

Abstract
Cancer immunotherapy using immune-checkpoint blockade (ICB) has created a paradigm shift in the treatment of advanced-stage cancers. The promising antitumour activity of monoclonal antibodies targeting the immune-checkpoint proteins CTLA-4, PD-1, and PD-L1 led to regulatory approvals of these agents for the treatment of a variety of malignancies. Patients might experience clinical benefits from treatment with these agents, despite unconventional patterns of tumour response that can be misinterpreted as disease progression, warranting a new, specific approach to evaluate responses to immunotherapy. In addition, biomarkers that can predict responsiveness to ICB are being extensively investigated to further advance precision immunotherapy. Herein, we review the biological mechanisms underlying the unconventional response patterns associated with ICB, describe strategies for the objective assessments of such responses, and also highlight the ongoing efforts to identify biomarkers, in order to guide treatment with ICB. We provide state-of-the-art knowledge of immune-related response evaluations, identify unmet needs requiring further investigations, and propose future directions to maximize the benefits of ICB therapy.
Cancer immunotherapy with immune-checkpoint blockade (ICB) is based on the inhibition of the tumour- mediated suppression of anticancer immune responses, in contrast with therapeutic strategies that exert direct cytotoxic effects on tumour cells1–4. T cells have a major role in immune defense mechanisms against cancer: they recognize tumour antigens, consequently become activated, disseminate and, ultimately, eliminate cancer cells1–4. In this context, T-cell activation is regulated by the interplay of the stimulatory and inhibitory ligand–receptor interactions between T cells, dendritic cells, tumour cells, and macrophages in the tumour microenvironment (TME), with tumour cells acting as critical mediators of immunosuppression2,5 (FIG. 1). Owing to their roles as regulators of T-cell activation, these receptor–ligand pairs are called ‘immune checkpoints’. In addition to the TME, important interactions between T cells and antigen-presenting cells expressing immune- checkpoint molecules occur in secondary lymphoid tissues5. Agents targeting these checkpoints — that is, ICB agents — have been identified as promising treatment options for patients with cancer2. Immune-checkpoint inhibitors (ICIs) include, among others, monoclonal antibodies to the receptor cytotoxic T-lymphocyte antigen-4 (CTLA-4) expressed on T cells; programmed cell death protein 1 (PD-1), also expressed on T cells; or the PD-1 ligand (PD-L1), which is expressed by a variety of cell types, including some tumour cells. In 2010, the results of studies with the anti-CTLA-4 antibody ipilimumab in patients with advanced-stage melanoma2,6 led to the regulatory approval of this agent 1 year after, and sparked a rapid increase of further studies of ICIs in patients with advanced-stage cancers. Subsequently, the anti-PD-1 antibodies nivolumab and pembrolizumab, and the anti-PD-L1 antibody atezolizumab, have shown marked therapeutic activity in various solid tumours and lymphomas, resulting in a number of regulatory approvals of these agents for the treatment of different malignancies.
Figure 1. Ligand–receptor interactions between tumour cells and immune cells in the tumour microenvironment.
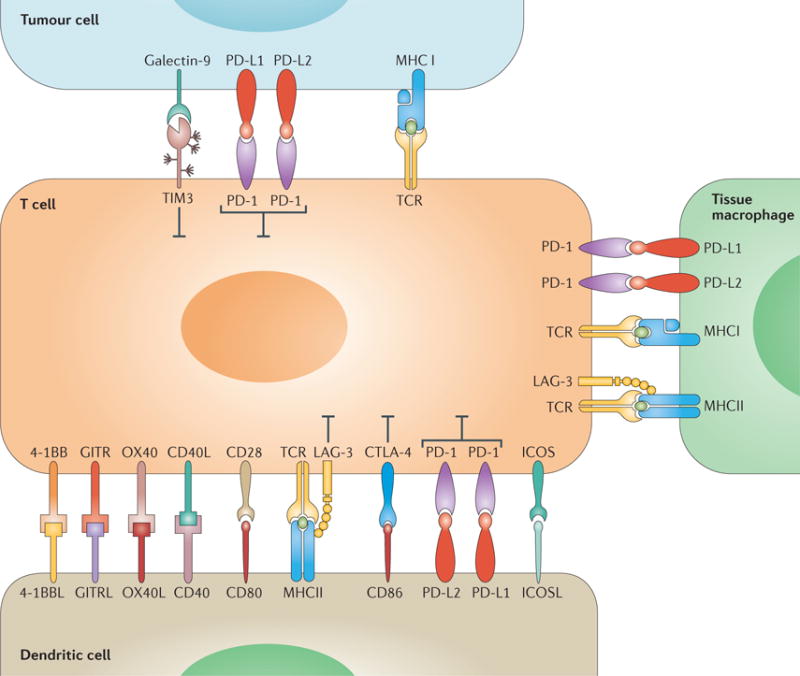
An overview of the immune-checkpoint molecules involved in the regulation of the antitumour immune response.
The scope of ICB is expanding rapidly in the clinical oncology practice, and is expected to continue to grow further as new agents become available in the clinical setting. The comprehensive understanding of the benefits and risks associated with ICB is essential for those involved in the care of patients with cancer. In this Review, we provide an overview of the mechanisms of action of these agents, examine strategies for evaluation of immune-related response to ICB, and discuss ongoing efforts to develop predictive biomarkers of responsiveness. The unmet needs that require urgent attention are emphasized to provide directions for further studies.
Immune-related response evaluation
Immune-related response criteria (irRC) — a key concept
Owing to the unique antitumour mechanisms elicited by ICB, patients treated with these agents can have tumour response patterns that are not adequately captured using the conventional tumour-response criteria3,4,7–10, such as the WHO criteria11 and Response Evaluation Criteria in Solid Tumours (RECIST)12,13. For example, in a small subset of patients treated with ICIs, a response is detected after an initial increase in tumour burden (FIG. 2), or during or after the appearance of new lesions3,4,10 (FIG. 3). According to the WHO criteria and RECIST, these events would be classified as tumour progression, and are therefore termed ‘pseudoprogression’. With pseudoprogression, the apparent tumour-size increase detected upon imaging is thought to be caused by T-cell infiltration as a result of immune activation, rather than by tumour-cell proliferation10. In order to accurately assess these immune-related response phenomena, a series of workshops were held among 200 oncologists, immunotherapists, and regulatory experts, which resulted in the proposal, in 2009, of the immune-related response criteria (irRC)3,4,10.
Figure 2. Response after initial increase in total tumour burden in a 77-year-old male with advanced-stage melanoma treated with ipilimumab.
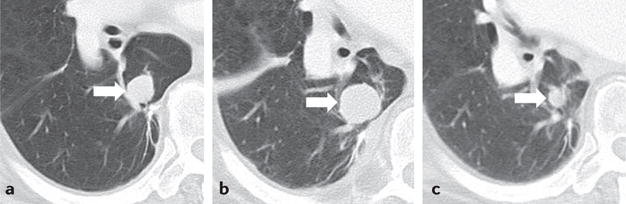
a | The baseline CT scan demonstrated a lung lesion (arrow) measuring 19 mm in the longest diameter. b | After 12 weeks of therapy, the lesion (arrow) measured 29 mm (53% increase compared with the baseline), indicating progressive disease by RECIST. c | The patient remained on therapy, and another follow-up CT scan (24 weeks after therapy initiation) showed a reduction in the size of the lesion (arrow), to 12 mm, indicative of an immune-related response to therapy. Reprinted from Nishino, M., Tirumani, S.H., Ramaiya, N.H. & Hodi, F.S. Cancer immunotherapy and immune-related response assessment: the role of radiologists in the new arena of cancer treatment, Eur. J. Radiol., 84, 1259–1268, Copyright (2015), with permission from Elsevier.
Figure 3. Response after appearance of a new lesion in a 56-year-old woman with metastatic melanoma treated with ipilimumab3.
