

Abstract
Background & aims
Macrophage migration inhibitory factor (MIF) is a multi-potent cytokine that contributes to the inflammatory response to injury. MIF is expressed by multiple cell types; however, the cellular source and actions of MIF in alcoholic liver disease (ALD) are not well known. Here we tested the hypothesis that non-myeloid cells, specifically hepatocytes, are an important cellular source of MIF in ALD.
Methods
MIF expression was measured in HuH7 and differentiated THP-1 cells in response to ethanol. Ethanol-induced liver injury was assessed in C57BL/6 (WT) and Mif−/− bone marrow chimeras. MIF was measured in peripheral and suprahepatic serum, as well as visualized by immunohistochemistry in liver biopsies, from patients with alcoholic hepatitis (AH).
Results
HuH7 hepatocytes, but not THP-1 macrophages, released MIF in response to challenge with ethanol in culture. In chimeric mice expressing MIF in non-myeloid cells (Mif−/− →WT), chronic ethanol feeding increased ALT/AST, hepatic steatosis, and expression of cytokine/chemokine mRNA. In contrast, chimeric mice not expressing MIF in non-myeloid cells (WT→ Mif−/−) were protected from ethanol-induced liver injury. Immunohistochemical staining of liver biopsies from patients with AH revealed a predominant localization of MIF to hepatocytes. Interestingly, the concentration of MIF in suprahepatic serum, but not peripheral serum, was positively correlated with clinical indicators of disease severity and with an increased risk of mortality in patients with AH.
Conclusions
Taken together, these data provide evidence that hepatocyte-derived MIF is critical to the pathogenesis of ALD in mice and likely contributes to liver injury in patients with AH.
Keywords: Alcoholic Liver Disease, inflammation, MIF, hepatocytes, innate immune system, translational research
Graphical abstract: Being developed
Introduction
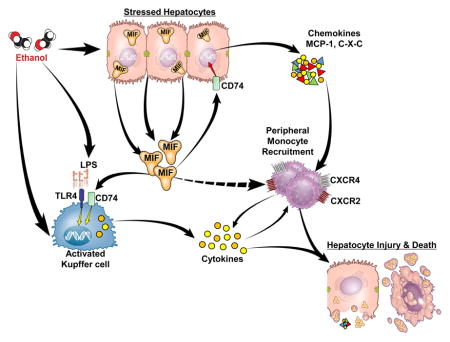
Excessive alcohol consumption is the primary cause of liver-related mortality in western countries[1]. Alcoholic hepatitis (AH) is the most severe form of alcoholic liver disease (ALD) and there is an urgent need to develop novel targeted therapies for severe AH[2]. Multiple molecular and cellular mechanisms contribute to the development of ALD. Prolonged alcohol abuse leads to an imbalance between intracellular antioxidant defense systems and the production of free radical species, promoting lipid peroxidation[3]. Injury to hepatocytes can result in the release of danger associated molecular patterns (DAMPs) which can activate pathways of sterile inflammation[3]. Chronic alcohol consumption also impairs the barrier function of the intestine and promotes bacterial dysbiosis, resulting in increased translocation of pathogen-associated molecular patterns (PAMPs) from the gastrointestinal lumen to the liver through the portal vein[1].
Recent studies have identified macrophage migration inhibitory factor (MIF), a pluripotent cytokine/chemokine, as a potential contributor to ethanol-induced liver injury in murine models of ALD[4,5]. MIF is constitutively expressed and stored in preformed intracellular pools in a wide variety of cell types including immune, endothelial, and epithelial cells. In the liver, MIF is produced by both hepatocytes and Kupffer cells[6]. MIF signals via the interaction with the CD74 receptor[7], as well as its co-receptors CXCR2, CXCR4 and CXCR7[8]. These receptors are expressed both on resident hepatic macrophages[9] and peripheral monocytes[10]; therefore, MIF release results in both activation of resident macrophages and recruitment of innate immune cells from the periphery.
The concentration of MIF is increased in the circulation of patients with ALD[11], as well as in mice in response to chronic ethanol feeding[5]. Since mice deficient in MIF are protected from ethanol-induced liver injury[5], it is important to determine the cellular source of MIF in response to ethanol exposure. Making use of cell culture and mouse models, as well as clinical samples from patients with AH, here we provide evidence that hepatocyte-derived MIF is important for the progression of ethanol-induced liver injury. Taken together, these data indicate that MIF released from injured hepatocytes likely serves as a DAMP in the progression of ALD, resulting in the recruitment of innate immune cells to the liver and activation of inflammatory pathways. Our results suggest that interfering with the release and/or signaling of MIF may be a viable therapeutic option in the prevention and/or treatment of ALD.
Patients/Materials and Methods
Cell lines and cell culture
HuH7 is a well-differentiated hepatocyte-derived carcinoma cell line, obtained from Japan Health Science Research Resources Bank (HSRRB, JCRB0403). THP-1 is an acute monocytic leukemia derived cell line (ATCC® TIB-202™). HuH7 and THP-1 cells were cultured as previously described[12]. Mycoplasma contamination has been excluded by testing the cultures periodically using fluorescent staining (Hoechst 33258). Cells were treated with or without 50mM ethanol or 200 ng/ml LPS (from E. coli serotype O127:B8), as detailed in the figure legends.
