

Abstract
The chemical versatility of the flavin coenzyme is nearly unparalleled in enzyme catalysis. An interesting illustration of this versatility can be found in the reaction catalyzed by the type II isopentenyl diphosphate:dimethylallyl diphosphate isomerase (IDI-2) – an enzyme that interconverts the two essential isoprene units (isopentenyl pyrophosphate and dimethylallyl pyrophosphate) that are needed to initiate the biosynthesis of all isoprenoids. Over the past decade, a variety of biochemical, spectroscopic, structural and mechanistic studies of IDI-2 have provided mounting evidence that the flavin coenzyme of IDI-2 acts in a most unusual manner – as an acid/base catalyst to mediate a 1,3-proton addition/elimination reaction. While not entirely without precedent, IDI-2 is by far the most extensively studied flavoenzyme that employs flavin-mediated acid/base catalysis. Thus, IDI-2 serves as an important mechanistic model for understanding this often overlooked, but potentially widespread reactivity of flavin coenzymes. This review details the most pertinent studies that have contributed to the development of mechanistic proposals for this highly unusual flavoenzyme, and discusses future experiments that may be able to clarify remaining uncertainties in the chemical mechanism of IDI-2.
Isoprenoid Biosynthesis
Isoprenoids (or terpenoids) constitute one of the largest and most structurally diverse classes of natural products that are found in all living systems. Collectively, isoprenoids serve many biological roles as vitamin precursors, hormones, photosynthetic pigments, electron transport mediators, antibiotics, and as anchors for membrane-associated proteins [1]. Two isoprenoid biosynthetic pathways exist: the mevalonate pathway and the non-mevalonate or methylerythritol phosphate (MEP) pathway. The mevalonate pathway [1–5] is employed by eukaryotes, archaea, and some bacteria. The MEP pathway is used in the chloroplasts of plants and algae, and by most bacteria [6, 7]. The MEP pathway has generated considerable interest because none of the biosynthetic enzymes present in this pathway have homologues in humans, making each enzyme a potential target for antimicrobial development [8–10]. Despite clearly distinct evolutionary histories, both pathways ultimately result in the production of the same two isoprene units: isopentenyl pyrophosphate (IPP, 1, Scheme 1) and dimethylallyl pyrophosphate (DMAPP, 2). These two isoprene units are then used to initiate the biosynthesis of all higher order isoprenoids (Scheme 2). The initiation and elongation reactions of isoprenoid biosynthesis proceed via a series of electrophilic alkylation reactions that are enabled by the unusual reactivity of the allylic pyrophosphate moiety present in DMAPP and other isoprenoid pyrophosphates (e.g. compounds 3 - 5). These compounds are susceptible to acid cleavage to form allylic cations that serve as the alkylating agents [11]. Indeed, the ability of isoprenoid biosynthetic enzymes to generate substrate carbocation intermediates is a common mechanistic feature of isoprenoid biosynthesis, and is largely responsible for the vast array of structurally diverse carbon skeletons observed in this class of natural products [12, 13].
Scheme 1.

Biosynthetic pathways for the ubiquitous isoprene units, isopentenyl pyrophosphate (IPP, 1) and dimethylallyl pyrophosphate (DMAPP, 2). These compounds can be interconverted by isopentenyl diphosphate:dimethylallyl diphosphate isomerase (IDI) enzymes.
Scheme 2.

Chain initiation and elongation in isoprenoid biosynthesis. Note that both IPP and DMAPP are required in the initial condensation event (mediated by farnesyl pyrophosphate synthase). Thus, IDI enzymes serve an essential function for the growth and survival of most organisms.
Isopentenyl Diphosphate:Dimethylallyl Diphosphate Isomerases
Considering their role in the conserved initial steps of long chain isoprenoid biosynthesis, all organisms must be able to produce both IPP and DMAPP. This is usually achieved by isopentenyl diphosphate:dimethylallyl diphosphate isomerase (IDI) enzymes, which catalyze the reversible interconversion of IPP and DMAPP via a formal 1,3-proton addition/elimination reaction (Scheme 1). Organisms that employ the MEP pathway for isoprenoid biosynthesis generate both IPP and DMAPP in the final enzymatic step of the pathway (via the intriguing metalloenzyme, IspH) [14]. However, these organisms also typically utilize an IDI enzyme, perhaps to balance the cytosolic pools of isoprene units for metabolic needs. IDI enzymes exist in two evolutionarily distinct forms, termed type I IDI (IDI-1) and type II IDI (IDI-2). IDI-1 enzymes are Zn-containing metalloenzymes, that were initially discovered by Lynen and co-workers during their attempts to understand squalene biosynthesis in baker’s yeast [15, 16]. Extensive mechanistic and structural studies over the ensuing decades has provided support for an acid/base chemical mechanism for IDI-1 involving conserved glutamate, tyrosine, and cysteine residues of the protein as well as an active site Zn ion (Scheme 3) [17–21]. The Zn ion assists catalysis by lowering the pKa of the Glu/Tyr dyad, which serves as the catalytic acid in the forward direction (IPP → DMAPP). The Cys residue abstracts the pro R proton at C2 of IPP. In co-crystal structures of IDI-1 with bound inhibitors [21, 22], the catalytic Glu/Tyr dyad and Cys residues are arranged on opposite sides of the inhibitor and appear to be positioned appropriately to effect an antarafacial 1,3-proton addition/elimination reaction. This reaction mechanism is further supported by a variety of stereochemical studies [17, 19, 23, 24].
Scheme 3.

Putative chemical mechanism of IDI-1 from Escherichia coli.
In 2001, Kuzuyama, Seto, and co-workers identified a distinct IDI during their studies of isoprenoid biosynthesis in Streptomyces sp. strain CL190 [25]. Interestingly, in addition to divalent metal ions, this isomerase (dubbed IDI-2) also required flavin mononucleotide (FMN) and a reducing agent (NADPH) for activity. Biochemical studies indicated that the pro S hydride of NADPH was transferred to the IDI-2 bound oxidized FMN (FMNox) to reduce the flavin to its 2e− reduced state (Scheme 4). This suggests that NADPH binds to the IDI-2:flavin complex in a specific orientation and is likely the physiological reductant [26]. Interestingly, only catalytic quantities of NADPH were required to activate the enzyme for multiple turnovers [26, 27]. This result firmly established IDI-2 a member of an unusual group of flavoenzymes that catalyze reactions involving no net redox change [28], and raised intriguing questions regarding the exact role of the flavin in the isomerization of IPP and DMAPP [29]. Other early mechanistic studies on IDI-2 established that solvent deuteria (derived from D2O) were incorporated into the C2 and C4 positions of IPP and into the (E)-methyl group of DMAPP (Scheme 4) [30], and that the pro R proton at C2 of IPP was removed during turnover [31]. Altogether, these observations are consistent with a flavin-dependent 1,3-proton addition/elimination mechanism.
Scheme 4.