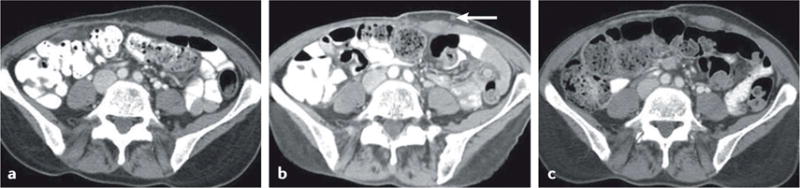
Contrast-enhanced CT scans of the abdomen a | before and b | 12 weeks after initiation of ipilimumab therapy revealed new subcutaneous nodule (arrow), suspected to be a new site of metastasis. c | A follow-up CT scan 24 weeks after initiation of therapy revealed resolution of the nodule, indicating response after appearance of a new lesion. Reprinted with permission from Nishino, M. et al. Personalized tumor response assessment in the era of molecular medicine: cancer-specific and therapy-specific response criteria to complement pitfalls of RECIST. AJR Am. J. Roentgenol. 198, 737–745 (2012).
The key features of irRC include: firstly, the requirement for confirmation of disease progression on two consecutive scans (performed at least 4 weeks apart), such that the initial increase in tumour burden alone does not immediately define progression; secondly, the inclusion of new lesions in the sum of lesion measurements is considered to better reflect the total tumour burden — as opposed to WHO criteria and RECIST that define progression at the appearance of new lesions3,4,10. The irRC were first used to define the end points of a phase II trial of ipilimumab plus paclitaxel and carboplatin as first-line therapy in stage IIIB/IV non-small-cell lung cancer (NSCLC)14; this concept has been applied in subsequent clinical trials of ICIs4,14–16.
The value of irRC was initially assessed retrospectively in patients with melanoma treated with ipilimumab in the first-line setting from whom post-progression scans were available, and did not initiate other anticancer therapies after disease progression10. Among 227 patients who received ipilimumab, 123 were described as having initial progression according to WHO criteria (which do not contemplate ‘pseudoprogression’) at the 12-week scan. Evidence of activity consistent with a response to this agent according to irRC was identified in 22 of 123 patients (18%; 10% of the whole cohort): five patients (23%) had an immune-related partial response, and 17 patients (77%) had immune-related stable disease10. These results indicate the irRC can be used to measure clinical benefit from ipilimumab for patients who have an initial increase in tumour burden and/or new detectable lesions. In those 22 patients with a response, overall survival was comparable to that of patients with a complete response, partial response, or stable disease defined using WHO criteria (median overall survival not reached (13.5 months–not reached) versus 31.2 months (27.8–31.2 months)), further emphasizing the importance of using specific criteria for the assessment of response to immunotherapy10. In a study published in 2016, the value of the irRC versus RECIST version 1.1 (RECIST1.1) in predicting patient outcome in response to the PD-1 inhibitor pembrolizumab was compared in 327 patients with advanced-stage melanoma15. The study reported that 24 (7%) of those 327 patients had atypical responses (defined as pseudoprogression by irRC). Importantly, overall survival was longer in the 84 patients (26%) who had progressive disease according to RECIST1.1, but non-progressive disease according to irRC, compared with that of 177 patients (54%) who had progressive disease according to both RECIST1.1 and irRC (median overall survival 22.5 months versus 8.4 months)15.
Pitfalls of irRC to evaluate immune-related responses
Clearly, the irRC describes an important concept necessary for the assessment of immune-related responses; however, several pitfalls and issues related to irRC remain to be solved. One of them is a methodological issue related to tumour measurements: irRC is based on the WHO criteria10,11, introduced in 1979, in which bidimensional measurements are performed and tumour burden is quantified as the product of the longest diameter and the longest perpendicular diameter11,17. Most clinical trials with patients with solid tumours conducted in the past decade, however, have followed RECIST measurement guidelines18 — introduced in 2000 and revised in 2009 — to define trial end points and provide a basis for regulatory approvals for novel agents12,13,17,19 (TABLE 1). In RECIST, unidimensional measurements are performed, using the longest diameters for non-nodal lesions, and the longest perpendicular diameters (short axis) for nodal lesions. Thus, the results from trials of ICIs evaluated using irRC (bidimensional measurements) cannot be compared directly with results from other trials that have used RECIST (unidimensional measurements)8,20. The comparison of irRC and RECIST-based assessments within the same trial is also challenging because divergent results might arise owing to the different measurement methods used, and not because of immune-related phenomena. Importantly, multiple prior studies have shown that bidimensional measurements are subject to larger measurement variability — and thus, have higher misclassification rates of response categories — than unidimensional measurements, resulting in inaccurate assessment of small changes in tumour burden8,21–24. Tumour response criteria were originally developed to serve as a common language to describe the outcomes of cancer treatment and thus, the above discussed methodological definition related to irRC requires further optimization8,20.
Table 1.
Summary of the response assessment strategy using different criteria
| Bidimensional approach | Unidimensional approach | ||||
|---|---|---|---|---|---|
| Strategy | WHO (1979)11 | irRC (2009)7 | RECIST1.1 (2009, revised based on the original RECIST published in 2000)12,13 | irRECIST (2013)4,7–9 | iRECIST (2017)31 |
| Measurement | LD × SD (cm2) | LD × SD (cm2) |
|
|
|
| Criteria for PR* | ≥50% decrease | ≥50% decrease | ≥30% decrease | ≥30% decrease | ≥30% decrease |
| Criteria for PD‡ | ≥25% increase, new lesion, or non-target PD | ≥25% increase | ≥20% and ≥5 mm increase, new lesion, or non-target PD | ≥20% and ≥5 mm increase, or non-target PD | ≥20% and ≥5 mm increase, or non-target PD |
| New lesions | Define PD |
|
Define PD |
|
|
| Confirmation of PD | Not needed | Required on a consecutive scan (at least 4 weeks later) | Not needed | Required on a consecutive scan (at least 4 weeks later) | Required at the next assessment (4–8 weeks later) |
The percent change is calculated in comparison with the measurements at baseline.
The percent change is calculated in comparison with the measurements at the nadir (smallest tumour burden since baseline). LD, longest diameter; PD, progressive disease; PR, partial response; SD, short-axis diameter (longest perpendicular diameter).
Further aspects of the definition of pseudoprogression remain to be clarified. For example, the degree of tumour regression after an initial size increase that has been observed in patients who display pseudoprogression is variable. Of note, in many studies — including the first report of irRC10 — patients did not need to meet the requirement of tumour reduction below the partial response threshold to be classified as having a response; instead, tumour regression in the range of stable disease (defined as neither sufficient shrinkage to qualify for partial response nor sufficient increase to qualify for progressive disease) was considered as evidence of therapeutic anticancer activity. This criterion is based, in part, on the concept that durable stable disease is a pattern of response to therapy. Indeed, results from a study in patients with melanoma treated with pembrolizumab demonstrated that increases in tumour burden of <20% from baseline (defined according to observations of tumour burden dynamics during ICB therapy using a spider plot) were associated with longer overall survival25. Prospective validations in other patient cohorts are needed but, notably, durable disease control (maintenance of a near baseline level of tumour burden measured at the time of initiation of ICB), even without a deep tumour reduction below the response threshold, can indicate benefit from treatment in the setting of ICB.
Moreover, in the irRC, pseudoprogression was defined mainly on the basis of tumour burden reductions after an initial increase observed around 12 weeks after initiation of therapy, a time point established according to the study design and follow-up intervals for patients with melanoma involved in trials of ICIs26,27. Results presented in 2017, however, indicate that pseudoprogression can occur later during the course of therapy and thus, a response after initial tumour size increase might be noted beyond the 4-week window between assessments defined in the irRC25. For example, in a study by Hodi and collaborators15, ‘delayed pseudoprogression’ (detected after 12 weeks of therapy) was not confirmed as progressive disease at the next assessment (at least 4 weeks later) in 3% (9/327) of patients with melanoma treated with pembrolizumab. In another study involving patients treated with pembrolizumab, after immune-related progressive disease was confirmed on a subsequent scan, a therapeutic response was detected in 3% (3/107) of patients (median time to detection of initial immune- related progressive disease: 2.7 months)25. Indeed, the authors of the 2015 report from the Response Assessment for Neuro-Oncology (RANO) Working Group point out the lack of scientific rationale of the 4-week window defined in the irRC and, instead, recommend a 3-month period for confirmation of progressive disease in neuro-oncology trials, to avoid excluding pseudoprogression too early in the therapeutic course28. This recommendation from RANO was not, however, based on scientific data from studies specifically designed to define an adequate timeframe of confirmatory scans and, rather, was based on observations of spider plots of a few early trials of ICIs28. Therefore, the identification of the most optimal timeframe for confirmation of progressive disease will require further investigations.