Generation of bone marrow chimeric mice and ethanol feeding
Female C57BL/6 (WT) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mif−/− mice on a C57BL/6 background were obtained from Dr. R. Bucala (Yale University, New Haven, CT, USA)[13] and a breeding colony was established at Cleveland Clinic. All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Bone marrow chimeras between WT and Mif−/− mice were generated as previously described[14] (see schematic of protocol in Supp Fig. S1). Four weeks post-transplant, chimeras were treated with clodronate-containing liposomes (Encapsula NanoSciences, Nashville, TN SKU # 8909) to deplete resident macrophages in the liver[15]. Seven days post-clodronate, chimeras were placed on the Lieber-DeCarli ethanol diet or pair-fed control diets for 25 days[16]. See supplemental information for additional details of the feeding protocol. Mouse body weight and food intakes are shown in Supp. Table S2.
Plasma ALT and AST and liver triglycerides
Plasma alanine aminotransferase (ALT) and plasma aspartate aminotransferase (AST) assay kits were purchased from Sekisui Diagnostics (Framingham, MA). Triglyceride assay kits were purchased from Pointe Scientific Inc. (Lincoln Park, MI).
RNA isolation and quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Cells/Mouse liver
RNA was isolated from cells or whole liver tissue using RNeasy Mini kits per the manufacturer’s instructions (Qiagen, Germantown, MD). 2–4μg of RNA was reverse transcribed and analyzed with Power SYBR qRT-PCR kits (Applied Biosystems) on an Mx3000P analyzer (Stratagene, La Jolla, CA). Relative messenger RNA (mRNA) expression was determined using gene-specific primers (Supp Table S1). Statistical analyses were performed on the ΔCt values (average Ct of gene of interest – average Ct of 18S)[17].
Human liver gene expression studies
RNA was extracted using TRIzol (Invitrogen, Life Technologies). Five hundred nanograms of total RNA were retro-transcribed and 200 ng of cDNA amplified (Applied Biosystems) in a final PCR volume of 10 μl using a StepOnePlus instrument (Applied Biosystems). TaqMan® Gene Expression Assay primers of MIF (Catalog number: 4331182, ID: Hs00236988_g1) were from Applied Biosystems. Results were normalized to 18S rRNA expression, and gene expression values were calculated based on the ΔΔCt method.
Patients
The study included patients admitted to the Liver Unit of the Hospital Clínic, Barcelona, between January 2000 and September 2007 with clinical, analytic, and histologic features of AH (Table 1). Inclusion criteria for patients with AH were as follows: patients with active alcohol abuse defined according to the Diagnostic and Statistical Manual of Mental Disorders IV[18] and excessive ethanol consumption (>60 g/day) for at least 3 months before admission; increased aminotransferase levels (aspartate aminotransferase [AST] > alanine aminotransferase [ALT]), high γ-glutamyltranspeptidase and bilirubin serum levels, and histologic diagnosis of AH characterized by the presence of hepatocellular damage (hepatocellular ballooning and presence of Mallory bodies), inflammatory infiltrate (neutrophils), and pericellular fibrosis. Patients with hepatocellular carcinoma or any other potential cause of liver disease were excluded from the study. Patients with autoimmune hepatitis were diagnosed by established criteria[7]. Additionally, as normal controls, we included fragments of healthy liver obtained during resection of liver metastases. A total of 66 patients with AH, 5 patients with autoimmune hepatitis and 7 for normal controls were included. Liver biopsy was obtained using a transjugular approach because most patients with AH have severe coagulation disorders. A fragment of liver tissue, serum and plasma were obtained at the time biopsy. All patients received nutritional and psychological support for achieving alcohol abstinence. The study was approved by the Ethics Committee of the Hospital Clínic of Barcelona, and all patients gave informed consent.
Table 1.
Baseline characteristics of patients.
| Characteristics | Median(25–75 IQR) |
|---|---|
| Age (years) | 52(46–56) |
| Male n (%) | 53(78) |
| Alcohol Consumption (g/day) | 100(80–160) |
| Laboratory and hemodynamic parameters | |
| Hemoglobin (g/dL) | 11.4(9.9–12.8) |
| Leukocyte count ×109/L | 8.5(6.3–12.5) |
| Platelet count ×109/L | 112(77–199) |
| AST (U/L) | 117(67–156) |
| ALT (U/L) | 39(24–60) |
| Serum albumin (g/dL) | 2.6(2.3–3.1) |
| Serum creatinine (mg/dL) | 0.9(0.7–1.1) |
| Serum bilirubin(mg/dL) | 6.2(2.7–19.3) |
| International normalized ratio | 1.6(1.4–1.8) |
| Alcoholic hepatitis severity scores at admission | |
| MELD score | 19(14–25) |
| ABIC score | 7.83(6.69–8.66) |
IQR: Interquartile range; AST: aspartate aminotransferase; ALT: alanine aminotransferase; MELD: Model for End-Stage Liver Disease; ABIC: Age, serum bilirubin, INR, and serum creatinine.
