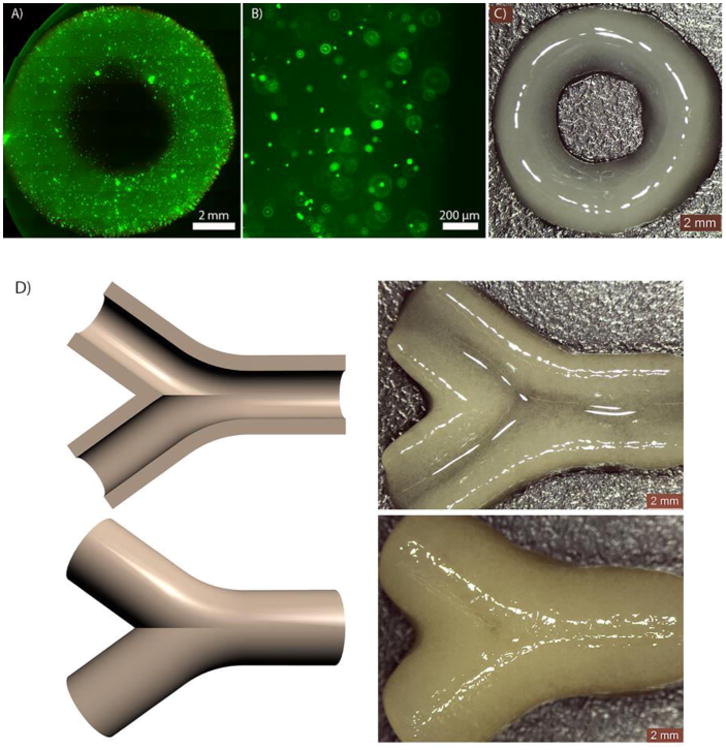
A-B) Fluorescence images and C) a photograph of a cell-laden hydrogel construct; D) CAD models of bifurcating vascular tubes and the resulting hydrogel vessels visualized by their cross-section and top view.
4. Discussion
Our goal in this work was to develop a new biodegradable photocrosslinkable polymer to expand the limited repertoire of polymers suitable for SLA-based 3D fabrication of cell-laden hydrogels. Subsequently, we used this polymer to increase the understanding of how light exposure time in SLA affects the key characteristics of the resulting hydrogel. As PEG is non-degradable polymer, it is not ideal for use in cell-laden hydrogels where cell growth requires increasing volume of void space inside the polymer matrix.9,10 To address the lack of degradation, we synthesized a biodegradable PEG-co-PDP oligomer by copolymerizing l-alanine-derived depsipeptide with a four-arm PEG polymer. The resulting oligomer was successfully functionalized with methacrylate end groups to obtain a photocrosslinkable macromer suitable for SLA.
1H NMR and FTIR analysis confirmed that the depsipeptide units introduced biodegradable ester and peptide bonds to the PEG chains and the methacrylate groups were successfully attached to the end of the copolymer chains. Thermal characterization revealed that copolymerization of PEG with depsipeptide decreased its melting temperature and enthalpy, indicating that the depsipeptide units interfered with the highly crystalline structure of PEG. For SLA-based hydrogel fabrication, a visible light sensitive photoinitiator, LAP, was synthesized, and a low concentration of LAP (0.25% w/v) was mixed with the aqueous macromer solution to generate a readily photocrosslinkable hydrogel solution. 3D modeled hydrogels were successfully photocrosslinked in a layer-by-layer manner leading to homogenously crosslinked hydrogel constructs. Cytocompatibility of PEG-co-PDP copolymer was evaluated by seeding HUVECs on the 3D fabricated hydrogels. Cells proliferated from day 4 to day 14 on both PEG-co-PDP and PEG-co-PDP/RGDS hydrogels. Immobilizing RGDS adhesion peptides to the hydrogels enhanced cell proliferation by providing them with integrin-specific attachment sites. Based on the encouraging proliferation results, we decided to combine PEG-co-PDP macromer with acryl-PEG-RGDS in further 3D fabrication of cell-laden hydrogels.
