

Summary
Effector T cells have the capability of recognizing and killing cancer cells. However, whether tumors can become immune-resistant through exclusion of effector T cells from the tumor microenvironment is not known. By using a tumor model resembling non-T cell-inflamed human tumors, we assessed whether adoptive T cell transfer might overcome failed spontaneous priming. Flow cytometric assays combined with intra-vital imaging indicated failed trafficking of effector T cells into tumors. Mechanistically, this was due to absence of CXCL9/10, which we found to be produced by CD103+ dendritic cells (DC) in T cell-inflamed tumors. Our data indicate that lack of CD103+ DC within the tumor microenvironment dominantly resists the effector phase of an anti-tumor T cell response, contributing to immune escape.
Keywords: Adoptive T cell transfer, immune escape, T cell-inflamed tumor microenvironment, non-T cell-inflamed tumor microenvironment, immunotherapy resistance
Introduction
Immunotherapy has emerged as a potent and effective treatment for multiple cancer types (Brahmer et al., 2012; Hodi et al., 2010; Larkin et al., 2015). The major modalities gaining broad clinical applicability are blockade of immune regulatory checkpoints, such as CTLA-4, PD-L1, and PD-1, as well as adoptive T cell therapy (Dudley et al., 2002; Morgan et al., 2006). Despite the fact that a large and growing number of cancer patients are benefiting from checkpoint blockade and other immunotherapies, a substantial fraction of patients fails to respond clinically (Hugo et al., 2016; Topalian et al., 2016; Tumeh et al., 2014), arguing that understanding and overcoming mechanisms of primary resistance is critical for advancing efficacy. Growing evidence supports the notion that the presence of CD8+ T cells in the tumor microenvironment (TME) at baseline is associated with clinical response to anti-PD-1 immunotherapies (Tumeh et al., 2014), which is consistent with preclinical data demonstrating that most of the therapeutic activity of checkpoint blockade can be mediated through reactivation of T cells already in the TME (Spranger et al., 2014).
Recent work suggests that innate immune sensing of tumors largely occurs through the host STING pathway, which leads to type I interferon (IFN) production, dendritic cell activation, cross-presentation of tumor-associated antigens to CD8+ T cells, and T cell recruitment into the TME (Woo et al., 2014). Furthermore, recent evidence suggests that the dominant chemokines for recruitment of effector CD8+ T cells are those that engage the chemokine receptor CXCR3 (Mikucki et al., 2015). It has been reported that the CXCR3 ligands CXCL9 and CXCL10 (CXCL11 in humans) are not expressed in tumors lacking a CD8+ T cell infiltrate (Harlin et al., 2009); however, the cellular source of these chemokines within the tumor has remained enigmatic. CXCL9 and CXCL10 expression is induced upon type I IFN production by antigen-presenting cells (APCs), and a type I IFN gene signature is also reduced in non-T cell-inflamed melanomas (Fuertes et al., 2013; Padovan et al., 2002). Initial cross-priming of anti-tumor CD8+ T cells has been shown to depend upon Batf3-driven DC, and rejection of immunogenic tumors is ablated in Batf3−/− mice (Broz et al., 2014; Edelson et al., 2010; Engelhardt et al., 2012; Fuertes et al., 2011; Roberts et al., 2016). However, whether Batf3-driven DC might also contribute to effector T cell recruitment into the TME has thus far not been investigated.
Recently, we uncovered a role for tumor cell-intrinsic Wnt/β-catenin pathway activation in mediating the lack of T cell priming against tumor-associated antigens in vivo (Spranger et al., 2015). The mechanism of this effect was mapped to defective recruitment of Batf3-dependent CD103+ DC into the TME, which led to defective early CD8+ T cell priming. Without endogenously generated effector T cells, there was no baseline T cell infiltration into tumors, which led to resistance to checkpoint blockade immunotherapy (Spranger et al., 2015). While these results demonstrate loss of immunotherapy efficacy when endogenous T cell priming is blunted through tumor cell-intrinsic β-catenin activation, they do not address what would occur if an effector T cell response were independently generated in vivo or established by T cell adoptive transfer. We carried out this study to address these important questions.
Results
Adoptively transferred tumor-specific effector T cells fail to control tumor growth in a non-T cell inflamed β-catenin-expressing tumor model
In order to address the question whether adoptive transfer of tumor-specific effector T cells into tumor bearing hosts would overcome the non-T cell inflamed tumor microenvironment we utilized previously established GEM melanoma models, mimicking the T cell-inflamed and non-T cell-inflamed TME (Spranger et al., 2015). In brief, melanomas are induced by a tamoxifen-regulated Cre driven by the Tyrosinase promoter, in concert with specific Cre-inducible oncogenic alterations. The basic model driven by active Braf and bi-allelic Pten deletion (Tyr:CreER;BrafLSL-V600E;Ptenfl/fl (designated BP mice) supports modest T cell infiltration and is responsive to checkpoint blockade therapy (Spranger et al., 2015). These mice have been intercrossed to mice additionally harboring a Cre-inducible SIYRYYGL (SIY) antigen (R26-LSL-SIY-Luc (designated BP-SIY mice) as well as a Cre-inducible stabilized β-catenin allele (LSL-CAT-STA; designated BPC and BPC-SIY mice, respectively). The introduction of the SIY-model antigen enabled us to use SIY-specific 2C T cells isolated from T cell receptor (TCR)-transgenic mice. Isolated 2C T cells were in vitro-activated in the presence of IL-2, IL-7 and IL-15 to generate primed effector cells, then administered at a therapeutic dose of 5×108 cells/kg (Rosenberg and Restifo, 2015) into BP-SIY, BP, BPC-SIY and BPC mice bearing a measurable tumor burden. As a primary readout we assessed tumor growth over time. Adoptive transfer of effector CD8+ T cells resulted in a significant reduction in tumor outgrowth in BP-SIY mice. However, no effect on tumor growth was observed BPC-SIY mice (Figure 1A and B). Similarly, mice bearing SIY-negative tumors exhibit no benefit from adoptive 2C transfer proving an antigen-specific therapeutic effect (Figure 1A and B, open symbols). Thus, T cell adoptive transfer was ineffective against these active β-catenin-expressing tumors.