Stereochemical studies of IDI-2.
Early Mechanistic Hypotheses for IDI-2
By analogy to other flavoenzymes that catalyze non-redox reactions [28], several potential chemical mechanisms involving the flavin can be envisioned for the reaction catalyzed by IDI-2. The first possibility is that the flavin serves a structural role, helping to keep the active site of IDI-2 folded in a catalytically competent conformation. This sort of role for flavins has been observed in the acetolactate synthases and YerE [32, 33], but is inconsistent with the inactivity of IDI-2 reconstituted either with FMNox [34] or with reduced 5-deaza-FMN (6, Scheme 5) [26, 35]. The inability of reduced 5-deaza-FMN to support IDI-2 catalysis is also at odds with a mode of flavin catalysis involving the electrostatic stabilization of a carbocation intermediate (Scheme 5). In contrast to these results with IDI-2, the reduced 5-deaza-FAD analogue has been shown to support catalysis by the lycopene synthase CrtY, a flavin-dependent isoprenoid biosynthetic enzyme that utilizes its reduced FAD for the electrostatic stabilization of a substrate carbocation [36]. These results suggest that chemical reactions mediated by the reduced flavin of IDI-2 are likely important for catalysis.
Scheme 5.

Early mechanistic models for IDI-2 and the structures of the deazaflavin analogues.
One seemingly reasonable hypothesis for the function of the reduced FMN coenzyme in IDI-2 catalysis was that the reduced FMN could transfer a single electron to IPP (or DMAPP) concomitant with or subsequent to double bond protonation to generate a flavin semiquinone/substrate radical pair (Scheme 5). This radical pair could then proceed to products by deprotonation of the substrate radical and single electron transfer back to the flavin semiquinone [26]. DNA photolyases are one well known example of flavoenzymes that employ similar cryptic 1e− redox cycles [37]. This early mechanistic proposal for IDI-2 was based largely on two sets of experimental observations [26]. First, a neutral FMN semiquinone (8) accumulated in redox titrations and photoreduction experiments conducted in the presence of IPP. However, in the absence of IPP, the IDI-2 bound FMN cycled directly between the 2e− reduced and fully oxidized forms, with no thermodynamic stabilization of a semiquinone. These experiments suggested that IPP binding to IDI-2:FMN perturbed the 1e− redox potentials of the flavin, and that the IDI-2 active site was capable of accommodating a semiquinone species in the presence of IPP. Second, IDI-2 was found to exhibit wild type levels of activity when the reduced FMN was replaced with reduced 1-deaza-FMN (7) [26], whereas IDI-2 reconstituted with reduced 5-deaza-FMN (6) was nearly devoid of activity [26, 35]. Previous studies of other flavoenzymes reconstituted with these coenzyme analogues demonstrated that 1-deaza-FMN is capable of both 1e− and 2e− transfers, whereas 5-deaza-FMN is only capable of 2e− transfers [38]. Thus, considering only canonical flavin coenzyme functions, these initial results appeared to support a mechanism involving single electron transfer chemistry mediated by the reduced FMN coenzyme of IDI-2.
The 1e− transfer mechanistic proposal was subsequently challenged by several experimental observations [39–41]. First, electron paramagnetic resonance (EPR) studies were unable to detect the expected IPP/FMN semiquinone radical pair in catalytically competent IDI-2 reaction mixtures [39, 40]. Moreover, no flavin semiquinone accumulation was detected by absorption spectroscopy in single turnover, pre-steady state stopped flow experiments [39]. As such, the kinetic competence of the semiquinone/substrate radical pair could not be validated. Studies with IPP substrate analogues provided additional strong evidence against a putative single electron transfer mechanism [41, 42]. Namely, the radical clock cyclopropyl analogue of IPP (9, Scheme 6) was converted to the corresponding DMAPP analogue (10) without the production of radical-induced ring-opened products (e.g. 12). These observations stipulate that if a catalytically competent radical pair is formed in the IDI-2 active site during turnover, then the substrate radical must be deprotonated and oxidized by 1e− faster than the known rate of radical fragmentation of the cyclopropyl radical intermediate (approximately 1.3 × 108 s−1 for 11→12, Scheme 6) [43]. Altogether, these mechanistic studies casted doubt on an isomerization mechanism involving a cryptic single electron transfer mechanism and suggested that the IDI-2 bound neutral semiquinone observed in potentiometric titration and photoreduction studies is likely not catalytically relevant.
Scheme 6.

The radical clock IPP analogue 3-cyclopropyl-3-buten-1-yl diphosphate (cIPP, 9) is converted by IDI-2 into the corresponding DMAPP product (10), suggesting that a radical intermediate (e.g. 11) is not generated by IDI-2 during turnover. These data argue strongly against a single electron transfer mechanism for IDI-2.
Kinetic and spectroscopic studies reveal key features of the IDI-2 catalyzed reaction
In light of these initial mechanistic studies, a chemical mechanism involving the reduced flavin as an acid/base catalyst emerged as a reasonable mechanistic alternative (Scheme 7). These acid/base chemical mechanisms strongly resemble the proposed mechanism for IDI-1 (Scheme 3), except that the flavin fulfills an integral role in the requisite proton transfers. To begin to validate this unusual putative mechanistic role for flavin, the chemical and kinetic mechanisms of IDI-2 enzymes were probed with a variety of biochemical experiments. At pH 7.0, the UV-visible absorption spectrum of the reduced IDI-2:FMN resting state (Figure 1A) is consistent with the flavin being in the anionic reduced state (i.e., FMNH−) [39, 40, 44]. Upon IPP (or DMAPP) binding, the anionic FMNH− is rapidly converted into a new species, whose absorption spectrum is most consistent with neutral reduced 1,5-dihydro-FMNH2 [39, 40, 44]. Likewise, IDI-2 bound, reduced 1- and 5-deaza-flavin are also converted from their anionic forms into their neutral forms upon IPP binding [39]. Thus, consistent with an acid/base catalytic role, the flavin in the substrate-bound IDI-2 Michaelis complex seems to be poised to donate a proton to the isoprene substrate to initiate a 1,3-proton addition/elimination reaction sequence. Unfortunately, studies of the pH-dependence of the steady state kinetic parameters of the Staphylococcus aureus IDI-2 enzyme did not provide any conclusive evidence for a kinetically relevant active site ionization in the pH-rate profiles [39]. Thus, direct evidence for a chemical mechanism involving general acid/base catalysis remains lacking.
Scheme 7.

