

Abstract
Endothelial dysfunction occurs in chronic kidney disease (CKD) and increases the risk for cardiovascular disease. The mechanisms of endothelial dysfunction seem to evolve throughout kidney disease progression culminating in reduced L-arginine transport and impaired nitric oxide bioavailability in advanced disease. This review examines the hypothesis that aerobic exercise may reverse endothelial dysfunction by improving endothelial cell L-arginine uptake in CKD.
Keywords: endothelial dysfunction, chronic kidney disease, nitric oxide, L-arginine, CAT-1, aerobic exercise
INTRODUCTION
Chronic kidney disease (CKD) is a major public health concern that results in progressive and irreversible loss of renal function, and a concomitant increase in cardiovascular disease (CVD) related mortality that accounts for more than half of all deaths in patients with CKD (38). Importantly, the increased incidence of CVD with CKD persists even in the absence of traditional CVD risk factors suggesting that the mechanisms involved are unique to CKD. Vascular endothelial dysfunction is emerging as an important mediator and “non-traditional” risk factor in the development of CVD and is thus an attractive target for interventions designed to reduce the risk and burden of CVD in patients with CKD (21). The mechanisms by which endothelial dysfunction occurs in CKD are multifaceted and appear to evolve throughout the progression of the disease. The early mechanisms likely involve a reduction in nitric oxide (NO) synthesis and bioavailability that is largely the result of oxidative stress and endogenous inhibition of endothelial NO synthase (eNOS) (21). Limited but compelling evidence suggests that these mechanisms become less reversible in later stages of CKD due to an overwhelming reduction in the endothelial cell uptake of L-arginine, the critical substrate for NO synthesis (2, 22, 36).
Superimposed on the cardiovascular consequences of CKD is a reduction in cardiorespiratory fitness and physical function that occurs during in the initial stages of CKD and continues throughout disease progression (14, 27). Physical deconditioning and CVD are linked in CKD; patients with lower cardiorespiratory fitness exhibit a higher CVD burden (13). Factors associated with both CKD and CVD exacerbate the decline in cardiorespiratory fitness, leading to a downward spiral of physical inactivity, deconditioning, and frailty that ultimately lead to decreased quality of life, increased hospitalization and increased mortality rates (Figure 1). Thus, interventions that increase physical activity and maintain cardiorespiratory fitness and physical function may be important for improving quality of life and reducing CVD mortality risk in CKD (16). Aerobic exercise training is an important intervention for modifying traditional CVD risk factors but may also improve endothelial function in CKD.
Figure 1.
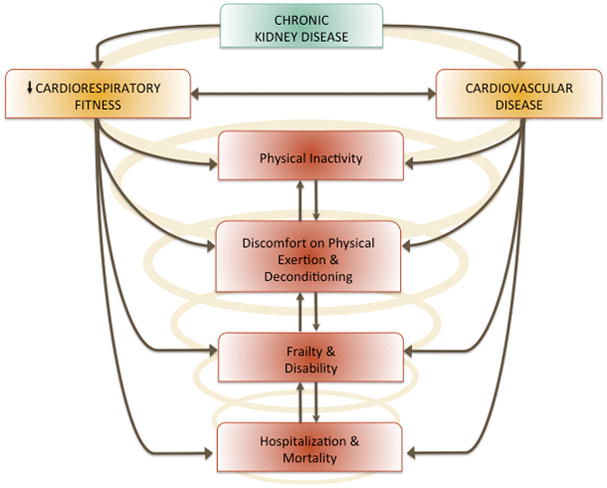
Chronic kidney disease is associated with both poor cardiorespiratory fitness and cardiovascular disease that have deleterious consequences including physical inactivity.
Regular aerobic exercise is a well-established stimulus for improving NO synthesis and is therefore an important “non-pharmacological” strategy for improving cardiorespiratory fitness and improving endothelial function in patients with CVD. Although previous findings have consistently reported increases in cardiorespiratory fitness with regular exercise in the CKD population (16), investigations determining the effects of regular aerobic exercise on endothelial function and the related mechanisms specific to CKD remain scarce. The mechanisms by which endothelial dysfunction occurs with advancing CKD may limit the efficacy of strategies such as antioxidant and L-arginine supplementation, but may be susceptible to modification by aerobic exercise training. We propose a novel hypothesis by which regular aerobic exercise may reverse endothelial dysfunction by improving endothelial cell L-arginine uptake in CKD; thus aerobic exercise training could represent a novel adjunctive therapy for lowering the burden of CVD and improving cardiorespiratory fitness across all stages of CKD. This review will focus primarily on the mechanisms of endothelial dysfunction in CKD and will introduce the novel mechanisms by which exercise might improve endothelial function in patients with CKD.
ENDOTHELIAL DYSFUNCTION
The vascular endothelium is a single monolayer of cells lining the interior lumen of all blood vessels. Among the many important functions of the endothelium is the synthesis and release of the vascular protective and vasodilatory molecule nitric oxide (NO), which maintains homeostasis within blood vessels by regulating leukocyte adhesion and infiltration, smooth muscle proliferation, inflammation, and vascular tone. Endothelium-derived NO is synthesized from the amino acid L-arginine by the enzyme endothelial nitric oxide synthase (eNOS), and diffuses into the surrounding vascular smooth muscle where it initiates a signaling cascade that ultimately results in smooth muscle relaxation and vasodilation. Endothelial dysfunction is typically characterized by a reduction in endothelium-dependent vasodilation, primarily due to a decline in endothelial-derived NO synthesis and/or bioavailability. Forearm reactive hyperemia, a measure of microvascular vasodilation that is mediated in part by NO production, is impaired in patients with end stage renal disease (ESRD) and is directly associated with increased mortality risk (20). Endothelial dysfunction also occurs in patients with more moderate CKD (18) suggesting that impairments in endothelial function serve as a key intermediary event linking CKD to increased CVD risk. Indeed, endothelial dysfunction has been shown to precede the development of atherosclerosis, and impairments in NO signaling have been linked to cardiac dysfunction (17), implicating a dysfunctional endothelium as a key mediator in pathogenesis of ischemic heart disease. Consequently, patients with CKD are more likely to die from CVD than to progress to ESRD (10). Thus, therapies aimed at improving endothelial function hold great promise for reducing the burden of CVD in patients with CKD.
