

Abstract
Photolyase, a flavoenzyme containing flavin adenine dinucleotide (FAD) molecule as a catalytic cofactor, repairs UV-induced DNA damage of cyclobutane pyrimidine dimer (CPD) and pyrimidine-pyrimidone (6-4) photoproduct using blue light. The FAD cofactor, conserved in the whole protein superfamily of photolyase/cryptochromes, adopts a unique folded configuration at the active site that plays a critical functional role in DNA repair. Here, we review our comprehensive characterization of the dynamics of flavin cofactor and its repair photocycles by different classes of photolyases on the most fundamental level. Using femtosecond spectroscopy and molecular biology, significant advances have recently been made to map out the entire dynamical evolution and determine actual timescales of all the catalytic processes in photolyases. The repair of CPD reveals seven electron-transfer (ET) reactions among ten elementary steps by a cyclic ET radical mechanism through bifurcating ET pathways, a direct tunneling route mediated by the intervening adenine and a two-step hopping path bridged by the intermediate adenine from the cofactor to damaged DNA, through the conserved folded flavin at the active site. The unified, bifurcated ET mechanism elucidates the molecular origin of various repair quantum yields of different photolyases from three life kingdoms. For 6-4 photoproduct repair, a similar cyclic ET mechanism operates and a new cyclic proton transfer with a conserved histidine residue at the active site of (6-4) photolyases is revealed.
Keywords: flavoprotein photolyase, DNA repair, ultrafast enzyme dynamics, intraprotein electron transfer, electron tunneling, electron hopping, photocycle
Graphical abstract
Introduction
The photolyase/cryptochrome superfamily is a diversified class of flavoproteins containing a non-covalently bound flavin adenine dinucleotide (FAD) molecule as the key cofactor [1, 2]. The superfamily contains eight major clades (Fig. 1A) [3]. Despite their remarkably high similarity of sequence and structure, photolyases (PLs) and cryptochromes (CRYs) perform different biological functions: photolyases repair UV-induced DNA lesions (Fig. 1B, C) [4, 5]; cryptochromes, being present mostly in higher plants and animals, have lost the DNA repair activity in evolution and act as blue-light photoreceptors to regulate various growth and adaptive functions [6–11].
Figure 1. Classes of photolyases and structures of CPD and 6-4 photolyases.
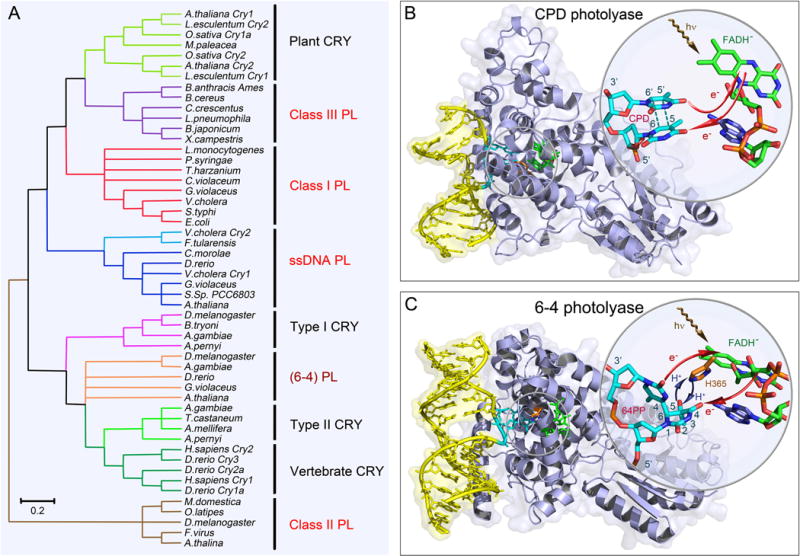
(A) The unrooted phylogenetic tree of the photolyase/cryptochrome protein family and representative members. Major classes in the family and their evolutionary relationships are shown. Photolyases (PLs) are divided into CPD PL and 6-4 PL based on their different DNA substrate. CPD PLs are further divided into class I, II, III and single-stranded DNA (ssDNA) PLs. The ssDNA PLs were previously classified as CRY with Cry-DASH designation. The class II PLs are distantly related to other classes. (B) X-ray complex structure of Anacystis nidulans CPD photolyase (PDB: 1TEZ) bound with double-stranded DNA (yellow) containing a thymine dimer after in situ repair. The thymine dimer is flipped out of DNA and inserts into the active site of protein. The close-up view shows the relative positions of the catalytic cofactor FADH− and the repaired substrate. The flavin cofactor is in the U-shaped configuration and the adenine moiety and the lumiflavin moiety have a short separation distance. The red arrows indicate the electron tunneling pathways in repair. (C) X-ray complex structure of Drosophila melanogaster (6-4) photolyase bound with double-stranded DNA (yellow) containing a (6-4) photoproduct (6-4PP). Like CPD, the 6-4PP is flipped out of DNA and inserts into the active site. The close-up view shows that the conserved histidine residue (orange) in the active site (H365 in D. melanogaster and H364 in Arabidopsis thaliana) is close to the 6-4PP. The blue arrow represents the proton transfer between the two during repair. Similar electron tunneling occurs in 6-4PLs as shown with the red arrows.
The ultraviolet (UV) irradiation in sunlight can cause damages of DNA by inducing formation of a cyclobutane pyrimidine dimer (CPD, ~80%) and a less-frequently pyrimidine-pyrimidone (6-4) photoproduct (6-4PP, ~20%) (Fig. 2A) [2]. In CPD, two adjacent thymine bases in the same DNA strand covalently connect to form a cyclobutane ring; in 6-4PP, a complicated chemical structure is formed, where the oxygen and hydrogen atoms in one base migrate to another base, in addition to the two bases being covalently linked. Both CPD and 6-4PP lead to mutagenesis, cell apoptosis and even to skin cancer [12–14]. Photolyase recognizes these damaged thymine dimers and restores these lesions [2, 15, 16] through direct absorption of blue light by FAD or through energy transfer from the excited antenna chromophore [17–19]. Two different kinds of photolyases, specifically repair CPD and 6-4PP and thus are usually classified as CPD photolyases and 6-4 photolyases, respectively, corresponding to their different substrates. The CPD and 6-4 photolyases share similar primary sequences and folding structures but a photolyase that repairs one photoproduct cannot repair another. Based on sequence analyses, CPD photolyases are highly diversified and are subdivided into three classes (I–III) [20–22] and single-stranded DNA (ssDNA) specific PLs [23, 24] (Fig. 1A). Crystal structures [19, 25–38] of photolyase/cryptochromes, including some enzyme-substrate complex structures of both CPD [28–30] and 6-4 [31] photolyases, have been solved. Throughout the protein family, the FAD cofactor is non-covalently incorporated at the active site in an unconventional U-shaped configuration, and its lumiflavin (Lf) moiety and adenine (Ade) moiety have a short separation distance; while in other flavoproteins, the FAD is usually in a stretched, open configuration. Both CPD and 6-4 photolyases contain a fully reduced flavin adenine dinucleotide (FADH−) molecule as the active cofactor. Fig. 1B and C shows the PL-substrate complex structures of the CPD photolyase from Anacystis nidulans (AnPL) and 6-4 photolyase from Drosophila melanogaster [Dm(6-4)PL], respectively. The close-up view shows that the substrate dimer flips out of the DNA double helix and inserts into the active site of PL. In both PLs, the catalytic flavin cofactor is folded and the Ade moiety is in close proximity of both the flavin isoalloxazine ring and the substrate dimer. The unique folded motif of flavin plays a critical functional role in initial photochemistry.
Figure 2. Chemical structures of the two types of substrates, and the catalytic cofactor in photolyases.
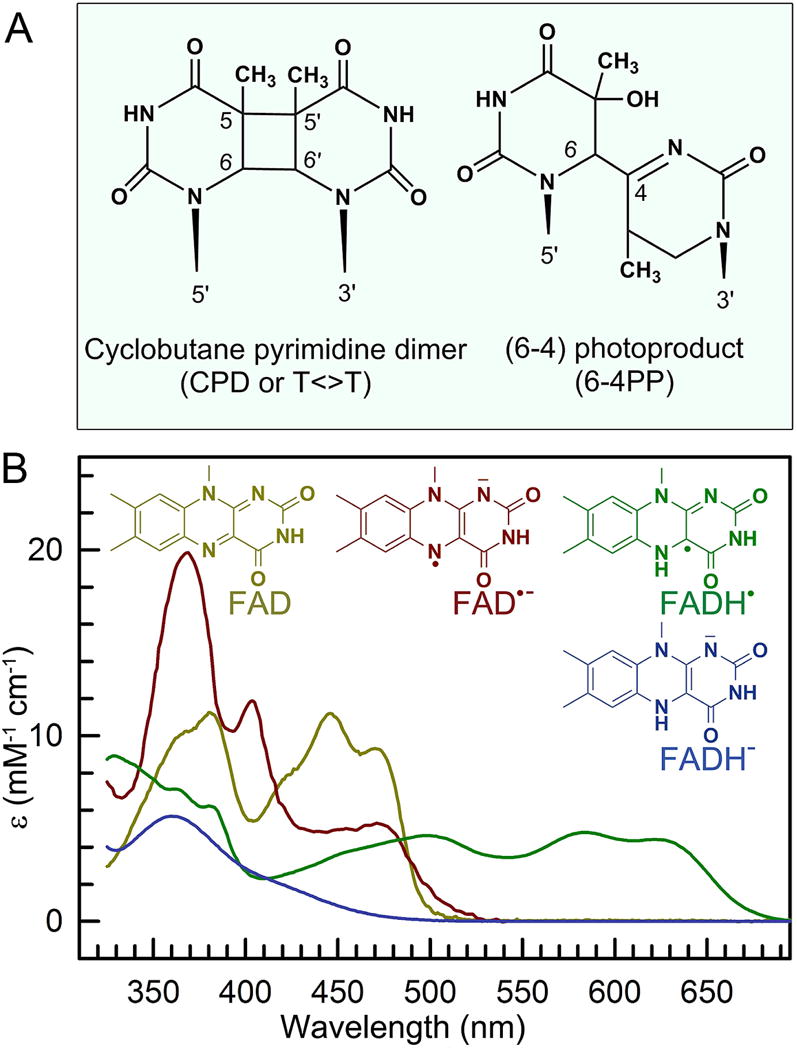
(A) Chemical structures of two UV-induced DNA photolesions, the more common cyclobutene pyrimidine dimer (CPD, ~80%), and the less common pyrimidine-pyrimidone (6-4) photoproduct (6-4PP, ~20%). (b) Absorption spectra of four redox states of flavin adenine dinucleotide (FAD) in photolyases and their corresponding structures.
The flavin cofactor, first discovered in the early 1930s, is essential and ubiquitously present throughout the biological kingdom [39–44]. In nature, the majority of flavin molecules are found in flavoproteins in the form of flavin adenine dinucleotide (FAD), or flavin mononucleotide (FMN) [45]. The flavin molecules have the basic structure of 7,8-dimethyl-10-alkylosialloxazine and are chemically versatile. They can exist in three different redox states: oxidized, one-electron reduced (semiquinone), and two-electron reduced (hydroquinone). Semiquinone and hydroquinone have pKa values of 8.3 and 6.7, respectively [46], and can be present in their neutral or anionic forms under physiological conditions. Due to their unique ability to participate in both one- and two-electron transfer processes, flavins are often involved in intermolecular electron transfer (ET) reactions, and flavoproteins are ubiquitous in biological systems and participate in a broad spectrum of key biological processes that rely on enzyme-mediated oxido-reduction reactions [42–44, 47–50]. Among the five redox forms, two redox pairs, oxidized flavin/anionic semiquinone (FAD/FAD•−) and neutral semiquinone/anionic hydroquinone (FADH•/FADH−), exist in photolyase/cryptochromes. The four states are convertible under physiological conditions through intraprotein ET and proton transfer (PT) [46] and can be spectrally differentiated by their distinct UV-vis absorptions (Fig. 2B). The steady-state spectroscopic properties of these redox flavins have been extensively studied, especially their absorption spectra in different proteins and solution [40, 46, 51–54].
For both CPD and 6-4 PLs, FADH− is the only catalytic state to repair DNA lesions [55, 56]. The mechanism of DNA photorepair has been proposed and examined in the past thirty years [57–72]. The dynamics and mechanism of CPD repair by class I PL from Escherichia coli (EcPL) has been investigated with great detail in recent years; a complete photocycle was resolved and critical cyclic electron-tunneling mechanism was revealed [60–67]. For PLs from other classes, the repair activities have also been reported [24, 68–71] but the overall understanding on their initial photochemistry and repair mechanisms lags behind the microbial class I members. Here we overview our recent key studies of photolyases and focus on the dynamics of the flavin cofactor and DNA repair of different CPD photolyases and the (6-4) photolyases. We have mapped out the intrinsic intramolecular ET cycles within the FAD cofactor of photolyases and subsequently, elucidated the various ET steps in DNA repair and finally unified a novel, universal ET repair mechanism for all different PLs in the superfamily [70]. A timely summary of the current field on DNA repair by photolyases was published recently by many excellent reviews in a special issue of Photochemistry and Photobiology dedicated to Dr. Aziz Sancar who received the Nobel Prize in Chemistry in 2015 with Tomas Lindahl and Paul Modrich for their “Mechanistic studies of DNA repair” [73].