General considerations for IDI-2 catalyzed acid/base chemical mechanisms. Various roles for the reduced flavin in acid/base catalysis can be envisioned. The anionic flavin (13) could work in concert with an IDI-2 derived amino acid side chain (top path), or FMNH2 could catalyze both proton transfers via the neutral 1,5-dihydro tautomer (14, middle path) or the zwitterionic 5,5-dihydro tautomer (15, bottom path). The reactions are drawn as occurring by stepwise mechanisms, but the paths involving FMNH− (top) and 1,5-dihydro-FMNH2 (middle) could also occur by concerted proton addition/elimination mechanisms.
Figure 1.

Biochemical studies of the Staphylococcus aureus IDI-2 enzyme. A) UV-visible absorption spectra of reduced IDI-2:FMN in the absence (blue) and presence (magenta) of IPP. The λmax value of ~ 425 nm in the IDI-2:FMN:IPP complex is consistent with the neutral 1,5-dihydro-FMNH2 tautomeric state of the reduced flavin. B) Pre-steady state accumulation and decay of the FMNH2 intermediate under single turnover conditions. After the rapid accumulation phase (kfast = 37 s−1), the intermediate decays to an equilibrium level at a kinetically competent rate (kslow = 2.1 s−1 > kcat = 1.3 s−1), suggesting that the FMNH2 species could be catalytically relevant. C) Pre-steady state burst experiment. The lack of a detectable pre-steady state burst in DMAPP formation suggests that the rate-determining step in the kinetic mechanism occurs prior to or concomitant with DMAPP formation, yielding a velocity (kss = 1.5 s−1) that is very similar to kcat (1.3 s−1). This observation is consistent with rate-limiting isomerization chemistry step(s). Please note that the theoretical maximum burst amplitude of 1.0 would not be expected for the isomerization reaction catalyzed by IDI-2 where IPP and DMAPP would be in equilibrium at the active site. The depicted curve is only meant to illustrate the diagnostic utility of a pre-steady state burst experiment. D) An 1H-NMR assay was used to measure a primary substrate deuterium kinetic isotope effect on the reaction velocity at saturating IPP concentrations as an approximation of Dkcat. The observed isotope effect (Dkcat = 2.2) is consistent with cleavage of the IPP pro-R C2-H/D bond in a partially rate-determining step.
The steady state kinetic parameters have been determined for IDI-2 enzymes from several sources [25, 26, 39, 40, 45–47]. These studies have consistently yielded kcat values of approximately 1 s−1, and Km values for IPP and reduced FMN in the low μM and nM ranges, respectively. Recent work by the Poulter group on the Streptococcus pneumoniae enzyme suggests an ordered sequential mechanism for IDI-2 with reduced FMN binding before IPP [45, 46]. This observation is in line with the observed binding affinities of IPP and FMN, as well as with the relative positioning of FMN and IPP in the IDI-2 active site as determined from the available X-ray crystal structures [48, 49]. Stopped-flow absorption experiments demonstrated that the putative FMNH2 intermediate forms rapidly in the pre-steady state upon the mixing of IPP (or DMAPP) with the S. aureus IDI-2:FMNH−. The observed rate of FMNH2 formation in these experiments is much greater than kcat, suggesting that the FMNH2 intermediate accumulates prior to a slower kinetic step in the pathway [39]. Moreover, single turnover stopped flow absorption studies performed with concentrations of IDI-2:FMNH− in molar excess over IPP, demonstrated a biphasic time-dependence of FMNH2 formation and decay (Figure 1B). Namely, the fast FMNH2 accumulation phase (kfast = 37 s−1) is followed by a slower phase where FMNH2 decays to an equilibrium level [39]. Importantly, the observed rate of the slow FMNH2 decay phase (kslow = 2.1 s−1) is faster than kcat (1.3 s−1) [39]. This observation validates the kinetic competence of the FMNH2 species and strongly suggests that FMNH2 is a true intermediate in the IDI-2 mediated conversion of IPP to DMAPP.
To more closely examine the nature of the rate-determining step(s) for the S. aureus IDI-2, a pre-steady state burst experiment was performed at saturating IPP concentration using a rapid-mix chemical quench-flow device (Figure 1C) [39]. These types of kinetic experiments are useful for determining the position of the rate-limiting step within the kinetic mechanism of a multistep enzyme-catalyzed reaction [50]. The observation of a burst indicates that product rapidly accumulates at the enzyme active site in the pre-steady state, and that the rate-limiting step in the mechanism occurs after product formation (e.g., a conformational change leading to product release). In contrast, if the chemistry step(s) leading to formation of product is rate-limiting, then no burst will be observed, and the rate of product formation measured in the burst experiment should be equivalent to kcat. For IDI-2, no pre-steady state burst was observed and the rate of DMAPP formation in the experiment (1.5 s−1) was very similar to the steady state kcat (1.3 s−1) [39]. This important result suggested that the isomerization chemistry mediated by IDI-2 could indeed be occurring in the rate-limiting step(s), and that additional steady state kinetic experiments might be useful in revealing the underlying chemical mechanism.
Probing the transition state of the IDI-2 catalyzed reaction with kinetic isotope and substituent effects
Following up on these observations, kinetic studies employing a number of substrate and coenzyme analogues were performed to investigate the nature of the transition state(s) involved in IDI-2 catalysis. Using the stereospecifically deuterated analogue, (R)-[2-2H]-IPP, a primary deuterium kinetic isotope effect of 2.2 was measured on kcat by direct comparison using an 1H-NMR assay (Figure 1D) [39]. This result confirmed the results of the burst experiment and conclusively demonstrated that the pro-R C2-H/D bond of IPP is breaking in a rate-limiting transition state of the IDI-2 catalyzed reaction. The modest magnitude of the 1° KIE suggests either an early/late transition state or a kinetic mechanism where the intrinsic rate of the isotope sensitive C2-H/D deprotonation step is masked in the steady state by an isotope-insensitive step. A similar 1° deuterium KIE of 2.3 was also measured with (R)-[2-2H]-IPP on the rate of FMNH2 decay in the slow phase of single turnover stopped flow studies [39]. This result demonstrates that the consumption of the FMNH2 species (as shown in the decay phase of Figure 1B) is kinetically coupled to C2-deprotonation of IPP and further validates the kinetic and catalytic competence of the FMNH2 intermediate.
In addition to deprotonation of IPP at C2, the isomerization of IPP to DMAPP also requires protonation at C4. To test if proton transfer to C4 of IPP was also contributing to the rate-limiting step(s), solvent kinetic isotope effects and proton inventory studies were conducted by measuring the steady state kinetic parameters of IDI-2 as a function of D2O concentration [51]. The solvent KIEs of D2Okcat/Km = 3.8 and D2Okcat = 1.4 measured in 100% D2O suggest that bonds to solvent exchangeable protons are breaking in the transition state. Moreover, the best fit of the proton inventory data (which in ideal circumstances can determine the contribution of individual solvent exchangeable protons to a solvent KIE) [52] was achieved with a model where a single solvent exchangeable proton is moving in a transition state that is only partially rate-limiting [51]. Altogether, these KIE studies are consistent with a chemical mechanism involving acid/base catalysis, perhaps involving proton transfers to and from the flavin (Scheme 7).
While the KIE studies clearly support the hypothesis that IDI-2 proceeds via a protonation/deprotonation mechanism facilitated by acid/base catalysis, these experiments do not provide much insight in the role of the flavin. Nevertheless, the fact that isomerization chemistry is at least partially rate-limiting suggested that steady state kinetic experiments could be employed to more precisely elucidate the role of the flavin in catalysis. Accordingly, a series of FMN analogues with altered electronic properties were chemoenzymatically synthesized, and their effects on the steady state kinetic parameters of the IDI-2 catalyzed reaction were investigated (Figure 2) [53]. These analogues were substituted at the 7- or 8-position of the isoalloxazine with various electron donating or withdrawing groups. The hypothesis guiding the design of the flavin analogues was that the substituents would alter the electronic properties of the N5 position of the flavin through inductive effects. If the N5 position of the flavin was participating directly in the proton transfers needed for isomerization, then the various flavin analogues should yield systematic perturbations of kcat and kcat/Km that could be correlated with the sum of the Hammett substituent constants (σ) [54]. Indeed, when the activity of IDI-2 reconstituted with the various analogues was tested, the expected linear free energy relationships (LFER) were observed on both kcat and kcat/Km. The negative slopes of the Hammett plots (ρ ~ −2.0) indicate that positive charge is developing (or that negative charge is being diminished) on the flavin in the vicinity of N5 in the transition state. The rate constants could also be correlated with the pKa of the N5 position of the flavin to yield Brønsted plots with slopes near 0.7, a result that suggested substantial proton transfer to the flavin in the transition state. A combined LFER-KIE study was then conducted by measuring the primary deuterium KIE on kcat with the (R)-[2-2H]-IPP analogue using IDI-2 reconstituted with each flavin analogue [53]. The Dkcat measured with each flavin analogue was very similar, strongly suggesting that the structure of the transition state is unperturbed when the flavin structure is altered. That is, the flavin analogues are not changing the isomerization mechanism. These data also suggest that the flavin analogues are not exerting any differential effects on the kinetics of other flavin-dependent steps in the kinetic mechanism that may alter the expression of the KIE in the steady state. These LFER studies provided the first direct evidence that the electronic properties of the flavin are being substantially altered in the transition state of the natural reaction (i.e., the conversion of IPP → DMAPP), and strongly suggested that the flavin is directly involved in the chemistry mediated by IDI-2.
Figure 2.