The microenvironment of cells encapsulated in 3D hydrogels comprises biophysical and biomechanical cues, including swelling capacity, material mass loss, and mechanical stiffness of the hydrogel. We aimed at studying how these properties depend on the crosslinking density of hydrogel, which is correlated to the light exposure time used for photocrosslinking the hydrogel. When the light exposure time in SLA was increased from 100 s to 160 s per layer, the gel content of hydrogels increased from 85% to 88%. The swelling degree decreased from 21 to 16 with increasing crosslinking time. As more double bonds reacted within the continued light exposure time, the crosslinking density and gel content of the hydrogels increased, resulting in decreased mesh size and more restricted water penetration into the hydrogel network. A similar decrease in the swelling capacity has been previously reported for PEG hydrogels, where the crosslinking density was increased by increasing polymer concentration27 or by decreasing the molecular weight of the polymer.9
Besides measuring the gel content and initial swelling capacity, we evaluated hydrogel mass loss and changes in swelling degree and mechanical stiffness as a measure of polymer degradation. The mass loss of PEG-co-PDP hydrogels followed a zero order curve trend within the 24-day incubation in PBS. Such similar linear mass loss has been previously reported for photocrosslinked PEG-co-polylactide (PEG-co-PLA) hydrogels.28 Total mass loss of our samples within 24 days in PBS ranged from 66% for 100 s samples to 35% for 160 s samples. When all the crosslinks of the multi-arm copolymer had broken, the copolymer unit was free to diffuse out of the hydrogel network, leading to hydrogel mass loss. The difference in mass loss rates of our SLA fabricated hydrogels can be explained by the difference in their crosslinking densities. Longer light exposure time increased the crosslinking degree, when more polymer arms became covalently bound to the hydrogel network, making the release of a polymer chain more difficult and slowing down the mass loss. The increased crosslinking degree significantly decreased the initial swelling degree from 21 to 16, and consequently, the increased water uptake of the hydrogels with shorter crosslinking time accelerated their hydrolytic mass loss. The degradation of polymer networks was also evidenced as an increase in the swelling degree of hydrogels during incubation in PBS. The increase in swelling degree was slow in the early stage of degradation, but accelerated in the later stage, indicating that the decrease in crosslinking density occurred in a non-linear fashion. This result was consistent with a similar exponential increase in swelling degree of previous biodegradable PEG-co-PLA hydrogels.28
In addition to the swelling capacity and mass loss, the control over mechanical properties of cell-laden hydrogels is important for cell viability and function.19 We used oscillatory rheometry to measure viscoelastic properties of bulk hydrogels and AFM to study local mechanical stiffness of the hydrogels both right after SLA fabrication and after 7-day incubation in PBS. The storage modulus describing the elastic properties of hydrogels increased with light exposure time and thus with the increased crosslinking density, being initially in the order of magnitude of 1–10 kPa. The Young's modulus of hydrogels ranged initially from 3 kPa to 38 kPa, being in the stiffness range of natural soft tissues regardless of the crosslinking time.29 Elsewhere, hydrogels composed of 10% w/v of PEG diacrylates with comparable chain lengths have shown bulk stiffness of 50 kPa30 and 170 kPa,20 indicating that our hydrogels were in the lower range of mechanical stiffness for corresponding PEG hydrogels. Within 7 d in PBS, the storage modulus of the gels decreased to the range of 0.1–1 kPa and the Young's modulus decreased to the range of 0.2 kPa to 1.5 kPa. Thus, the one order magnitude decrease was consistent both in the bulk storage modulus and the local stiffness of hydrogels. As the mechanical elasticity and strength of photocrosslinked hydrogel are caused by retractive forces of the crosslinked polymer chains,28 the cleavage of biodegradable PEG-co-PDP chains decreased the total forces, and together with the increased swelling of the gels, resulted in decreased mechanical stiffness of degraded hydrogels. The hydrolysis of degradable bonds in PEG-co-PDP chains resulted in broken crosslinks, first weakening the stiffness of hydrogel and subsequently leading to mass loss after every crosslink of the multi-arm macromer had broken. Even though the mechanical stiffness of the hydrogels decreased by one order magnitude within 7-day incubation in PBS, the samples retained their structural integrity.