FAD in photolyase: conserved folded structure
Electron shuttling and functional state
Among the four redox states of FAD, the anionic hydroquinone FADH− is the only functional state in vivo to repair the UV-induced DNA lesion, CPD or 64PP, which is an unusual redox state in flavoenzymes. In principle, the substrate thymine dimer can accept one electron to be reduced or donate one electron to be oxidized and then the ionic dimer subsequently splits into two bases [74, 75]. The two anionic states of flavin, anionic semiquinone FAD•− and anionic hydroquinone FADH−, could donate one electron to the substrate, and the two neutral states, oxidized FAD and neutral semiquinone FADH• can accept one electron from the substrate. For the anionic flavin, the critical question is: why does photolyase utilizes FADH−, not FAD•− as the active state? For the two neutral states, the excited flavin can obtain one electron and get reduced on the ultrafast time scale from the neighboring aromatic residues (tryptophan or tyrosine) [76–78]. One challenging question is whether the neutral flavins can accept one electron from the substrate if the active site becomes inert by mutations of aromatic residues.
Using femtosecond (fs)-resolved spectroscopy and site-directed mutagenesis, we investigated dynamics of class I PL from Escherichia coli (EcPL) in four redox states. For the two anionic states, a cyclic ET between the Lf and Ade moieties of the conserved folded FAD cofactor was observed and Ade behaves as an electron acceptor (Fig. 3A). For FAD•−, the forward ET from Lf•− to Ade takes 12 ps and the back ET from Ade− to Lf is even faster at 2 ps. Significantly, for FADH−, the forward ET is slow at 2 ns, followed by ultrafast back ET at 25 ps. The slow forward ET in the FADH− state is a result of unfavorable driving force (+0.04 eV) [79, 80] and thus ensures the fast, direct electron tunneling from the Lf moiety to the substrate in 250 ps [62]. While in FAD•−, the electron quickly shuttles between Lf and Ade within 12 ps, eliminating the further tunneling to the substrate. Thus, as the electron donor, the active state of the cofactor flavin favors FADH−, and only FADH− functions as the active state.
Figure 3. Intrinsic intramolecular electron transfer dynamics of the folded flavin cofactor in photolyases.
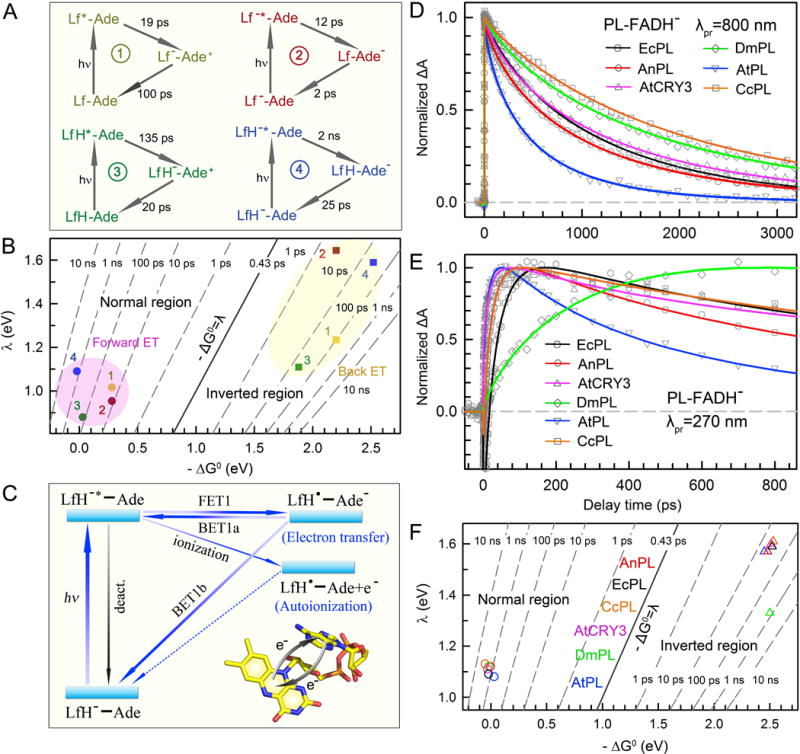
(A) Reaction times and mechanisms of the cyclic electron transfer (ET) in EcPL between the lumiflavin (Lf) and adenine (Ade) moieties of FAD in four redox states. All dot symbols (•) representing radicals are not shown. 1, FAD state; 2, FAD•− state; 3, FADH• state; 4, FADH− state. (B) Two-dimensional (2D) contour plot of the ET times relative to free energy (ΔG0) and reorganization energy (λ) for the ET processes in EcPL of four redox states. All the forward ET reactions (dot) are in the Marcus normal region (−ΔG0≤λ) while all the back ET steps (square) are in the Marcus inverted region (−ΔG0>λ). (C) The universal intramolecular ET cycles of the folded flavin cofactor at FADH− state in photolyases. FET1, forward ET to adenine; BET1a, back ET from anionic adenine to original flavin excited state. BET1b, back ET from anionic adenine to flavin ground state. Upon excitation, the cyclic ET between Lf and Ade occurs intrinsically. The LfH−* energy lies above the ionization level, leading to an autoionization process. The excited and charge-separated states also interconvert because the free energy of the charge separation is close to zero. (D) Absorption transients of the pure excited-state (LfH−*) decay in various photolyases probed at 800 nm. The LfH−* dynamics is a summation of the effective deactivation lifetime (kLT), the forward ET to adenine (kFET1), and the back ET from adenine (kBET1a). (E) Absorption transients of various photolyases probed at 270 nm mainly represent the intermediate flavin radical (LfH•). The slow rise in DmPL transient (green) indicates a significantly slow back ET to the ground state (BET1b). (F) Two-dimensional (2D) contour plot of the ET times relative to free energy (ΔG0) and reorganization energy (λ) for various PLs at the functional state FADH−. The forward ET (circle) is in the Marcus normal region (−ΔG0≤ λ) and the back ET (triangle) is in the Marcus inverted region (−ΔG0> λ).
For the two neutral flavin states, an inert environment was created with mutation of the neighboring tryptophan, and the cyclic ET dynamics between Lf and Ade were also observed on the picosecond time scale with Ade as an electron donor. For FAD and FADH•, ET occurs in 19 and 135 ps for forward ET and 100 and 20 ps for back ET, respectively, preventing further tunneling from the substrate. Using the semi-classical Marcus ET expression [81–83], the reorganization energies (λ) of these four cyclic ET reactions are evaluated and plotted with respect to the reaction free energies (ΔG0) as shown in Fig. 3B.
Thus, a cyclic electron transfer occurs intrinsically between Lf and Ade in four redox states of EcPL with the folded structure. To repair damaged DNA in photolyase, the ET must be from the anionic flavin cofactor and the intramolecular ET dynamics unambiguously revealed that only FADH− functions as the active state in photolyases to allow efficient electron injection into CPD to initiate repair with the intervening Ade in the middle [83].
Intrinsic intramolecular electron transfer
We have systematically studied the intramolecular ET dynamics of FADH− in all classes of PLs, including: class I EcPL and class I PL from Anacystis nidulans (AnPL); class III PL from Caulobacter crescentus (CcPL); ssDNA PL from Arabidopsis thaliana (AtCRY3); and class II PLs from Drosophila melanogaster (DmPL) and Arabidopsis thaliana (AtPL). This unified a complete cyclic ET scheme in all PLs (Fig. 3C). Fig. 3D and E show the absorption transients of the six PLs probed at 800 nm for the pure excited state LfH−* and at 270 nm for mainly the intermediate state LfH•, respectively, after excitation at 400 nm. With the conserved folded flavin structure, an electron readily hops from LfH−* to Ade. Due to the unfavorable driving forces (ΔG0~0–+0.1 eV) of the forward ET reactions [79, 80], we have a back ET (BET) process directly to the initial excited state (BET1a) in addition to the BET process to the original ground state (BET1b). In some PLs, we also observed a very small fraction of long-lived LfH• due to the autoionization process of LfH−* that has been recently proposed by theory [84]. Together with the transients obtained from the visible range that detect both LfH−* and LfH•, we globally analyzed these dynamics and derived the average forward ET (FET1) times of 1450, 1340, 1800, 1590, 564 and 2980 ps, the average BET1a times of 730, 1230, 800, 1029, 1505 and 690 ps, and the average BET1b times of 28, 26, 20, 715, 17 and 22 ps for EcPL, AnPL, AtCRY3, DmPL, AtPL and CcPL, respectively, with a nearly constant deactivation lifetime of 6 ns. The autoionization time scale is estimated to be larger than 100 ns in all PLs here. In most PLs, the BET1a process is much slower than BET1b, and thus the reaction promptly returns to the ground state. However, DmPL has a remarkably slow BET1b process (green curve, Fig. 3E) which allows more feedback to the excited state via BET1a and leads to an apparent slower decay at 800 nm compared to AnPL, even though these two enzymes have similar FET1 times. Similarly, the reorganization energies (λ) and the reaction free energies (ΔG0) of FET1 and BET1b for these PLs are derived and plotted in Fig. 3F.
Thus, using femtosecond spectroscopy, we described the universal intramolecular ET cycles of FADH− in PLs after blue-light excitation. With the conserved folded flavin, a cyclic ET between LfH− and Ade occurs intrinsically with a relatively slow forward ET from hundreds of picoseconds to few nanoseconds, and a fast back ET to the ground state (BET1b) in tens of picoseconds except for DmPL (715 ps). The excited state and charge-separated state will also interconvert, leading to a non-exponential decay behavior of the LfH−* dynamics [85]. These dynamics are critical to determining the actual repair pathways and therefore the quantum yields of different classes of PLs.
CPD repair: bifurcating electron-transfer pathways determine repair quantum yield
The enzymatic reaction of UV-damaged DNA repair involves many elementary steps. With femtosecond temporal resolution and single-residue spatial resolution, the entire evolution of the CPD repair process has been mapped out by probing the dynamics from all initial reactants, to various intermediates, and to the final products. In the past decade, the dynamics and mechanism of CPD repair by class I EcPL have been extensively characterized and a critical cyclic electron-tunneling mechanism was revealed [60–64]. The complete CPD repair photocycles of PLs from different classes have also been dissected recently; a unified repair photocycle with bifurcating electron-transfer pathways through the folded flavin in the conserved active-site structure is unraveled (Fig. 4A). For all PLs, the initial electron injection manifests a bifurcation of direct electron tunneling and two-step electron hopping. Both pathways are operative in CPD repair with different efficiencies, which determine the final repair quantum yield [70]. CPD photolyase, to our knowledge, is the first enzyme system to have the complete catalytic dynamics mapped out in real time at the molecular level.
Figure 4. Femtosecond-resolved dynamics and mechanisms of CPD repair by class I EcPL.
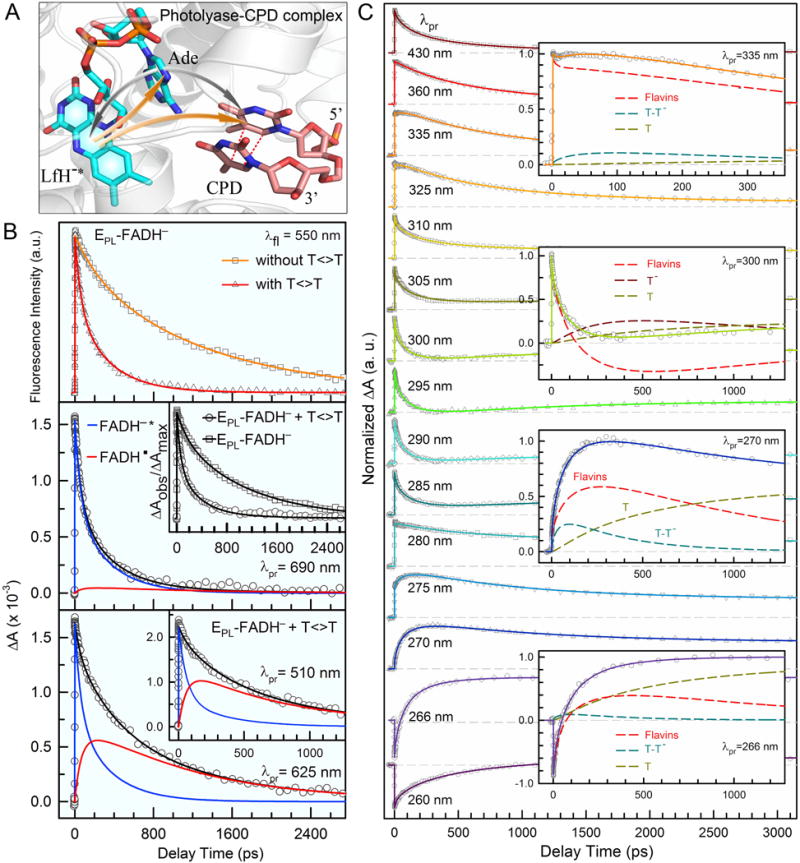
(A) Active-site structure of photolyase-CPD complex showing the initial electron injection in CPD repair. The repair photocycle is initiated by electron injection from lumiflavin to CPD via bifurcated electron-transfer pathways. In class I EcPL, an electron dominantly tunnels to CPD through the intervening adenine via a superexchange mechanism. The two-step hopping pathway is bridged by the intermediate adenine and is minor in EcPL. (B) Top: The fluorescence transients at 550 nm showing the dynamics of FADH−* with and without the substrate. Middle: The absorption transient probed at 690 nm, showing a dominant contribution of FADH−* decay (95%) with a minor signal from FADH•. Inset shows the drastically different dynamics with and without the CPD substrate. Bottom: Absorption transients probed at 625 and 510 nm (Inset) show both FADH−* and the intermediate FADH• dynamics. (C) Femtosecond-resolved transient absorption dynamics of reactants, various intermediates and products probed at visible and UV regions. Insets: Transient absorption signals probed at 335, 300, 270 and 266 nm, respectively. These dynamics are systematically fitted and deconvoluted by total flavin-related species (FADH−*+FADH•+FADH−, dashed red), thymine dimer intermediate T-T− (dashed cyan), repaired thymine anion (dashed dark red) and final thymine products (dashed dark yellow)
Class I photolyase: electron tunneling and high repair efficiency
We have performed fs-resolved fluorescence and absorption measurements and proved that photolyase repairs CPD with a cyclic ET mechanism. In EcPL, the initial electron injection from LfH− to CPD occurs ultrafast via dominant direct tunneling with the Ade as a mediator through a superexchange mechanism [60, 62]. During the whole repair cycle, various reactants, intermediates, and products were detected. We mapped out the catalytic evolution of CPD repair by EcPL, using probing wavelengths from 800 nm to UV-light region to follow the changes of different species during repair. The repair evolution was successfully mapped by first following the changes of the excited state FADH−* and the intermediate FADH• in the visible-light region, which avoided the absorption of thymine-related species of various intermediates and final products which absorb in the UV region. Fig. 4B shows the stretched dynamics of electron injection from the excited LfH−* to the substrate in 170 ps with a stretched parameter of 0.71 (the average time ) and the electron return from the repaired thymine to the intermediate FADH• in 560 ps to restore FADH− and close the entire photocycle. This observation was significant to show that the entire repair is ultrafast and occurs in less than one nanosecond and the reported turnover times in milliseconds to seconds [86] was masked by the recognition process. Given the FET1 time (1450 ps) determined from the enzyme only measurements, we obtained the average direct electron tunneling (FET2) time of 236 ps, and a branching quantum yield (ΦFET2) of 0.83. The two-step electron hopping is a minor channel in EcPL and contributes 12% of the total repair quantum yield [70, 71]. Fig. 4C shows a striking pattern of the transient-absorption signals of the PL-CPD complex, probed at fifteen wavelengths in the UV region. Knowing the overall flavin dynamics probed at the visible region, the dimer splitting is completely solved [62]. The C5–C5′ and C6–C6′ bonds brake sequentially: the C5–C5′ bond splits spontaneously in less than a few picoseconds, consistent with the theoretical predictions [87–90]; the C6–C6′ bond cleavage takes 90 ps after the energy redistribution in the radical intermediate. We successfully detected the dynamics of the intermediates T-T− after the first-bond C5–C5′ cleavage and T− after the complete dimer splitting and the formation of the final product T as the electron returns to the cofactor (Fig. 4C insets). The decay dynamics of T-T− in ~80 ps mainly represents the splitting time of second-bond C6–C6′. Given the total repair quantum yield of 0.82 [57, 91, 92], the branching quantum yields of the two ET pathways, and the yield of C5–C5′ breaking of 1.0, the branching quantum yield of C6–C6′ splitting of 0.87 was obtained. Therefore, the second-bond breakage finishes in 90 ps, and the futile BET without the second-bond splitting occurs in 570 ps. After the second-bond C6–C6′ cleavage, the signal of T− forms and then decays in 700 ps, as the electron returns from T− to FADH• that is completely decoupled from the second-bond breakage.