Linear free energy relationship (LFER) studies of IDI-2. A) Hammett plots correlating the steady state kinetic parameters of IDI-2 enzymes reconstituted with the flavins shown at the bottom of the figure. LFERs were observed when the rate constants were plotted vs. either the sum of the Hammett σm and σp substituent constants or the estimated pKa of the N5 position of the flavin (data not shown). The negative slopes in the Hammett plot (ρ = −2.1) are consistent with the accumulation of positive charge on the flavin N5 atom in the transition state(s) that limit steady state turnover. B) Combined KIE/LFER study. The steady state reaction velocities for IDI-2 reconstituted with each flavin analogue were measured at saturating concentrations of either IPP or (R)-[2-2H]-IPP. The KIE was found to be approximately constant across the series, suggesting that the flavin analogues are neither perturbing the transition state structure nor altering the expression of the KIE by exerting effects on the energetic barriers of other steps in the mechanism.
Finally, the transition state structure of the isoprene unit has also been probed with fluoromethyl analogues of IPP and DMAPP (16-18, Scheme 8) [42]. Using a sensitive GC-based assay, Poulter and co-workers showed that, while these fluoromethyl analogues were isomerized by IDI-2, they were processed at much slower rates. The monofluorinated IPP and DMAPP analogues suffered approximate drops in activity of 50- and 100-fold, respectively, whereas the activity with the difluorinated DMAPP analogue was reduced over 3000-fold. These rate reductions are in line with the ~ 106–fold decrease in the solvolysis rate of the corresponding trifluoromethyl methanesulfonate compounds (19), which presumably proceeds via SN1 chemistry and an allylic carbocation intermediate (Scheme 8) [55]. The strong electron withdrawing effects of fluorine clearly indicate that positive (or partial positive) charge is developing on the isoprene substrate in the IDI-2 catalyzed reaction. Thus, the IDI-2 catalyzed reaction appears to mechanistically parallel IDI-1 and many other reactions in isoprenoid biosynthesis [11, 13], where the unusual stability of the isoprenoid 3° carbocation plays an integral role in the chemical mechanism of the enzyme-catalyzed reaction. Mechanistic models for IDI-2 that incorporate the observations discussed in this section will be presented below in Scheme 11.
Scheme 8.

Mechanistic studies with fluoromethyl IPP/DMAPP analogues. Compounds 16-18 are isomerized by IDI-2 at rates much slower than the native substrate (IPP) – paralleling the reactivity trends seen in the hydrolysis of 19, which is believed to proceed by an SN1 mechanism through a 3°cationic intermediate. This data is consistent with the formation of a carbocation intermediate or cation-like transition state in the IDI-2 catalyzed reaction.
Scheme 11.