MECHANISMS OF ENDOTHELIAL DYSFUNCTION IN CKD
Endothelial dysfunction is evident in patients with moderate-to-severe CKD (18) and can be modeled in rodents that have undergone surgical renal mass reduction or infarction (12, 22). The specific mechanisms by which endothelial dysfunction occur in CKD are not completely understood; however, limited evidence from animal models and human studies point to reduced NO synthesis and/or bioavailability secondary to oxidative stress, and a disruption in the synthesis, delivery, and utilization of L-arginine, the primary substrate for NO synthesis. The relative contribution of these mechanisms appears to change throughout the progression of CKD culminating in endothelial dysfunction that is less reversible under severe uremic conditions (Figure 2). These mechanisms are reviewed below.
Figure 2.
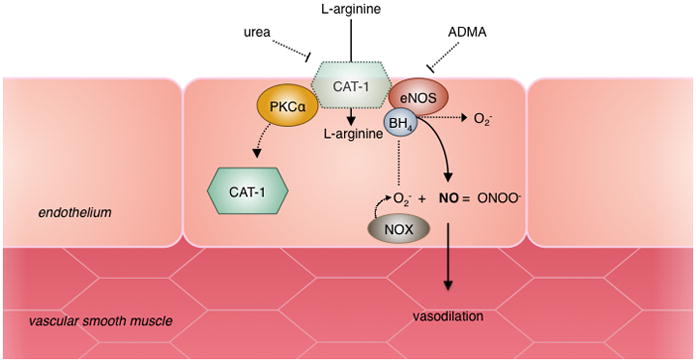
Nitric oxide (NO) is synthesized from L-arginine by endothelial nitric oxide synthase (eNOS) and diffuses into the vascular smooth muscle where it initiates vasodilation. Elevated levels of ADMA and oxidative stress contribute to endothelial dysfunction in early stages of CKD. ADMA inhibits NO synthesis through competitive inhibition of eNOS. Superoxide (O2−) is produced by NADPH oxidase (NOX) enzymes and combines with NO, forming peroxynitrite (ONOO−) leading to reduced NO bioavailability. Oxidation of the eNOS cofactor BH4 can also lead to uncoupling of eNOS resulting in further O2− production. In later stages of CKD, increased levels of circulating urea inhibit L-arginine transport through the cationic amino acid transporter (CAT-1) and post-translational modifications of CAT-1 by protein kinase C alpha (PKCα) result in decreased transport activity and internalization of CAT-1 to the cytosol.
Oxidative stress
Oxidative stress is characterized by a disproportionate increase in cellular reactive oxygen species (ROS) relative to endogenous antioxidant defenses, and is a key mechanism in the early pathogenesis of endothelial dysfunction in patients with mild-to-moderate CKD. Our lab has used laser Doppler flowmetry in the human skin microvasculature (a representative model of systemic microcirculatory function) coupled with intradermal microdialysis, to determine the role of oxidative stress in endothelial dysfunction of patients with mild-to-moderate CKD (8, 9). Local heating of the skin results in a vasodilatory response and marked increase in blood flow within the cutaneous circulation that is primarily mediated by an increase in NO synthesis. Compared with healthy, age-matched control subjects, this measure of microcirculatory function is attenuated in patients with stage 3–4 CKD; however, acute local perfusion of the nonspecific antioxidant ascorbic acid within the cutaneous microvasculature ameliorates the impaired dilatory response in CKD patients to levels that resemble healthy control subjects, suggesting that excess ROS contribute to the suppression of vascular function in CKD (8). Pathological levels of the free radical superoxide (O2−) are capable of rapidly reacting with NO to form the secondary radical peroxynitrite (ONOO−), reducing the overall bioavailability of NO available for vasodilation. This mechanism appears to contribute to the oxidative stress mediated suppression of endothelial function in CKD, as local perfusion the skin microvasculature with the superoxide dismutase (SOD) mimetic Tempol, restores microvascular function in patients with CKD (9).
Elevated levels of ROS have been shown to oxidize the essential eNOS cofactor, tetrahydrobiopterin (BH4). Not only does this result in destabilization and uncoupling of the eNOS dimer, but can also convert eNOS itself into a generator of superoxide, further perpetuating oxidative stress within the endothelium. Chronic supplementation with exogenous BH4 has been shown to prevent eNOS uncoupling and preserve endothelium-dependent relaxation in rats with CKD (40); however, a recent clinical trial with the oral drug sapropterin (a synthetic form of BH4), failed to demonstrate similar improvements in patients with CKD (28), thus, the role of reduced BH4 in mediating oxidative stress and endothelial dysfunction in CKD remains inconclusive.