Figure 1. Adoptively transferred SIY-specific TCR-transgenic effector T cells only control BP-SIY tumor and not BPC-SIY tumors.
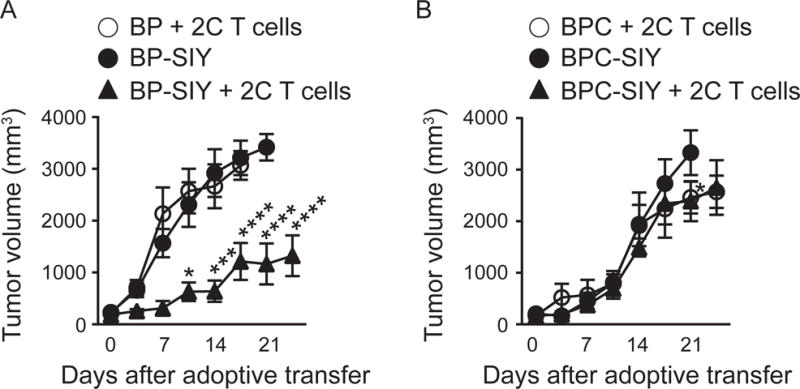
(A, B) Tumor outgrowth curves of untreated BP-SIY (A) and BPC-SIY (B) tumor (filled circle) or after adoptive transfer of 10×106 2C T cells (filled triangle). As controls, antigen-negative BP and BPC mice were treated with the same number of 2C T cells (open symbols). Adoptive transfer was performed at day 21 post-TAM application. Shown are mean with SEM, significance was assessed using a two-sided Anova, n = 2, 8, 8 (BP+2C, BP-SIY, BP-SIY+2C) and n = 4, 8, 6 (BCP+2C, BCP-SIY, BCP-SIY+2C), data are pooled out of two independent experiments. Significance was assumed with p ≤ 0.05, with * ≤ 0.05, *** ≤ 0.001 **** ≤ 0.0001.
Non-T cell-inflamed β-catenin-expressing tumors are resistant to endogenous memory CD8+ T cells
As an alternative approach to generate primed anti-tumor T cells but from the endogenous polyclonal repertoire, we generated a spontaneous antigen-specific anti-tumor memory T cell response by utilizing the immunogenic sarcoma cell line MC57, genetically engineered to express the model antigen SIY (MC57.SIY). These tumors are spontaneously rejected within ten days following implantation into syngeneic C57BL/6 immune-competent mice (Spiotto et al., 2002). Following complete rejection of MC57.SIY tumors in cohorts of GEM bearing each genotype, mice were rested for an additional two months to establish a memory T cell response prior to topical 4OH-tamoxifen (TAM) application for autochthonous tumor induction (Figure 2A). Analysis of SIY-specific CD8+ T cell responses showed comparable effector and memory phases prior to tumor induction in all genotypes of mice (Figure S1A). As a control we confirmed that non-immunized BP-SIY tumors contained CD3+ T cells, while BPC-SIY tumors lacked the infiltration of endogenous T cells (Figure S1B) (Spranger et al., 2015). Following induction of the autochthonous tumor, significantly delayed tumor outgrowth was observed in BP-SIY mice that had been immunized against the SIY epitope (Figure 2B) whereas no protection was observed with BPC-SIY mice (Figure 2C). As expected, no protection was observed with control BP mice that lacked expression of the SIY antigen (Figure 2D). These results further indicate that tumor cell-intrinsic β-catenin activation can result in resistance to antigen-specific effector/memory T cells.
Figure 2. Pre-existing immunological memory increases tumor control in BP-SIY mice but not in BPC-SIY mice in an antigen-dependent manner.
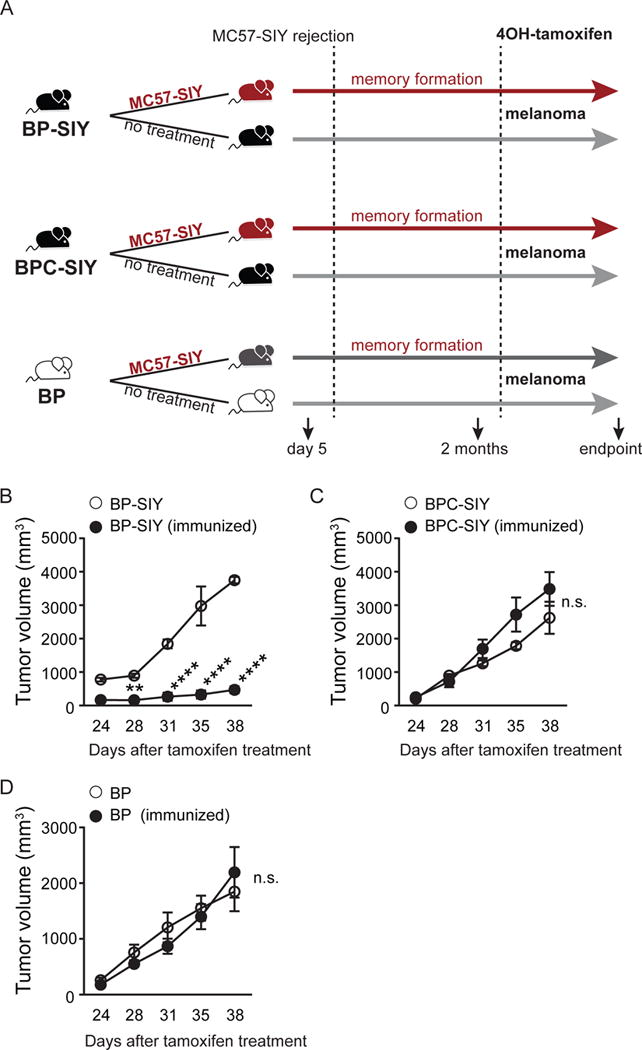
(A) Schematic of experimental procedure to induce immunological memory against tumor-derived SIY. All mice were housed for equal times and were 6–8 weeks of age at the beginning of the experiment. Mice were treated with TAM 2 months after complete rejection of the primary MC57-SIY tumor. At day 24 post-TAM treatment, tumor growth of the autochthonous tumor was assessed until the experimental endpoint was reached. (B–D) Growth of tumors in naive or MC57-SIY immunized BP-SIY (B), BPC-SIY (C), and BP (D) mice as shown in (A). Given are mean with SEM, significance was determined using a two-sided Anova test, n = (B) 4 and 7, (C) 4 and 8, (D) 4 and 4 (naive/immunized) pooled out of two independent experiments. Significance was assumed with p ≤ 0.05, with ** ≤ 0.01; **** ≤ 0.0001. See also Figure S1.