Overall, the photocycle of CPD repair utilizes a radical ET mechanism with no net change in the redox state of the flavin cofactor. Class I EcPL uses the direct electron tunneling as the dominant repair channel, and the six major elementary reactions involved in repair are completely characterized. Among them, two consecutive competitions make key contributions to the final repair efficiency. The first competition is the tunneling FET1 from LfH−* to CPD in an average time of 236 ps against the excited-state deactivation process without substrate in 1.3 ns. The second one is the C6–C6′ splitting in 90 ps against the BET without repair in 570 ps. The photorepair machinery of photolyase utilizes the blue-light energy and the released energy from biotransformation of the substrate to repair UV-damaged cyclobutane dimer in DNA. The fast electron tunneling with the mediation of Ade ensures efficient repair of class I PLs in a synergistic way, optimizing the forward ET, back ET, and ring splitting to finally achieve high repair quantum yield of close to unity. Mutations at the active site that modulates the active-site reduction potentials and ET reorganization energies always break the dedicated synergy of the optimization for the four elementary reactions in the two competing pairs, and thus leads to lower repair efficiency than the wild-type enzyme. [64]
As seen in the free energy profile along the reaction coordinate (Fig. 5), the first forward ET from the excited cofactor to the substrate has a small negative free energy and is in the Marcus normal region. The first charge-recombination channel (intact BET) has a very large negative free energy and is usually in the Marcus inverted region. Therefore, after charge separation, the reaction dominantly proceeds to the barrierless and energetically favorable C5–C5′ bond cleavage, eliminating the intact BET to the original ground state without splitting the dimer. After the C5–C5′ bond breaking, the reaction encounters a small barrier for the C6–C6′ bond splitting and the reaction bifurcates. The second charge-recombination channel (BET) competes with the productive second C6–C6′ bond cleavage, leading to a loss of the repair yield. The intermediate with the flavin cofactor has a small negative free energy and the BET is in the Marcus normal region again. After the dimer completely splits, the electron return (ER), a third charge-recombination channel that restores the active flavin cofactor and closes the repair photocycle, should not be too slow to avoid new damage of repaired DNA by the extra electron [93]. The ER has a large negative free energy and is in the Marcus inverted region. With the determined ET rates, these ET driving forces and related reorganization energies can also be calculated [63].
Figure 5. Reaction free energy profile along the reaction coordinate for EcPL CPD repair.
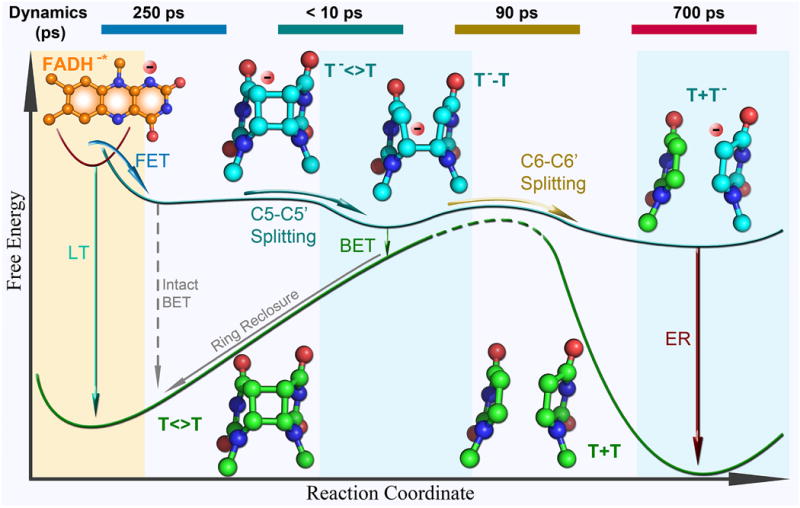
Shown are six major reaction steps (solid lines) with the structures of the excited flavin (orange) and all anionic thymine-related intermediates (blue) and products (green). The relative energy between each state is mainly determined from dynamics measurements. The intact back electron transfer (BET) (dashed grey) is slow and does not occur during repair because of the ultrafast C5–C5′ bond splitting. The reaction times are shown at the top of the diagram. LT, deactivation lifetime; FET, forward electron transfer; ER, electron return.
Adenine mediation in electron tunneling pathways
In photolyases, the FAD cofactor has an unusual folded configuration with the isoalloxazine and adenine rings in close proximity (Fig. 4A). The crystal structure of A. nidulans photolyase with CPD complex shows that the Ade moiety of FADH− is at van der Waals distances with both base moieties of CPD, 3.1 Å to the 5′ side and 3.2 Å to 3′. The first carbon atom is linked to the isoalloxazine ring at 3.6 Å [28]. In class I EcPL, the initial electron injection adopts dominant tunneling pathways directly from LfH− to CPD. A central question is how electrons tunnel between the donor (LfH−) and the acceptor (CPD) and whether a specific pathway exists for such a long-distance ET through the protein [94–96]. The role of Ade in repair and the electron tunneling directionality are a matter of some debate. One view is that the electron tunnels through the intervening Ade to the 5′ side of CPD with a total distance of about 8 Å [97, 98]. An alternative model suggests that the electron directly travels through space from the o-xylene ring of FADH− to the 3′ side of CPD over a shorter distance of 4.3 Å [99, 100]. Given the different electron affinity of thymine and uracil [79], we designed different dimer substrates that consist of thymine and (deoxy)uracil, U<>T, U<>U, T<>U, and T<>T (chemical structures in Fig. 6A), to examine the electron tunneling directionality.
Figure 6. Femtosecond-resolved transient-absorption dynamics of DNA repairs with CPD substrates of different combinations of pyrimidine bases.
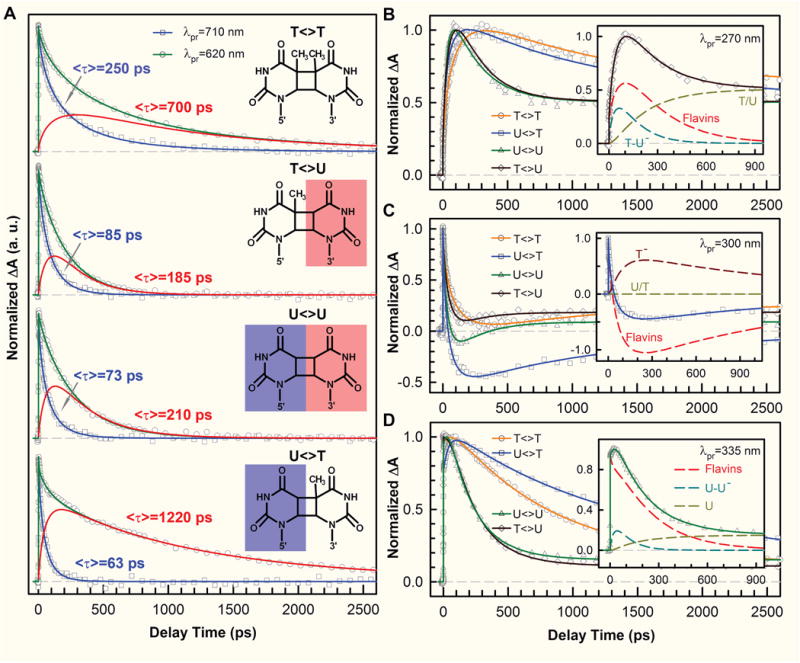
(A) Transient-absorption signals of repair with T<>T, T<>U, U<>U, and U<>T probed at 710 and 620 nm. The dynamics of FADH− (blue) was probed at 710 nm. The signal at 620 nm (green) is the combination of FADH− and intermediate FADH• (red) contributions. The chemical structures of various CPD substrates are also shown with the uracil sides highlighted. The blue shading of U indicates the forward electron tunneling to the 5′ side of DNA and the red shading for electron return starting at the 3′ side after the complete two-bonds splitting. (B–D) Repair dynamics of T<>T (orange), U<>T (blue), U<>U (green), and T<>U (dark red) probed at 270 nm (B), 300 nm (C) and 335 nm (D). Insets: The deconvolution of total flavin-related species (dashed red), substrate intermediate anions T−-U/U−-U (dashed cyan), and T− (dashed dark red), and the products of T/U (dashed dark yellow) of repair with T<>U, U<>T, and U<>U in (B), (C), and (D), respectively.
At 710 nm, only FADH−* was probed and we observed the dynamics of forward ET for T<>U, U<>T and U<>U in 85, 63, and 73 ps, respectively, which are much faster than that of T<>T in 236 ps (Fig. 6A), and lead to even lower contributions from the two-step hopping channel of less than 5% for the three substrates. The ET rates from FADH−* to the four CPD substrates vary in a decreasing order of 5′-U<>T-3′ > U<>U > T<>U > T<>T. This observation is significant and indicates that the electron tunnels toward ending at the 5′ side, not the 3′ side, of the dimers. The reduction potential of the uracil base is more positive than thymine by ~0.11 V [97]. Although the electronic interactions between T and U in the covalent CPD species delocalize in the CPD states and result in the similar redox properties of U<>T and T<>U, the U moiety at the 5′ side should have a larger electron affinity than the T moiety. Clearly, the forward ET to U<>T is faster than that to T<>U, indicating the electron from the excited flavin cofactor tunnels to the 5′ side of damaged DNA through the intervening Ade, even though the distances from Ade to the two sides are nearly equal. Theoretical studies showed similar binding configurations for U<>T and T<>U and thus the observed differences in ET rates should not come from the changes in electronic couplings [28, 97]. The larger ET rate of U<>T relative to T<>U also excludes the electron tunneling pathway directly from the o-xylene ring of the flavin to the 3′ side of the dimer.
With the transients probed at 620 nm (Fig. 6A) and at the UV region (Fig. 6B–D), the flavin intermediate FADH•, and various substrate-related intermediates and products were detected and deconvoluted. At wavelengths shorter than 360 nm, the dynamics of T−-U, U−-U and U−-T decay in 64, 31 and 30 ps, respectively, represent the total dynamics (rates) of both the C6–C6′ bond breakage of anionic CPD intermediates and futile BET after the C5–C5′ splitting. Given their splitting branching yields [63], the bond splitting times of T−-U in 75 ps, U−-U and U−-T both in 35 ps, are obtained. Clearly, the second C6–C6′ bond has a longer breakage time with T and a shorter breakage time with U at the 5′ side, which further supports the tunneling pathway to the 5′ side through Ade mediation. The stabilization by the methyl group on the tertiary C5 position of thymine at the 5′ side results in a longer C6–C6′ cleavage time (90 ps in T<>T and 75 ps in T<>U) while the uracil intermediate without stabilization (a secondary carbon at the C5 position) splits in 35 ps, showing that the electron ends at the 5′ side. We also obtained the futile BET time scales of 1175, 315 and 260 ps for T−-U, U−-T and U−-U, respectively (Fig. 7A). Similarly, the intermediates with thymine at the 5′ side are stabilized by the methyl group on the thymine C5 position, resulting in a smaller driving force and thus a slower BET of T<>T and T<>U in the Marcus normal region, while the other intermediates with uracil at the 5′ side have less stabilization without the methyl group and thus a faster BET of U<>T and U<>U in the normal region as well. After repair, the electron returns to flavin to complete the photocycle. We found that the electron-return dynamics for the four substrates vary in a decreasing order of 5′-T+U−-3′ (185 ps) > U+U− (210 ps) > T+T− (700 ps) > U+T− (1220 ps) (Fig. 7A). The back electron tunneling from T−/U− passing the Ade-mediated pathway to FADH• has a large driving force and is in the Marcus inverted region. The fact that the ER was faster with uracil at the 3′ side of DNA suggests that the electron migrates from the 5′ to 3′ side during the dimer splitting and stays at the 3′ side of DNA after ring splitting, due to a more polar environment [28–30] around the 3′ side to solvate the electron and stabilize the system.