One potential therapeutic approach to combat vascular oxidative stress is to target the cellular sources of superoxide production. NADPH oxidases have been identified as a major enzymatic source of superoxide generation and likely play a role in endothelial dysfunction in CKD. Our lab has recently demonstrated that acute local perfusion of the NADPH oxidase inhibitor apocynin, restores NO-mediated vasodilation in the skin microvasculature (9). The mechanisms by which NADPH oxidases are activated in CKD are not completely known; however, angiotensin II has been shown to increase superoxide production through activation of NADPH oxidases (11) and may be an important contributor to oxidative stress in CKD. In support of this hypothesis, short-term ACE inhibition has been shown to improve endothelial function in humans with stage 1 diabetic CKD (41). Thus, interventions that target the cellular sources of superoxide production within the endothelium have the potential to improve endothelial function and reduce CVD risk in patients with CKD.
Endogenous NO Inhibition by ADMA
In addition to oxidative stress, increased formation of the endogenous NOS inhibitor asymmetric dimethylarginine (ADMA) is independently associated with CVD risk in patients with CKD (21). ADMA is produced as the result of post-transcriptional methylation of L-arginine residues within proteins by arginine methyltransferase enzymes (PRMTs) and is released in its free form following protein hydrolysis. The chemical structure of ADMA is close enough to that of L-arginine that it prevents NO production through competitive inhibition of eNOS. ADMA is elevated in patients with stage 3–4 CKD who exhibit endothelial dysfunction (8) and elevated ADMA contributes to a reduction in the L-arginine/ADMA ratio (9), suggesting a relative reduction in L-arginine availability for NO production in CKD. Local perfusion with exogenous L-arginine in patients with mild-to-moderate CKD restores NO-mediated vasodilation to levels of healthy control subjects (8) presumably by restoring the levels of L-arginine relative to increased ADMA.
The clearance of ADMA by the kidney is impaired in CKD and contributes to elevated plasma concentrations; however, elevated ADMA in CKD is not explained by impaired urinary clearance alone (2) and is likely associated with increased ADMA synthesis. The mechanism by which ADMA formation is increased in CKD is likely related to increased expression and activity of PRMT’s, and reduced degradation of ADMA by dimethylarginine dimethylaminohydrolase (DDAH) (2). PRMT expression is elevated in an animal model of CKD (24) and may be a potential therapeutic target to restore endothelial function in humans. PRMT expression and activity is increased in the presence of oxidized LDL cholesterol and results in increased production of ADMA in endothelial cells (2) suggesting a potentiation of ADMA formation by oxidative stress in CKD. Treatment with exogenous antioxidants has been shown to reduce ADMA levels in patients with CKD and in patients on hemodialysis (35) and may be an effective strategy for restoring endothelial function in CKD. In addition, suppression of the renin-angiotensin system with short-term ACE inhibition or angiotensin II receptor inhibition has been shown to reduce ADMA levels and improve flow mediated dilation (FMD) in patients with non-diabetic CKD (41).
Impaired L-arginine Availability
Although the intracellular concentration of L-arginine typically exceeds what is necessary for NO synthesis, treatment with exogenous L-arginine has repeatedly been shown to augment NO production in a variety of clinical pathologies (5, 6, 31). This phenomenon is referred to as the “L-arginine Paradox” and suggests that NO production relies, at least in part, on an adequate supply of extracellular L-arginine. Decreases in the availability, production or delivery of L-arginine all appear to play important roles in the pathogenesis of endothelial dysfunction in CKD; however, the relative contributions of these mechanisms appear to be different throughout disease progression.
L-arginine is a semi-essential amino acid that can either be obtained through dietary sources or synthesized by the proximal tubule cells of the kidney. In addition to acting as a substrate for NO synthesis, L-arginine is also metabolized by the enzyme arginase as part of the urea cycle. Increased arginase activity is thought to contribute to endothelial dysfunction in CVD through competition with eNOS for L-arginine. Systemic arginase inhibition slows the early progression of renal dysfunction in animals with CKD (4); however, vascular arginase expression and activity do not appear to be elevated once renal dysfunction is established (4, 22) and arginase inhibition does not reverse endothelial dysfunction in animals with more advanced CKD (22). The accumulation of circulating urea that results from impaired renal function may act in a negative feedback manner to suppress the urea cycle, including arginase activity. This has been demonstrated in the liver of animals with CKD (26) and may also explain why arginase does not contribute to endothelial dysfunction during later stages of CKD.
An alternative mechanism by which impaired L-arginine availability may occur in CKD is through decreased synthesis. L-arginine is synthesized from the precursor, L-citrulline, in the proximal tubule cells of the kidney (3). Protein expression of the key enzymes responsible for L-arginine synthesis is diminished in the kidneys of animals with CKD, and plasma levels of L-citrulline levels are elevated, suggesting less conversion of L-citrulline to L-arginine (3). To compensate for the decrease in endogenous L-arginine synthesis, treatment with exogenous L-arginine has been explored in the context of endothelial function; however, this approach has been met with mixed results. Dietary supplementation with L-arginine has been shown to prevent the development of endothelial dysfunction in an animal model of CKD when initiated immediately following disease induction (40); however, we have recently demonstrated that oral supplementation with L-arginine does not reverse endothelial dysfunction in animals once CKD has already been established (23). These findings are mirrored by acute infusion studies in humans in which exogenous L-arginine improved vasodilation in the skin microvasculature of patients with moderate CKD (8) but did not restore endothelial function in the forearm microvasculature of patients with severe renal failure (7). Much like the role of arginase, the ability of exogenous L-arginine to compensate for reduced L-arginine availability appears to be diminished by the progression of CKD and limits its ability to restore endothelial function.