Secondary challenge with antigen is typically associated with re-expansion of antigen-specific T cell populations (Klebanoff et al., 2006). We therefore examined the pattern of secondary immune responses in the respective GEM models that had tumors induced following establishment of SIY-specific memory. As expected, a substantially expanded (6–7-fold) SIY-specific T cell response was detected in the spleen of immunized BP-SIY mice following tumor induction as assessed by IFN-γ ELISPOT (Figure 3A). However, no secondary expansion was observed in the BPC-SIY cohorts (Figure 3A). Similar results were observed using a SIY-Kb pentamer and flow cytometric analysis as a measure of T cell frequencies (Figure 3B–D). When assessing the frequency of SIY-reactive T cells in the spleen as well as the tumor-draining lymph node (TdLN), we failed to observe any meaningful differences with the exception of an increase in pentamer-positive cells in TdLN of immunized BP-SIY tumor- bearing mice (Figure S2A–S2C). These data indicate that β-catenin-positive tumors (BPC-SIY) fail to support re-expansion of a tumor-specific memory T cell response.
Figure 3. Increased tumor control in BP-SIY tumor is associated with reactivation of a peripheral memory T cell response and increased infiltration of antigen-specific T cells into the TME.
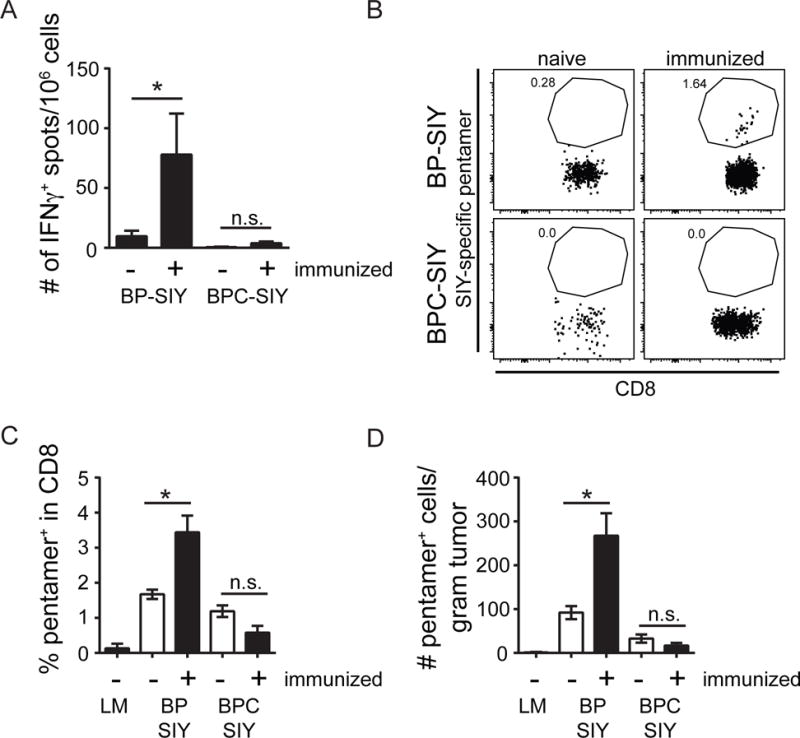
(A) IFN-γ ELISpot assessing the number of SIY-specific IFN-γ production by splenocytes isolated at the end point of the experiment shown in Figure 2. (B–D) The number of antigen-specific cells within the TME assessed though SIY-specific pentamer staining. A representative example of the pentamer staining (B), the percent within the CD8 T cell compartment (C), and the absolute number of SIY-reactive T cells per gram tumor (D) in immunized and non-immunized BP-SIY and BPC-SIY tumors at the endpoint of the experiment. Shown are mean with SEM and n numbers are corresponding to the experiment shown in Figure 2. The tumor weight at time of analysis was 1.65 g ± 0.13 g for BP, 0.9 g ± 0.56 g for BP-SIY and 1.76 g ± 0.19 g for BPC-SIY (mean ± SD). Significance was assumed with p ≤ 0.05, with * ≤ 0.05. See also Figure S2.