Current models for IDI-2 mediated acid/base catalysis. See text for details.
Structural studies support a direct catalytic role for the flavin
X-ray crystal structures of IDI-2 have provided additional support for a direct role of the flavin in IDI-2 catalysis [45, 48, 49, 56–58]. The first structure of IDI-2 from Bacillus subtilis was solved in 2003 in the presence of oxidized FMN and revealed that the enzyme adopts a TIM-barrel fold [56], which is structurally distinct from the α/β fold adopted by IDI-1 [22]. Even though this structure was not solved in the presence of IPP/DMAPP, several key conserved interactions between the active site and the coenzyme could be discerned. These interactions included putative hydrogen bonds between Thr68, Ser96, Lys193, and His155 (Sulfolobus shibatae numbering) with the N5, C4=O, N1, and C2=O groups of the flavin, respectively. Mutations at these positions were subsequently shown to affect catalysis by IDI-2 [48], but none of these residues are absolutely required for catalysis and the precise effects of these mutations have not yet been elucidated in detail. In 2009, the Hemmi group published the first fully-complexed structure of the S. shibatae IDI-2 in the presence of reduced FMN, IPP/DMAPP, and Mg2+ (Figure 3) [48]. This structure revealed a very close association of the isoprenoid substrate with the flavin. Importantly, no amino acid derived acid/base functional groups were present in vicinity of the bound substrate, leaving the flavin coenzyme as the only reasonable candidate for mediating the 1,3-proton addition/elimination needed to isomerize IPP and DMAPP. In particular, the N5-atom of the reduced flavin is positioned only 3.19 and 3.52 Å, respectively, from the C2 and C4 positions of IPP/DMAPP (Figure 3A). The observed IPP binding mode is consistent with stereochemical studies in that the pro-R C2 proton of IPP (which is removed during turnover) [31] is oriented towards the flavin. Moreover, in the observed binding mode, protonation at C4 of the substrate by the flavin would result in a suprafacial 1,3-proton migration – an observation that is consistent with the stereochemical outcome observed in a chiral methyl analysis of the IDI-2 catalyzed reaction [51]. In this study, deuterium (from D2O) was incorporated into (E)- and (Z)-[4-3H]-IPP analogues (20 and 21, Scheme 9) by IDI-2 under single turnover conditions to yield DMAPP products with a chiral (E)-methyl group [51]. Each IPP analogue yielded the distinct enantiomeric configuration at the (E)-methyl group of DMAPP that is consistent with protonation/deprotonation mediated by the flavin. Altogether, the mechanistic and structural data are most consistent with a chemical mechanism involving the flavin N5 atom as the key acid/base catalyst. Other potential flavin-derived acid/base groups are the N1 atom and C4=O group (Figure 3B), but deviations in the observed binding mode of the substrate would be needed in order for these groups to be positioned optimally for acid/base chemistry.
Figure 3.

Active site views of IDI-2 from Sulfolobus shibatae (PDB ID 2ZRY). Panel A shows the constellation of conserved amino acid residues in the immediate vicinity of the flavin and bound isoprene. Mutations of these residues perturb IDI-2 catalysis, but none of them are suitably positioned to mediate proton transfers to and from IPP. In contrast, the flavin N5 atom appears to be optimally positioned to both C2 (3.19 Å) and C4 (3.52 Å) to mediate the suprafacial 1,3-proton addition/elimination suggested by stereochemical studies. Panel B shows an alternative view of the same structure to illustrate the putative π-stacking between the flavin and isoprene. The flavin N1 and O4′ are also in the vicinity of IPP C4 and C2, respectively. Roles for these residues in catalysis cannot yet be ruled out, but they are not positioned as optimally as N5.
Scheme 9.

Suprafacial protonation/deprotonation by IDI-2 revealed by chiral methyl analysis. IPP analogues 20 and 21 yield DMAPP products with distinct enantiomeric configurations at the (E)-methyl group, suggesting that protonation at C4 and deprotonation at C2 occur from the face of the isoprene molecule that is exposed to the flavin in the crystal structure.
Studies with mechanistic probes support a direct catalytic role for the flavin
In a series of studies, the Eguchi and Poulter groups [41, 42, 59] demonstrated that a variety of electrophilically activated IPP and DMAPP analogues (such as oIPP, Scheme 10) irreversibly inactivate IDI-2 by forming covalent adducts with the flavin. The mechanism-based inactivation likely involves protonation of the electrophilic moiety of the inhibitor (by the flavin), followed by nucleophilic addition of the flavin to the incipient carbocation [42]. The covalent adducts were initially proposed to form at N5 of the flavin [42]; however, subsequent more detailed studies of the aerobic stability and pH-dependence of the UV-visible absorption of the flavin adducts formed in these assays are more consistent with alkylation at the C4a position of the flavin [49]. The covalent linkage between the inhibitor and the flavin in the IDI-2 active site was also visualized by X-ray crystallography; however, the resolution of these structures was not sufficient to unambiguously assign the site of adduct formation [49]. Apparently, the resulting adducts cannot be deprotonated by the IDI-2 active to form isomerized products or to regenerate the reduced flavin. While the inhibitors used in these studies are likely artificially activated (relative to IPP) towards nucleophilic addition of the flavin, these observations nevertheless suggest that the electron density on the flavin may not be strongly delocalized into other portions of the isoalloxazine. The enhanced electron density in the vicinity of N5-C4a could therefore be available in the wild type reaction to facilitate isomerization of the IPP/DMAPP double bond by other means – such as by serving as a base to catalyze proton transfers to and from the substrate.
Scheme 10.