A key mechanism that may explain why L-arginine is effective in earlier stages of CKD but is less effective during advanced disease progression is impaired cellular uptake of L-arginine. The plasma concentration of L-arginine is paradoxically maintained in patients with CKD, despite a decrease in endogenous production suggesting that plasma L-arginine may become “trapped” within the vascular space at some point throughout the disease progression. The uptake of L-arginine into cells, including endothelial cells, occurs through the cationic amino acid transporter CAT-1, and has been shown to be impaired in endothelial cells cultured in uremic plasma (2). CAT-1 co-localizes with eNOS within the endothelial cell membrane (25) creating a convenient mechanism by which L-arginine can be directly delivered to the endothelium to facilitate NO synthesis. Our lab and others have shown that the abundance and transport activity of CAT-1 within the vasculature is impaired in animal models of CKD (22, 36) and is likely an important contributor to endothelial dysfunction (22). The mechanisms regulating the decrease in L-arginine transport in CKD have not been completely elucidated; however, expression and activation of protein kinase C alpha (PKCα) has been shown to occur within the vasculature of uremic rats and is associated with a reduction in CAT-1 transport activity (15). The exact mechanisms by which PKCα attenuates L-arginine transport remain to be fully understood but appear to influence the subcellular localization of CAT-1 as activation of PKCα has been shown to promote the internalization of CAT-1 into the cytosol (34). Additionally, PKCα may exert direct effects on L-arginine transport via post-translational phosphorylation of CAT-1 (15). Taken together, these findings suggest that L-arginine transport can be altered via changes in both CAT-1 activity and protein abundance within the cell membrane. Treatments aimed at restoring CAT-1 expression and activity may therefore have a positive effect on vascular function by improving substrate delivery and NO production.
Endothelial Dysfunction in Advanced CKD: Evidence for a “Uremic Switch”
The stimulus for decreased L-arginine transport appears to be related to an increase in circulating urea and other uremic toxins that occurs with CKD. CAT-1 transport activity has been shown to be inhibited in endothelial cells cultured in uremic plasma and in cells cultured in a synthetic solution containing uremic levels of urea (2). The influx of extracellular urea through endothelial urea transporters (UT) prevents L-arginine transport in cells treated with high levels of urea (2). Thus, the progressive increase in circulating urea and other uremic toxins that accompanies advancement of CKD may trigger a “uremic switch” that results in endothelial dysfunction that is less reversible by exogenous treatment with L-arginine due to inefficient transport through CAT-1 (Figure 3).
Figure 3.
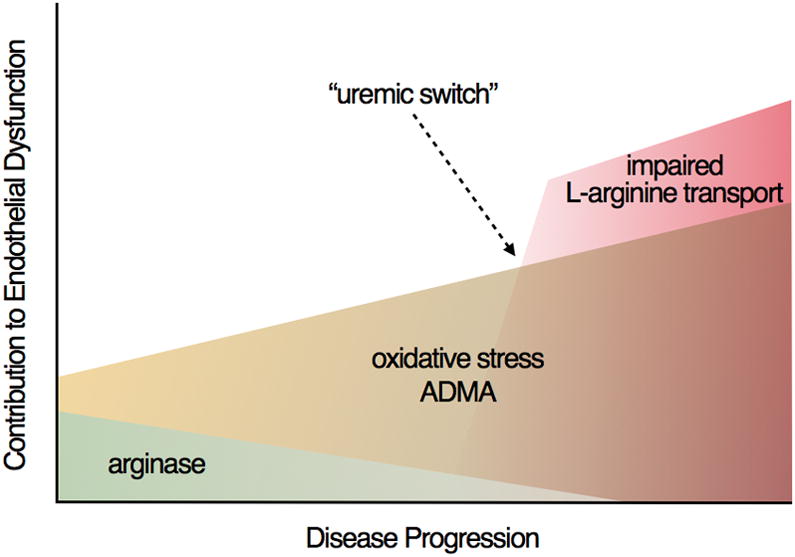
The mechanisms of endothelial dysfunction appear to change throughout the progression of CKD resulting in endothelial dysfunction that is increasingly less reversible. Competition for L-arginine by the enzyme arginase may play a limited role in early CKD but appears to play less of a role throughout disease progression. Oxidative stress and increased levels of ADMA are important contributors to endothelial dysfunction in mild-to-moderate CKD. The accumulation of uremic toxins that occurs throughout the progression of CKD contributes to impaired transport of L-arginine into the endothelium. During advanced stages of CKD, this uremic toxin burden becomes so severe that a “uremic switch” occurs in which reduced L-arginine transport becomes the rate limiting step for NO production.
The point at which a uremic switch may occur during kidney disease progression is presently unknown; however, in addition to limiting the efficacy of L-arginine, the consequences appear to overshadow the contribution of other important mechanisms of endothelial dysfunction such as oxidative stress. Evidence for a uremic switch is supported by our own studies in which acute scavenging of superoxide with the antioxidant Tempol, was unable to reverse endothelial dysfunction in isolated arteries from animals with advanced CKD that exhibited impaired L-arginine transport (22) as it did in our studies of patients with moderate CKD (9). Collectively, studies in animal models and in patients with CKD suggest that oxidative stress, ADMA, and impaired L-arginine transport contribute to endothelial dysfunction that is increasingly less reversible by conventional approaches as the disease progresses. As such, there is a critical need for interventions that target these mechanisms and reduce CVD risk in patients with CKD.
EXERCISE AND ENDOTHELIAL FUNCTION IN CKD
Aerobic exercise is a well-known stimulus for improving endothelial function in CVD and limited but compelling evidence from animal studies suggests that exercise may have similar beneficial effects on endothelial function in CKD (1, 23, 37). The mechanisms by which aerobic exercise training improves endothelial function in CVD are multifaceted, but are largely associated with an increase in endothelial NO bioavailability. Recent evidence from our own work supports the novel hypothesis that aerobic exercise training improves endothelial function in CKD by reversing impairments to the L-arginine transport system, thus facilitating substrate delivery for NO production (23). Due to the body of literature suggesting that endothelial dysfunction becomes increasingly more difficult to reverse in advanced CKD, aerobic exercise training may represent a novel adjunctive therapy to treating endothelial dysfunction in CKD.