Importantly, the generally low incidence of pseudoprogression should be acknowledged when monitoring patients treated with ICIs18,20. In studies of patients with melanoma receiving both CTLA-4 and PD-1 inhibitors, the incidence rate of pseudoprogression is approximately 10% (or often less)10,15,25. A lower incidence (5%; 6/129) was reported in patients with advanced-stage NSCLC in a phase I study of nivolumab; the incidence might be even lower in patients treated with standard-of-care immunotherapies9,29. Despite an increasing awareness of the pseudoprogression phenomenon, accumulating data indicate that this is a relatively uncommon event, and an increase of tumour burden is more likely to reflect true progression than pseudoprogression in most patients, raising a caution for optimism among health-care providers treating patients with tumour burden that increases in the setting of ICB.
Outstanding issues and future directions
The future directions of immune-related response evaluations are currently under active debate, with growing efforts to use standardized criteria that can improve upon the pitfalls and limitations of irRC. One proposed approach was to modify irRC to use unidimensional measurements and the same response categories as RECIST15. This modification was originally proposed in a study that evaluated the response to ipilimumab of 57 patients with advanced-stage melanoma in a phase II trial8. The study compared response assessments using the original, bidimensional irRC with those obtained using unidimensional measurement methods and adopting the threshold to define response categories according to RECIST. The key features of irRC (inclusion of new lesions and confirmation of pseudoprogression) were incorporated in both criteria. The response assessments were highly concordant: the evaluation of best immune-related response according to two measurements using both criteria was identical in 53 of 57 patients, whereas the estimated progression-free rate at 6 months was 70% for bidimensional irRC versus 81% for unidimensional irRC8. Estimates of the 25th percentile (time point at which a 75% progression-free survival (PFS) is reached) were 5.3 months (3.5 months–∞) by bidimensional assessment versus 9.1 months (3.7 months–∞) by unidimensional assessment; on the basis of the almost identical confidence intervals for this percentile, no evidence supports a difference in time to progression between both assessment methods8. Importantly, evaluation of the reproducibility of both measurement methods showed that the 95% limits of agreement of bidimensional measurements were twice as wide compared with unidimensional measurements (−31.3% to 19.7% versus −16.1% to 5.8%)8. This study provided a rationale to use a new method, termed immune-related RECIST (irRECIST), that incorporates RECIST-based measurement methods while maintaining the key features of irRC4,7,20,30. The results of these studies also provided a basis for the development of the iRECIST guidelines31,32 for the evaluation of response in trials testing immunotherapeutics, which are largely based on RECIST1.1, but require confirmation of progression and do not define progression when new lesions appear. While iRECIST mostly follows the guidelines proposed by irRECIST, this method mandates new lesions to be measured and recorded separately instead of being added to the sum, and increase of new lesions or appearance of additional new lesions on the subsequent scan confirms progression (TABLE 1). Further efforts are ongoing to collect sufficient data to validate both irRECIST and iRECIST, which will contribute to further advance the immune-related response evaluation strategy.
Advances in the knowledge of immune-related responses have been challenged by the fact that only a few clinical trials have used the irRC or irRECIST as the primary criteria to define their end points14,16,18. Most trials continue to use RECIST1.1 to define the end points, mostly because regulatory agencies continue to base the approvals of novel agents on RECIST-defined outcomes. Of note, however, some trials have included irRC, irRECIST or similar modified criteria as secondary response criteria14,16,33,34. Whether the introduction of iRECIST contributes to wider and uniform application of immune-related response criteria in trials with ICIs remains to be confirmed.
Immuno-oncologists also face a dilemma in terms of treatment decision-making when tumour progression occurs during therapy18,20. In trials conducted in the past 9 years, patients have often been allowed to continue therapy beyond progression when they are deemed by investigators to be deriving continued therapeutic benefit3. Similarly, clinical providers base their treatment decisions on the overall assessments of clinical improvements and treatment tolerance. These approaches are subject to clinician’s discretion, and objective guidelines establishing when to continue therapy or otherwise consider alternate options do not exist. Criteria for the evaluation of immune-related responses, including whether increases in tumour burden indicate progression and whether progression is confirmed, can provide information to guide treatment decisions; however, these criteria have essentially been developed for use in clinical trials of novel agents to describe the treatment results, and are not designed primarily to assist therapeutic decision-making in the clinical setting.
A preliminary observation25 indicates that factors such as younger age might be associated with pseudoprogression (the median age of patients defined to undergo pseudoprogression is 46 years compared with 63 years for all other patients), but the contribution of this factor remains to be validated25. A possible approach to validation would be to evaluate clinical, biological, and radiographic features at baseline in large databases of patients treated with ICB, and identify predisposing factors for pseudoprogression. Information about the likelihood of disease progression could then be used by health-care providers to guide treatment decisions. Serial evaluations of serum factors (including cytokines) during therapy and how they correlate with radiographic tumour kinetics might also help to distinguish pseudoprogression from true progression. The use of radiographic strategies (combining both serial anatomic imaging and functional imaging) in immune-related evaluations should advance together with these efforts.
Biomarkers of immunotherapy response
Despite the remarkable success of clinical applications of immunotherapy reported in the past decade, the efficacy and effectiveness of these therapies varies greatly across individual patients and among different tumour types. A substantial unmet need is the development of biomarkers of response to immunotherapeutic agents, in order to identify, before initiation of treatment, which patients are likely to experience a response to and clinical benefit from such treatments. This aspect is particularly important in the management of tumours with low response rates, such as NSCLC (response rate ≤20%)29,33,35–37, renal-cell carcinoma (RCC) and urothelial carcinoma (UCC), both with response rates ≤30%38–42. Herein, we present the immune-response- associated biomarkers that have been studied to date, discuss their strengths and weaknesses, and highlight ongoing efforts of biomarker development for use in precision cancer immunotherapy (FIG. 4).
Figure 4. Key elements in biomarker development for immune-checkpoint inhibitor therapy.
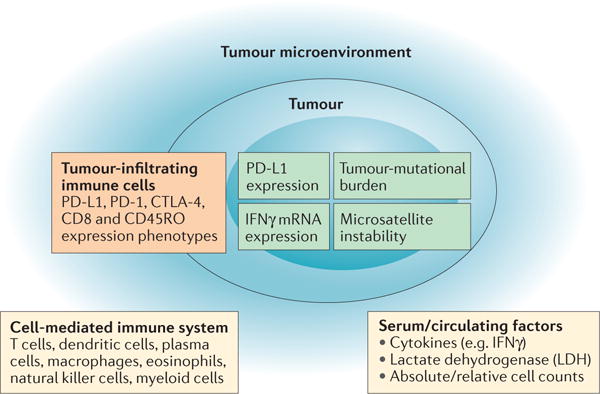
Consideration of the tumour, tumour microenvironment, and immune system must be incorporated in the ongoing efforts in biomarker development for immune-checkpoint inhibitor therapy.
PD-L1 and other immune-checkpoint molecules
Expression levels of PD-L1 on tumour cells (assessed by immunohistochemistry (IHC)) has been studied as a potential biomarker of response to ICB since the early phase of the development of these agents. In a phase I trial of nivolumab in patients with advanced-stage melanoma, NSCLC, RCC, castration-resistant prostate cancer, or colorectal cancer43, the overall response rate (ORR) of patients with PD-L1-positive tumours (defined as ≥5% tumour cells expressing PD-L1) was higher overall than that of those with PD-L1-negative tumours43 (TABLE 2). Subsequent trials of ICB, however, yielded contradictory results regarding the role of PD-L1 expression as a marker of response to treatment and clinical outcome because of the different criteria used to define PD-L1 positivity in order to determine the study cohorts26,27,29,35–37,44–46 (TABLE 2). Of note, although the results support a role for PD-L1 expression as a predictive biomarker, some patients with PD-L1-negative melanoma can also derive durable clinical benefit from PD-1 blockade; thus, PD-L1 expression alone is not used for patient selection in this setting45.
Table 2.