Irreversible covalent inactivation of flavin by oIPP (3-oxiranyl-3-buten-1-yl pyrophosphate).
Working models and unresolved questions
The accumulated data on IDI-2 is consistent with one of several potential mechanisms for the 1,3-allylic isomerization (Scheme 11). In line with the absence of suitable active site amino acid residues observed in the X-ray crystal structures, each of these mechanisms employs the flavin as the sole general acid/base catalyst. Moreover, each mechanism involves the accumulation of positive charge on both the flavin and the substrate at some point in the reaction sequence. The mechanisms begin with the binding of IPP/DMAPP to the IDI-2:FMNH− resting state, which triggers pre-steady state formation of a spectroscopically distinct flavin intermediate (assigned as 1,5-dihydro-FMNH2, 14) [39, 40]. While the N1 position of the 1,5-dihydro-FMNH2 species could conceivably be used to protonate the substrate directly, this pathway seems unlikely given the observed IPP/DMAPP binding mode (Figure 3B) and the likelihood that the N1-H proton would be fixed in the plane of the ring and not oriented towards the substrate. Thus, the 1,5-dihydro-FMNH2 tautomer (14) may first isomerize to the zwitterionic 5,5-dihydro-FMNH2 (15). The chemical precedence for 15 stems from studies of the pH dependence of free flavin 15N-NMR signals by Macheroux and co-workers [60], who estimated an N5 pKa of ~ 4.0 for anionic FMNH− – a value that is clearly accessible to enzymes under physiological conditions. This species has never been detected in an enzyme-catalyzed reaction, but has been proposed to play a catalytic role in NikD, an enzyme involved in the biosynthesis of nikkomycin antibiotics [61].
If the species that accumulates in the pre-steady state upon IPP binding is indeed 1,5-dihydro-FMNH2 (14), then the isomerization of 14 → 15 must be partially rate limiting (otherwise, 14 wouldn’t accumulate). A rate-limiting tautomerization of 14 → 15 could account for both the solvent KIE and the negative slope observed in the Hammett plots derived from the flavin analogues (Figure 2). Alternatively, IPP/DMAPP may bind directly to the flavin zwitterion-bound form of IDI-2, which would represent only a small fraction of the total enzyme present in solution at pH 7.0 and, hence, would not be spectroscopically detectable in the absence of IPP/DMAPP. In this case, the species that accumulates in the pre-steady state would be the zwitterion (15), rather than the neutral FMNH2 (14). Spectroscopic experiments could help to clarify the true identity of the flavin intermediate. One possible approach would be to conduct NMR studies on IDI-2 reconstituted with uniformly 15N-labeled flavin analogues. Previous work has shown that the magnitude of 15N-1H J-coupling constants are related to the hybridization state of the N nucleus [62, 63]. Thus, if the flavin zwitterion indeed accumulates in the presence of IPP/DMAPP, an 15N-1H coupling constant that is consistent with sp3 hybridization (~72 Hz) should be observed. Similar types of studies have proven useful in unraveling the structural and electronic properties of other flavoenzyme active sites [64–69] and could significantly advance our understanding of the role of FMN in IDI-2 catalysis.
If the reduced flavin zwitterion 15 is involved in IDI-2 catalysis, either stepwise or concerted protonation/deprotonation mechanisms can be envisioned. In a stepwise transformation, proton transfer from the zwitterionic FMNH2 to IPP/DMAPP would yield a tertiary carbocation/FMNH− pair. The protonation step could account for the solvent KIE, and the formation of the 3° carbocation could account for the poor activity of the fluoromethyl IPP/DMAPP analogues. The preponderance of hydride shifts, methyl shifts, and C-C bond rearrangements observed in the reactions catalyzed by isoprenoid biosynthetic enzymes suggest that tertiary carbocation intermediates are common in isoprenoid biosynthesis [11, 13], and the IDI-2 catalyzed reaction may be no exception to this general phenomenon. However, if the carbocation/FMNH− state is generated during turnover (as would be required in this obligate stepwise single base protonation/deprotonation mechanism), it is not clear how covalent inactivation of the flavin at C4a is avoided, or how a flavin C4a-IPP/DMAPP adduct would be resolved if it indeed forms. Detection of a flavin C4a-adduct in the wild type reaction would provide strong support for a stepwise protonation/deprotonation mechanism; however, to date, these adducts have only been detected with irreversible inhibitors (Scheme 10). Finally, in order to account for the measured Dkcat in the stepwise mechanism, the substrate deprotonation step would also have to be partially rate-limiting, despite the extremely low pKa expected for the carbocation intermediate. Because the deprotonation step also involves formation of positive charge on the flavin at N5, this step would contribute to the LFERs measured with the flavin analogues.
A concerted mechanism involving the flavin zwitterion resonance form 15b can also be envisioned. The oxygen atom of the C4=O carbonyl is only 3.68 Å removed from C2 of IPP/DMAPP in the S. shibatae crystal structure (Figure 3), and a similar intermediate has been proposed to mediate proton transfers in the reaction catalyzed by NikD [61]. Once again, additional spectroscopic evidence for the identity of the flavin intermediate could help to validate or refute this mechanistic proposal. In this scenario, the solvent KIE, Dkcat, and flavin LFER would all originate from the same step in the mechanism. If the proton transfers are asynchronous in the transition state, then partial positive charge will also be developing on the isoprene moiety, thus accounting for the decreased activity of the fluoromethyl IPP/DMAPP analogues. It should be noted that due to the expected high pKa of the allylic protons, protonation of the alkene is expected to precede deprotonation in the 1,3-proton addition/elimination sequence; thus an asynchronous transition state carrying partial positive charge at C3 of IPP/DMAPP is possible. The widespread existence of concerted, asynchronous rearrangement reactions in isoprenoid biosynthesis has been supported by extensive quantum mechanical calculations [70].