Figure 7. Reaction times in repair of various CPDs and electron-tunneling directionality.
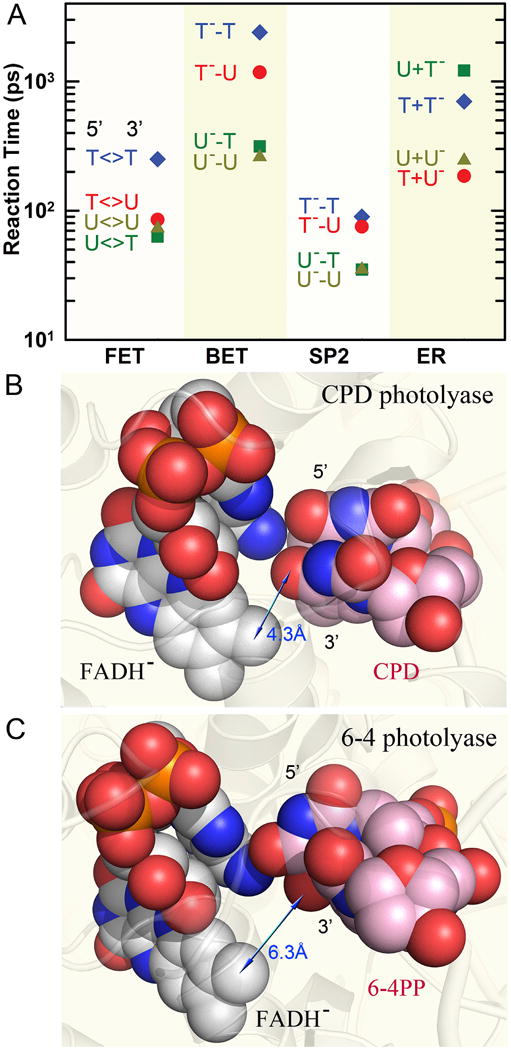
(A) Reaction times of each elementary step observed in repair of various substrates, including the forward electron transfer (FET), futile back electron transfer (BET), C6–C6′ bond splitting (SP2) and final electron return (ER) to complete the photocycle. (B–C) The space-filled representation of the relative positions of the active cofactor FADH− with repaired dimer substrate in A. nidulans CPD photolyase (B) and with 6-4PP in D. melanogaster 6-4 photolyase (C). The adenine-mediated tunneling pathways are completely filled with atom contacts while the through-space tunneling routes have gaps in the pathways that cause slower ET due to the weak electronic coupling. The blue arrows show the approximate distance of the tunneling route in the two enzymes.
As shown in Fig. 7B, the local structure of CPD photolyase with substrate and the atoms along the Ade-mediated tunneling pathway are all at van der Waals contacts. Such a hybrid tunneling pathway mediated by Ade is also observed in the 6-4 photoproduct (6-4PP) repair by 6-4 photolyase [63, 101]. Both CPD and 6-4PP photolyases have a similar structural motif [31] for the Ade-mediated tunneling pathway to the 5′side (Fig. 7C) and the forward ET dynamics in 6-4PP repair occurs with a similar average reaction time of 255 ps [101]. The distance between the flavin cofactor and the 3′ side of the damaged DNA in 6-4PL is about 6.3 Å and the forward ET would take more than nanoseconds to the 3′ side. Thus, both photolyases take the same strategy to utilize the critical intervening Ade moiety of the unusual bent flavin cofactor to inject one electron through a superexchange mechanism to initiate damaged-DNA repair. A recent quantum mechanical calculation reported a similar mechanism but involving the Ade moiety and five water molecules for an electron to tunnel through an Ade-mediated proton wire to reach the 5′ side [98].
Other PLs: various repair QYs and bifurcating ET pathways
The diverse subfamily of CPD photolyases consists of classes I, II and III, and ssDNA PLs (Fig. 1A). Crystal structures of class II [30, 32], III [38] and ssDNA PLs [25–27, 29] have been solved, including the enzyme-substrate complex structure of class II PL from Methanosarcina mazei (MmPL) [30] and ssDNA AtCRY3 [29]. These crystal structures show overall similarities as class I PLs in protein folding and cofactor binding motif with several critical changes. In AtCRY3, two conserved residues, M353 and W392 in AnPL that interact with the flipped CPD and the complementary dsDNA strand, are replaced with hydrophilic residues Q395 and Y434. The same replacements are conserved in all ssDNA PLs. An increased number of water molecules at the active site is also observed in these proteins [29]. The M and W residues are also replaced in plant CRYs by V and Y, and the M residue is replaced by H in the (6-4) PLs, suggesting a possible role of these two residues in differentiating substrates for each protein subfamily. For the class II PLs, amino acid alignment analyses show that they are distantly related to other members of the protein family with sequence identities of less than 20% [102], suggesting that they had arisen early in evolution [20]. In addition, the conserved asparagine residue (N378 in EcPL) at the redox-active N5 position of flavin is from a different α-helix in class II PLs, and is located at the opposite face of the isoalloxazine ring [30, 32]. An alternative tryptophan electron-transfer pathway (Trp triad) is also found in class II PLs to transfer electrons to reduce oxidized or neutral semiquinone flavin as in other PLs. Class II PLs also have the replacement of several key residues at the active site: The G and Y (or F) residues near the substrate replaced the N and R residues, respectively, in the other PLs. The replacement of G in place of N also introduces several water molecules to fill in the empty space [30].
The steady-state measurements of enzyme activities show that the repair quantum yields (QYs) of different PLs vary from 0.32 to 0.88 [70]. The class I EcPL and AnPL have the highest QYs of 0.82 and 0.88, respectively. The class II, III and ssDNA PLs have relatively low QYs ranging from 0.32 to 0.62. The wide distribution of repair QYs in different classes of PLs led us to examine whether the molecular repair mechanism obtained from class I EcPL [65, 66] can apply to all other members, especially to the distantly related class II PLs. The subtle changes in the active-site environment discussed above could lead to distinct reaction dynamics, which would provide unprecedented insight into the structure-function relationships of enzymes at a molecular level.
Fig. 8 presents the transient absorption dynamics of three PL enzymes, class I AnPL, ssDNA PL AtCRY3 and class II AtPL, and their enzyme-substrate complexes. At 800 nm (Fig. 8A), we observed a fast decay of LfH−* in 140 ps with a stretched parameter β of 0.71 for the AnPL complex (solid black). Compared to the average 1.1-ns decay dynamics (dashed black) probed without the substrate, the LfH−* is significantly quenched due to the direct electron tunneling from the LfH−* to the substrate in an average of 209 ps, indicating a similar ET mechanism as the class I EcPL. Given the ET time from LfH− to Ade (FET1) obtained from the enzyme only measurements, the direct electron-tunneling step (FET2) leads to a high branching quantum yield ΦFET2 of 0.84 for AnPL (0.83 for EcPL). For other PLs, the excited-state quenching probed at 800 nm in the presence of substrate is much less drastic. In AtCRY3, the average decay time of LfH−* changes from 1383 ps (dashed magenta) without the substrate to 732 ps (solid magenta) with the substrate. In class II AtPL, the excited flavin dynamics of the complex (solid blue) is surprisingly similar as that without the substrate (dashed blue). Together with the intramolecular ET dynamics, we derived the FET2 times of 1555 ps for AtCRY3, and 6500 ps for AtPL. In class I EcPL and AnPL, the direct tunneling is much faster than FET1 and is therefore dominant in the repair cycle. However, in AtCRY3, the direct tunneling FET2 time of 1555 ps is only slightly faster than the FET1 time of 1800 ps. In AtPL, FET2 occurs in 6.5 ns and is one order of magnitude slower than FET1 in 564 ps. Yet these PLs still maintain decent repair quantum yields, which in some cases, such as AtPL with a repair QY of 0.49, cannot be achieved by the slow electron-tunneling pathway. Apparently, the direct tunneling pathway loses its dominance in the repair of these PLs and since FET1 occurs intrinsically in all PLs, the initial electron injection from the excited state LfH−* bifurcates (Fig. 8B). The alternative ET channel, a two-step hopping pathway bridged by Ade in the folded flavin motif, must be operative in these PLs to compensate the low quantum yield from the direct electron tunneling. The second-step hopping from Ade to CPD (FET3) is instantaneous, given a short separation distance of about 3 Å and a favorable, negative free energy, and therefore, the two-step hopping pathway is feasible, and thus is also universal in all PLs.
Figure 8. Bifurcating electron-transfer pathways and transient-absorption dynamics of CPD repair by three types of photolyases.
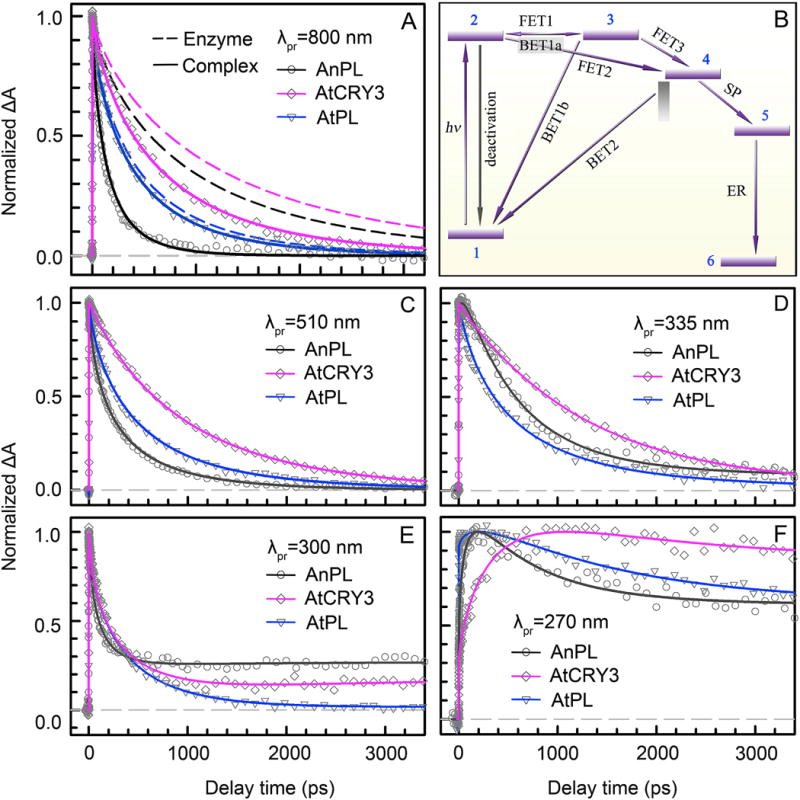
(A) Absorption transients of AnPL, AtCRY3 and AtPL enzymes (dashed lines) and their enzyme-CPD complexes (solid lines) probed at 800 nm for the pure excited-state (LfH−*) decay. In the presence of substrate, the LfH−* dynamics of AnPL becomes drastically faster due to fast direct electron tunneling to the substrate; the change of the LfH−* dynamics is much less in ssDNA PL AtCRY3 and is almost negligible in class II AtPL. (B) The universal repair scheme of CPD by photolyases with ten resolved elementary steps, including seven electron-transfer reactions and dimer splitting processes. The initial electron injection is bifurcated into two routes: the direct tunneling to CPD through the intervening adenine via a superexchange mechanism (FET2), and the two-step hopping pathway (FET1, FET3) bridged by the intermediate adenine. 1, LfH−-Ade+T<>T(CPD); 2, LfH−*-Ade+T<>T; 3, LfH•-Ade−+T<>T; 4, LfH•-Ade+T<>T−; 5, LfH•-Ade+T+T−; 6, LfH−-Ade+T+T. (C) Absorption transients of AnPL, AtCRY3 and AtPL complexes probed at 510 nm for the detection of LfH−* and flavin intermediate LfH•. (D–F) Absorption transients of the three complexes probed in the UV region at 335 nm, 300 nm and 270 nm for the detection of all flavin-related species, the thymine intermediates (T-T− and T−) and final products (T).
After determining the initial bifurcation of electron injection, we probed the repair dynamics systematically from visible to the deep UV region to resolve various reaction intermediates and products. Fig. 8C to F shows the absorption transients of these distinct dynamic profiles probed at four typical wavelengths. Similar as that in EcPL, after the initial charge separation the anionic CPD could follow two routes: splitting of the dimer by sequentially breaking the C5–C5′ and C6–C6′ bonds (SP), or returning to the original ground state through the futile back ET (BET2). The first C5–C5′ bond breaking is nearly barrierless and occurs ultrafast in less than 10 ps [87] and the second C6–C6′ bond cleavage usually occurs in tens of picoseconds [62]. Finally, the electron returns to LfH• to restore the flavin ground state and complete the entire photocycle. In the visible region at 510 nm (Fig. 8C), the flavin intermediate LfH• and the excited state LfH−* were detected. Note that with the initial electron bifurcation, the overall LfH• dynamics is a summation of three channels [70]. In the UV region, the transients contain all the flavin signals, including the excited state LfH−*, the immediate states LfH• and Ade−, and the final states of LfH− and Ade. It also contains the overall substrate signals of the intermediate T-T− after the first C5–C5′ bond breakage, T− after the second C6–C6′ cleavage, and final product of repaired base T. With systematic analyses, we dissected each transient and obtained the ultrafast electron hopping of FET3 in 6, 11 and 15 ps, the second C6–C6′ cleavage in 87, 187 and 36 ps and the electron return (ER) after repair in 437, 525, and 819 ps for AnPL, AtCRY3, and AtPL, respectively. Knowing the total repair quantum yields and the branching yields of the three forward ETs, we also derived BET2 times of 1138, 143 and 527 ps, respectively, for the three PLs.