The Role of Shear Stress in L-arginine Transport
Aerobic exercise results in a systemic increase in blood flow that produces tangential and circumferential shear forces against the endothelial surface. Shear stress is the primary endogenous stimulus for endothelial NO production, which is largely attributable to increased eNOS expression and activation, and is thought to be among the most important mechanisms by which regular aerobic exercise training improves endothelial function in CVD. Endothelial cell L-arginine uptake occurs in a stimulus-dependent manner in response to increasing levels of shear stress in cultured endothelial cells (32). This mechanism likely plays an important role in maintaining endothelial function by ensuring that an appropriate supply of substrate is available for NO synthesis in response to increasing demands for blood flow. Importantly, shear-stress mediated NO production is attenuated when L-arginine transport is impaired (32) suggesting that CAT-1 may be important for transducing the effects of shear stress to increase NO synthesis. The ability of shear stress to augment L-arginine transport activity suggests that regular aerobic exercise training may be an effective therapy to counteract the impairments in L-arginine transport that occur with CKD. In support of this hypothesis, 8 weeks of aerobic exercise training has been shown to improve L-arginine uptake by the forearm microvasculature in patients with congestive heart failure (29). We have recently extended these findings to an animal model of CKD in which we demonstrated improved L-arginine transport activity in response to 8 weeks of very low volume voluntary wheel running, a model of regular aerobic exercise training (23).
The precise mechanism by which elevated shear stress increases L-arginine transport in CKD is not known. Evidence from our own work suggests that the increase in L-arginine transport by voluntary wheel running is not associated with an increase in CAT-1 abundance, but rather through an increase in L-arginine transport activity (23). Thus, the regulation of L-arginine transport by aerobic exercise training likely occurs at the post-translational level, presumably through modification of CAT-1. One potential mechanism by which exercise may improve CAT-1 activity is through a decrease in the expression or activity of PKCα. As mentioned previously, vascular PKCα expression is increased in CKD and is thought to play a role in the post-translational regulation of L-arginine transport via direct phosphorylation and/or by promoting the internalization of CAT-1 to the cytosol. Our recent work demonstrated that 4 weeks of voluntary wheel running reverses the increase in aortic PKCα protein expression that occurs with CKD and that this is associated with a robust increase in L-arginine transport activity, independent of changes in total aortic CAT-1 abundance (23). Thus, we propose a novel hypothesis by which aerobic exercise may target the vascular endothelium in CKD and augment endothelial cell L-arginine transport by reducing PKCα expression, ultimately resulting in improved endothelial function (Figure 4). Future work is necessary to determine whether this mechanism increases L-arginine transport predominately by increasing CAT-1 transport activity, or by maintaining the abundance of CAT-1 within the endothelial cell membrane.
Figure 4.
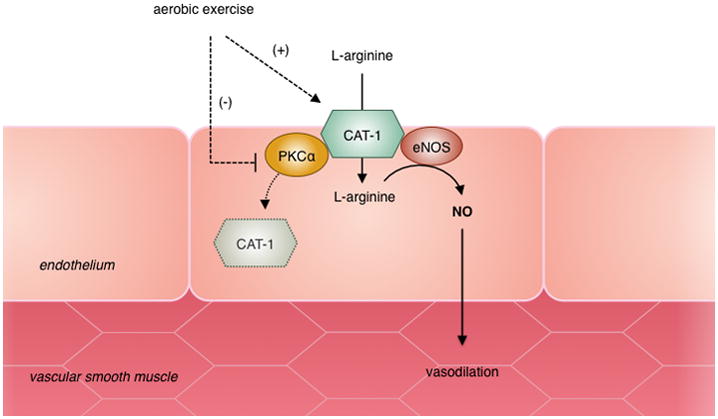
Novel hypothesis by which exercise improves endothelial function in CKD. Impairments in L-arginine uptake are largely mediated by activation of PKCα resulting in decreased CAT-1 transport activity and internalization of CAT-1 to the cytosol. Exercise improves L-arginine transport activity in part by decreasing PKCα expression and preventing the decline in CAT-1 activity and/or membrane localization.
Flipping the Switch: Exercise & L-arginine to Improve Endothelial Function in CKD
The ability of exogenous L-arginine to augment nitric oxide production, despite a seemingly sufficient intracellular concentration, has long been a topic of interest in vascular physiology. The fact that this phenomenon appears to be absent in patients and animals with advanced CKD provides a unique window into the role of the “L-arginine Paradox” in CKD. The finding that exercise improves L-arginine transport in CKD demonstrates the potential benefit of increased physical activity on vascular function in CKD; however, the association between exercise and L-arginine transport may be most effectively exploited when exercise and L-arginine are used in combination. We have recently demonstrated that dietary supplementation with L-arginine alone does not reverse endothelial dysfunction in rats with CKD; presumably due to impaired L-arginine transport activity (23). Importantly, wheel running significantly improved L-arginine transport activity and augmented NO-dependent endothelial function; however, the greatest improvement was seen when voluntary wheel running and L-arginine supplementation were combined (Figure 5). These findings suggest that while L-arginine itself may not be an effective therapy to treat vascular dysfunction in CKD, aerobic exercise may be capable of overcoming the “uremic switch” thus facilitating improved delivery and utilization of L-arginine. The use of aerobic exercise to flip the uremic switch may increase the efficacy of L-arginine supplementation for improving endothelial function in CKD.