Several circumstantial observations seem to provide indirect support for the concerted mechanism. First, the quaternary ammonium center of 2-(dimethylamino)ethyl-pyrophosphate (NIPP, Scheme 12) is believed to mimic a tertiary carbocation. Interestingly, NIPP is a weak competitive inhibitor of IDI-2, with a Ki value in the low μM range – very similar to the Km value for IPP [40]. In contrast, this same inhibitor exhibits a Ki value in the pM range for IDI-1 – a full 4–5 orders of magnitude tighter than it binds to IDI-2. Thus, the transition state for the IDI-1 catalyzed reaction appears to involve a substantially higher degree of cationic character on the substrate isoprene moiety than the transition state for the IDI-2 catalyzed reaction – an observation that is consistent with a mechanism that is more stepwise in the case of IDI-1 and more concerted in the case of IDI-2. Of course, it could be argued that the weak Ki of NIPP for IDI-2 is a result of unfavorable steric interactions induced by the quaternary ammonium center. This hypothesis would be consistent with the drastic reductions in the steady state kinetic parameters induced by mutation of Gln160 [53], a residue that appears to help position the planar isoprene unit over the flavin in the IDI-2 active site (Figure 3). Another interesting difference between IDI-1 and IDI-2 involves the pattern of solvent deuterium incorporation [19, 30]. In IDI-1, all of the protons (except those at C1) are exchanged with solvent deuterium, perhaps indicating the presence of a reactive carbocation intermediate that can be deprotonated by multiple (weak) bases present in the IDI-1 active site. In contrast, IDI-2 only catalyzes the exchange of the pro-R proton at C2 of IPP and the (E)-methyl protons of DMAPP (Scheme 4). This strict regio- and stereochemistry of solvent deuterium incorporation is expected for a concerted reaction, where no formal substrate carbocation is formed. Thus, an experiment that definitively distinguishes stepwise and concerted mechanisms is needed. One interesting potential experiment would be to perform KIE studies with a doubly-labeled IPP analogue containing a fluorine label on the methyl group and a deuterium in the pro-R position at C2. If a concerted mechanism is operative, the KIE would still be measurable and would be approximately the same magnitude as the KIE measured using isotopologues of the native substrate (i.e. Dkcat ~ 2.2) [39], even if the overall rate of the reactions with the fluoromethyl analogues is severely reduced. In contrast, if the protonation/deprotonation sequence is stepwise, the deuterium KIE on the deprotonation step may be completely masked by a preceding protonation step that has become fully rate limiting because of the fluorine substitution.
Scheme 12.
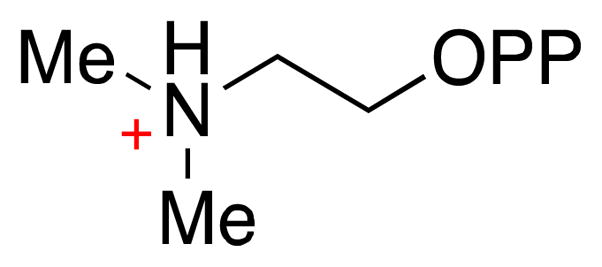
Chemical structure of 2-(dimethylamino)ethyl-pyrophosphate.
Concluding Remarks
As illustrated throughout this review, despite over a decade of intense research on IDI-2, significant questions regarding the chemical mechanism of this interesting and unique flavoenzyme remain. Clearly, the reaction catalyzed by IDI-2 extends beyond the classical paradigms of flavoenzyme function. The current evidence in favor of a flavin-mediated protonation/deprotonation sequence is substantial; however, exact details regarding the precise identity of the acid/base groups still need to be clarified. In particular, a more definitive demonstration of the involvement of the reduced flavin zwitterion in the catalytic cycle, and additional kinetic studies aimed at distinguishing among stepwise and concerted reaction mechanisms should be the focus of future mechanistic work on IDI-2.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (GM040541) and the Welch Foundation (F-1511) to HWL. CJT also acknowledges start up funding from McGill University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Porter JW, Spurgeon SL. In: Biosynthesis of isoprenoid compounds. Porter JW, Spurgeon SL, editors. John Wiley and Sons; New York: 1981. pp. 1–93. [Google Scholar]
- 2.Chaykin S, Law J, Phillips AH, Tchen TT, Bloch K. Proc Natl Acad Sci USA. 1958;44:998–1004. doi: 10.1073/pnas.44.10.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloch K, Chaykin S, Phillips AH, de Waard A. J Biol Chem. 1959;234:2595–2604. [PubMed] [Google Scholar]
- 4.Lynen F, Eggerer H, Henning U, Kessel I. Angew Chem. 1958;70:738–742. [Google Scholar]
- 5.Beytia ED, Porter JW. Annu Rev Biochem. 1976;45:113–142. doi: 10.1146/annurev.bi.45.070176.000553. [DOI] [PubMed] [Google Scholar]
- 6.Rohmer M. Nat Prod Rep. 1999;16:565–574. doi: 10.1039/a709175c. [DOI] [PubMed] [Google Scholar]
- 7.Laupitz R, Hecht S, Amslinger S, Zepeck F, Kaiser J, Richter G, Schramek N, Steinbacher S, Huber R, Arigoni D, Bacher A, Eisenreich W, Rohdich F. Eur J Biochem. 2004;271:2658–2669. doi: 10.1111/j.1432-1033.2004.04194.x. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Concepcion M. Curr Pharm Des. 2004;10:2391–2400. doi: 10.2174/1381612043384006. [DOI] [PubMed] [Google Scholar]
- 9.Singh N, Cheve G, Avery MA, McCurdy CR. Curr Pharm Des. 2007;13:1161–1177. doi: 10.2174/138161207780618939. [DOI] [PubMed] [Google Scholar]
- 10.Thibodeaux CJ, Liu HW. Chimia. 2009;63:334–339. [Google Scholar]
- 11.Kellogg BA, Poulter CD. Curr Opin Chem Biol. 1997;1:570–578. doi: 10.1016/s1367-5931(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 12.Thibodeaux CJ, Chang WC, Liu HW. Chem Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christianson DW. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 14.Chang WC, Song H, Liu HW, Liu P. Curr Opin Chem Biol. 2013;17:571–579. doi: 10.1016/j.cbpa.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agranoff BW, Eggerer H, Henning U, Lynen F. J Am Chem Soc. 1958;81:1254–1255. [Google Scholar]
- 16.Agranoff BW, Eggerer H, Henning U, Lynen F. J Biol Chem. 1960;235:326–332. [PubMed] [Google Scholar]
- 17.Shah DH, Cleland WW, Porter JW. J Biol Chem. 1965;240:1946–1956. [PubMed] [Google Scholar]
- 18.Lee S, Poulter CD. J Am Chem Soc. 2006;128:11545–11550. doi: 10.1021/ja063073c. [DOI] [PubMed] [Google Scholar]
- 19.Street IP, Christensen DJ, Poulter CD. J Am Chem Soc. 1990;112:8577–8578. [Google Scholar]
- 20.Street IP, Coffman HR, Baker JA, Poulter CD. Biochemistry. 1994;33:4212–4217. doi: 10.1021/bi00180a014. [DOI] [PubMed] [Google Scholar]
- 21.Wouters J, Oudjama Y, Barkley SJ, Tricot C, Stalon V, Droogmans L, Poulter CD. J Biol Chem. 2003;278:11903–11908. doi: 10.1074/jbc.M212823200. [DOI] [PubMed] [Google Scholar]
- 22.Durbecq V, Sainz G, Oudjama Y, Clantin B, Bompard-Gilles C, Tricot C, Caillet J, Stalon V, Droogmans L, Villeret V. EMBO J. 2001;20:1530–1537. doi: 10.1093/emboj/20.7.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Popjak G, Conforth JW. Biochem J. 1966;101:553–568. [PMC free article] [PubMed] [Google Scholar]