Intra-vital imaging reveals differential motility and localization of adoptively transferred T cells depending on the tumor genotype
For control of tumor growth to occur via adoptively transferred 2C T cells or endogenous SIY-specific effector CD8+ T cells, we envisioned that direct tumor cell contact would be required. In order to pursue this question, an intra-vital imaging approach (Schietinger et al., 2013) was adapted to the melanoma GEM model. BP-SIY and BPC-SIY mice were intercrossed with YFP reporter mice, which allowed visualization of transformed cells. TAM application to the flank of the mice was performed such that surgical implantation of the window apparatus could be installed over the tumor. The secure implantation of the frame allowed sequential imaging of the same tumor lesions over the course of multiple days.
Red-fluorescently-labeled effector 2C T cells were adoptively transferred 21 days after TAM application. Transferred effector 2C T cells (red) and tumor cells (green) could be identified in the context of tumor development in vivo (Figure 4A and 4B). While we observed strong infiltration of BP-SIY tumors with effector 2C T cells, we only occasionally observed infiltration of transferred T cells into BPC-SIY tumor lesions (8-fold lower) (Figure 4C). Nonetheless this occasional infiltration of T cell into BPC-SIY tumors allowed us to evaluate the migratory dynamics of the T cells in both tumors. In BP-SIY tumors, the CD8+ effector T cells made direct contact with tumor cells, dynamically screening the tumor through directional migration. In contrast, we failed to observe direct contact between T cells and BPC-SIY tumor cells (Movie S1 and Figure 4D and 4E). Quantitative analysis of individual movement tracks revealed quite distinct overall displacements (Figure S3A and S3B, Movies S2, representative examples corresponding to Movie S1). Quantification over multiple lesions and individual mice revealed a highly significant reduction in T cell velocity, displacement (total traveled distance) and net displacement (directed traveled distance) in BPC-SIY tumors (Figure 4F and 4G and Figure S3C). In addition, the mean distance between T cells and tumor cells was measurably greater in BPC-SIY tumors (Figure 4H and Figure S3D). The measured velocity in BP-SIY tumors is consistent with published rates for settings in which TCR engagement by antigen generates T cell slowing (Mempel et al., 2004). In contrast, the complete lack of T cell movement in BPC-SIY tumors was striking and has not typically been observed in other tumor model systems. Although it is conceivable that the minimal motility observed might be attributed to T cell death, it is noteworthy that the fluorescent label remained intact within the plasma membrane, and flow cytometric analysis in parallel experiments included a live/dead gate supporting viability of these cells (see below Figure 4I). While the observed entry and dynamic motility of effector CD8+ T cells in BP-SIY tumors was ultimately associated with eradication of tumor lesions, it is noteworthy that the pace of tumor cell elimination was slow. Despite the dynamic and direct contact between T cells and tumor cells that was quickly initiated in BP-SIY tumors, tumor eradication took several days (represented in Movie S3 and Figure S3E), indicating that the kinetics of tumor elimination in vivo was much slower than what one might expect from in vitro cytotoxicity assays, as has been proposed by others (Halle et al., 2016).
Figure 4. Effector T cells associated with BP-SIY tumors show enhanced motility and interaction with tumor cells compared to T cells infiltrating BPC-SIY tumors.
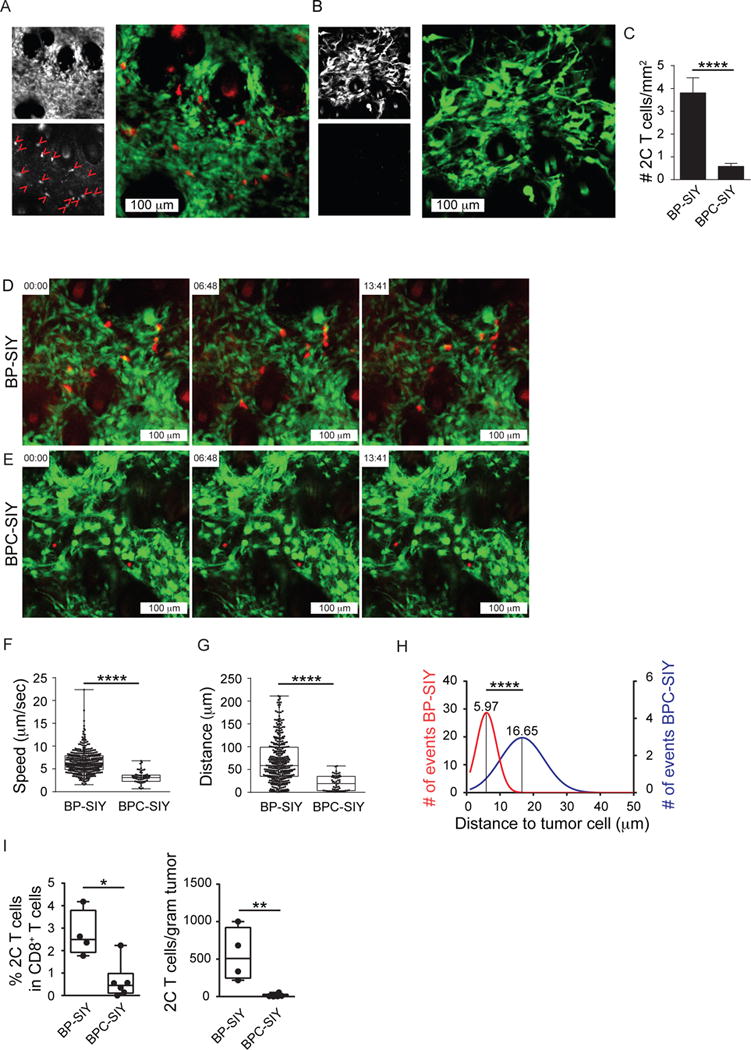