Cytokine ELISAs
Cell culture media/Human serum
ELISA was performed in cell culture media and in patient serum following the manufacturer’s instruction (Human MIF Quantikine ELISA Kit, R&D System; Human TNF-α Instant ELISA, eBioscience).
Immunohistochemistry (IHC)
Staining for H&E was performed in paraffin-embedded mouse liver sections. Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) positive staining in liver was analyzed using ApopTag plus In Situ Apoptosis Detection Kit (S7111, Millipore, Billerica, MA). F4/80 (MCA497G) and Ly6C (MCA239) (Bio-Rad, Hercules, CA) positive cells were visualized by IHC in mouse liver and counterstained with DAPI mounting medium (H-1200, Vector Laboratories, Burlingame, CA). IHC for MIF was conducted in liver specimens from patients with AH and subjects with healthy livers using the Bond fully-automated slide staining system (Leica Microsystems). See supplemental information for additional details.
Statistics
Values are reported as means ± standard error of the mean (SEM). Data were analyzed by analysis of variance (ANOVA) using general linear models procedure (SAS, Cary, NC). If data were not normally distributed, data were log transformed. Multiple comparisons were analyzed using least square means. For human data, correlations between variables were evaluated using Spearman’s rho or Pearson’s r, when appropriate.
Results
Hepatocytes (HuH7), but not monocyte-derived cells (THP-1), release MIF in response to challenge with ethanol
To understand the cellular sources of MIF in the context of ethanol exposure, HuH7 hepatocytes and differentiated THP-1 monocytes were treated with or without ethanol or LPS. Challenge of HuH7 with ethanol for 4–24 h increased MIF mRNA, while LPS only increased MIF mRNA after 24 h (Fig. 1A). Ethanol induced the release of MIF (but not TNF-α) from HuH7 starting after 8 h (Fig. 1B,C). In contrast, challenge of THP-1 cells with ethanol did not increase MIF mRNA (Fig. 1D) or release of MIF (Fig. 1E). However, LPS modestly increased MIF mRNA at 24 h (Fig. 1D) and stimulated release of MIF and TNFα as early as 4 h (Fig. 1E, F). These data suggested that hepatocytes might be an important source of MIF in the liver in response to ethanol exposure.
Fig. 1. MIF release from HuH7 hepatocytes, but not THP-1 macrophages, in response to ethanol.

(A,D) Expression of MIF mRNA in HuH7 and differentiated THP-1 macrophages was measured by qRT-PCR.
MIF and TNF-α accumulation was detected by ELISA in the cell culture medium of (B, C) HuH7 cells and (E, F) differentiated THP-1 macrophages after challenge with 50mM EtOH or 200 ng/mL LPS. Values represent mean ± SEM. * p< 0.05 compared to basal, n= 3.
MIF expression in non-myeloid cells contributed to ethanol-induced liver injury in chimeric mice
Bone marrow transplants were carried out to generate the following chimeric mice: WT → WT (expressing MIF in both myeloid and non-myeloid cells), WT → Mif−/− (expressing MIF only in myeloid cells) and Mif−/− → WT (expressing MIF only in non-myeloid cells). MIF mRNA in livers of WT → Mif−/− mice was substantially lower than in WT → WT and Mif−/− → WT mice (Suppl Fig. 2), suggesting that non-myeloid cells are the predominant cell type expressing MIF in liver.
Chronic ethanol feeding increased ALT and AST, as well as hepatic triglycerides, in both WT → WT and Mif−/− → WT mice (Fig. 2A, B, and C). Hepatic steatosis and inflammatory foci were also observed in H&E sections of liver from ethanol-fed WT → WT and Mif−/− → WT mice (Fig. 2D). Chronic ethanol feeding to WT → WT and Mif−/− → WT mice also increased TUNEL positive nuclei in the liver (Fig. 2E). In contrast, WT→ Mif−/− mice were resistant to chronic ethanol-induced increases in ALT/AST, hepatic steatosis, inflammatory infiltrates and apoptosis (Fig. 2A–E).
Fig. 2. MIF-deficiency in non-myeloid cells protected mice from chronic ethanol-induced hepatic injury, inflammation and steatosis.

WT→WT, Mif−/−→WT and WT→Mif −/− chimeric mice were allowed free access to diets with increasing concentrations of ethanol or pair-fed a control diet for 25 days. Enzyme activities of (A) ALT and (B) AST were measured in plasma. (C) Hepatic triglyceride content was measured in whole liver homogenates. Values with different alphabetical superscripts were significantly different from each other (p< 0.05). (D) Paraffin-embedded liver sections were stained with hematoxylin and eosin and (E) Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining was quantified using ImagePro. All images were acquired using a 10X objective. Values represent means ± SEM, n = 4 pair-fed and n = 6 ethanol-fed.