- 24.Clifford K, Conforth JW, Mallaby R, Phillips GT. J Chem Soc D. 1971:1599–1600. [Google Scholar]
- 25.Kaneda K, Kuzuyama T, Takagi M, Hayakawa Y, Seto H. Proc Natl Acad Sci USA. 2001;98:932–937. doi: 10.1073/pnas.020472198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kittleman W, Thibodeaux CJ, Liu YN, Zhang H, Liu HW. Biochemistry. 2007;46:8401–8413. doi: 10.1021/bi700286a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita S, Hemmi H, Ikeda Y, Nakayama T, Nishino T. Eur J Biochem. 2004;271:1087–1093. doi: 10.1111/j.1432-1033.2004.04010.x. [DOI] [PubMed] [Google Scholar]
- 28.Bornemann S. Nat Prod Rep. 2002;19:761–772. doi: 10.1039/b108916c. [DOI] [PubMed] [Google Scholar]
- 29.Mansoorabadi SO, Thibodeaux CJ, Liu HW. J Org Chem. 2007;72:6329–6342. doi: 10.1021/jo0703092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barkley SJ, Desai SB, Poulter CD. Org Lett. 2004;6:5019–5021. doi: 10.1021/ol0477273. [DOI] [PubMed] [Google Scholar]
- 31.Kao C-l, Kittleman W, Zhang H, Seto H, Liu H-w. Org Lett. 2005;7:5677–5680. doi: 10.1021/ol0524050. [DOI] [PubMed] [Google Scholar]
- 32.Barak CDZ, Schloss JV. Biochim Biophys Acta. 1998;1385:401–419. doi: 10.1016/s0167-4838(98)00083-1. [DOI] [PubMed] [Google Scholar]
- 33.Chen HW, Guo ZH, Liu HW. J Am Chem Soc. 1998;120:11796–11797. [Google Scholar]
- 34.Kaneda K, Kuzuyama T, Takagi M, Hayakawa Y, Seto H. Proc Natl Acad Sci USA. 2001;98:932–937. doi: 10.1073/pnas.020472198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hemmi H, Ikeda Y, Yamashita S, Nakayama T, Nishino T. Biochem Biophys Res Commun. 2004;322:905–910. doi: 10.1016/j.bbrc.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Yu Q, Schaub P, Ghisla S, Al-Babili S, Krieger-Liszkay A, Beyer P. J Biol Chem. 2010;285:12109–12120. doi: 10.1074/jbc.M109.091843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sancar A. Chem Rev. 2003;103:2203–2237. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 38.Hersh LB, Walsh C. Methods Enzymol. 1980;66:277–287. doi: 10.1016/0076-6879(80)66469-6. [DOI] [PubMed] [Google Scholar]
- 39.Thibodeaux CJ, Mansoorabadi SO, Kittleman W, Chang W-c, Liu H-w. Biochemistry. 2008;47:2547–2558. doi: 10.1021/bi701467g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothman SC, Helm TR, Poulter CD. Biochemistry. 2007;46:5437–5445. doi: 10.1021/bi0616347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnston JB, Walker JR, Rothman SC, Poulter CD. J Am Chem Soc. 2007;129:7740–7741. doi: 10.1021/ja072501r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rothman SC, Johnston JB, Lee S, Walker JR, Poulter CD. J Am Chem Soc. 2008;130:4906–4913. doi: 10.1021/ja7108954. [DOI] [PubMed] [Google Scholar]
- 43.Griller D, Ingold KU. Acc Chem Res. 1980;13:317–323. [Google Scholar]
- 44.Ghisla S, Massey V, Lhoste JM, Mayhew SG. Biochemistry. 1974;13:589–597. doi: 10.1021/bi00700a029. [DOI] [PubMed] [Google Scholar]
- 45.de Ruyck J, Janczak MW, Neti SS, Rothman SC, Schubert HL, Cornish RM, Matagne A, Wouters J, Poulter CD. Chembiochem. 2014;15:1452–1458. doi: 10.1002/cbic.201402046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janczak MW, Poulter CD. Biochemistry. 2016;55:2260–2268. doi: 10.1021/acs.biochem.6b00087. [DOI] [PubMed] [Google Scholar]
- 47.Barkley SJ, Desai SB, Poulter CD. J Bacteriol. 2004;186:8156–8158. doi: 10.1128/JB.186.23.8156-8158.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unno H, Yamashita S, Ikeda Y, Sekiguchi SY, Yoshida N, Yoshimura T, Kusunoki M, Nakayama T, Nishino T, Hemmi H. J Biol Chem. 2009;284:9160–9167. doi: 10.1074/jbc.M808438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagai T, Unno H, Janczak MW, Yoshimura T, Poulter CD, Hemmi H. P Natl Acad Sci USA. 2011;108:20461–20466. doi: 10.1073/pnas.1115749108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson KA. In: Wiley Encyclopedia of Chemical Biology. Begley TP, editor. 2009. pp. 605–613. [Google Scholar]
- 51.Calveras J, Thibodeaux CJ, Mansoorabadi SO, Liu HW. Chembiochem. 2012;13:42–46. doi: 10.1002/cbic.201100694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schowen KB, Schowen RL. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 53.Thibodeaux CJ, Chang WC, Liu HW. J Am Chem Soc. 2010;132:9994–9996. doi: 10.1021/ja104090m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 55.Poulter CD, Satterwhite DM. Biochemistry. 1977;16:5470–5478. doi: 10.1021/bi00644a012. [DOI] [PubMed] [Google Scholar]
- 56.Steinbacher S, Kaiser J, Gerhardt S, Eisenreich W, Huber R, Bacher A, Rohdich F. J Mol Biol. 2003;329:973–982. doi: 10.1016/s0022-2836(03)00527-8. [DOI] [PubMed] [Google Scholar]
- 57.de Ruyck J, Rothman SC, Poulter CD, Wouters J. Biochem Biophys Res Commun. 2005;338:1515–1518. doi: 10.1016/j.bbrc.2005.10.114. [DOI] [PubMed] [Google Scholar]
- 58.de Ruyck J, Pouyez J, Rothman SC, Poulter D, Wouters J. Biochemistry. 2008;47:9051–9053. doi: 10.1021/bi801159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoshino T, Tamegai H, Kakinuma K, Eguchi T. Bioorg Med Chem. 2006;14:6555–6559. doi: 10.1016/j.bmc.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 60.Macheroux P, Ghisla S, Sanner C, Ruterjans H, Muller F. BMC Biochem. 2005;6:26. doi: 10.1186/1471-2091-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kommoju PR, Bruckner RC, Ferreira P, Jorns MS. Biochemistry. 2009;48:6951–6962. doi: 10.1021/bi9006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Binsch G, Lambert JB, Roberts BW, Roberts JD. J Am Chem Soc. 1964;86:5564–5570. [Google Scholar]
- 63.Bourn AJR, Randall EW. Mol Phys. 1964;8:567–579. [Google Scholar]
- 64.Franken HD, Ruterjans H, Muller F. Eur J Biochem. 1984;138:481–489. doi: 10.1111/j.1432-1033.1984.tb07942.x. [DOI] [PubMed] [Google Scholar]
- 65.Vervoort J, Mueller F, Mayhew SG, Van den Berg WAM, Moonen CTW, Bacher A. Biochemistry. 1986;25:6789–6799. doi: 10.1021/bi00370a010. [DOI] [PubMed] [Google Scholar]
- 66.Moonen CTW, Vervoort J, Muller F. Biochemistry. 1984;23:4859–4867. doi: 10.1021/bi00316a007. [DOI] [PubMed] [Google Scholar]
- 67.Beinert WD, Ruterjans H, Muller F. Eur J Biochem. 1985;152:573–579. doi: 10.1111/j.1432-1033.1985.tb09234.x. [DOI] [PubMed] [Google Scholar]
- 68.Chang FC, Swenson RP. Biochemistry. 1999;38:7168–7176. doi: 10.1021/bi982203u. [DOI] [PubMed] [Google Scholar]
- 69.Fleischmann G, Lederer F, Muller F, Bacher A, Ruterjans H. Eur J Biochem. 2000;267:5156–5167. doi: 10.1046/j.1432-1327.2000.01584.x. [DOI] [PubMed] [Google Scholar]
- 70.Tantillo DJ. Nat Prod Rep. 2011;28:1035–1053. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]