(A, B) Representative images depicting tumor (YFP) and adoptively transferred 2C T cells (deep red) as single channel and merge of BP-SIY tumor (A) and BPC-SIY tumor (B). T cells are highlighted in the red channel with red arrowheads (16 in BP-SIY vs. 0 in BPC-SIY). (C) Numbers of adoptively transferred effector 2C T cells assessed in BP-SIY and BCP-SIY tumors 72 hr post transfer (total of 20 field of view/ genotype were analyzed from 3 independent experiments). Data are shown as mean with SEM. (D, E) Representative images selected from Movie 1 showing 2C effector T cell migrating in BP-SIY (D) or BPC-SIY (E) tumors. (F, G) Velocity (speed, μm/sec) (F) and displacement (distance, μm) (G) of tumor-infiltrating effector 2C T cells assessed 48–72 hr post adoptive transfer (n= 357 (BP-SIY) and 55 (BPC-SIY), pooled out of 20 movies obtained from 3 independent experiments, shown are mean with 95th percentile). (H) Histogram of mean distance between T cell (center) and nearest tumor cell (edge), interaction larger the 50 μm were disregarded (n= 126 (BP-SIY) and 96 (BPC-SIY), pooled out of 40 images obtained from 3 independent experiments). (I) The amount of detectable 2C T cells within the tumor 3 days post adoptive transfer of 1×106 in vitro activated T cells. Left panel shown percent within CD8 T cells and right panel depicts amount total 2C T cells per gram tumor. Box plots show median with 95th percentile, maximal deviation shown by error bars, n = 4 and 6 for BP-SIY and BPC-SIY, respectfully. The tumor weight at time of analysis was 1.65 g ± 0.16 g for BP-SIY and 1.46g ± 0.2 g for BPC-SIY (mean ± SD). Significance was assumed with p ≤ 0.05, with * ≤ 0.05; ** ≤ 0.01; **** ≤ 0.0001. See also Figure S3 and Movies S1–S3.
As an alternative approach to intra-vital microscopy we enumerated the number of effector 2C T cells migrated into the TME of tumor-bearing BP-SIY and BPC-SIY mice using flow cytometry. Whereas 2C effector T cells were found within BP-SIY tumors, almost none were detected in BPC-SIY tumors (Figure 4I). Greater accumulation of 2C T cells was also observed in the TdLN of BP-SIY mice compared to BPC-SIY mice, likely due to secondary re-expansion, whereas the number in the spleen was comparable (Figure S3F). These results suggest a defect at the level of effector T cell trafficking into non-T cell-inflamed tumors.
Together, these data indicate that, whereas β-catenin-negative tumors allow effector T cell entry and dynamic cell contact with antigen-expressing tumor cells ultimately leading to tumor cell killing, β-catenin-positive tumors fail to support meaningful effector T cell entry and dynamics within the TME.
Immune selection of antigen loss variants only occurs within the T cell-inflamed TME
The observed tumor eradication and the significantly delayed tumor outgrowth induced through MC57.SIY immunization suggested that BP-SIY tumors might be immune edited in the presence of a strong anti-tumor immune response, while tumors comprising a non-T cell inflamed TME might not be subject to classical editing. Previous studies have shown that immune editing can lead to the outgrowth of less immunogenic tumor cell clones either lacking antigen or the class I MHC presenting molecule (DuPage et al., 2011; DuPage et al., 2012; Matsushita et al., 2012). Autochthonous tumors were harvested from several mice at the endpoint (Figure 2 B–C) and cells lines were generated (BP-SIY: 3/10; BPC-SIY: 2/8). As readout of immunogenicity the obtained tumor cell lines were co-cultured with CFSE-labeled SIY-specific 2C T cells for 72 hr. As a control we used tumor cells pulsed exogenous SIY-peptide as well as tumor cells established from SIY-negative animals but transduced with a SIY-GFP expression vector. While both cell lines isolated from BPC-SIY mice showed similar capacities to activate 2C T cells, only one BP-SIY tumor cell line (BP-SIY3) showed this ability (Figure 5A–B). Analysis of the BP-SIY1 cell line revealed loss of the SIY mRNA transcript, while all other cell lines retained SIY mRNA expression (Figure 5C). This was not associated with loss of the gene locus (Figure 5D) arguing that epigenetic silencing is likely the mechanism of antigen loss, as has been described previously (DuPage et al., 2011). One of the BP-SIY cells lines (BP-SIY2) was incapable of stimulating T cells even in the presence of exogenous peptide (Figure 5B) and we failed to detect expression of MHC-I, indicating that down-regulation of MHC-I expression is the dominant mechanism in this cells line (Figure 5E). Thus, within our limited sample set, a non-T cell-inflamed TME can result in lack of immune-mediated selection for antigen- or class I MHC-loss variant tumor cells.
Figure 5. Immune surveillance results in a loss of immunogenicity in β-catenin-negative tumor cells but not in β-catenin-positive tumor cells.