PD-L1 expression as a biomarker for response and outcome in trials of anti-PD-1 agents
| Study and phase | Tumour type and agents tested | Tissue specimen used and requirement for enrolment | Cell type with IHC-stained surface | Cut-off to define PD-L1 positivity | Percentage of PD-L1 positivity | Association with response and outcome | |
|---|---|---|---|---|---|---|---|
| Topalian et al. (2012)43 Phase I |
|
|
Tumour cells | ≥5% | 59.5% (25/42) | PD-L1-positive versus PD-L1 negative:
|
|
| Robert et al. (2015)26 Phase III |
|
|
Tumour cells | ≥5% | 35.4% (148/418) | Nivolumab versus dacarbazine:
|
|
| Robert et al. (2015)27 Phase III |
|
|
Tumour cells | ≥1% | 80.5% (671/834) | Pembrolizumab (every 2 weeks or every 3 weeks) versus ipilimumab:
|
|
| Daud et al. (2016)45 Phase Ib |
|
|
Tumour and tumour-associated immune cells | ≥1%§ | 76% (344/451) | PD-L1-positive versus PD-L1-negative:
|
|
| Brahmer et al. (2015)44 Phase III |
|
|
Tumour cells | ≥1%, ≥5%, or ≥10% |
|
Nivolumab versus docetaxel:
|
|
| Gettinger et al. (2015)29 Phase I |
|
|
Tumour cells | ≥5% | 49% (33/68) | PD-L1 positive versus PD-L1-negative:
|
|
| Rizvi et al. (2015)35 Phase II |
|
|
Tumour cells | ≥5% | 33% (25/76) | PD-L1-positive versus PD-L1-negative PD-L1:
|
|
| Borghaei et al.(2015)37 Phase III |
|
|
Tumour cells | ≥1%, ≥5%, or ≥10% |
|
Nivolumab versus docetaxel:
|
|
| Garon et al. (2015)36 Phase I |
|
|
Tumour cells | ≥50%║ |
|
PD-L1-positive versus PD-L1-negative (1–49% and <1%):
|
|
| Reck et al. (2016)46 Phase III |
|
|
Tumour cells | ≥50% |
|
Pembrolizumab versus chemotherapy in PD-L1 positive patients:
|
|
18 patients with melanoma, ten patients with NSCLC, seven patients with colorectal cancer, five patients with renal-cell cancer, and two patients with prostate cancer.
Percentage of patients with PD-L1 expression tested in total cohort.
The study also used MEL score (MEL)45: 0 (no membrane staining), 1 (>0%–<1%), 2 (≥1–<10%), 3 (≥10%–<33%), 4 (≥33%–<66%), 5 (≥66%).
PD-L1 expression status evaluated using ‘proportion score’ (REF. 36): <1%, 1–49%, and ≥50%; initially PD-L1 positivity was defined as ≥1%, but ≥50% was used to compare outcomes.
PD-L1 positivity (membrane staining ≥50%) was part of trial eligibility criteria. HR, hazard ratio; m, median; NA, not assessed; NSCLC, non-small-cell lung cancer; ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
The predictive and prognostic values of PD-L1 expression in studies in patients with NSCLC have also been debated. In the exploratory analyses of some trials29,44, no association was reported between PD-L1 expression and ORR, PFS, or overall survival; however, positive correlations have been reported in other trials35,37 (TABLE 2). One of the limitations of these studies is the assessment of PD-L1 expression in archival tumour tissue, which might not reflect the PD-L1 expression status at the time of therapy initiation29. To this end, a phase I study of pembrolizumab in patients with advanced-stage NSCLC required contemporaneous tumour biopsy samples for the biomarker evaluation; a higher response rate and longer PFS and overall survival were observed in patients with PD-L1-positive tumours (defined as ≥50% of neoplastic cells showing membranous staining of PD-L1) compared with those with PD-L1-negative tumours, regardless of whether they had previously received treatment36. This observation was subsequently confirmed in a phase III study in treatment-naive patients with NSCLC receiving first-line pembrolizumab who were required to have ≥50% PD-L1 expression on tumour tissue samples46 (TABLE 2). On the basis of the positive results of these studies, the FDA approved pembrolizumab for previously untreated metastatic NSCLC with PD-L1 expression ≥50%47, expanding the application of PD-1 inhibitors for the first-line treatment of lung cancer. This approval also highlights the role of PD-L1 expression as a biomarker that enables an effective selection of the patients with advanced-stage NSCLC who are most likely to benefit from first-line ICB; the role of PD-L1 expression as a predictive biomarker in patients with tumours other than NSCLC remains to be established.
Of note, the use of PD-L1 expression on IHC as a biomarker is associated with some issues. Most studies evaluated PD-L1 expression as the percentage of tumour cells showing cell-surface and/or membranous PD-L1 staining in a section containing at least 100 evaluable tumour cells; however, different assays and antibodies are currently used to detect PD-L1 staining, without standardization48,49. Moreover, variable cut-off values and different scoring methods have been used to define PD-L1 positivity on IHC (TABLE 2). The lack of standardized methods makes it difficult to collectively analyse the results from individual studies in order to reach robust overall consensus. Additionally, most anti-PD-L1 antibodies currently in use are directed to the extracellular domain of PD-L1 and their use results in a mixture of both cytoplasmic and membranous staining of tumour tissue on IHC50. Cytoplasmic staining obscures the interpretation of a positive result on the tumour-cell membrane, thus affecting the accuracy of the analysis50.
A study using tissue samples from patients with NSCLC reported poor concordance in the assessment of PD-L1 protein expression with two different PD-L1 antibodies on conventional IHC (Cohen κ range: 0.124–0.340)51. Quantitative immunofluorescence showed an interassay discordance rate of 26.6%, demonstrating the heterogeneity of PD-L1 expression in serial sections of whole tissue from the same patient51. In the Blueprint PD-L1 IHC Assay Comparison Project52, an industrial– academic collaborative partnership, four different PD-L1 IHC assays that have been used in clinical trials were evaluated. Using 39 NSCLC tumour samples, the study showed that the percentage of PD-L1-stained tumour cells was comparable between three of the four assays, but consistently lower with the fourth assay method. For all assays, greater variability in PD-L1 staining was found in immune cells than in tumour cells52. The study indicated that interchanging assay methods and cutoff values for PD-L1 positivity can lead to inconsistent classifications of PD-L1 status in some patients52, highlighting the limitations of the current IHC approach to assessing PD-L1 expression in terms of reproducibility, as well as in sampling variability49.
Tumour heterogeneity and sampling variability are inherent limitations of assessment approaches using tissue samples. Tumour heterogeneity exists both within the same tumour lesions and among different lesions within the same patient. Because of the invasiveness of tissue sampling, only one of multiple lesions is usually selected for sampling based on the accessibility, and only small samples might be obtained when needle biopsy is performed. Thus, a tissue sample might not necessarily reflect the major immune phenotype of the tumour or the patient. Additionally, PD-L1 expression levels can change over time and thus, specific requirements (such as the time of treatment with ICB or other agents) need to be clearly defined49,53. Understanding the utility of changes in PD-L1 expression in response to ICB during the course of therapy is of great interest; however, serial assessments of PD-L1 expression can be difficult because repeated tissue sampling is required. A substantial number of patients with PD-L1 positivity (at least 40–50%) do not achieve objective response to anti-PD-1–PD-L1 therapies; in addition, approximately 15% of patients negative for PD-L1 expression experience objective responses, in contrast to the initial report53. While some of the limitations discussed might, at least partially, explain the discrepancies observed in the assessment of PD-L1, further efforts are needed to refine the use of PD-L1 expression status as a robust biomarker for immunotherapy. In addition, genetic and non-genetic factors enabling response prediction should be incorporated in the process of patient selection for ICB in order to establish a paradigm of precision immunotherapy53.