Consistent with the histological evidence for increased inflammatory cells after chronic ethanol feeding to WT → WT and Mif−/− → WT mice, expression of mRNA of the chemokines MCP-1, CXCL10, CXCL1 and CXCL2 in liver was also higher in Mif−/− → WT chimeric mice compared to WT→MIF−/− chimeric mice (Fig. 3A). Immunohistochemical analysis of cells positive for F480, a marker of resident macrophages, and Ly6C, a marker of infiltrating monocytes, revealed that while populations of F4/80+ (Fig. 3B) and Ly6C+ (Fig. 3C) cells were maintained at control values in livers of WT→ Mif−/− chimeric mice after chronic ethanol feeding, both populations were increased by chronic ethanol in Mif−/− → WT mice (Fig. 3B/C). Ly6C+ cells also clustered into inflammatory foci, as observed on H&E stained liver section (Fig. 2D).
Fig. 3. MIF-deficiency in non-myeloid, but not myeloid cells protected mice from chronic ethanol-induced expression of mRNA for immune cells and cytokines/chemokines.

MIF−/−→WT and WT→MIF −/− chimeric mice were allowed free access to diets with increasing concentrations of ethanol or pair-fed a control diet for 25 days. Expression of (A) MCP-1, CXCL10, CXCL1, CXCL2 mRNA was detected in mouse livers using qRT-PCR. Gene expression was normalized to 18S (B/C) Paraffin-embedded liver sections were stained with antibody against (B) F4/80 and (C) Ly6C. Images were acquired with a 20X objective. F4/80 positive staining and Ly6C+ cells were quantified using ImagePro. Values represent means ± SEM, n = 4 pair-fed and n = 6 ethanol-fed.
Values with different superscripts are significantly different from each other (p< 0.05).
Taken together, these data from chimeric mice suggest that bone marrow-derived cells do not contribute to MIF release after ethanol feeding in mice. Instead, non-myeloid cells represent the critical cell type generating MIF in response to chronic ethanol feeding. Importantly, the absence of MIF in non-myeloid cells protected mice from chronic ethanol-induced liver injury to a similar extent as in global MIF-deficient mice[5].
MIF expression in liver of alcoholic hepatitis patients
We next investigated the expression and localization of MIF in liver biopsies from patients with AH, autoimmune hepatitis (disease control) and non-diseased controls. Increased MIF staining was detected in liver biopsies from patients with AH, as well as autoimmune hepatitis, compared with controls (Fig. 4A). Expression of MIF mRNA was increased 1.4 fold in patients with AH compared to healthy controls (Fig. 4B). However, the cellular localization differed between AH and autoimmune hepatitis. MIF was predominantly localized to hepatocytes and ductular cells in AH cases, while MIF was primarily localized in non-parenchymal cells in autoimmune hepatitis. Hepatocytes and non-parenchymal cells were identified according to their cellular and nuclear morphology. Although the majority of MIF staining was localized in the cytosol of hepatocytes, nuclear staining was also detected in a few cells in sections from cases with AH.
Fig. 4. Expression of MIF in livers and peripheral and suprahepatic serum of patients with alcoholic hepatitis.

(A) Paraffin-embedded liver sections were stained for MIF in liver biopsies obtained from patients with alcoholic hepatitis, auto-immune hepatitis or healthy liver controls. All images were acquired using a 40X objective. Images are representative of 5 samples for AH, 5 autoimmune hepatitis and 4 for healthy liver controls. (B) qRT-PCR analysis of MIF mRNA in livers of patients with alcoholic hepatitis and healthy liver controls. Values represent means ± SEM, n = 17 AH and n = 7 healthy liver controls. (C–G) MIF concentration in suprahepatic serum was correlated by regression analysis with (C) peripheral serum, (D) bilirubin, (E) AST, (F) triglycerides and (G) GGT. (H/I) MIF concentration in (H) suprahepatic and (I) peripheral serum was measured by ELISA. p<0.05.
Positive correlation between MIF in suprahepatic serum and biochemical markers of liver disease in patients with alcoholic hepatitis
If hepatocytes are an important source of MIF in response to ethanol, then the concentration of MIF in suprahepatic serum should be correlated with the extent of liver disease in patients with AH. To test this hypothesis, MIF was quantified in peripheral and suprahepatic serum. The concentration of circulating MIF correlated with parameters indicative of disease severity. Peripheral serum levels positively correlated with suprahepatic serum levels. A positive correlation was found between MIF concentration in the suprahepatic, but not peripheral, serum including bilirubin, AST, circulating triglycerides and GGT (Table 2 and Fig. 4C–G). Increased MIF in the suprahepatic serum (Fig. 4H), but not peripheral serum (Fig. 4I), was also associated with an increased risk of mortality.
Table 2.