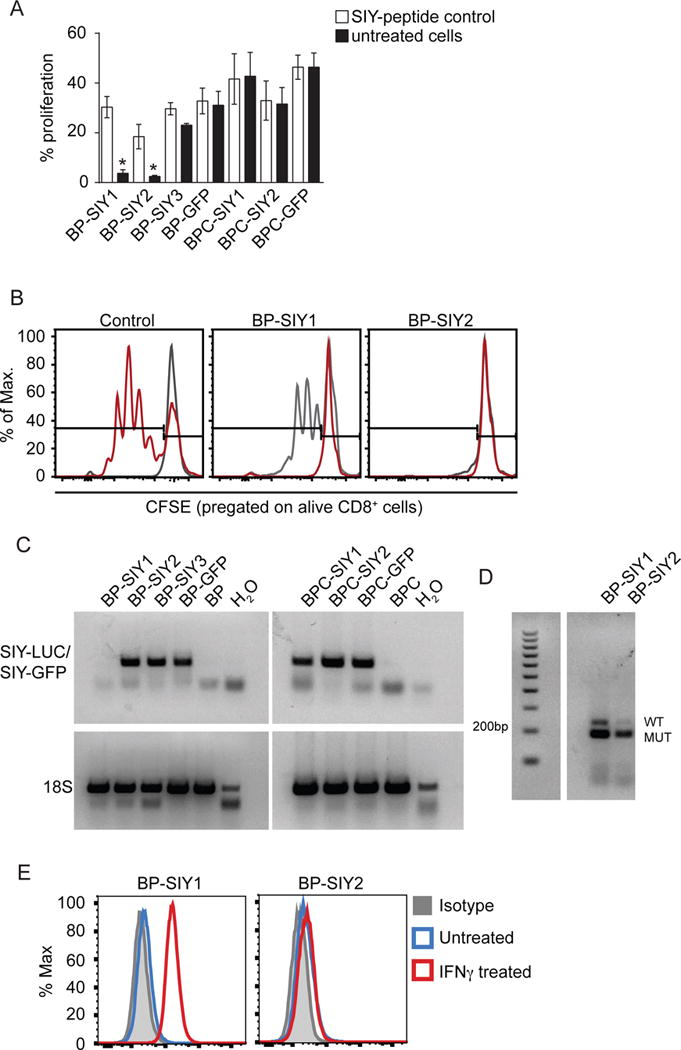
(A) The immune-stimulatory capacity (proliferation) of tumor cell lines isolated from MC57-SIY immunized BP-SIY and BPC-SIY tumor bearing mice. Immune stimulatory potential was assessed by measuring T cell proliferation in a co-culture assay of CFSE-labeled 2C TCR transgenic T cells and untreated or SIY-peptide pulsed tumor cells. We used tumor cells isolated from antigen negative mice transduced with GFP-SIY constructs (here shown as BP-/BPC-GFP) as positive control (data are given as mean with SEM and significance was assumed with p ≤ 0.05, with * ≤ 0.05, n = 8, pooled from two independent experiments). (B) Representative examples of histograms depicting CFSE dilution for control (gray unstimulated, red transduced BP cell lines) and the two cell lines shown defective T cell stimulation. (C) RT-PCR analysis assessing the SIY-mRNA levels in the cell lines used in (A) (representative for two independent experiments). (D) Genotyping PCR confirming the presence of the SIY-LUC transgene in the BP-SIY1 cell line (WT band 235 base pairs, SIY-mutation 190 base pairs). (E) Representative histogram assessing the expression profile of MHC-I on the surface of untreated (blue) or IFN-γ-treated (red) BP-SIY1 and BP-SIY2 cell lines.
Defective migration of effector CD8+ T cells into β-catenin-expressing tumors is associated with alteration in the CXCR3-CXCL9/10 chemokine axis
To investigate whether the accumulation of effector T cells within tumors was chemokine receptor-dependent, in vitro-activated effector 2C T cells were treated for 12h with pertussis toxin (PTX) prior to injection into BP-SIY tumor-bearing hosts. PTX inhibits G-protein-coupled receptors, including chemokine receptors and thereby inhibits chemotaxis-induced migration (Cyster and Goodnow, 1995). As expected, PTX treatment completely blocked the accumulation of effector 2C T cells into BP-SIY tumors (Figure S4A), while transferred cells were still detected in the spleen and TdLN (Spleen: 2C 1.1% ± 0.1 SEM; 2C-PTX 1.4% ± 0.5 SEM; TdLN: 2C 2.5% ± 0.17 SEM; 2C-PTX 2% ± 0.4 SEM). Additionally, we transferred in vitro-activated TCR-transgenic T cells (P14) with an irrelevant specificity (LCMV-gp33 KAVYNFATC), which failed to accumulate in BP-SIY tumor bearing mice, indicating antigen specificity is required for accumulation or retention of tumor specific T cells within the TME. In order to elucidate the chemokines responsible for the recruitment of effector T cells into the TME, the spectrum of chemokine receptors expressed on the endogenous effector T cells infiltrating BP and BPC tumors was assessed. Consistent with the literature (Groom and Luster, 2011; Mikucki et al., 2015) we observed that CD3+ T cells isolated from BP tumors expressed CXCR3 (Figure 6A). Expression of CCR5 was not observed on tumor-infiltrating T cells, which excluded CCL4 as the driving chemokine for T cell infiltration, known to be differentially expressed between BP and BPC tumors (Spranger et al., 2015). Among the few T cells found in BPC tumors, we failed to detect expression of either CCR5 or CXCR3 (Figure 6A).
Figure 6. CXCR3-CXCL9/10 chemokine axis and the presence of CD103+ dendritic cells are associated with the presence of T cells in the TME.