Tumour-infiltrating lymphocytes
A non- negligible amount of non-neoplastic cells are found within tumours; these cells include immune cells, which are probably of biological significance54. In particular, tumour-infiltrating lymphocytes (TILs) are increasingly recognized as important players in the immune response against cancer. A number of studies have demonstrated that increased TIL numbers are associated with better outcomes and longer survival for patients with a variety of malignancies55. For example, in 46 patients with melanoma treated with pembrolizumab, higher numbers of cells expressing CD8, PD-1, or PD-L1 at the invasive tumour margin and inside tumours were detected in pretreatment samples from responding patients, compared with patients who did not respond56. In addition, a greater increase in CD8+ cells in serial tumour samples during therapy correlated with a greater tumour-size decrease on imaging (Spearman’s correlation coefficient = −0.75; P = 0.0002)56. Herbst et al.57 also highlighted the importance of PD-L1 expression assessment, not only on tumour cells but also on TILs. In their study, IHC assessment of pretreatment tissue samples from patients with advanced-stage cancer treated with atezolizumab revealed PD-L1 positivity on both tumour cells and TILs, with a higher percentage of PD-L1-positive TILs than PD-L1-positive tumour cells. PD-L1-positive TILs included myeloid cells (macrophages and dendritic cells) and T cells, whereas B cells were negative for PD-L1 expression57. The study also demonstrated that the likelihood of response to atezolizumab was significantly associated with higher levels of PD-L1 expression on TILs (ORR according to the percentage of PD-L1 positive cells per area: 46% (in 15/33 patients), 17% (4/23), 21% (7/34), 13% (8/60), and 8% (2/25), for ≥10%, ≥5%–<10%, 1%–<5%, <1%, and unknown, respectively; P = 0.007), but not with PD-L1 expression on tumour cells (P = 0.079)57. Serial tissue sampling during therapy showed that lesions regressing after treatment had a dense immune infiltrate and extensive tumour-cell necrosis. Nevertheless, in most patients with disease progression, no PD-L1 upregulation in tumour cells or TILs was detected, with three patterns observed: little or no TIL infiltration (‘immunological ignorance’); the presence of intratumour immune infiltrates with minimal to no expression of PD-L1 (‘non-functional immune response’); or the presence of an immune infiltrate solely around the outer edge of the tumour cell mass (‘excluded infiltrate’)57.
A study using multiparameter flow cytometry assessment of freshly isolated pretreatment tumour samples from patients with metastatic melanoma showed that an increase in the fraction of tumour-infiltrating CD8+ T cells with high expression of both PD-1 and CTLA-4 (PD-1hi/CTLA-4hi) strongly correlated with response to pembrolizumab or nivolumab58. In both the discovery and validation cohorts, patients with >20% PD-1hi/CTLA-4hi cytotoxic T lymphocytes (CTLs) had higher response rates and longer PFS durations than those with ≤20% PD-1hi/CTLA-4hi CTLs (ORR: 85.7% versus 0%, PFS: 31.6 months versus 9.6 months in the discovery cohort; ORR: 78.6% versus 0%, PFS: 15.9 months versus 9.9 months in the validation cohort). Functional analyses of PD-1hi/CTLA-4hi CTLs demonstrated a partially exhausted T-cell phenotype, characterized by the ability to produce IFNγ and the inability to produce TNFα and IL-2 (REF.58). The mechanism of action of ICB involves the interaction between tumour cells and their TME, and the cellular immune reaction against tumours; therefore, increased awareness and further studies of the role of TILs are needed for comprehensive biomarker development to predict response to immunotherapy.
The ‘immunoscore’ as a biomarker for immunotherapy
Immune cells are scattered in both the core and in the invasive margin of tumours54. The analysis of the presence of TILs in both tumour regions improved the accuracy of prediction of survival for the different groups of patients with early-stage colorectal cancer, compared with that of single-region analysis59, providing a rationale for conducting a systematic evaluation of immune cell infiltration in both the tumour core and the invasive margin. This approach, termed immunoscore, has been proposed as a method to characterize the immune contexture of the TME, and has been studied as a tool for tumour classification, prognostication, and prediction of response to therapy54,59,60.
The immunoscore reflects the density of two lymphocyte populations (cytotoxic (CD8+) and memory (CD45RO+) T cells) in the core and in the invasive margin regions of tumours54,61, resulting in a four-point scale score ranging from immunoscore 0 (I0; low densities of both cell types in both regions), to immunoscore 4 (I4; high densities of both cell types in both regions)54,61. The prognostic and predictive values of the immunoscore have been addressed mostly in the setting of colorectal cancer60,62. Mlecnik et al.63 assessed the immunoscore of 599 specimens of stage I–IV colorectal tumours, and studied its relationship with the degree of extension of the primary tumour (according to the American Joint Committee on Cancer–International Union Against Cancer (AJCC–UICC) TNM classification system) and in relation to the recurrence rate63. The study demonstrated that the immunoscore was significantly associated with differences in disease-free, disease-specific, and overall survival, with a better prognosis for those patients classified as having a coordinated T-cell response. Multivariate analyses also showed the superiority of the immunoscore over the AJCC–UICC TNM classification system in predicting recurrence as well as survival63. Pages et al.62 demonstrated that the immunoscore is an independent predictor of tumour recurrence and survival in patients with early-stage colorectal cancer, confirming that the extent of the immune reaction at the tumour site is directly correlated with better outcomes62. On the basis of these results62,63, the value of the immunoscore is currently being validated internationally in routine clinical settings.
Given the association between the presence of CD8+ T cells in the invasive margin regions and the expression of PD-1 and PD-L1 in the tumour and TME, the value of the immunoscore as a possible marker of response to ICB therapy is of great interest54,56,64. The application of the immunoscore in melanoma is currently under investigation; however, in this setting, the definition of immunoscore is a challenge because of the complex intratumoural immune reaction60. To date, the evaluation of the immunoscore in melanoma has been performed on tissue samples from metastatic lymph nodes, which are the most accessible and available malignant tissues in patients with melanoma. The immunoscore might facilitate treatment decisions, especially for patients with stage III disease who can benefit from adjuvant systemic therapy after tumour resection and lymph-node dissection60. Concerns exist regarding the validity of the approach that focuses on lymph nodes alone60, because lymph nodes are enriched in CD3+ and CD20+ lymphocytes and, in addition, lymph-node metastases can have different immune infiltration patterns compared with other metastatic lesions60. Further studies are ongoing to evaluate different immune populations and their relationships with response and benefit to ICB in patients with melanoma60,64.
NSCLC is another tumour type in which the development of an effective biomarker is needed. The immunoscore remains to be prospectively evaluated in patients with NSCLC; however, in one study, tumour tissue samples from 536 patients with stage I–IIIA NSCLC were evaluated, revealing that a high density of PD-L1-positive immune cells in the stromal compartment, and of PD-1-positive intraepithelial TILs was associated with favourable disease-specific survival65. Conversely, low PD-L1-positive immune-cell density in the stromal compartment and low PD-1-positive intraepithelial TILs were associated with poor disease-specific survival across all disease stages (HR = 1.81; 95% CI 1.37–2.40; P <0.001). The contribution of both factors was shown to be significant in multivariate analyses, indicating that they are independent prognostic factors65. The utility of the immunoscore to predict tumour response to therapy of patients with advanced-stage NSCLC remains to be further investigated.
In addition to the scientific knowledge derived from ongoing research about immunoscore, technical barriers need to be overcome in order to translate this promising approach into routine clinical pathology and oncology practices. Robust software solutions are needed to enable automated detection of TILs in tumours, faster processing and turnaround, and accurate and efficient interpretation of a large volume of quantitative data on immune-cell densities54,60. As the concept of immunoscore becomes widely applied in different clinical contexts, the importance of developing and maintaining internationally agreed consensus of the definitions and strategies related to the immunoscore approach will increase.
IFNγ
IFNγ is a cytokine that plays a key part in immune regulation and anticancer immunity. IFNγ is mainly produced by natural killer cells and natural killer T cells in innate immune response, and by activated T cells in the setting of antigen-specific immunity56,66. The expression of PD-L1 on most tumour cells is induced by interferons, predominantly IFNγ; IFNγ signalling is necessary for an adaptive response to endogenous antitumour immunity5,55,66,67. In a phase I/II study of the PD-L1 inhibitor durvalumab68, tumour samples were profiled for the expression of 100 preselected genes involved in immune activation68. The highest correlation with response was found for baseline IFNγ mRNA expression levels, with a response rate of 33% (14/43) in patients positive for IFNγ and 8% (6/79) in those negative for IFNγ expression. When combined with PD-L1 positivity, the highest response rate (46%, 10/22) was noted in those patients positive for both IFNγ and PD-L1 expression68. In a phase I study of the PD-L1 inhibitor atezolizumab, pretreatment tumour samples from patients with melanoma who responded to therapy had elevated expression of IFNγ and IFNγ-inducible genes (such as IDO1 and CXCL9); however, this association was weak in patients with NSCLC or RCC57. Given the important role of IFNγ in the adaptive immune response, further studies are needed to determine whether the signalling status of this interferon is a reliable biomarker for response to ICB across different cancer types.