We have followed the entire repair photocycle of the PLs and identified ten elementary steps including seven ET reactions and measured their actual timescales. We obtained a unified repair mechanism for all CPD PLs with the critical, bifurcating electron-transfer pathways through the folded flavin cofactor in the conserved active-site structure (Fig. 9A and B). It indicated the critical role of the intervening Ade in initial photochemistry at the unique position between Lf and the substrate. Two complete photocycles of class I AnPL and class II AtPL, are shown in Fig. 9 C and D, respectively, with all the elementary reaction times. The timescales of the elementary steps involved in repair, especially the forward tunneling ET (FET2) and futile back ET (BET2), differ greatly in PLs due to different reaction driving forces resulting from structural differences at the active sites. The various timescales of FET1 and FET2 lead to different branching yields for the two electron pathways, and synergistically modulate the whole repair pattern with other elementary steps, and determine the total repair QYs.
Figure 9. Complete photocycles of CPD repair and repair quantum yields with electron bifurcation in different photolyases.
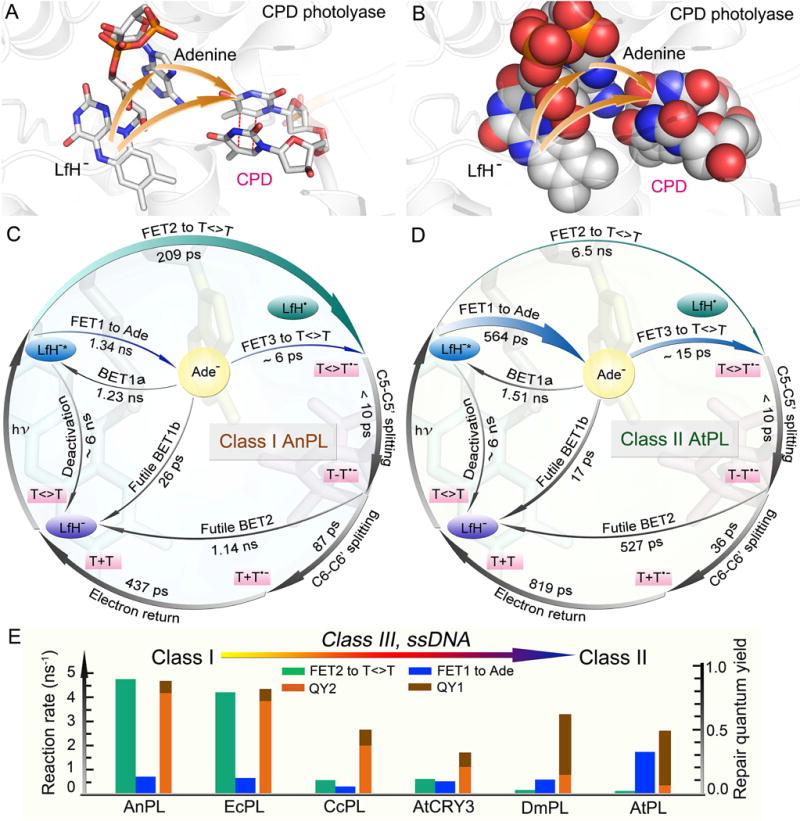
(A) Active-site structure of enzyme-substrate complex showing relative positions of lumiflavin, adenine, and repaired substrate in CPD photolyase (A. nidulans). The orange arrows represent the initial bifurcating electron-transfer pathways. (B) The space-filled representation of the active-site structure in (A). The adenine-mediated tunneling pathway and the two-step hopping pathway are filled with atom contacts. (C–D) Repair photocycles of class I AnPL and class II AtPL with ten elementary steps including seven electron-transfer reactions. In AnPL, the direct electron-tunneling channel (FET2) is dominant and much faster than the FET1. In AtPL, the electron-hopping to adenine (FET1) is much faster than FET2 and the two-step hopping pathway is dominant. (E) Changes in bifurcated ET rates for FET2 and FET1 and the resulting quantum yields of the two respective ET pathways, QY2 and QY1, ordered from microbial to eukaryotic PLs.
In all PLs, both direct electron tunneling and two-step electron hopping pathways are operative in repair with divergent efficiencies depending on distinct reduction potentials in different protein environments. Fig. 9E shows the total repair QYs of six PLs from the microbial class I to the eukaryotic class II with initial electron bifurcation into the direct tunneling path FET2 and the hopping path FET1 and their corresponding branching quantum yields QY2 and QY1. With fast direct tunneling, the two-step hopping pathway in both class I EcPL and AnPL has minor contribution to the overall repair quantum yield. However, the two-step hopping pathway is critical to the observed repair quantum yield in ssDNA AtCRY3 (0.32), class III CcPL (0.50), and class II DmPL (0.62), and becomes dominant in class II AtPL (0.49), a situation opposite of that in class I AnPL and EcPL. Clearly, the class I PLs have the highest repair QYs because the direct tunneling pathway is more efficient and yields a higher repair efficiency, while the two-step hopping pathway has a slow first-step hopping, due to a small positive free energy, followed by the second-step hopping from Ade (FET3), which is further branched by the fast BET1b to the ground state and the charge recombination to the original excited state (BET1a). With the decrease in the rates of direct tunneling, the electron branching through the hopping channel increases and reaches dominance in AtPL. Other than the extreme cases in which one of the two pathways dominates the repair, many photolyases from different classes, such as ssDNA AtCRY3, adopt a parallel electron injection pattern in which both channels make substantial contributions to the overall repair efficiency. These PLs could not reach the high repair QYs as observed in the class I PLs. However, decent repair QYs are maintained by these PLs through adopting the two-step hopping channel as a compensation of the slow electron tunneling channel. From lower microbes to higher eukaryotes, the electron exploits from mainly direct tunneling to dominant two-step hopping with the same conserved active-site structure through evolution to maintain decent repair efficiency and thus genome stability.
Among the ten elementary steps, the seven ET reactions involved in CPD repair by PLs are essential in modulating the whole repair cycle. With the determined timescales of the ET reactions, the branching quantum yields of the elementary steps are obtained (Fig. 10A). Various reorganization energies λ and their relevant reduction potentials, i.e., the standard free energy of the reaction, ΔG0, are derived using the semi-classical Marcus ET expression [81, 82] (Fig. 10B). For all the PLs studied, we use 2.48 eV for the S1←S0 transition of LfH− for 500-nm excitation, and assume that the reduction potential of LfH•/LfH− remains at +0.08 V vs. NHE [80] since no significant conformation variation in the active site for Lf is observed from the crystal structures. For the ET between the LfH− and substrate, a similar electron coupling J = 3.0 meV in all the PLs for the forward tunneling, and J = 2.6 meV for all the returning tunneling are used [63, 64, 70]. Due to the ultrafast C5–C5′ bond breakage and the localized electron near the 5′ site [87], we also assume the futile back ET (BET2) has the same J and λ values as FET2. The FET2 and BET2 processes are in the Marcus normal region (−ΔG0≤ λ), and the electron return is in the Marcus inverted region (−ΔG0> λ).
Figure 10. Reaction times, quantum yields and electron-transfer reorganization energies in CPD repair by photolyases.
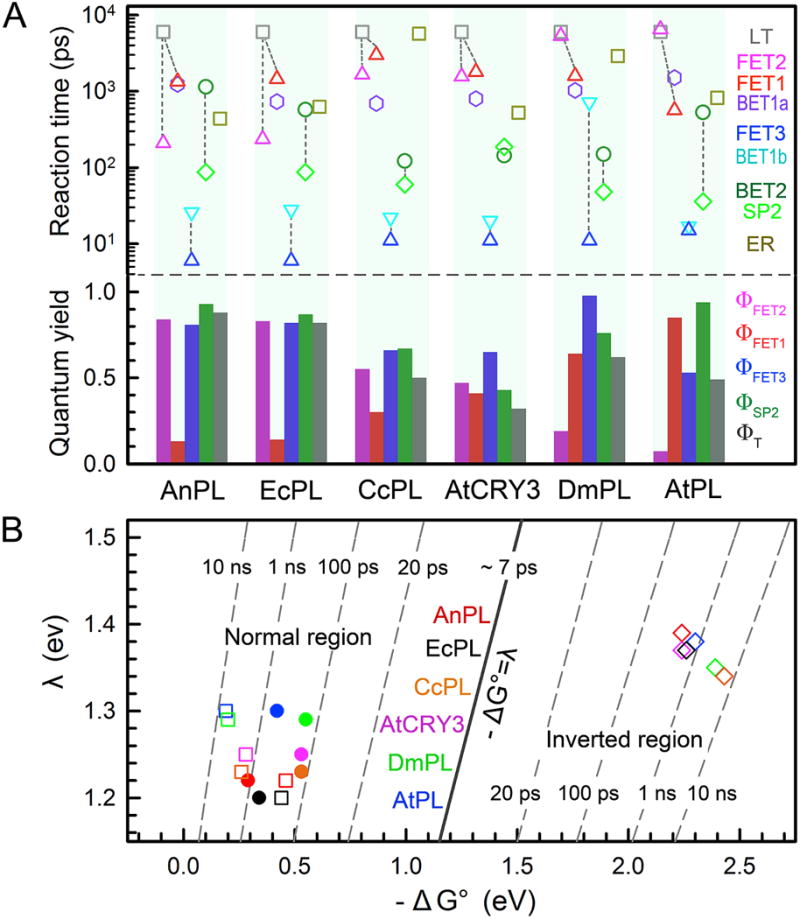
(A) Top: Reaction times of all elementary steps involved in repair by different photolyases. LT, deactivation lifetime; FET1, forward electron transfer (ET) to adenine; FET2, forward ET directly to substrate; FET3, forward ET from anionic adenine to substrate; BET1b, back ET from anionic adenine to flavin ground state; BET2, back ET from anionic substrate to flavin ground state without repair; SP2, the C6–C6′ bond splitting; ER, electron return after repair. The C5–C5′ bond cleavage is ultrafast and is not shown here. The dashed lines link three sets of competing branching channels responsible for the repair efficiencies. Bottom: Elementary quantum yields (QYs) of three forward ETs and SP2, and the total repair quantum yield ΦT for each photolyase. The four elementary QYs are calculated as follows: ΦFET1=<τFET1>−1/(<τLT−1+<τFET1>−1 +<τFET2 >−1; ΦFET2+<τFET2>−1/(τLT−1+ +<τFET1>−1 +<τFET2>−1); ΦFET3=+<τFET3>−1/(<τFET3>−1+<τBET1a>−1+<τBET1b>−1); ΦSP2=<τSP2>−1/(<τSP2>−1+<τBET2>−1). Because the ET between LfH−* and adenine interconverts, the overall repair QY is calculated as ΦT= (ΦFET2+ ΦFET1× ΦFET3)× ΦSP2×(1+Φ′+Φ′2 +Φ′3…), where Φ′=ΦFET1×[<τBET1a>−1/(<τFET3>−1+ <τBET1a>−1+ <τBET1b>−1)]. In all photolyases studied here, Φ′ is less than 0.01 due to a much faster τFET3 than τBET1a. (B) Two-dimensional (2D) contour plot of the ET dynamics in repair relative to free energy (ΔG0) and reorganization energy (λ) for FET2 (filled circle), BET2 (open square) and ER (open diamond). The FET2 is in the Marcus normal region (−ΔG0≤ λ) and the ER is in the Marcus inverted region (−ΔG0> λ). BET2 is a dissociative ET process and is in the normal region again.
(6-4) photoproduct repair and photocycle
For 6-4 photoproduct repair, various hypothetical repair models [31, 33, 103–109] including formation of an oxetane intermediate in the ground state before the photochemical reaction [103, 105] and, more recently, a model that includes formation of a water molecule following the primary photochemical reaction [31] have been proposed to rationalize the bond breakages and group arrangements for complete repair. Also, a two-photon repair mechanism has been proposed [110, 111]. Various theoretical calculations were carried out and several repair models have been suggested [104, 107–109, 112, 113]. Most of these proposed models invoke one proton transfer from a neighboring histidine residue at the active site (Fig. 11A).
Figure 11. Dynamics and mechanisms of damaged DNA repair by (6-4) photolyase.