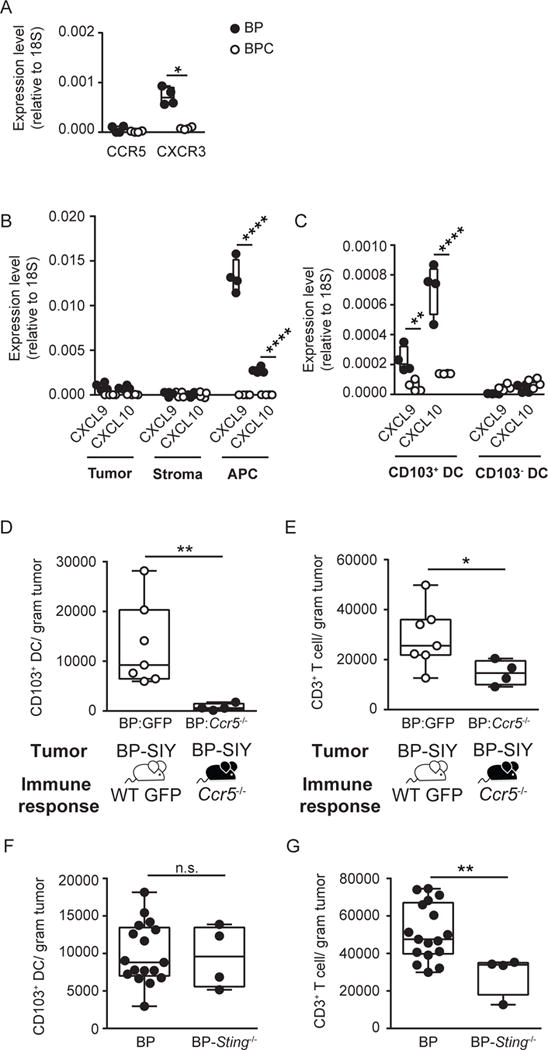
(A) Expression level of chemokine receptors CCR5 and CXCR3 on tumor-infiltrating T cells isolated from BP (filled) and BPC (open) tumors. Expression was assessed on T cells isolated from two tumors per data point via quantitative real-time PCR and normalized to 18S (n = 4; data are pooled out of two independent experiments). The tumor weight at time of analysis was 1.5 g ± 0.15 g for BP and 1.8 g ± 0.28 g for BPC (mean ± SD). (B, C) Expression level of CXCL9 and CXCL10 in tumor (CD45−, YFP+), stroma (CD45−, YFP−) and ACP (CD45+, MHCII+, CD11c+) (B) and on CD103+ and CD103− DC (MHCII+, CD11c+, CD103/CD8α−) (C) isolated from BP (filled) and BPC (open) tumors (n =4; data are pooled out of two independent experiments). The tumor weight at time of analysis across all experiments was 1.4 g ± 0.1 g for BP and 2 g ± 0.14 g for BPC (mean ± SD). (D, E) Ccr5−/− (black circles) and control (GFP, grey circles) bone marrow chimeras with BP hosts were generated and following tumor induction the amount of tumor infiltrating CD103+ DC (D) and CD3+ T cells (E) were assessed and are depicted as number /gram tumor (n = 7 and 4; data are pooled out of two independent experiments). The tumor weight at time of analysis was 0.7 g ± 0.08 g for control chimeras and 0.8 g ± 0.2 g for Ccr5−/− chimeras. (F, G) Absolute amount of CD103+ DC (F) and CD3+ T cells (G) detected in BP (WT) and BP-Sting−/− tumors. The tumor weight at time of analysis was 1.8 g ± 0.5 g for BP and 2.3 g ± 0.6 g for BP-Sting−/− (mean ± SD). Box plots show median with 95th percentile, maximal deviation shown by error bars; significance was assumed with p ≤ 0.05, with * ≤ 0.05, ** ≤ 0.01, **** ≤ 0.0001. See also Figure S4.
We therefore investigated the cellular source of the CXCR3-engaging chemokines, CXCL9 and CXCL10. To distinguish tumor cells from non-tumor host cells, BP and BPC mice were crossed to YFP-reporter mice (Spranger et al., 2015). Following tumor induction, we separated YFP-positive tumor cells, non-hematopoietic stroma cells (CD45−, YFP−) and antigen-presenting cells (CD45+, MHCII+, CD11c+, CD11b+), and performed quantitative RT-PCR for CXCL9 and CXCL10. Tumor-infiltrating APCs produced higher levels of CXCL9 and CXCL10 than did tumor cells. In contrast, no expression of CXCL9 or CXCL10 was detected from any cell type in BPC tumors (Figure 6B). We previously had observed a profound defect in accumulation of CD103+ DC in BPC tumors (Spranger et al., 2015). We therefore evaluated whether CXCL9 and CXCL10 were predominantly made by CD103+ DC. Indeed, the predominant source of CXCL9 and CXCL10 was CD103+ DC, and CXCL10 appeared to be expressed at higher levels than was CXCL9 (Figure 6C). Consistently, the few CD103+ DC from BPC tumors failed to produce those chemokines (Figure 6C). To analyze distinct APC subtypes with higher resolution, we isolated the four major APC sub-types (CD103+ DC: CD45+, MHCII+, CD11c+, CD11bdim, CD103/CD8a+; cDC: CD45+, MHCII+, CD11c+, CD11bdim, CD103/CD8a−; monocyte-derived DC (moDC): CD45+, MHCII+, CD11c+, CD11bhigh; macrophages (MΦ): CD45+, MHCII+, CD11c−, CD11b+) from the tumor microenvironment using flow cytometric sorting and assessed the expression level of the chemokines. Indeed, CD103+ DC were found to be the predominant source for CXCL10, with lesser expression observed in moDC. Similarly, CXCL9 was expressed highest in CD103+ DC but was also expressed in moDC and cDC. Macrophages seemed to express the lowest level of either chemokine (Figure S4B). Additional assessment of CXCL10 protein expression in CD103+ DC by intracellular flow cytometry confirmed CXCL10 protein expression consistent with the mRNA results (Figure S4C and D).