Additionally, in a study of mutations associated with acquired resistance to pembrolizumab therapy69, loss-of-function mutations in the genes encoding the IFN-receptor-associated tyrosine kinases JAK1 and JAK2 were identified as relevant. Truncating mutations in JAK1 and JAK2 resulted in a lack of response to IFNγ in cell lines derived from tumours that were progressing69. These observations indicate an additional role of IFNγ in the development of acquired resistance to ICB, which remains to be further studied.
Mutational burden, neoantigens and microsatellite instability
Marked efficacy of ICB has been noted in patients with melanoma or squamous NSCLC, tumours that are known to have higher numbers of somatic mutations than other tumours70. These mutations in tumour cells can result in the generation of novel antigens, termed neoantigens, which can be recognized as non-self-epitopes by the immune system, thereby enhancing T-cell reactivity against the tumour, thus facilitating the efficacy of ICB53,66,71. Additionally, a deficiency in DNA-repair mechanisms can lead to a high mutational burden in tumours, which can lead to increased response to ICB53,66,72.
In 2014, Snyder and collaborators had demonstrated the association between tumour mutational burden and the degree of clinical benefit in patients with melanoma treated with the CTLA-4 inhibitors ipilimumab or tremelimumab, and they identified a neoantigen landscape specific to tumours with a strong response to CTLA-4 inhibitors73. In this study, long-term clinical benefit was defined by radiographic evidence of freedom from disease, or evidence of stable or decreased volume of disease lasting >6 months. The mutational load (determined by the number of nonsynonymous mutations per exome) was significantly higher in patients with long-term clinical benefit in both the discovery set and validation set (P = 0.01 and P = 0.009, respectively). In a study by Rizvi et al.74, a higher burden of somatic nonsynonymous mutations was associated with clinical efficacy in response to pembrolizumab in patients with NSCLC. In the discovery cohort (n = 16), the number of nonsynonymous mutations was higher in patients who derived durable clinical benefit (defined as partial or stable response lasting >6 months), compared with those without durable benefit: median of 302 versus 148 mutations; P = 0.02 (REF.74). Both ORR and PFS were higher in patients with an ‘elevated’ nonsynonymous mutational burden — defined as above 209, the median mutational burden of the cohort — compared with those with a ‘non-elevated’ burden (ORR: 63% versus 0%, P = 0.03; median PFS: 14.5 months versus 3.7 months, P = 0.01)74. In a validation cohort of 18 samples from patients with NSCLC treated with pembrolizumab, a higher rate of durable clinical benefit and longer PFS durations were noted in patients with a nonsynonymous mutational burden above 200 (median mutational burden of this cohort) compared with other patients (durable clinical benefit rate: 83% versus 22%, P = 0.04; median PFS: not reached versus 3.4 months, P = 0.006)74.
The relationship between the ability of tumour cells to correct intrinsic DNA errors and a response to immunotherapies is another important concept that has been under active investigation in the past few years. Intrinsic errors in DNA replication occur at a rate of 1/1 × 104–105, and are corrected by the mismatch- repair (MMR) machinery53. Mutations in MMR genes in tumour cells therefore result in a large number of mutations that affect, among others, DNA microsatellite motifs53. Thus, microsatellite instability (MSI) is inversely correlated with the ability to repair DNA errors, and has been demonstrated to have a strong association with response to ICB53,66. The role of MMR deficiency (MSI) as a marker for clinical benefit from ICB has been studied in a phase II trial in patients with progressive metastatic carcinoma treated with pembrolizumab72. Both ORR and PFS were higher in patients with colorectal cancer with MMR deficiency than in those without (ORR: 40% (4/10) versus 0% (0/18); 20-week PFS: 78% (7/9) versus 11% (2/18))72. Similar ORR and PFS rates were noted in patients with MMR-deficient noncolorectal cancer (ORR: 71% (5/7); 20-week PFS: 67% (4/6))72. Whole-exome sequencing revealed a significantly higher tumour mutational burden in patients with MMR-deficient tumours compared with those with MMR-proficient tumours (mean number of somatic mutations per tumour: 1,782 versus 73; P = 0.007); a higher somatic mutational burden was associated with longer PFS (P = 0.02)72.
These studies present promising evidence for the use of tumour mutational burden as a biomarker; however, several limitations should be noted, including the small size of patient cohorts, the inclusion of patients with variable treatment history, and the use of tumour samples obtained at various time points73. Important exceptions were also noted in these studies, including patients with high mutational burden who did not respond to ICB, and those with very low mutational burden with a good response to these agents53. Additionally, logistical barriers should be solved to widely apply tumour mutational burden as a biomarker using advanced genomic analysis techniques (such as whole-exome sequencing) in the clinical setting.
Moreover, results published in 2017 (REF.75), indicate that the importance of neoantigen load in predicting response to ICB is not as straightforward as had been initially anticipated. In this study75, previously published cancer exome data from patients with melanoma treated with the CTLA-4 inhibitors ipilimumab and tremelimumab were re-analysed and combined with additional RNA-sequencing data. Both the total neoantigen and expressed neoantigen load were associated with a response to CTLA-4 blockade; however, neither was more predictive than somatic mutational burden75. In addition, the association between mutational burden and response to treatment was found only when samples obtained before treatment were analysed75, whereas no association between mutation burden and treatment response was noted in patients whose tumour samples had been collected after initiating anti-CTLA-4 therapy. These findings reinforce the importance of multiple factors — ranging from IFNγ signalling to systemic factors — to the likelihood of a response to ICB treatment; these factors need to be analysed in an integrated way to improve the understanding of the immune-related response activated by those therapies and therapeutic resistance.
Serum markers
Although the assessment of the tumour and TME profiles is essential to identify robust biomarkers, clinical markers and morphological phenotypes of patients and tumours can also be useful to stratify patients into subpopulations. Serum markers, such as lactate dehydrogenase (LDH) and immune-cell counts, on routine blood analyses can be useful predictors of response to ICB. Indeed, these markers have shown potential utility for the prediction of response to ipilimumab. In patients treated with ipilimumab, high absolute-lymphocyte counts, low absolute-monocyte counts, and high relative-lymphocyte counts at baseline, and increasing absolute-lymphocyte counts during treatment were indicative of a favourable prognosis76–78. High relative eosinophil counts at baseline indicated favourable overall survival76, and an early increase of the abundance of this cell type in the peripheral blood taken 18–19 days after the first ipilimumab infusion was associated with improved clinical responses to CTLA-4 blockade79. An increase of the absolute lymphocyte counts at 7 weeks of therapy after administering two doses of ipilimumab given 3 weeks apart was also shown to be associated with clinical benefit2,80. A study with results published in 2017 reported that, in patients with melanoma treated with ipilimumab, the baseline levels of CD45RO+/CD8+ T cells were significantly lower in patients without a response to therapy compared with those who had a response (P <0.01)81. In 80% (12/15) of patients with no response, the baseline levels of CD45RO+/CD8+ T cells were ≤25%, whereas all (6/6) patients with a response had ≥30% CD45RO+/CD8+ T cells81. In addition, elevated pretreatment levels of LDH were shown to be negatively associated with overall survival in patients with melanoma treated with ipilimumab or pembrolizumab76–78,82,83.
The predictive value of combined assessment of these serum markers has been evaluated in 616 patients with melanoma treated with pembrolizumab83; high relative eosinophil counts and high relative lymphocyte counts, low LDH levels, and the absence of metastases in organs other than soft-tissue or lung were identified as independent baseline characteristics associated with favourable overall survival. The presence of all four favourable factors was associated with an excellent prognosis, with an ORR of 58.3% and a 1-year overall survival of 83.9%83. These serum markers have the advantage of being readily assessable in tests used routinely in clinical practice, and are also suitable for serial sampling over the course of treatment.
Radiographic markers
Radiographic assessments during therapy provide quantitative data on tumour dynamics that have been found to be associated with the response to and outcomes of precision cancer therapies84–86, and might be useful to classify patients into subgroups according to different immune-related response patterns, with different prognostic implications25. Importantly, noninvasive functional imaging strategies need to be developed in the future to visualize and quantify target cells and molecules in vivo. For example, immuno-PET with the 89Zr-desferrioxamine-labelled anti-CD8 cys-diabody (89Zr-malDFO-169 cDb) enabled the noninvasive tracking of cytotoxic T cells in murine models of cancer immunotherapy87. Another study reported the use of small non-antibody therapeutics (the PD-1 ectodomain) as a high-affinity (110 pM) competitive antagonist of PD-L1; the ectodomain was radiolabelled and used as a PET-imaging tracer to distinguish PD-L1-positive tumours from PD-L1-negative tumours in mice88. Further development and application of these in vivo functional imaging approaches will enable investigators to address the issue of tumour heterogeneity and perform serial biomarker assessments over time, eventually overcoming the shortcomings of the current biomarkers, mostly derived from tissue samples.