Figure 5.
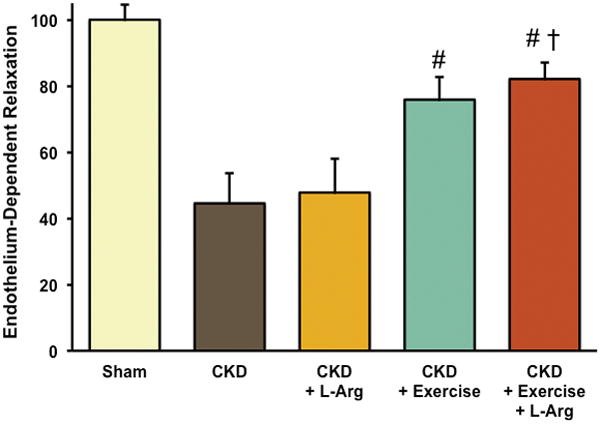
Endothelial function is impaired in rats with CKD relative to sham control rats. Supplementation with L-arginine alone does not improve endothelium-dependent relaxation (EDR); however 4 weeks of aerobic exercise (through voluntary wheel running) improves EDR in rats with CKD and the greatest benefit is observed when aerobic exercise and L-arginine supplementation are performed in combination. Data represent the area under the dose response curve of EDR in response to increasing doses of the endothelium-dependent vasodilator acetylcholine. * p<0.05 vs. Sham; # p<0.05 vs. CKD; † p<0.05 vs. CKD+ L-arg. [Adapted from (23). Copyright © 2014 The American Physiological Society. Used with permission.]
Reversing the impairment in endothelial L-arginine transport may also lead to direct improvements in cardiac function, presumably through increased NO production within coronary arteries. In support of this hypothesis, endothelial-specific overexpression of CAT-1 has recently been reported to prevent pressure overload-induced cardiac hypertrophy in mice (33) and we have recently demonstrated preserved cardiac function in a rodent model of CKD subjected to 4-weeks of voluntary wheel running (19). While the combined effect of exercise and L-arginine supplementation on cardiac function in CKD has not yet been tested, these findings collectively suggest that aerobic exercise and sufficient endothelial L-arginine transport are important for the maintenance of cardiac function. Improved L-arginine transport and utilization by the endothelium would be particularly beneficial to patients with CKD and exercise might be a novel therapeutic approach to treat endothelial dysfunction and reduce CVD risk in CKD.
SUMMARY & PERSPECTIVES
In summary, CVD remains the leading cause of death in CKD, with patients more likely to die from CVD than progress to end stage renal failure. Endothelial dysfunction is characteristic of CKD and is thought be a major non-traditional risk factor mediating the CVD burden in CKD. As renal function worsens with progression of CKD, the mechanisms of endothelial dysfunction appear to change and conventional therapies to restore endothelial health become less effective. While oxidative stress and increased levels of ADMA contribute to impaired NO production and bioavailability in the earlier stages of CKD, reduced endothelial transport of L-arginine appears to be a limiting factor for NO production in severe CKD. Recent findings from our laboratory demonstrate that L-arginine uptake is increased following relatively low volumes of aerobic exercise in an animal model of severe CKD, improving the efficacy of L-arginine supplementation. Although the mechanisms remain to be fully elucidated, it is hypothesized that exercise induced endothelial shear stress prevents the phosphorylation and internalization of the L-Arginine transporter CAT-1, thus maintaining adequate L-arginine delivery for NO production.
Despite the benefits of aerobic exercise training on L-arginine transport, NO production, and overall vascular function in animal models, the influence of exercise training on vascular function in humans with CKD is less established (16). To date, the only known trial to investigate the effects of aerobic exercise on endothelial function showed no significant improvements; however the exercise intervention consisted of a low-volume, intermittent home-based training program that may not have provided an adequate stimulus to reverse endothelial dysfunction (39). While moderate intensity aerobic exercise training does appear to positively influence some known mediators of endothelial dysfunction such as oxidative stress in CKD patients (30), there is a need for more randomized controlled trials investigating the effects of aerobic exercise on vascular health in CKD patients to ascertain the most effective exercise doses and guide evidence based exercise prescription. In addition, further research is needed to determine whether the mechanisms by which aerobic exercise improves endothelial function in animal models of CKD translate to CKD patients. The novel mechanisms by which aerobic exercise targets the vascular endothelium in animal models of CKD provide a strong rationale for translational investigations in human patients. Regular aerobic exercise has the potential to serve as an important adjunctive therapy in patients with CKD to ameliorate endothelial dysfunction and reduce the high risk of CVD making this an important area for future research.
SUMMARY.
Exercise reverses impairments in endothelial L-arginine transport and improves endothelial function in chronic kidney disease by increasing nitric oxide bioavailability.
Acknowledgments
This work was supported by National Institutes of Health grants DK080469, GM103446, HL113514, and the ACSM Foundation.