Correlation analysis between peripheral and suprahepatic MIF and biochemical parameters in AH patients.
| Peripheral Blood MIF (n=65) | Suprahepatic Blood MIF (n=58) | |||
|---|---|---|---|---|
| MIF ng/mL (mean±SEM) | 37.0 ± 5.0 | 60.8 ± 9.4 | ||
| Biochemical parameters | Pearson’s r coefficient | p-value | Pearson’s r coefficient | p-value |
| Leucocyte count (109) | −0.06 | 0.6 | 0.16 | 0.2 |
| Bilirubin serum levels (mg/dl) | 0.008 | 0.9 | 0.30 | 0.02 |
| AST (IU/l) | −0.13 | 0.2 | 0.28 | 0.03 |
| ALT (IU/l) | −0.10 | 0.4 | 0.1 | 0.9 |
| Cholesterol serum levels (mg/dL) | −0.12 | 0.3 | 0.25 | 0.8 |
| Triglycerides serum levels(mg/dl) | 0.1 | 0.9 | 0.27 | 0.04 |
| GGT (IU/l) | −0.13 | 0.3 | 0.40 | 0.002 |
| Albumin (g/dl) | −0.05 | 0.6 | −0.05 | 0.6 |
| Alkaline Phosphatase (IU/l) | −0.08 | 0.5 | 0.05 | 0.7 |
| HVPG (mmHg) | −0.12 | 0.3 | −0.10 | 0.4 |
Discussion
Chronic, heavy alcohol consumption results in injury to hepatocytes, at least in part due to the oxidative stress resulting from ethanol metabolism, as well as the development of inflammation in the liver[19]. However, the complex mechanisms linking hepatocyte injury to inflammation are not completely understood. Release of DAMPs from injured hepatocytes activates the resident Kupffer cells in the liver[20], leading to production of chemokines and further recruitment of immune cells to the liver[19]. Recent data also suggests that hepatocytes release chemokines in response to injury, thus adding to the recruitment of more immune cells[21]. MIF, a potent chemokine, is released by both immune cells[22] and hepatocytes[11,23] in other disease models. Since MIF is a critical mediator of ethanol-induced liver injury, here we sought to identify the predominant cellular source of MIF in response to ethanol exposure. Challenge of HuH7 hepatocytes, but not THP-1 macrophages, with ethanol resulted in the accumulation of MIF in the cell culture media. Importantly, chimeric mice deficient in MIF in non-myeloid cells were protected from chronic ethanol-induced liver injury. Finally, patients with AH had increased expression of MIF in hepatocytes and accumulation in the circulation. The concentration of MIF in the suprahepatic serum correlated with multiple clinically-relevant parameters indicative of disease severity, as well as the risk for death from AH. Taken together, these data suggest that hepatocytes are an important source of MIF in response to chronic ethanol feeding in mice and patients with AH and that the release of MIF from hepatocytes likely provides an important link between hepatocyte injury from ethanol and an exacerbation of inflammation in the liver.
Despite its name, MIF has significant chemotactic activity and is a potent enhancer of macrophage activity, increasing phagocytosis, as well as expression of inflammatory cytokines and iNOS[24]. MIF up-regulates the expression of MHC-II molecules, co-stimulatory and adhesion molecules, as well as cytokines, in a wide variety of cell types including Kupffer cells, peritoneal macrophages and dendritic cells[25]. MIF also maintains macrophage viability by suppressing activation-induced macrophage apoptosis by inhibiting p53[26]. Thus, the ability of MIF to act as a chemokine, as well as to prevent activation-induced apoptosis, increases expression of inflammatory mediators and contributes to MIF’s profound pro-inflammatory effects.
MIF plays an important role in the liver in response to acute and chronic stress[27], contributing to T-cell mediated injury in murine ethanol-induced liver damage[5]. In contrast, MIF can also have protective effects at specific phases of liver disease. For example, MIF is required for the recruitment of scar-associated macrophages to the liver; these macrophages are critical for the resolution of fibrosis after an injury[28]. This recruitment likely contributes to the protective function of MIF and its receptor CD74 in response to fibrotic insults[29]. Taken together, these studies suggest a complex interaction between MIF in the progression of liver disease.
Given this complex role, here we first investigated the direct effects of ethanol on HuH7 cells and differentiated THP-1 macrophages, as cellular models of the primary cell types expressing MIF in the liver. When HuH7 cells were challenged with ethanol, MIF mRNA increased by 4 h and MIF accumulated in the cell culture media over 8–24 h, consistent with previous reports that hepatocyte injury in response to CCl4 increased expression of MIF[23]. In contrast, MIF was not released in differentiated THP-1 macrophages, nor did MIF mRNA increase, in response to ethanol. The differences in the response of these two cell types to ethanol may be related to the ability of HuH7 cells to metabolize ethanol[30]; ethanol metabolism likely contributed to injury of the hepatocytes and release of MIF[31].
Based on these cell culture studies, we posited that if hepatocytes were the primary source of MIF in response to ethanol, then chimeric mice deficient in MIF in non-myeloid cells should be protected from ethanol-induced liver injury. Indeed, when bone marrow chimeric mice expressing WT or Mif−/− bone marrow in a WT background were exposed to chronic ethanol feeding, liver injury developed, characterized by increased ALT/AST in the circulation and hepatic steatosis. In contrast, bone marrow chimeric mice expressing WT bone marrow on a Mif−/− background were protected from ethanol-induced liver injury. While these studies cannot exclude a potential contribution of MIF in other hepatic cell types, such as endothelial cells[32] or hepatic stellate cells[33], they are consistent with a likely role of hepatocytes as an important cellular source of MIF in the liver.