3D fabrication of cell-laden hydrogels with biomimetic complexity and enhanced functionality is particularly promising in tissue engineering and drug screening; it provides more physiologically relevant data on cell behavior and response to stimuli than can be obtained by seeding cells on a surface of prefabricated hydrogels. We fabricated cell-laden hydrogels by SLA using the same crosslinking parameters as for the cell-free hydrogels. Cell activity studies revealed that the initially non-porous PEG-co-PDP/RGDS hydrogels supported proliferation of encapsulated HUVECs within the 10-day cell culturing period regardless of crosslinking time. Between day 7 and 10, cell metabolic activity slightly decreased in the hydrogel group with the longest crosslinking time. This most likely resulted from their high crosslinking degree and slow mass loss so that the dense polymer network restricted cell proliferation within the hydrogels. However, no statistically significant difference was observed between day 7 and 10 in any group. A previous study elsewhere showed that non-degradable, non-porous PEG hydrogels prepared by SLA had no cell proliferation within 7-day cell culturing, while the addition of small channels to the hydrogel improved cell proliferation.10 In our study, we attributed successful cell proliferation to polymer degradation and subsequent mass loss and decreased mechanical stiffness of the hydrogel matrix. This is consistent with previous studies showing that high stiffness of PEG hydrogels tends to decrease the proliferation of encapsulated cells due to physical barrier caused by its non-degradable polymer network.9,15 By using new biodegradable polymer, we achieved hydrogels that degrade simultaneously with cell growth and can continuously provide the cells with more space and a softer environment to grow inside the hydrogel matrix.
Our long-term goal is to engineer biodegradable cell-laden hydrogel-based vasculature using visible light SLA. In our current work, we successfully demonstrated the use of our new PEG-co-PDP macromer for SLA-based fabrication of 3D modeled vascular grafts. Though HUVECs proliferated within the hydrogels, we understand that the initial cell density, cell culture period, and biomimetic incubation conditions such as pulsatile perfusion may need to be optimized to achieve endothelium and cell network formation within the hydrogel. The polymer degradation rate can be further tuned to fit in with the rate of cell growth and appropriate mechanical change over time by changing the amount of depsipeptide monomer in the biodegradable PEG-co-PDP copolymer. In the future, more studies are needed to evaluate in vivo biodegradation and biocompatibility of the biodegradable hydrogels developed in this study.
5. Conclusions
We successfully synthesized a biodegradable, photocrosslinkable PEG-co-PDP macromer and demonstrated its use in SLA-based fabrication of cell-laden hydrogels for vascular applications. The depsipeptide units introduced biodegradable bonds to the PEG backbone, and by adjusting the light exposure time in the SLA, we were able to tune the swelling capacity, degradation rate, and mechanical stiffness of the resulting hydrogels without need for changing the parameters of hydrogel solution. The good printability and cell encapsulation capacity of our new biodegradable photocrosslinkable polymer makes it an attractive extension to the extremely limited repertoire of biodegradable polymers currently available for SLA-based fabrication of cell-laden tissue engineering grafts.
Acknowledgments
Graduate School in Chemical Engineering (LE), American-Scandinavian Foundation (LE), Research Foundation of Helsinki University of Technology (LE), Emil Aaltonen Foundation (LE), Alfred Kordelin Foundation (LE), NIH R01AR057837 (NIAMS, YY), NIH R01DE021468 (NIDCR, YY), DOD W911NF-14-1-0545 (YY), DOD W81XWH-10-1- 0966 (PRORP, YY), Coulter Translational Research Award (YY), and Stanford Nano/Biomedical Seed Grant (YY) are gratefully acknowledged for the funding of this work. Alexander Stahl (Stanford University) is acknowledged for running the 1H NMR of acryl-PEG samples. Stanford Nano Shared Facilities were used for part of the experiments in this work.