Footnotes
Disclosures: CRM: None; DLK: None; DGE: None
References
- 1.Adams G, Zhan C, Haddad F, Vaziri N. Voluntary exercise during chronic renal failure in rats. Med Sci Sports Exerc. 2005;37(4):557–62. doi: 10.1249/01.mss.0000159006.87769.67. [DOI] [PubMed] [Google Scholar]
- 2.Baylis C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat Clin Pract Nephrol. 2006;2(4):209–20. doi: 10.1038/ncpneph0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen G, Baylis C. In vivo renal arginine release is impaired throughout development of chronic kidney disease. Am J Physiol -Renal Physiol. 2010;298(1):F95–F102. doi: 10.1152/ajprenal.00487.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen G, Moningka NC, Sasser JM, et al. Arginine and Asymmetric Dimethylarginine in Puromycin Aminonucleoside-Induced Chronic Kidney Disease in the Rat. Am J Nephrol. 2012;35(1):40–8. doi: 10.1159/000334740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarkson P, Adams MR, Powe AJ, et al. Oral L-arginine improves endothelium-dependent dilation in hypercholesterolemic young adults. J Clin Invest. 1996;97(8):1989–94. doi: 10.1172/JCI118632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooke JP, Andon NA, Girerd XJ, Hirsch AT, Creager MA. Arginine Restores Cholinergic Relaxation of Hypercholesterolemic Rabbit Thoracic Aorta. Circulation. 1991;83(3):1057–62. doi: 10.1161/01.cir.83.3.1057. [DOI] [PubMed] [Google Scholar]
- 7.Cross JM, Donald AE, Kharbanda R, Deanfield JE, Woolfson RG, MacAllister RJ. Acute administration of L-arginine does not improve arterial endothelial function in chronic renal failure. Kidney Int. 2001;60(6):2318–23. doi: 10.1046/j.1523-1755.2001.00059.x. [DOI] [PubMed] [Google Scholar]
- 8.DuPont JJ, Farquhar WB, Townsend RR, Edwards DG. Ascorbic acid or L-arginine improves cutaneous microvascular function in chronic kidney disease. J Appl Physiol. 2011;111(6):1561–7. doi: 10.1152/japplphysiol.00419.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DuPont JJ, Ramick MG, Farquhar WB, Townsend RR, Edwards DG. NAD(P)H Oxidase-Derived Superoxide Contributes to Impaired Cutaneous Microvascular Function in Chronic Kidney Disease. Am J Physiol Renal. 2014;306(12):F1499–506. doi: 10.1152/ajprenal.00058.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foley R, Murray A, Li S, et al. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States medicare population, 1998 to 1999. J Am Soc Nephrol. 2005;16(2):489–95. doi: 10.1681/ASN.2004030203. [DOI] [PubMed] [Google Scholar]
- 11.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91(1–3):21–7. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 12.Hasdan G, Benchetrit S, Rashid G, Green J, Bernheim J, Rathaus M. Endothelial dysfunction and hypertension in 5/6 nephrectomized rats are mediated by vascular superoxide. Kidney Int. 2002;61(2):586–90. doi: 10.1046/j.1523-1755.2002.00166.x. [DOI] [PubMed] [Google Scholar]
- 13.Howden EJ, Weston K, Leano R, et al. Cardiorespiratory fitness and cardiovascular burden in chronic kidney disease. J Sci Med Sport. 2015;18(4):492–7. doi: 10.1016/j.jsams.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Howden EJ, Fassett RG, Isbel NM, Coombes JS. Exercise Training in Chronic Kidney Disease Patients. Sports Medicine. 2012;42(6):473–88. doi: 10.2165/11630800-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.Ingbir M, Schwartz IF, Shtabsky A, et al. Rosiglitazone improves aortic arginine transport, through inhibition of PKC alpha, in uremic rats. Am J Physiol -Renal Physiol. 2008;295(2):F471–7. doi: 10.1152/ajprenal.00619.2007. [DOI] [PubMed] [Google Scholar]
- 16.Kirkman DL, Lennon-Edwards S, Edwards DG. The Importance of Exercise for Chronic Kidney Disease Patients. J Renal Nutr. 2014;24(6):E51. doi: 10.1053/j.jrn.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuczmarski JM, Martens CR, Lennon-Edwards SL, Edwards DG. Cardiac function and tolerance to ischemia-reperfusion injury in chronic kidney disease. Nephrol Dial Transplant. 2014;29(8):1514–24. doi: 10.1093/ndt/gft336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuczmarski JM, Darocki MD, DuPont JJ, et al. Effect of moderate-to-severe chronic kidney disease on flow-mediated dilation and progenitor cells. Exp Biol Med. 2011;236(9):1085–92. doi: 10.1258/ebm.2011.011008. [DOI] [PubMed] [Google Scholar]
- 19.Kuczmarski JM, Martens CR, Kim J, Lennon-Edwards SL, Edwards DG. Cardiac function is preserved following 4 weeks of voluntary wheel running in a rodent model of chronic kidney disease. J Appl Physiol. 2014;117(5):482–91. doi: 10.1152/japplphysiol.00344.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.London GM, Pannier B, Agharazii M, Guerin AP, Verbeke FHM, Marchais SJ. Forearm reactive hyperemia and mortality in end-stage renal disease. Kidney Int. 2004;65(2):700–4. doi: 10.1111/j.1523-1755.2004.00434.x. [DOI] [PubMed] [Google Scholar]
- 21.Martens CR, Edwards DG. Peripheral vascular dysfunction in chronic kidney disease. Cardiol Res Pract. 2011;2011:267257. doi: 10.4061/2011/267257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martens CR, Kuczmarski JM, Lennon-Edwards S, Edwards DG. Impaired L-arginine uptake but not arginase contributes to endothelial dysfunction in rats with chronic kidney disease. J Cardiovasc Pharmacol. 2014;63(1):40–8. doi: 10.1097/FJC.0000000000000022. [DOI] [PubMed] [Google Scholar]