Future directions: multidisciplinary approach to establish robust biomarkers
A careful design of biomarker exploratory studies, based on a consensus from multiple parties, is essential to develop effective approaches to identify and validate robust markers for response and outcome of ICB. As evidenced by the exploratory analyses of PD-L1 expression as a possible marker, unified approaches and clear-cut guidelines are needed for the collection and processing of tissue specimens; as are detection methods (IHC or other bio chemical approaches) and evaluation strategies; and cut-off values to define positive results. A solution to the current controversies will be best found when such consensus and standardization are widely applied in prospective studies.
In order to achieve this goal and establish robust biomarkers for response and outcome of cancer immunotherapy, the US National Cancer Institute (NCI) has introduced important initiatives that encourage multidisciplinary approaches and public–private partnerships: the Partnership to Accelerate Cancer Therapies (PACT)89 will bring multiple public and private stakeholders together to accelerate cancer research as part of the Cancer Moonshot Initiatives, focusing on the identification and validation of biomarkers of response and resistance to cancer therapies, with a special emphasis on immunotherapies. Related opportunities include the creation of Cancer Immune Monitoring and Analysis Centers (CIMACs)90 and Cancer Immunologic Data Commons (CIDC)91, which together will constitute the CIMAC–CIDC Network for correlative studies in clinical trials involving immunotherapy to identify immunotherapy biomarkers for optimizing the therapeutic strategies for patients. This network is expected to provide the infrastructure to perform correlative studies in clinical trials of cancer immunotherapy sponsored by the NCI90,91.
As advocated in these initiatives, data sharing related to the collection and analysis of promising biomarkers among investigators will contribute to establishing large-scale databases, enabling validation of individual results, and subsequently translating biomarker discoveries into the clinical setting. Clinical trials going forward can then be designed in a biomarker-driven manner, and patients can be selected on the basis of biomarker findings, as has been successfully done for PD-L1 expression in the setting of first-line therapy for advanced-stage NSCLC. The establishment of a robust biomarker for patient selection, and the monitoring and management of decisions in ICB require a cohesive approach based on the active communication and collaboration among investigators from multiple disciplines, health-care providers, pharmaceutical industry and patients.
Conclusions
The advent of ICB has brought about a paradigm shift in the landscape of advanced-stage cancer treatment. ICB using these agents can result in unconventional response patterns and, thus, special strategies are required for the accurate evaluation of tumour responses and clinical outcomes. The development of robust biomarkers to monitor response to immunotherapy is a key next step for the further progress of this field towards precision immuno-oncology approaches.
Key points.
A subset of patients receiving immune-checkpoint inhibitor therapy develop unconventional response patterns (termed ‘pseudoprogression’), in which tumour burden decreases after an initial increase, or during or after the appearance of new lesions
The evaluation of pseudoprogression provides new challenges in treatment monitoring and therapeutic decision-making because it cannot be evaluated with the existing response-evaluation criteria
The establishment of a standardized strategy to evaluate immune-related responses in patients receiving immune-checkpoint inhibitors is extremely important
In addition, the development of robust biomarkers to assist prediction of response and clinical benefits of immune-checkpoint inhibitor therapy is essential to further advance the field as precision immuno-oncology
The therapeutic activity of immune-checkpoint inhibitors is the result of a complex interplay between multiple factors in the tumour, tumour microenvironment, and immune system, requiring a collaborative approach to translate the emerging knowledge into the clinical context
Acknowledgments
The work of M.N. has been supported by grant 1K23CA157631 from the National Cancer Institute.
M.N. is a consultant for Bristol-Myers Squibb, Toshiba Medical Systems and WorldCare Clinical, and has received research grants from Canon and the Merck Investigator Studies Program, and honoraria from Bayer. H.H. is a consultant for Toshiba Medical Systems, and has received research support from Canon, Konica-Minolta and Toshiba Medical Systems. F.S.H. has served as a non-paid consultant for Bristol-Myers Squibb, has received clinical trial support from Bristol-Myers Squibb, is an adviser and receives clinical trial support from Genentech and Merck, is a consultant for Amgen, EMD Serono and Novartis, and has a patent relating to tumour antigens (issued), and a patent related to the institution of major histocompatibility complex (MHC) class I polypeptide-related sequence A (MICA) as a target (licensed).
Footnotes
Author contributions
All authors researched data for the article, contributed to discussing the content of the article, and wrote, reviewed, and edited the manuscript before submission.
Competing interests statement
N.H.R. declares no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 2.Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300–5309. doi: 10.1158/1078-0432.CCR-13-0143. [DOI] [PubMed] [Google Scholar]
- 3.Nishino M, et al. Personalized tumor response assessment in the era of molecular medicine: cancer-specific and therapy-specific response criteria to complement pitfalls of RECIST. AJR Am J Roentgenol. 2012;198:737–745. doi: 10.2214/AJR.11.7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishino M, Tirumani SH, Ramaiya NH, Hodi FS. Cancer immunotherapy and immune-related response assessment: the role of radiologists in the new arena of cancer treatment. Eur J Radiol. 2015;84:1259–1268. doi: 10.1016/j.ejrad.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishino M, Gargano M, Suda M, Ramaiya NH, Hodi FS. Optimizing immune-related tumor response assessment: does reducing the number of lesions impact response assessment in melanoma patients treated with ipilimumab? J Immunother Cancer. 2014;2:17. doi: 10.1186/2051-1426-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishino M, et al. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin Cancer Res. 2013;19:3936–3943. doi: 10.1158/1078-0432.CCR-13-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishino M, et al. Immune-related response assessment during PD-1 inhibitor therapy in advanced non-small-cell lung cancer patients. J Immunother Cancer. 2016;4:84. doi: 10.1186/s40425-016-0193-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolchok JD, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 11.Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer. 1981;47:207–214. doi: 10.1002/1097-0142(19810101)47:1<207::aid-cncr2820470134>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 12.Therasse P, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 13.Eisenhauer EA, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Lynch TJ, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 15.Hodi FS, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol. 2016;34:1510–1517. doi: 10.1200/JCO.2015.64.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishino M, Jagannathan JP, Ramaiya NH, Van den Abbeele AD. Revised RECIST guideline version 1.1: what oncologists want to know and what radiologists need to know. AJR Am J Roentgenol. 2010;195:281–289. doi: 10.2214/AJR.09.4110. [DOI] [PubMed] [Google Scholar]
- 18.Chiou VL, Burotto M. Pseudoprogression and immune-related response in solid tumors. J Clin Oncol. 2015;33:3541–3543. doi: 10.1200/JCO.2015.61.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishino M, Hatabu H, Johnson BE, McLoud TC. State of the art: response assessment in lung cancer in the era of genomic medicine. Radiology. 2014;271:6–27. doi: 10.1148/radiol.14122524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishino M. Immune-related response evaluations during immune-checkpoint inhibitor therapy: establishing a “common language” for the new arena of cancer treatment. J Immunother Cancer. 2016;4:30. doi: 10.1186/s40425-016-0134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erasmus JJ, et al. Interobserver and intraobserver variability in measurement of non-small-cell carcinoma lung lesions: implications for assessment of tumor response. J Clin Oncol. 2003;21:2574–2582. doi: 10.1200/JCO.2003.01.144. [DOI] [PubMed] [Google Scholar]
- 22.Nishino M, et al. CT tumor volume measurement in advanced non-small-cell lung cancer: performance characteristics of an emerging clinical tool. Acad Radiol. 2011;18:54–62. doi: 10.1016/j.acra.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oxnard GR, et al. Variability of lung tumor measurements on repeat computed tomography scans taken within 15 minutes. J Clin Oncol. 2011;29:3114–3119. doi: 10.1200/JCO.2010.33.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao B, et al. Evaluating variability in tumor measurements from same-day repeat CT scans of patients with non-small cell lung cancer. Radiology. 2009;252:263–272. doi: 10.1148/radiol.2522081593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishino M, et al. Immune-related tumor response dynamics in melanoma patients treated with pembrolizumab: identifying markers for clinical outcome and treatment decisions. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-17-0114. http://dx.doi.org/10.1158/1078-0432.CCR-17-0114. [DOI] [PMC free article] [PubMed]