References
- 1.Melchels FPW, Feijen J, Grijpma DW. Biomaterials. 2010;31:6121–6130. doi: 10.1016/j.biomaterials.2010.04.050. [DOI] [PubMed] [Google Scholar]
- 2.Elomaa L, Kokkari A, Närhi T, Seppälä JV. Compos Sci Technol. 2013;74:99–106. [Google Scholar]
- 3.Seck TM, Melchels FPW, Feijen J, Grijpma DW. J Control Release. 2010;148:34–41. doi: 10.1016/j.jconrel.2010.07.111. [DOI] [PubMed] [Google Scholar]
- 4.Ikehata H, Ono T. J Radiat Res. 2011;52:115–125. doi: 10.1269/jrr.10175. [DOI] [PubMed] [Google Scholar]
- 5.Grogan SP, Chung PH, Soman P, Chen P, Lotz MK, Chen S, D'Lima DD. Acta Biomater. 2013;9:7218–7226. doi: 10.1016/j.actbio.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gauvin R, Chen YC, Lee JW, Soman P, Zorlutuna P, Nichol JW, Bae H, Chen S, Khademhosseini A. Biomaterials. 2012;33:3824–3834. doi: 10.1016/j.biomaterials.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arcaute K, Mann BK, Wicker RB. Ann Biomed Eng. 2006;34:1429–1441. doi: 10.1007/s10439-006-9156-y. [DOI] [PubMed] [Google Scholar]
- 8.Dhariwala B, Hunt E, Boland T. Tissue Eng. 2004;10:1316–1322. doi: 10.1089/ten.2004.10.1316. [DOI] [PubMed] [Google Scholar]
- 9.Chan V, Zorlutuna P, Jeong JH, Kong H, Bashir R. Lab Chip. 2010;10:2062–2070. doi: 10.1039/c004285d. [DOI] [PubMed] [Google Scholar]
- 10.Lin H, Zhang D, Alexander PG, Yang G, Tan J, Cheng AW, Tuan RS. Biomaterials. 2013;34:331–339. doi: 10.1016/j.biomaterials.2012.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arcaute K, Ochoa L, Medina F, Elkins C, Mann B, Wicker R. Mater Res Soc Symp Proc. 2005;874:191–198. [Google Scholar]
- 12.Lin L, Zhu J, Kottke-Marchant K, Marchant RE. Tissue Eng Part A. 2014;20:864–873. doi: 10.1089/ten.tea.2013.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao H, Munoz-Pinto D, Qu X, Hou Y, Grunlan MA, Hahn MS. Acta Biomater. 2008;4:1161–1171. doi: 10.1016/j.actbio.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gombotz WR, Guanghui W, Horbett TA, Hoffman AS. J Biomed Mater Res. 1991;25:1547–1562. doi: 10.1002/jbm.820251211. [DOI] [PubMed] [Google Scholar]
- 15.Bott K, Upton Z, Schrobback K, Ehrbar M, Hubbell JA, Lutolf MP, Rizzi SC. Biomaterials. 2010;31:8454–8464. doi: 10.1016/j.biomaterials.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 16.Bryant SJ, Bender RJ, Durand KL, Anseth KS. Biotechnol Bioeng. 2004;86:747–755. doi: 10.1002/bit.20160. [DOI] [PubMed] [Google Scholar]
- 17.Mironi-Harpaz I, Wang DY, Venkatraman S, Seliktar D. Acta Biomater. 2012;8:1838–1848. doi: 10.1016/j.actbio.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 18.Nicodemus GD, Bryant SJ. Tissue Eng Part B. 2008;14:149–165. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dikovsky D, Bianco-Peled H, Seliktar D. Biophys J. 2008;94:2914–2925. doi: 10.1529/biophysj.107.105841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peyton SR, Raub CB, Keschrumrus VP, Putnam AJ. Biomaterials. 2006;27:4881–4893. doi: 10.1016/j.biomaterials.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 21.Elomaa L, Kang Y, Seppälä JV, Yang Y. J Polym Sci Part A. 2014;52:3307–3315. [Google Scholar]
- 22.Ohya Y, Yamamoto H, Nagahama K, Ouchi T. J Polym Sci Part A. 2009;47:3892–3903. [Google Scholar]
- 23.Zhao Y, Li J, Yu H, Wang G, Liu W. Int J Pharm. 2012;430:282–291. doi: 10.1016/j.ijpharm.2012.03.043. [DOI] [PubMed] [Google Scholar]
- 24.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lendlein A, Schmidt AM, Schroeter M, Langer R. J Polym Sci Part A. 2005;43:1369–1381. [Google Scholar]
- 26.Lin H, Kai T, Freeman BD, Kalakkunnath S, Kalika DS. Macromolecules. 2005;38:8381–8393. [Google Scholar]
- 27.Andreopoulos FM, Beckman EJ, Russell AJ. Biomaterials. 1998;19:1343–1352. doi: 10.1016/s0142-9612(97)00219-6. [DOI] [PubMed] [Google Scholar]
- 28.Metters AT, Anseth KS, Bowman CN. Polymer. 2000;41:3993–4004. [Google Scholar]
- 29.Levental I, Georges PC, Janmey PA. Soft Matter. 2007;3:299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- 30.Lee S, Tong X, Yang F. Acta Biomater. 2014;10:4167–4174. doi: 10.1016/j.actbio.2014.05.023. [DOI] [PubMed] [Google Scholar]