- 23.Martens CR, Kuczmarski JM, Kim J, et al. Voluntary wheel running augments aortic L-arginine transport and endothelial function in rats with chronic kidney disease. American Journal of Physiology-Renal Physiology. 2014;307(4):F418–26. doi: 10.1152/ajprenal.00014.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuguma K, Ueda S, Yamagishi S, et al. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J Am Soc Nephrol. 2006;17(8):2176–83. doi: 10.1681/ASN.2005121379. [DOI] [PubMed] [Google Scholar]
- 25.McDonald K, Zharikov S, Block E, Kilberg M. A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the “arginine paradox”. J Biol Chem. 1997;272(50):31213–6. doi: 10.1074/jbc.272.50.31213. [DOI] [PubMed] [Google Scholar]
- 26.Moradi H, Kwok V, Vaziri ND. Effect of chronic renal failure on arginase and argininosuccinate synthetase expression. Am J Nephrol. 2006;26(3):310–8. doi: 10.1159/000094344. [DOI] [PubMed] [Google Scholar]
- 27.Padilla J, Krasnoff J, DaSilva M, et al. Physical functioning in patients with chronic kidney disease. J Nephrol. 2008;21(4):550–9. [PubMed] [Google Scholar]
- 28.Park J, Liao P, Sher S, Lyles RH, Deveaux DD, Quyyumi AA. Tetrahydrobiopterin lowers muscle sympathetic nerve activity and improves augmentation index in patients with chronic kidney disease. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2015;308(3):R208–18. doi: 10.1152/ajpregu.00409.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parnell M, Holst D, Kaye D. Augmentation of endothelial function following exercise training is associated with increased L-arginine transport in human heart failure. Clin Sci. 2005;109(6):523–30. doi: 10.1042/CS20050171. [DOI] [PubMed] [Google Scholar]
- 30.Pechter U, Ots M, Mesikepp S, et al. Beneficial effects of water-based exercise in patients with chronic kidney disease. International Journal of Rehabilitation Research. 2003;26(2):153–6. doi: 10.1097/00004356-200306000-00013. [DOI] [PubMed] [Google Scholar]
- 31.Pieper G, Peltier B. Amelioration by L-Arginine of a Dysfunctional Arginine Nitric-Oxide Pathway in Diabetic Endothelium. J Cardiovasc Pharmacol. 1995;25(3):397–403. doi: 10.1097/00005344-199503000-00008. [DOI] [PubMed] [Google Scholar]
- 32.Posch K, Schmidt K, Graier W. Selective stimulation of L-arginine uptake contributes to shear stress-induced formation of nitric oxide RID B-7052-2008. Life Sci. 1999;64(8):663–70. doi: 10.1016/s0024-3205(98)00608-0. [DOI] [PubMed] [Google Scholar]
- 33.Rajapakse NW, Johnston T, Kiriazis H, Chin-Dusting J, Du XJ, Kaye DM. Augmented endothelial L-arginine transport ameliorates pressure overload induced cardiac hypertrophy. LID - 10.1113/EP085250 [doi] Exp Physiol. 2015 May 11; doi: 10.1113/EP085250. (1469-445X (Electronic); 0958-0670 (Linking)) [DOI] [PubMed] [Google Scholar]
- 34.Rotmann A, Stand D, Martine U, Closs E. Protein kinase C activation promotes the internalization of the human cationic amino acid transporter hCAT-1 - A new regulatory mechanism for hCAT-1 activity. J Biol Chem. 2004;279(52):54185–92. doi: 10.1074/jbc.M409556200. [DOI] [PubMed] [Google Scholar]
- 35.Saran R, Novak JE, Desai A, et al. Impact of vitamin E on plasma asymmetric dimethylarginine (ADMA) in chronic kidney disease (CKD): a pilot study. Nephrology Dialysis Transplantation. 2003;18(11):2415–20. doi: 10.1093/ndt/gfg406. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz I, Ayalon R, Chernichovski T, et al. Arginine uptake is attenuated through modulation of cationic amino-acid transporter-1, in uremic rats. Kidney Int. 2006;69(2):298–303. doi: 10.1038/sj.ki.5000067. [DOI] [PubMed] [Google Scholar]
- 37.Shelkovnikov S, Summers SM, Elahimehr R, Adams G, Purdy RE, Vaziri ND. Effect of exercise training on aortic tone in chronic renal insufficiency. Am J Hypertens. 2008;21(5):564–9. doi: 10.1038/ajh.2008.24. [DOI] [PubMed] [Google Scholar]
- 38.Tonelli M, Wiebe N, Culleton B, et al. Chronic kidney disease and mortality risk: A systematic review. Journal of the American Society of Nephrology. 2006;17(7):2034–47. doi: 10.1681/ASN.2005101085. [DOI] [PubMed] [Google Scholar]
- 39.Van Craenenbroeck AH, Van Craenenbroeck EM, Van Ackeren K, et al. Effect of Moderate Aerobic Exercise Training on Endothelial Function and Arterial Stiffness in CKD Stages 3–4: A Randomized Controlled Trial. Am J Kidney Dis. 2015;66(2):285–96. doi: 10.1053/j.ajkd.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 40.Yamamizu K, Shinozaki K, Ayajiki K, Gemba M, Okamura T. Oral administration of both tetrahydrobiopterin and L-arginine prevents endothelial dysfunction in rats with chronic renal failure. J Cardiovasc Pharmacol. 2007;49(3):131–9. doi: 10.1097/FJC.0b013e31802f9923. [DOI] [PubMed] [Google Scholar]
- 41.Yilmaz MI, Axelsson J, Sonmez A, et al. Effect of Renin Angiotensin System Blockade on Pentraxin 3 Levels in Type-2 Diabetic Patients With Proteinuria. Clin J Am Soc Nephrol. 2009;4(3):535–41. doi: 10.2215/CJN.04330808. [DOI] [PMC free article] [PubMed] [Google Scholar]