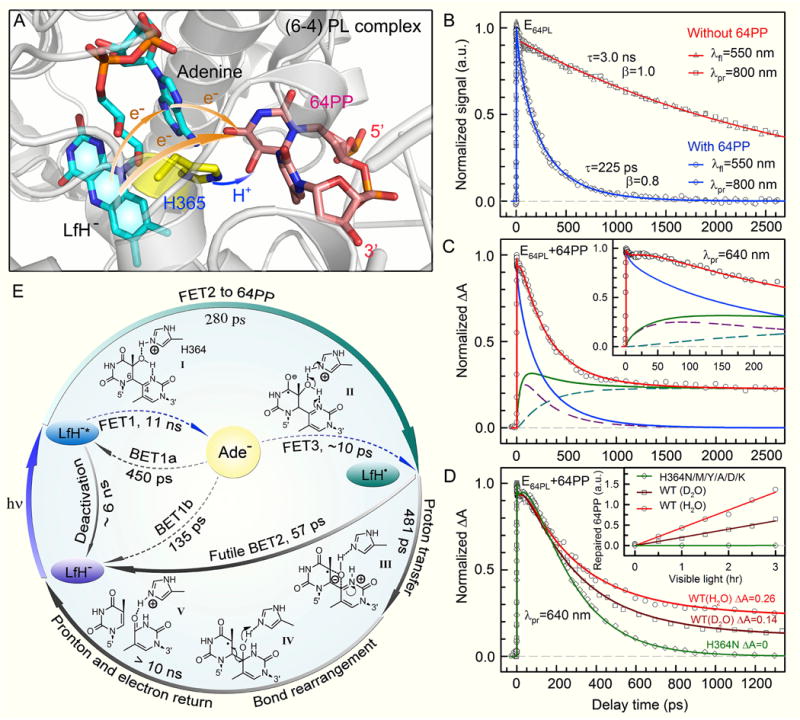
(A) Active-site structure of the (6-4) photolyase (D. melanogaster) complex showing the catalytic cofactor FADH−, the conserved histidine residue (H365 in D. melanogaster and H364 in A. thaliana 6-4 PL), and the pyrimidine-pyrimidone (6-4) photoproduct (6-4PP). The orange arrows represent the bifurcating electron-transfer (ET) pathways of the initial electron injection, and the blue arrow represents the proton transfer from the conserved histidine residue to 6-4PP. (B) Femtosecond-resolved dynamics detected by both fluorescence up-conversion (gated around the emission peak of 550 nm) and transient absorption (probed at 800 nm) methods without (red) and with (blue) 6-4PP substrate in the active site, showing the same lifetime and forward ET decays. (C) Absorption transient probed at 640 nm with detection of both FADH−* (blue) and intermediate FADH• (green). The total FADH• signal is contributed by two channels, the initially formed FADH•, and the branched FADH• in the repair channel. Inset: The flat signal in tens of picoseconds, reflecting an apparent fast rise signal. (D) Absorption transients probed at 640 nm of the H364N mutant in H2O (green) and the wild-type (WT) enzymes in D2O (dark red), compared with the WT in H2O (light red). Inset: The corresponding relative steady-state quantum yield measurements. (E) The repair photocycle of 6-4PP by (6-4) photolyase. The initial electron injection is dominated by direct electron tunneling directly to 6-4PP (FET2) in 280 ps (I to II) and the two-step hopping is negligible. After charge separation, the catalytic proton transfer between the enzyme [H364 in At(6-4) PL] and the substrate (II to III), induced by the initial electron transfer, occurs in 481 ps, which is branched by the back electron transfer (BET2) occurring in 57 ps without any repair (II to I). The subsequent repair reactions involve a series of atom arrangements with bond breaking and forming (III to IV), and final proton and electron return (to the H364 residue and flavin cofactor, respectively) to convert the 6-4PP to two thymine bases on timescales longer than 10 ns (IV to V).
We have examined the repair dynamics and mechanism of 6-4PP by At(6-4) PL [101], following a strategy similar to the one we used to examine CPD repair. We first characterized the dynamics of forward ET. By capturing the fluorescence emission at 550 nm from excited state FADH−* with the substrate, the fs-resolved transient of the complex can be best represented by a stretched-single-exponential decay with a time constant of 225 ps and β=0.8 (Fig. 11B). Considering the effective lifetime of FADH−* in 3.0 ns (Fig. 7B) with an intramolecular FET1 in 11 ns, a deactivation time of 6 ns and an autoionization process in 14 ns (Fig. 11E) (unpublished data), the resulting tunneling FET2 takes 280 ps. When the potential active-site proton donor, H364, was replaced by charged (K and D), polar (N and Y) and hydrophobic (A and M) residues, we observed the similar ET dynamics with time constants of 165–379 ps. This finding, along with the observation that the 6-4PP is in its standard form in the structure of enzyme-substrate complex [31, 33] excludes the early repair model that required a mixture of oxetane precursor and ground state 6-4PP before photoexcitation [103, 105]. We conclude that after one electron injection, the repair of 6-4PP takes place completely in the anionic ground state.
After the photoinduced charge separation (FADH•+6-4PP•−), the reaction can evolve along two pathways: futile BET2 or 6-4PP repair (Fig. 11E). The bifurcating branching of FET1 pathway is negligible in this case due to the long ET time of 11 ns. Knowing the forward ET dynamics of FADH−*, we can map out the temporal evolution of FADH• by probing at wavelengths from 500 to 700 nm. In Fig. 11C, the transient probed at 640 nm shows drastically different behavior from that probed at 800 nm, owing to the detection of the flavin intermediate FADH•. We observed an apparent rise signal of FADH• in 51 ps (initial flat part, Fig. 11C inset) and a long plateau, indicating that complete 6-4PP repair takes longer than several nanoseconds. By deconvolution, we obtained the futile BET2 in 57 ps and the repair channel in 481 ps to form a 6-4PP•− related intermediate. From these rate constants, we obtained a repair branching constant of 0.097, a value that is in excellent agreement with the reported steady-state repair quantum yield of 0.1, suggesting that after this branched repair step, all subsequent reactions proceed to the final 6-4PP repair without any back ET that would lead to a futile cycle.
These findings revealed the underlying reason for the low repair quantum yield of (6-4) photolyase (0.1). Compared with class I CPD photolyases, the ultrafast rate of futile BET2 from 6-4PP•− to FADH• in At(6-4)PL repair, relative to the parallel repair channel of 481 ps, causes great loss in the overall repair QY, although the tunneling FET2 is comparatively efficient and the branching of FET2 is high (0.8). For the series of mutants (H364N/M/Y/A/D/K) designed to examine the proposed reaction mechanism, we observed that all transients probed in the 500–700 nm region show similar BET2 dynamics in the range of 80–295 ps [101], but decay to zero without any long plateaus (shown in Fig. 11D for the H364N mutant). This observation is critical to the proposed repair mechanism. Even though the ET from FADH−* to 6-4PP is essentially intact, the repair channel is completely shut off and all FADH• formed by initial charge separation follows a futile ET cycle back to flavin by charge recombination without repair. These results are also consistent with our steady-state quantum yield measurements, which revealed lack of repair in these mutants (Fig. 11D inset). Collectively, our data, in agreement with an earlier report [105], indicate that H364 in the active site is a functional residue that is irreplaceable in the repair channel, and a proton transfer from H364 to the 6-4PP is conceivably the rate-limiting branching step in our repair photocycle (Fig. 11E).
We examined the enzymatic reaction of the proton transfer from H364 during repair using D2O. As shown in Fig. 11D for wild-type photolyase, we observed a different transient with an obviously lower plateau, reflecting a slower repair process (1.25 ns) but with the similar forward (240 ps) and back (68 ps) ET dynamics as observed in H2O. The X-ray structure shows a hydrogen-bond distance of 2.7 Å between the amine group of H364 and the hydroxyl group at the C5 position of the 5′ base (Fig. 11A); hence, a proton transfer from H364 to the hydroxyl group is feasible. The lower plateau of the transient in D2O is about half of that in H2O (Fig. 11D), which corresponds to a halving in the repair branching and is in good agreement with the steady-state measured quantum yield ratio of 1:2 for D2O:H2O (Fig. 11D inset). We also studied the repair dynamics and the steady-state enzyme activity over a pH range from 7 to 9 and did not observe any changes, consistent with the observation that H364 remains protonated over a wide basic pH range. All these results are consistent with the proton transfer from H364 to 6-4PP to generate a protonated neutral radical 6-4PPH• as a key step in the repair cycle (Fig. 11A and E). This critical proton-transfer process, facilitated by the initial photoinduced electron transfer, completely blocks the futile back ET from 6-4PP•− and allows the reaction to proceed to repair with 100% efficiency after this step.
Based on these findings, a catalytic photocycle for the repair of thymine (6-4) photoproduct was proposed (Fig. 11E). In this scheme, the primary reactions are the initial electron transfer (I to II in Fig. 11E) and the subsequent proton transfers (II to III in Fig. 11E). The ET-induced proton transfer from the H364 residue in photolyase to the 6-4PP substrate is a key step in the repair photocycle, like the ‘dividing line’ in the transition state, and makes the subsequent reactions ‘downhill’ without the possibility of back reaction. This critical step competes with the futile BET process, resulting in an overall repair quantum yield of about 0.1, which is probably the maximum value that could be achieved for such a structurally and chemically challenging reaction. The successive elementary steps naturally proceed to an intramolecular proton transfer from the hydroxyl group on the C5 of the 5′ base to the N3 at the 3′ base to form a transient zwitterion, and then an oxygen-atom attack of the C4 position at the 3′ base to form a transient oxetane-type structure (III in Fig. 11E). The transient-water-molecule-formation model, which proposes direct breakage of the C–O bond at the 5′ base after the initial proton transfer, seems unlikely because it necessitates a series of proton-transfer reactions, including the protonation of the carbonyl group at the 3′ base, and there are no potential proton donors in proximity to this carbonyl group. In addition, any interruption in such a complicated scheme proposed by this water model would be expected to give rise to permanently damaged DNA; this is not observed in the repair reaction by (6-4) photolyase. Our scheme, in which a simple transient-oxetane formation facilitates the oxygen-atom transfer from the 5′ to 3′ base followed by C6–C4 bond split (IV in Fig. 11E), would be less prone to mutagenic side reactions because, after the oxygen-atom transfer and C–C bond cleavage, the proton returns to the essential H364 residue and the electron returns to FADH•, restoring the enzyme to its active form and the 6-4PP to two thymine bases (V in Fig. 11E).
The observed dynamics and resulting photocycle in Fig. 11E are triggered by the excitation of only one initial photon, which has also been supported by theoretical calculations [104, 107, 108, 113]. Recently, a two-photon repair mechanism of 6-4PP was suggested [111] by experiment [110] and some theoretical work [109]. However, we recently examined the laser power dependence of 6-4PP repair and observed a linear relationship between the photon energy and repaired products. We also carefully examined the existence of the long-lived intermediate (1.8 minutes in ref. [110]) and did not observe the recovery of the 6-4PP at 325 nm after certain laser irradiation. More studies are needed to further clarify the different observations and ultimately reveal the complete repair mechanism with more details of various intermediates.
Conclusions
The rich photochemistry of flavins has drawn significant interest for decades. This review summarizes our recent efforts and the advances on the understanding of the dynamics and molecular mechanisms of an important flavoprotein family, photolyases. These delicate photomachines are powerful tools in nature that reverse the lethal damages on DNA, cyclobutane pyrimidine dimer and pyrimidine-pyrimidone (6-4) photoproduct, caused by excessive ultraviolet irradiation. For CPD repair, we have examined various species of PLs from prokaryotes to eukaryotes, including their intrinsic intramolecular ET dynamics of the folded cofactor structure, bifurcating intermolecular ET reactions, and sequential bond-splitting steps. Ten elementary steps in the catalytic repair process are resolved with their actual timescales and the complete photocycle is determined. A unified bifurcating-ET strategy with a cyclic radical mechanism for the whole diverse superfamily is established. Although individual PL favors either the direct tunneling channel, the two-step hopping channel, or both, the two pathways are operative and the initial electron would always bifurcate. The electron bifurcation also reveals the molecular origin of various repair quantum yields of many PLs and signifies the critical functional role of the intervening Ade moiety in the conserved folded flavin cofactor. For 6-4PP repair, we have also revealed the ET-induced proton-transfer mechanism in 6-4PLs and proposed a key repair framework. With the conserved folded flavin (FAD) structure, the initial bifurcating ET pathways are operative as in CPD PLs, and the subsequent reaction to complete such a challenging repair requires a critical proton transfer from an active-site histidine. With the key reactions determined, future studies are to capture various intermediates, especially after the proton transfer, to complete characterization of all involved elementary steps.
These mechanistic studies unify our understanding of the well characterized class I PL and other PLs, including the most distant class II PLs. Photolyases are the first enzyme system in which the entire catalytic evolution is completely mapped out, with each elementary step determined with the actual reaction time. Since the 1980s, femtosecond spectroscopy has been used to study many chemical and biological systems and has provided deep insights into molecular behaviors. By integrating fs-resolved spectroscopies and molecular biology techniques, we successfully dissected ultrafast enzyme dynamics of photolyases on the most fundamental level with unprecedented details, and will tackle more complex biological systems to explore the dynamics and mechanisms of the uncharted territories in nature.
Recent advances on the dynamics and mechanisms of photolyases are reviewed.
The complete DNA repair photocycles of photolyases are solved with actual timescales.
The universal bifurcating electron-transfer pathways determine the repair quantum yield.