- 26.Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 27.Robert C, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 28.Okada H, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 2015;16:e534–e542. doi: 10.1016/S1470-2045(15)00088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gettinger SN, et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;33:2004–2012. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohnsack O, Hoos A, Ludajic K. Adaptation of the immune-related response criteria: irRECIST [abstract. Ann Oncol. 2014;25:iv369. [abstract 1070P] [Google Scholar]
- 31.Seymour L, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–e152. doi: 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.[No authors listed.]; iRECIST. RECIST. 2017 http://www.eortc.org/recist/irecist/
- 33.Herbst RS, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 34.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rizvi NA, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16:257–265. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 37.Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg JE, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDermott DF, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase Ia study. J Clin Oncol. 2016;34:833–842. doi: 10.1200/JCO.2015.63.7421. [DOI] [PubMed] [Google Scholar]
- 40.Motzer RJ, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol. 2015;33:1430–1437. doi: 10.1200/JCO.2014.59.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Motzer RJ, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDermott DF, et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol. 2015;33:2013–2020. doi: 10.1200/JCO.2014.58.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brahmer J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daud AI, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. 2016;34:4102–4109. doi: 10.1200/JCO.2016.67.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reck M, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 47.U.S. Food and Drug Administration. Pembrolizumab (KEYTRUDA) checkpoint inhibitor. FDA; 2016. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm526430.htm. [Google Scholar]
- 48.Hansen AR, Siu LL. PD-L1 testing in cancer: challenges in companion diagnostic development. JAMA Oncol. 2016;2:15–16. doi: 10.1001/jamaoncol.2015.4685. [DOI] [PubMed] [Google Scholar]
- 49.Sacher AG, Gandhi L. Biomarkers for the clinical use of PD-1/PD-L1 inhibitors in non-small-cell lung cancer: a review. JAMA Oncol. 2016;2:1217–1222. doi: 10.1001/jamaoncol.2016.0639. [DOI] [PubMed] [Google Scholar]
- 50.Mahoney KM, et al. PD-L1 antibodies to its cytoplasmic domain most clearly delineate cell membranes in immunohistochemical staining of tumor cells. Cancer Immunol Res. 2015;3:1308–1315. doi: 10.1158/2326-6066.CIR-15-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLaughlin J, et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non-small-cell lung cancer. JAMA Oncol. 2016;2:46–54. doi: 10.1001/jamaoncol.2015.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirsch FR, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 IHC Assay Comparison Project. J Thorac Oncol. 2017;12:208–222. doi: 10.1016/j.jtho.2016.11.2228. [DOI] [PubMed] [Google Scholar]
- 53.Mandal R, Chan TA. Personalized oncology meets immunology: the path toward precision immunotherapy. Cancer Discov. 2016;6:703–713. doi: 10.1158/2159-8290.CD-16-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galon J, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol. 2014;232:199–209. doi: 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Remon J, Chaput N, Planchard D. Predictive biomarkers for programmed death-1/programmed death ligand immune checkpoint inhibitors in nonsmall cell lung cancer. Curr Opin Oncol. 2016;28:122–129. doi: 10.1097/CCO.0000000000000263. [DOI] [PubMed] [Google Scholar]
- 56.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daud AI, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. 2016;126:3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 60.Galon J, et al. Immunoscore and Immunoprofiling in cancer: an update from the melanoma and immunotherapy bridge 2015. J Transl Med. 2016;14:273. doi: 10.1186/s12967-016-1029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bindea G, Mlecnik B, Angell HK, Galon J. The immune landscape of human tumors: implications for cancer immunotherapy. Oncoimmunology. 2014;3:e27456. doi: 10.4161/onci.27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pages F, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol. 2009;27:5944–5951. doi: 10.1200/JCO.2008.19.6147. [DOI] [PubMed] [Google Scholar]
- 63.Mlecnik B, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29:610–618. doi: 10.1200/JCO.2010.30.5425. [DOI] [PubMed] [Google Scholar]
- 64.Ascierto PA, et al. The additional facet of immunoscore: immunoprofiling as a possible predictive tool for cancer treatment. J Transl Med. 2013;11:54. doi: 10.1186/1479-5876-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paulsen EE, et al. Assessing PDL-1 and PD-1 in non-small cell lung cancer: a novel immunoscore approach. Clin Lung Cancer. 2017;18:220–233.e8. doi: 10.1016/j.cllc.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 66.Shukuya T, Carbone DP. Predictive markers for the efficacy of anti-PD-1/PD-L1 antibodies in lung cancer. J Thorac Oncol. 2016;11:976–988. doi: 10.1016/j.jtho.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taube JM, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Higgs BW, et al. High tumoral IFNg mRNA, PD-L1 protein, and combined IFNγ mRNA/PD-L1 protein expression associates with response to durvalumab (anti-PD-L1) monotherapy in NSCLC patients [abstract] European Cancer Congress. 2015 [Google Scholar]
- 69.Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 72.Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nathanson T, et al. Somatic mutations and neoepitope homology in melanomas treated with CTLA-4 blockade. Cancer Immunol Res. 2017;5:84–91. doi: 10.1158/2326-6066.CIR-16-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martens A, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22:2908–2918. doi: 10.1158/1078-0432.CCR-15-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Delyon J, et al. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol. 2013;24:1697–1703. doi: 10.1093/annonc/mdt027. [DOI] [PubMed] [Google Scholar]
- 78.Kelderman S, et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol Immunother. 2014;63:449–458. doi: 10.1007/s00262-014-1528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gebhardt C, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res. 2015;21:5453–5459. doi: 10.1158/1078-0432.CCR-15-0676. [DOI] [PubMed] [Google Scholar]
- 80.Ku GY, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer. 2010;116:1767–1775. doi: 10.1002/cncr.24951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tietze JK, et al. The proportion of circulating CD45RO+CD8+ memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur J Cancer. 2017;75:268–279. doi: 10.1016/j.ejca.2016.12.031. [DOI] [PubMed] [Google Scholar]
- 82.Diem S, et al. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br J Cancer. 2016;114:256–261. doi: 10.1038/bjc.2015.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weide B, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res. 2016;22:5487–5496. doi: 10.1158/1078-0432.CCR-16-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nishino M, et al. Tumor volume decrease at 8 weeks is associated with longer survival in EGFR-mutant advanced non-small-cell lung cancer patients treated with EGFR TKI. J Thorac Oncol. 2013;8:1059–1068. doi: 10.1097/JTO.0b013e318294c909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nishino M, et al. Volumetric tumor growth in advanced non-small cell lung cancer patients with EGFR mutations during EGFR-tyrosine kinase inhibitor therapy: developing criteria to continue therapy beyond RECIST progression. Cancer. 2013;119:3761–3768. doi: 10.1002/cncr.28290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nishino M, et al. Volumetric tumor response and progression in EGFR-mutant NSCLC patients treated with erlotinib or gefitinib. Acad Radiol. 2016;23:329–336. doi: 10.1016/j.acra.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tavare R, et al. An effective immuno-PET imaging method to monitor CD8-dependent responses to immunotherapy. Cancer Res. 2016;76:73–82. doi: 10.1158/0008-5472.CAN-15-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maute RL, et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Natl Acad Sci USA. 2015;112:E6506–E6514. doi: 10.1073/pnas.1519623112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.National Institutes of Health. New drug formulary will help expedite use of agents in clinical trials. NIH. 2017 https://www.nih.gov/news-events/news-releases/new-drug-formulary-will-help-expedite-use-agents-clinical-trials.
- 90.Department of Health and Human Services. Cancer Immune Monitoring and Analysis Centers (CIMACs) NIH. 2017 https://grants.nih.gov/grants/guide/rfa-files/RFA-CA-17-005.html.
- 91.Department of Health and Human Services. Cancer Immunologic Data Commons (CIDC) NIH. 2017 https://grants.nih.gov/grants/guide/rfa-files/RFA-CA-17-006.html.