Because of its potent chemokine activity, the absence of MIF should prevent the recruitment of immune cells to the liver in response to ethanol. Indeed, the number of both F4/80+ and Ly6C+ cells in the liver were increased by ethanol in chimeric mice on a WT background, but not in mice on a Mif−/− background. This finding is consistent with published reports that indicate a role of MIF in macrophage/monocyte replenishment and recruitment during ethanol exposure[5,34]. Specifically, Barnes et al.[5] found that the number of hepatic F4/80+ cells is reduced in ethanol-fed Mif−/− mice compared to WT mice, suggesting that MIF was critical for the maintenance of the hepatic macrophage population during ethanol exposure. Similarly, the recruitment of Ly6C+ monocytes was also reduced in MIF-deficient mice after ethanol feeding and challenge with LPS[5], as well as after challenge with CCl4[34].
Although MIF has direct chemokine activity, it can also indirectly influence immune cell recruitment via modulation in the expression of other chemokines. For example, MIF upregulates MCP-1 expression in an autocrine mechanism in mouse liver or hepatocytes following acute CCl4 exposure[23]. MCP-1 contributes to progression of ethanol-induced liver injury, particularly related to its ability to act both as a chemokine and steatokine[30]. Here we find that in WT→ Mif−/−, expression of mRNA for some chemokines, including MCP-1, CXCL1, CXCL2 and CXCL10 after ethanol feeding was lower than in Mif −/− → WT (Fig. 3). Interestingly, CXC chemokines, mainly derived from hepatocytes, are known to correlate with mortality in AH[31]. Taken together, these data indicate that MIF released from non-myeloid cells in response to ethanol-induced liver injury acted as a signal regulating the expression of a network of chemokines known to be important for the progression of ethanol-induced liver injury.
Our cell culture and mouse studies suggest that MIF is an important danger signal released from hepatocytes in response to ethanol-induced injury. Here we also report that MIF expression, both at the level of mRNA and protein, was increased in livers of patients with AH. Immunohistochemistry revealed a predominant expression of MIF in hepatocytes in AH patients compared to both healthy liver controls and patients with autoimmune hepatitis. Kumagi, et al.[11] reports that MIF concentrations in the circulation were increased in relation to disease severity in patients with ALD. If this increase in circulating MIF was derived from release from injured hepatocytes and/or other liver cell types, then MIF concentration in suprahepatic serum should be correlated with the extent of hepatocyte injury in patients with AH. This hypothesis is supported by our finding that suprahepatic, but not peripheral, MIF correlated with bilirubin, AST, GGT and circulating triglycerides, as well as with short-term mortality in AH patients. It will also be important in future experiments to determine if there are post-translational modifications to MIF in patients with AH, such as the redox-dependent conformational isoforms reported in other inflammatory diseases[35].
It is interesting to note that MIF has protective effects on hepatocytes in the face of high-fat diet-induced injury[34]. Clearly, our data suggest that this potential hepatoprotective effect of MIF is not sufficient to protect hepatocytes in the context of ethanol exposure. Instead, it is likely that the pro-inflammatory, chemotactic properties of MIF are predominant in AH. These differential effects of MIF are likely related to its interactions with multiple receptors. The cytokine activity of MIF, as well as its protective effects on hepatocytes, is largely due to binding to the CD74/CD44 receptor complex[9,25]. In contrast, its chemokine activity is exerted through the formation of different homo-and hetero-dimers of the CXCR2, CXCR4 and CXCR7 receptors, possibly also in coordination with CD74[10,36,37]. These differences in MIF function will be important to keep in mind during the development of potential therapeutic interventions to inhibit MIF action.
In conclusion, our present findings reveal that MIF was released from non-myeloid cells during the progression of ethanol-induced liver injury. Our data are consistent with a predominant role for hepatocyte injury in driving the release of MIF. It is interesting to note that MIF release from injured hepatocytes functions in a similar manner to classic DAMPs, signaling sterile inflammation and the recruitment of inflammatory cells to the site of injury. Strategies to specifically dampen the chemotactic, rather than the potential hepatoprotective, effects of MIF are likely to be a relevant therapeutic approach to treatment of AH.
Supplementary Material
Lay summary.
Alcoholic liver disease is a major cause of preventable mortality worldwide, and lacks specific pharmacological therapies. Recent studies have recognized that macrophage migration inhibitor factor (MIF) has a critical role in the inflammatory response to liver damage. However, the cells that produce this protein are still unknown. Our present findings reveal that hepatocytes, the main cell type in the liver, are primarily responsible for MIF production in response to alcohol, which promotes liver injury. Our study suggests that drugs inhibiting MIF production could be beneficial to treat patients with liver disease due to excessive alcohol consumption.
Highlights.