Acknowledgments
We thank all group members, past and current, who involved in the photolyase project. Their work has been cited here. This work is supported in part by the National Institute of Health (Grant GM118332).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sancar A. Mechanisms of DNA repair by photolyase and excision nuclease (Nobel Lecture) Angew Chem Int Ed Engl. 2016;55(30):8502–8527. doi: 10.1002/anie.201601524. [DOI] [PubMed] [Google Scholar]
- 2.Sancar A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev. 2003;103(6):2203–2237. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 3.Ozturk N, Song SH, Ozgur S, Selby CP, Morrison L, Partch C, Zhong D, Sancar A. Structure and function of animal cryptochromes. Cold Spring Harb Symp Quant Biol. 2007;72:119–131. doi: 10.1101/sqb.2007.72.015. [DOI] [PubMed] [Google Scholar]
- 4.Rupert CS, Goodgal SH, Herriott RM. Photoreactivation in vitro of ultraviolet-inactivated hemophilus influenzae transforming factor. J Gen Physiol. 1958;41(3):451–471. doi: 10.1085/jgp.41.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sancar A. Structure and function of DNA photolyase. Biochemistry. 1994;33(1):2–9. doi: 10.1021/bi00167a001. [DOI] [PubMed] [Google Scholar]
- 6.Chaves I, Pokorny R, Byrdin M, Hoang N, Ritz T, Brettel K, Essen LO, van der Horst GT, Batschauer A, Ahmad M. The cryptochromes: Blue light photoreceptors in plants and animals. Annu Rev Plant Biol. 2011;62:335–364. doi: 10.1146/annurev-arplant-042110-103759. [DOI] [PubMed] [Google Scholar]
- 7.Cashmore AR. Cryptochromes: Enabling plants and animals to determine circadian time. Cell. 2003;114(5):537–543. [PubMed] [Google Scholar]
- 8.Ahmad M, Cashmore AR. Hy4 gene of A. thaliana encodes a protein with characteristics of a blue-light photoreceptor. Nature. 1993;366(6451):162–166. doi: 10.1038/366162a0. [DOI] [PubMed] [Google Scholar]
- 9.Guo H, Yang H, Mockler TC, Lin C. Regulation of flowering time by Arabidopsis photoreceptors. Science. 1998;279(5355):1360–1363. doi: 10.1126/science.279.5355.1360. [DOI] [PubMed] [Google Scholar]
- 10.Gegear RJ, Casselman A, Waddell S, Reppert SM. Cryptochrome mediates light-dependent magnetosensitivity in drosophila. Nature. 2008;454(7207):1014–1018. doi: 10.1038/nature07183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dodson CA, Hore PJ, Wallace MI. A radical sense of direction: Signalling and mechanism in cryptochrome magnetoreception. Trends Biochem Sci. 2013;38(9):435–446. doi: 10.1016/j.tibs.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Taylor JS. Unraveling the molecular pathway from sunlight to skin cancer. Accounts of Chemical Research. 1994;27(3):76–82. [Google Scholar]
- 13.Daya-Grosjean L, Dumaz N, Sarasin A. The specificity of p53 mutation spectra in sunlight induced human cancers. Journal of Photochemistry and Photobiology B: Biology. 1995;28(2):115–124. doi: 10.1016/1011-1344(95)07130-t. [DOI] [PubMed] [Google Scholar]
- 14.Lima-Bessa KM, Menck CF. Skin cancer: Lights on genome lesions. Curr Biol. 2005;15(2):R58–61. doi: 10.1016/j.cub.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 15.Husain I, Carrier WL, Regan JD, Sancar A. Photoreactivation of killing in E. Coli K-12 phr-cells is not caused by pyrimidine dimer reversal. Photochem Photobiol. 1988;48(2):233–234. doi: 10.1111/j.1751-1097.1988.tb02814.x. [DOI] [PubMed] [Google Scholar]
- 16.Todo T, Takemori H, Ryo H, Ihara M, Matsunaga T, Nikaido O, Sato K, Nomura T. A new photoreactivating enzyme that specifically repairs ultraviolet light-induced (6-4)photoproducts. Nature. 1993;361(6410):371–374. doi: 10.1038/361371a0. [DOI] [PubMed] [Google Scholar]
- 17.Selby CP, Sancar A. The second chromophore in Drosophila photolyase/cryptochrome family photoreceptors. Biochemistry. 2012;51(1):167–171. doi: 10.1021/bi201536w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiontke S, Gnau P, Haselsberger R, Batschauer A, Essen LO. Structural and evolutionary aspects of antenna chromophore usage by class II photolyases. J Biol Chem. 2014;289(28):19659–19669. doi: 10.1074/jbc.M113.542431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park HW, Kim ST, Sancar A, Deisenhofer J. Crystal structure of DNA photolyase from Escherichia coli. Science. 1995;268(5219):1866–1872. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 20.Kanai S, Kikuno R, Toh H, Ryo H, Todo T. Molecular evolution of the photolyase-blue-light photoreceptor family. J Mol Evol. 1997;45(5):535–548. doi: 10.1007/pl00006258. [DOI] [PubMed] [Google Scholar]
- 21.Öztürk N, Kao YT, Selby CP, Kavaklı IH, Partch CL, Zhong D, Sancar A. Purification and characterization of a type III photolyase from Caulobacter crescentus. Biochemistry. 2008;47(39):10255–10261. doi: 10.1021/bi801085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lucas-Lledo JI, Lynch M. Evolution of mutation rates: Phylogenomic analysis of the photolyase/cryptochrome family. Mol Biol Evol. 2009;26(5):1143–1153. doi: 10.1093/molbev/msp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daiyasu H, Ishikawa T, Kuma K, Iwai S, Todo T, Toh H. Identification of cryptochrome DASH from vertebrates. Genes Cells. 2004;9(5):479–495. doi: 10.1111/j.1356-9597.2004.00738.x. [DOI] [PubMed] [Google Scholar]
- 24.Selby CP, Sancar A. A cryptochrome/photolyase class of enzymes with single-stranded DNA-specific photolyase activity. Proc Natl Acad Sci U S A. 2006;103(47):17696–17700. doi: 10.1073/pnas.0607993103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brudler R, Hitomi K, Daiyasu H, Toh H, Kucho K, Ishiura M, Kanehisa M, Roberts VA, Todo T, Tainer JA, Getzoff ED. Identification of a new cryptochrome class. Structure, function, and evolution. Mol Cell. 2003;11(1):59–67. doi: 10.1016/s1097-2765(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Baxter R, Smith BS, Partch CL, Colbert CL, Deisenhofer J. Crystal structure of cryptochrome 3 from Arabidopsis thaliana and its implications for photolyase activity. Proc Natl Acad Sci U S A. 2006;103(47):17701–17706. doi: 10.1073/pnas.0608554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klar T, Pokorny R, Moldt J, Batschauer A, Essen LO. Cryptochrome 3 from Arabidopsis thaliana: Structural and functional analysis of its complex with a folate light antenna. Journal of Molecular Biology. 2007;366(3):954–964. doi: 10.1016/j.jmb.2006.11.066. [DOI] [PubMed] [Google Scholar]
- 28.Mees A, Klar T, Gnau P, Hennecke U, Eker AP, Carell T, Essen LO. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science. 2004;306(5702):1789–1793. doi: 10.1126/science.1101598. [DOI] [PubMed] [Google Scholar]
- 29.Pokorny R, Klar T, Hennecke U, Carell T, Batschauer A, Essen LO. Recognition and repair of UV lesions in loop structures of duplex DNA by DASH-type cryptochrome. Proc Natl Acad Sci U S A. 2008;105(52):21023–21027. doi: 10.1073/pnas.0805830106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiontke S, Geisselbrecht Y, Pokorny R, Carell T, Batschauer A, Essen LO. Crystal structures of an archaeal class II DNA photolyase and its complex with UV-damaged duplex DNA. Embo J. 2011;30(21):4437–4449. doi: 10.1038/emboj.2011.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maul MJ, Barends TR, Glas AF, Cryle MJ, Domratcheva T, Schneider S, Schlichting I, Carell T. Crystal structure and mechanism of a DNA (6-4) photolyase. Angew Chem Int Ed Engl. 2008;47(52):10076–10080. doi: 10.1002/anie.200804268. [DOI] [PubMed] [Google Scholar]
- 32.Hitomi K, Arvai AS, Yamamoto J, Hitomi C, Teranishi M, Hirouchi T, Yamamoto K, Iwai S, Tainer JA, Hidema J, Getzoff ED. Eukaryotic class II cyclobutane pyrimidine dimer photolyase structure reveals basis for improved ultraviolet tolerance in plants. J Biol Chem. 2012;287(15):12060–12069. doi: 10.1074/jbc.M111.244020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glas AF, Schneider S, Maul MJ, Hennecke U, Carell T. Crystal structure of the T(6-4)C lesion in complex with a (6-4) DNA photolyase and repair of UV-induced (6-4) and Dewar photolesions. Chemistry. 2009;15(40):10387–10396. doi: 10.1002/chem.200901004. [DOI] [PubMed] [Google Scholar]
- 34.Hitomi K, DiTacchio L, Arvai AS, Yamamoto J, Kim ST, Todo T, Tainer JA, Iwai S, Panda S, Getzoff ED. Functional motifs in the (6-4) photolyase crystal structure make a comparative framework for DNA repair photolyases and clock cryptochromes. Proc Natl Acad Sci U S A. 2009;106(17):6962–6967. doi: 10.1073/pnas.0809180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zoltowski BD, Vaidya AT, Top D, Widom J, Young MW, Crane BR. Structure of full-length Drosophila cryptochrome. Nature. 2011;480(7377):396–399. doi: 10.1038/nature10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xing W, Busino L, Hinds TR, Marionni ST, Saifee NH, Bush MF, Pagano M, Zheng N. Scf(fbxl3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature. 2013;496(7443):64–68. doi: 10.1038/nature11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brautigam CA, Smith BS, Ma Z, Palnitkar M, Tomchick DR, Machius M, Deisenhofer J. Structure of the photolyase-like domain of cryptochrome 1 from Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2004;101(33):12142–12147. doi: 10.1073/pnas.0404851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheerer P, Zhang F, Kalms J, von Stetten D, Krauss N, Oberpichler I, Lamparter T. The class III cyclobutane pyrimidine dimer photolyase structure reveals a new antenna chromophore binding site and alternative photoreduction pathways. J Biol Chem. 2015;290(18):11504–11514. doi: 10.1074/jbc.M115.637868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walsh C. Flavin coenzymes: At the crossroads of biological redox chemistry. Accounts of Chemical Research. 1980;13(5):148–155. [Google Scholar]
- 40.Heelis PF. The photophysical and photochemical properties of flavins (isoalloxazines) Chemical Society Reviews. 1982;11(1):15–39. [Google Scholar]
- 41.Ghisla S, Massey V. Mechanisms of flavoprotein-catalyzed reactions. Eur J Biochem. 1989;181(1):1–17. doi: 10.1111/j.1432-1033.1989.tb14688.x. [DOI] [PubMed] [Google Scholar]
- 42.Massey V. Activation of molecular oxygen by flavins and flavoproteins. J Biol Chem. 1994;269(36):22459–22462. [PubMed] [Google Scholar]
- 43.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 44.Fraaije MW, Mattevi A. Flavoenzymes: Diverse catalysts with recurrent features. Trends Biochem Sci. 2000;25(3):126–132. doi: 10.1016/s0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- 45.Massey V. The chemical and biological versatility of riboflavin. Biochemical Society Transactions. 2000;28(4):283–296. [PubMed] [Google Scholar]
- 46.Müller F. The flavin redox-system and its biological function, Radicals in biochemistry. Springer Berlin Heidelberg; Berlin, Heidelberg: 1983. pp. 71–107. [DOI] [PubMed] [Google Scholar]
- 47.Senda T, Senda M, Kimura S, Ishida T. Redox control of protein conformation in flavoproteins. Antioxid Redox Signal. 2009;11(7):1741–1766. doi: 10.1089/ars.2008.2348. [DOI] [PubMed] [Google Scholar]
- 48.Fitzpatrick PF. Substrate dehydrogenation by flavoproteins. Accounts of Chemical Research. 2001;34(4):299–307. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- 49.Dagley S. Lessons from biodegradation. Annu Rev Microbiol. 1987;41:1–23. doi: 10.1146/annurev.mi.41.100187.000245. [DOI] [PubMed] [Google Scholar]
- 50.Dong C, Flecks S, Unversucht S, Haupt C, van Pee KH, Naismith JH. Tryptophan 7-halogenase (prna) structure suggests a mechanism for regioselective chlorination. Science. 2005;309(5744):2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Müller F. Chemistry and biochemistry of flavoenzymes. CRC Press; Boca Raton: 1991. [Google Scholar]
- 52.Ghisla S, Massey V, Lhoste JM, Mayhew SG. Fluorescence and optical characteristics of reduced flavines and flavoproteins. Biochemistry. 1974;13(3):589–597. doi: 10.1021/bi00700a029. [DOI] [PubMed] [Google Scholar]
- 53.Sakai M, Takahashi H. One-electron photoreduction of flavin mononucleotide: Time-resolved resonance raman and absorption study. Journal of Molecular Structure. 1996;379(1):9–18. [Google Scholar]
- 54.Massey V, Palmer G. On the existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones*. Biochemistry. 1966;5(10):3181–3189. doi: 10.1021/bi00874a016. [DOI] [PubMed] [Google Scholar]
- 55.Kim ST, Sancar A, Essenmacher C, Babcock GT. Time-resolved EPR studies with DNA photolyase: Excited-state FADH• abstracts an electron from Trp-306 to generate FADH−, the catalytically active form of the cofactor. Proc Natl Acad Sci U S A. 1993;90(17):8023–8027. doi: 10.1073/pnas.90.17.8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kavakli IH, Sancar A. Analysis of the role of intraprotein electron transfer in photoreactivation by DNA photolyase in vivo. Biochemistry. 2004;43(48):15103–15110. doi: 10.1021/bi0478796. [DOI] [PubMed] [Google Scholar]
- 57.Kim ST, Sancar A. Effect of base, pentose, and phosphodiester backbone structures on binding and repair of pyrimidine dimers by Escherichia coli DNA photolyase. Biochemistry. 1991;30(35):8623–8630. doi: 10.1021/bi00099a019. [DOI] [PubMed] [Google Scholar]
- 58.Langenbacher T, Zhao X, Bieser G, Heelis PF, Sancar A, Michel-Beyerle ME. Substrate and temperature dependence of DNA photolyase repair activity examined with ultrafast spectroscopy. J Am Chem Soc. 1997;119(43):10532–10536. [Google Scholar]
- 59.MacFarlane AWt, Stanley RJ. Cis-syn thymidine dimer repair by DNA photolyase in real time. Biochemistry. 2003;42(28):8558–8568. doi: 10.1021/bi034015w. [DOI] [PubMed] [Google Scholar]
- 60.Kao YT, Saxena C, Wang L, Sancar A, Zhong D. Direct observation of thymine dimer repair in DNA by photolyase. Proc Natl Acad Sci U S A. 2005;102(45):16128–16132. doi: 10.1073/pnas.0506586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thiagarajan V, Byrdin M, Eker AP, Muller P, Brettel K. Kinetics of cyclobutane thymine dimer splitting by DNA photolyase directly monitored in the UV. Proc Natl Acad Sci U S A. 2011;108(23):9402–9407. doi: 10.1073/pnas.1101026108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Z, Tan C, Guo X, Kao YT, Li J, Wang L, Sancar A, Zhong D. Dynamics and mechanism of cyclobutane pyrimidine dimer repair by DNA photolyase. Proc Natl Acad Sci U S A. 2011;108(36):14831–14836. doi: 10.1073/pnas.1110927108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Z, Guo X, Tan C, Li J, Kao YT, Wang L, Sancar A, Zhong D. Electron tunneling pathways and role of adenine in repair of cyclobutane pyrimidine dimer by DNA photolyase. J Am Chem Soc. 2012;134(19):8104–8114. doi: 10.1021/ja2105009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan C, Liu Z, Li J, Guo X, Wang L, Sancar A, Zhong D. The molecular origin of high DNA-repair efficiency by photolyase. Nat Commun. 2015;6 doi: 10.1038/ncomms8302. [DOI] [PubMed] [Google Scholar]
- 65.Liu Z, Wang L, Zhong D. Dynamics and mechanisms of DNA repair by photolyase. Physical Chemistry Chemical Physics. 2015;17(18):11933–11949. doi: 10.1039/c4cp05286b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhong D. Electron transfer mechanisms of DNA repair by photolyase. Annu Rev Phys Chem. 2015;66:691–715. doi: 10.1146/annurev-physchem-040513-103631. [DOI] [PubMed] [Google Scholar]
- 67.Brettel K, Byrdin M. Reaction mechanisms of DNA photolyase. Curr Opin Struct Biol. 2010;20(6):693–701. doi: 10.1016/j.sbi.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 68.Saxena C, Wang H, Kavakli IH, Sancar A, Zhong D. Ultrafast dynamics of resonance energy transfer in cryptochrome. J Am Chem Soc. 2005;127(22):7984–7985. doi: 10.1021/ja0421607. [DOI] [PubMed] [Google Scholar]
- 69.Okafuji A, Biskup T, Hitomi K, Getzoff ED, Kaiser G, Batschauer A, Bacher A, Hidema J, Teranishi M, Yamamoto K, Schleicher E, Weber S. Light-induced activation of class II cyclobutane pyrimidine dimer photolyases. DNA Repair (Amst) 2010;9(5):495–505. doi: 10.1016/j.dnarep.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 70.Zhang M, Wang L, Shu S, Sancar A, Zhong D. Bifurcating electron-transfer pathways in DNA photolyases determine the repair quantum yield. Science. 2016;354(6309):209–213. doi: 10.1126/science.aah6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang M, Wang L, Zhong D. Photolyase: Dynamics and mechanisms of repair of sun-induced DNA damage. Photochemistry and Photobiology. 2017;93(1):78–92. doi: 10.1111/php.12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sancar A, Zhong D. It is chemistry but not your grandfather’s chemistry. Biochemistry. 2017;56(1):1–2. doi: 10.1021/acs.biochem.6b01262. [DOI] [PubMed] [Google Scholar]
- 73.Zhong D, Cadet J. Introduction. Photochemistry and Photobiology. 2017;93(1):5–6. doi: 10.1111/php.12722. [DOI] [PubMed] [Google Scholar]
- 74.Kim ST, Li YF, Sancar A. The third chromophore of DNA photolyase: Trp-277 of Escherichia coli DNA photolyase repairs thymine dimers by direct electron transfer. Proc Natl Acad Sci U S A. 1992;89(3):900–904. doi: 10.1073/pnas.89.3.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vicic DA, Odom DT, Núñez ME, Gianolio DA, McLaughlin LW, Barton JK. Oxidative repair of a thymine dimer in DNA from a distance by a covalently linked organic intercalator. J Am Chem Soc. 2000;122(36):8603–8611. [Google Scholar]
- 76.Byrdin M, Lukacs A, Thiagarajan V, Eker APM, Brettel K, Vos MH. Quantum yield measurements of short-lived photoactivation intermediates in DNA photolyase: Toward a detailed understanding of the triple tryptophan electron transfer chain. The Journal of Physical Chemistry A. 2010;114(9):3207–3214. doi: 10.1021/jp9093589. [DOI] [PubMed] [Google Scholar]
- 77.Liu Z, Tan C, Guo X, Li J, Wang L, Sancar A, Zhong D. Determining complete electron flow in the cofactor photoreduction of oxidized photolyase. Proc Natl Acad Sci U S A. 2013;110(32):12966–12971. doi: 10.1073/pnas.1311073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Z, Tan C, Guo X, Li J, Wang L, Zhong D. Dynamic determination of active-site reactivity in semiquinone photolyase by the cofactor photoreduction. The Journal of Physical Chemistry Letters. 2014;5(5):820–825. doi: 10.1021/jz500077s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seidel CAM, Schulz A, Sauer MHM. Nucleobase-specific quenching of fluorescent dyes. 1. Nucleobase one-electron redox potentials and their correlation with static and dynamic quenching efficiencies. The Journal of Physical Chemistry. 1996;100(13):5541–5553. [Google Scholar]
- 80.Gindt YM, Schelvis JP, Thoren KL, Huang TH. Substrate binding modulates the reduction potential of DNA photolyase. J Am Chem Soc. 2005;127(30):10472–10473. doi: 10.1021/ja051441r. [DOI] [PubMed] [Google Scholar]
- 81.Hopfield JJ. Electron transfer between biological molecules by thermally activated tunneling. Proceedings of the National Academy of Sciences. 1974;71(9):3640–3644. doi: 10.1073/pnas.71.9.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402(6757):47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 83.Liu Z, Zhang M, Guo X, Tan C, Li J, Wang L, Sancar A, Zhong D. Dynamic determination of the functional state in photolyase and the implication for cryptochrome. Proc Natl Acad Sci U S A. 2013;110(32):12972–12977. doi: 10.1073/pnas.1311077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harbach PH, Schneider M, Faraji S, Dreuw A. Intermolecular coulombic decay in biology: The initial electron detachment from FADH(−) in DNA photolyases. J Phys Chem Lett. 2013;4(6):943–949. doi: 10.1021/jz400104h. [DOI] [PubMed] [Google Scholar]
- 85.Zimmt MB, Waldeck DH. Exposing solvent’s roles in electron transfer reactions: Tunneling pathway and solvation. The Journal of Physical Chemistry A. 2003;107(19):3580–3597. [Google Scholar]
- 86.Jorns MS, Sancar GB, Sancar A. Identification of oligothymidylates as new simple substrates for Escherichia coli DNA photolyase and their use in a rapid spectrophotometric enzyme assay. Biochemistry. 1985;24(8):1856–1861. doi: 10.1021/bi00329a008. [DOI] [PubMed] [Google Scholar]
- 87.Hassanali AA, Zhong D, Singer SJ. An AIMD study of the CPD repair mechanism in water: Reaction free energy surface and mechanistic implications. The Journal of Physical Chemistry B. 2011;115(14):3848–3859. doi: 10.1021/jp107722z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hassanali AA, Zhong D, Singer SJ. An AIMD study of CPD repair mechanism in water: Role of solvent in ring splitting. The Journal of Physical Chemistry B. 2011;115(14):3860–3871. doi: 10.1021/jp107723w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tachikawa H, Kawabata H. Interaction between thymine dimer and flavin–adenine dinucleotide: A DFT and direct ab initio molecular dynamics study. The Journal of Physical Chemistry B. 2008;112(24):7315–7319. doi: 10.1021/jp801564t. [DOI] [PubMed] [Google Scholar]
- 90.Masson F, Laino T, Tavernelli I, Rothlisberger U, Hutter J. Computational study of thymine dimer radical anion splitting in the self-repair process of duplex DNA. J Am Chem Soc. 2008;130(11):3443–3450. doi: 10.1021/ja076081h. [DOI] [PubMed] [Google Scholar]
- 91.Payne G, Wills M, Walsh C, Sancar A. Reconstitution of Escherichia coli photolyase with flavins and flavin analogues. Biochemistry. 1990;29(24):5706–5711. doi: 10.1021/bi00476a010. [DOI] [PubMed] [Google Scholar]
- 92.Ramsey AJ, Alderfer JL, Jorns MS. Energy transduction during catalysis by Escherichia coli DNA photolyase. Biochemistry. 1992;31(31):7134–7142. doi: 10.1021/bi00146a015. [DOI] [PubMed] [Google Scholar]
- 93.Huels MA, Boudaiffa B, Cloutier P, Hunting D, Sanche L. Single, double, and multiple double strand breaks induced in DNA by 3–100 eV electrons. J Am Chem Soc. 2003;125(15):4467–4477. doi: 10.1021/ja029527x. [DOI] [PubMed] [Google Scholar]
- 94.Stubbe J, Nocera DG, Yee CS, Chang MC. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem Rev. 2003;103(6):2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 95.Prytkova TR, Kurnikov IV, Beratan DN. Coupling coherence distinguishes structure sensitivity in protein electron transfer. Science. 2007;315(5812):622. doi: 10.1126/science.1134862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlcek A, Jr, Richards JH, Winkler JR, Gray HB. Tryptophan-accelerated electron flow through proteins. Science. 2008;320(5884):1760–1762. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 97.Antony J, Medvedev DM, Stuchebrukhov AA. Theoretical study of electron transfer between the photolyase catalytic cofactor FADH− and DNA thymine dimer. J Am Chem Soc. 2000;122(6):1057–1065. [Google Scholar]
- 98.Wang H, Chen X, Fang W. Excited-state proton coupled electron transfer between photolyase and the damaged DNA through water wire: A photo-repair mechanism. Physical Chemistry Chemical Physics. 2014;16(46):25432–25441. doi: 10.1039/c4cp04130e. [DOI] [PubMed] [Google Scholar]
- 99.Prytkova TR, Beratan DN, Skourtis SS. Photoselected electron transfer pathways in DNA photolyase. Proc Natl Acad Sci U S A. 2007;104(3):802–807. doi: 10.1073/pnas.0605319104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Acocella A, Jones GA, Zerbetto F. What is adenine doing in photolyase? The Journal of Physical Chemistry B. 2010;114(11):4101–4106. doi: 10.1021/jp101093z. [DOI] [PubMed] [Google Scholar]
- 101.Li J, Liu Z, Tan C, Guo X, Wang L, Sancar A, Zhong D. Dynamics and mechanism of repair of ultraviolet-induced (6-4) photoproduct by photolyase. Nature. 2010;466(7308):887–890. doi: 10.1038/nature09192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yasui A, Eker AP, Yasuhira S, Yajima H, Kobayashi T, Takao M, Oikawa A. A new class of DNA photolyases present in various organisms including aplacental mammals. Embo j. 1994;13(24):6143–6151. doi: 10.1002/j.1460-2075.1994.tb06961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhao X, Liu J, Hsu DS, Zhao S, Taylor JS, Sancar A. Reaction mechanism of (6-4) photolyase. J Biol Chem. 1997;272(51):32580–32590. doi: 10.1074/jbc.272.51.32580. [DOI] [PubMed] [Google Scholar]
- 104.Faraji S, Dreuw A. Proton-transfer-steered mechanism of photolesion repair by (6-4)-photolyases. The Journal of Physical Chemistry Letters. 2012;3(2):227–230. [Google Scholar]
- 105.Hitomi K, Nakamura H, Kim ST, Mizukoshi T, Ishikawa T, Iwai S, Todo T. Role of two histidines in the (6-4) photolyase reaction. J Biol Chem. 2001;276(13):10103–10109. doi: 10.1074/jbc.M008828200. [DOI] [PubMed] [Google Scholar]
- 106.Yamamoto J, Hitomi K, Hayashi R, Getzoff ED, Iwai S. Role of the carbonyl group of the (6-4) photoproduct in the (6-4) photolyase reaction. Biochemistry. 2009;48(39):9306–9312. doi: 10.1021/bi900956p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Borg OA, Eriksson LA, Durbeej B. Electron-transfer induced repair of 6-4 photoproducts in DNA: A computational study. J Phys Chem A. 2007;111(12):2351–2361. doi: 10.1021/jp0676383. [DOI] [PubMed] [Google Scholar]
- 108.Domratcheva T, Schlichting I. Electronic structure of (6-4) DNA photoproduct repair involving a non-oxetane pathway. J Am Chem Soc. 2009;131(49):17793–17799. doi: 10.1021/ja904550d. [DOI] [PubMed] [Google Scholar]
- 109.Sadeghian K, Bocola M, Merz T, Schutz M. Theoretical study on the repair mechanism of the (6-4) photolesion by the (6-4) photolyase. J Am Chem Soc. 2010;132(45):16285–16295. doi: 10.1021/ja108336t. [DOI] [PubMed] [Google Scholar]
- 110.Yamamoto J, Martin R, Iwai S, Plaza P, Brettel K. Repair of the (6-4) photoproduct by DNA photolyase requires two photons. Angew Chem Int Ed Engl. 2013;52(29):7432–7436. doi: 10.1002/anie.201301567. [DOI] [PubMed] [Google Scholar]
- 111.Yamamoto J, Plaza P, Brettel K. Repair of (6-4) lesions in DNA by (6-4) photolyase: 20 years of quest for the photoreaction mechanism. Photochem Photobiol. 2017;93(1):51–66. doi: 10.1111/php.12696. [DOI] [PubMed] [Google Scholar]
- 112.Faraji S, Dreuw A. Physicochemical mechanism of light-driven DNA repair by (6-4) photolyases. Annu Rev Phys Chem. 2014;65:275–292. doi: 10.1146/annurev-physchem-040513-103626. [DOI] [PubMed] [Google Scholar]
- 113.Faraji S, Dreuw A. Insights into light-driven DNA repair by photolyases: Challenges and opportunities for electronic structure theory. Photochemistry and Photobiology. 2017;93(1):37–50. doi: 10.1111/php.12679. [DOI] [PubMed] [Google Scholar]