The source of MIF in Alcoholic Liver Disease (ALD) has been investigated
In vitro cultured hepatocytes released MIF upon ethanol challenge
Chimeric mice not expressing MIF in hepatocytes were protected from ethanol
MIF was detected in liver biopsies from patients with AH, localized in hepatocytes
The study identified hepatocytes as source of MIF in ALD
Acknowledgments
Financial support: This work was supported in part by NIH grants U01AA020821(LEN), U01AA021890 (LEN), F32AA024955 (KP) and U01AA021908 (RB); by the Italian Liver Foundation (NR, CT) and grants from Instituto de Salud Carlos III (FIS PI14/00320) and Miguel Servet (CP11/00071) co-financed by FondoEuropeo de Desarrollo Regional (FEDER), Unión Europea, ‘Unamanera de hacer Europa’ (PS-B).
JA wishes to express his gratitude to the Mexican National Council of Science and Technology (CONACyT, Mexico City, Mexico) for partially supporting his predoctoral stay at IDIBAPS.
Abbreviations
- ALD
Alcoholic Liver disease
- AH
Alcoholic Hepatitis
- BMC
bone marrow cells
- BMT
bone marrow transplant
- KC
Kupffer cells
- MCP1
Monocyte chemoattractant protein-1
- MIF
Macrophage Migration Inhibitory Factor
- PAMPs
Pathogen-associated molecular patterns
- TLRs
Toll-like receptors
- TUNEL
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
- WT
Wild-type
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
Authors’ contributions
Study concept and design: LE Nagy, R Bataller, N Rosso
Acquisition of data; analysis and interpretation of data: V Marin, K Poulsen, G Odena, MR McMullen, J. Altamirano, P Sancho-Bru, CTiribelli, J. Caballeria, N Rosso, LE Nagy, R Bataller
Drafting of the manuscript: V Marin, K Poulsen, N Rosso, LE Nagy
Critical revision of the manuscript for important intellectual content: V Marin, K Poulsen, G Odena, MR McMullen, P Sancho-Bru, C Tiribelli, N Rosso, R Bataller and LE Nagy
Statistical analysis: V Marin, K Poulsen, J. Altamirano, G Odena, J. Caballeria, LE Nagy
Obtained funding: LE Nagy, R Bataller, K Poulsen, N Rosso, C Tiribelli
Technical support: MR McMullen
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–42. doi: 10.1038/nrgastro.2015.35. [DOI] [PubMed] [Google Scholar]
- 2.Orman ES, Odena G, Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol. 2013;28(Suppl 1):77–84. doi: 10.1111/jgh.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albano E. Alcohol, oxidative stress and free radical damage. Proc Nutr Soc. 2006;65:278–90. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- 4.Nanji AA, Lau GK, Tipoe GL, Yuen ST, Chen YX, Thomas P, et al. Macrophage migration inhibitory factor expression in male and female ethanol-fed rats. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res. 2001;21:1055–62. doi: 10.1089/107999001317205187. [DOI] [PubMed] [Google Scholar]
- 5.Barnes MA, McMullen MR, Roychowdhury S, Pisano SG, Liu X, Stavitsky AB, et al. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatol Baltim Md. 2013;57:1980–91. doi: 10.1002/hep.26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, et al. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol. 1997;150:235–46. [PMC free article] [PubMed] [Google Scholar]
- 7.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF Signal Transduction Initiated by Binding to CD74. J Exp Med. 2003;197:1467–76. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Vorst EPC, Döring Y, Weber C. Chemokines and their receptors in Atherosclerosis. J Mol Med Berl Ger. 2015;93:963–71. doi: 10.1007/s00109-015-1317-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maubach G, Lim MCC, Kumar S, Zhuo L. Expression and upregulation of cathepsin S and other early molecules required for antigen presentation in activated hepatic stellate cells upon IFN-gamma treatment. Biochim Biophys Acta. 2007;1773:219–31. doi: 10.1016/j.bbamcr.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 11.Kumagi T, Akbar F, Horiike N, Onji M. Increased serum levels of macrophage migration inhibitory factor in alcoholic liver diseases and their expression in liver tissues. Clin Biochem. 2001;34:189–93. doi: 10.1016/s0009-9120(01)00214-4. [DOI] [PubMed] [Google Scholar]
- 12.Chavez-Tapia NC, Rosso N, Tiribelli C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012;12:20. doi: 10.1186/1471-230X-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci. 2003;100:9354–9. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCullough RL, Saikia P, Pollard KA, McMullen MR, Nagy LE, Roychowdhury S. Myeloid Mixed Lineage Kinase 3 Contributes to Chronic Ethanol-Induced Inflammation and Hepatocyte Injury in Mice. Gene Expr. 2016;17:61–77. doi: 10.3727/105221616X691730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emmanouilidis NHA, Adams AB. Adoptive Immunotherapy: Methods and Protocols. Springer Science & Business Media; 2002. [Google Scholar]
- 16.McCullough RL, McMullen MR, Das D, Roychowdhury S, Strainic MG, Medof ME, et al. Differential contribution of complement receptor C5aR in myeloid and non-myeloid cells in chronic ethanol-induced liver injury in mice. Mol Immunol. 2016;75:122–32. doi: 10.1016/j.molimm.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–72. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasin Deborah. Classification of Alcohol Use Disorders. Natl Inst Alcohol Abuse Alcohol; 2003. https://pubs.niaaa.nih.gov/publications/arh27-1/5-17.htm. [PMC free article] [PubMed] [Google Scholar]
- 19.Nagy LE. The role of innate immunity in alcoholic liver disease. Alcohol Res Curr Rev. 2015:37. [PMC free article] [PubMed] [Google Scholar]
- 20.Nagy LE, Ding W-X, Cresci G, Saikia P, Shah VH. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology. 2016;150:1756–68. doi: 10.1053/j.gastro.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MALHI H, GUICCIARDI ME, GORES GJ. Hepatocyte Death: A Clear and Present Danger. Physiol Rev. 2010;90:1165–94. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lolis E, Bucala R. Macrophage migration inhibitory factor. Expert Opin Ther Targets. 2003;7:153–64. doi: 10.1517/14728222.7.2.153. [DOI] [PubMed] [Google Scholar]
- 23.Xie J, Yang L, Tian L, Li W, Yang L, Li L. Macrophage Migration Inhibitor Factor Upregulates MCP-1 Expression in an Autocrine Manner in Hepatocytes during Acute Mouse Liver Injury. Sci Rep. 2016;6:27665. doi: 10.1038/srep27665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morand EF. New therapeutic target in inflammatory disease: macrophage migration inhibitory factor. Intern Med J. 2005;35:419–26. doi: 10.1111/j.1445-5994.2005.00853.x. [DOI] [PubMed] [Google Scholar]
- 25.Stavitsky AB, Xianli J. In vitro and in vivo regulation by macrophage migration inhibitory factor (MIF) of expression of MHC-II, costimulatory, adhesion, receptor, and cytokine molecules. Cell Immunol. 2002;217:95–104. doi: 10.1016/s0008-8749(02)00516-6. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99:345–50. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohkawara T, Nishihira J, Takeda H, Asaka M, Sugiyama T. Pathophysiological roles of macrophage migration inhibitory factor in gastrointestinal, hepatic, and pancreatic disorders. J Gastroenterol. 2005;40:117–22. doi: 10.1007/s00535-004-1526-3. [DOI] [PubMed] [Google Scholar]
- 28.Barnes MA, McMullen MR, Roychowdhury S, Madhun NZ, Niese K, Olman MA, et al. Macrophage migration inhibitory factor is required for recruitment of scar-associated macrophages during liver fibrosis. J Leukoc Biol. 2015;97:161–9. doi: 10.1189/jlb.3A0614-280R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinrichs D, Knauel M, Offermanns C, Berres M-L, Nellen A, Leng L, et al. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc Natl Acad Sci U S A. 2011;108:17444–9. doi: 10.1073/pnas.1107023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plumlee CR, Lazaro CA, Fausto N, Polyak SJ. Effect of ethanol on innate antiviral pathways and HCV replication in human liver cells. Virol J. 2005;2:89. doi: 10.1186/1743-422X-2-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes MA, Roychowdhury S, Nagy LE. Innate immunity and cell death in alcoholic liver disease: role of cytochrome P4502E1. Redox Biol. 2014;2:929–35. doi: 10.1016/j.redox.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu C-T, Guo L-L, Feng N, Zhang L, Zhou N, Ma L-L, et al. MIF, secreted by human hepatic sinusoidal endothelial cells, promotes chemotaxis and outgrowth of colorectal cancer in liver prometastasis. Oncotarget. 2015;6:22410–23. doi: 10.18632/oncotarget.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Copple BL, Bai S, Burgoon LD, Moon J-O. Hypoxia-inducible Factor-1α Regulates Expression of Genes in Hypoxic Hepatic Stellate Cells Important for Collagen Deposition and Angiogenesis. Liver Int Off J Int Assoc Study Liver. 2011;31:230–44. doi: 10.1111/j.1478-3231.2010.02347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinrichs D, Berres M-L, Coeuru M, Knauel M, Nellen A, Fischer P, et al. Protective role of macrophage migration inhibitory factor in nonalcoholic steatohepatitis. FASEB J. 2014 doi: 10.1096/fj.14-256776. fj.14-256776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiele M, Kerschbaumer RJ, Tam FWK, Völkel D, Douillard P, Schinagl A, et al. Selective Targeting of a Disease-Related Conformational Isoform of Macrophage Migration Inhibitory Factor Ameliorates Inflammatory Conditions. J Immunol Baltim Md 1950. 2015;195:2343–52. doi: 10.4049/jimmunol.1500572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tarnowski M, Grymula K, Liu R, Tarnowska J, Drukala J, Ratajczak J, et al. Macrophage migration inhibitory factor is secreted by rhabdomyosarcoma cells, modulates tumor metastasis by binding to CXCR4 and CXCR7 receptors and inhibits recruitment of cancer-associated fibroblasts. Mol Cancer Res MCR. 2010;8:1328–43. doi: 10.1158/1541-7786.MCR-10-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatterjee M, Borst O, Walker B, Fotinos A, Vogel S, Seizer P, et al. Macrophage migration inhibitory factor limits activation-induced apoptosis of platelets via CXCR7-dependent Akt signaling. Circ Res. 2014;115:939–49. doi: 10.1161/CIRCRESAHA.115.305171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.