We next took advantage of the observation that the CCL4:CCR5 interaction appeared to be restricted to the recruitment of CD103+ DC (Figure S4E), and generated bone marrow chimeras of Ccr5−/− versus wildtype BM into BP hosts (Spranger et al., 2015). Upon tumor induction, a marked reduction in in the number of CD103+ DC infiltrating into the tumor site was observed, whereas conventional DC appeared to be unperturbed (Figure 6D, Figure S4F). Concomitantly, a significant reduction in the number of tumor-infiltrating T cells was also observed, supporting the notion that the CD103+ DC compartment is necessary for effector T cell accumulation (Figure 6E). We also wanted to examine whether the dendritic cells needed to be activated in order to recruit effector T cells into the TME. Previous work using transplantable tumor models has indicated that the STING pathway of innate immune sensing is necessary within host APCs for optimal IFN-β production and tumor antigen cross-presentation (Woo et al., 2014). CXCL9 and CXCL10 are also produced by DCs in a STING-dependent fashion (Woo et al., 2014). Our previous work had indicated that APCs isolated from BP tumors were producing IFN-β ex vivo, suggesting a similar pathway of DC activation in this GEM model (Spranger et al., 2015). In order to address whether failed DC activation via STING would result in reduced T cell infiltration, we intercrossed BP mice with Sting−/− mice (Ishikawa and Barber, 2008). Analysis of tumors from BP-Sting−/− mice indeed revealed a significant reduction of T cell infiltration compared to wildtype BP mice, despite equal accumulation of CD103+ DC (Figure 6F and G). Taken together, these data strongly support the notion that migration of effector T cells into the TME depends on the production of CXCR3-engaging chemokines by activated DC103+ DC.
Batf3-dependent DC within the TME are required for the recruitment of CD8+ effector T cells
To investigate a direct role for Batf3-lineage CD103+ DC in effector T cell recruitment, we generated CD11c-DTR/Batf3−/− or CD11c-DTR/WT mixed bone marrow chimeras into BP-SIY host mice, as Batf3 is known to be required for development of CD103+ DC (Broz et al., 2014; Edelson et al., 2010). Depletion of DTR-positive DC was performed 21 days post-tumor initiation in order to allow normal development of an anti-tumor immune response. Five days following depletion, in vitro-activated 2C T cells, engineered to express cerulean fluorescent protein, were adoptively transferred intravenously, and homing to the TME was assessed 48h later. Indeed, effector 2C cells were detected within tumors in BP-SIY mice reconstituted with WT bone marrow but not in mice reconstituted with Batf3−/− bone marrow (Figure 7A, Figure S5A). A significant reduction in CD103+ DC was observed in the tumor and periphery in Batf3−/− reconstituted mice compared to WT reconstituted mice (Figure S5B). These results indicate that Batf3-lineage CD103+ DC are necessary for recruitment of effector CD8+ T cells within the TME.
Figure 7. Batf3-driven dendritic cells are sufficient and required for recruitment of effector T cells into the TME.
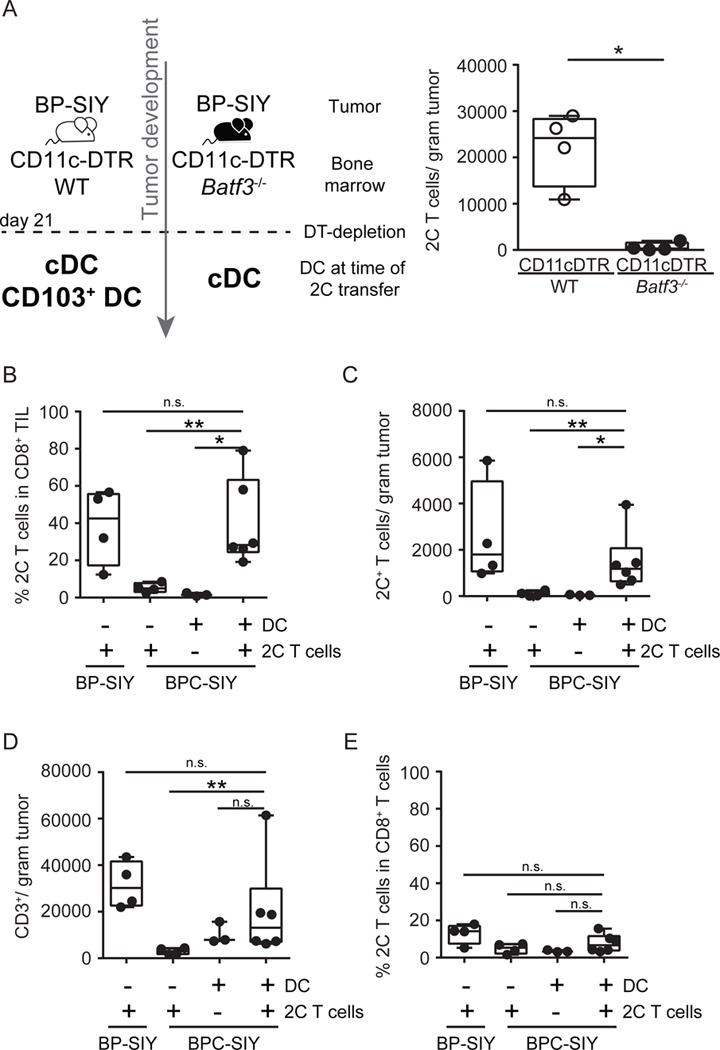
(A) CD11c-DTR/WT (grey circles) and CD11c-DTR/Batf3−/− (black circles) bone marrow chimeras were generated and 21 days following tumor induction diphtheria toxin was given to deplete DTR expression DC. Day 27 post-tumor induction, 1×106 effector T cells were transferred and their accumulation in the tumor was assessed 48h later. Shown are number of 2C T cells detected in the tumor (n = 4). The tumor weight at time of analysis was 0.8 g ± 0.1 g for WT chimeras and 0.6 g ± 0.1 g for Batf3−/− chimeras (mean ± SD). (B–E) BP-SIY and BPC-SIY tumor-bearing mice were injected twice intra-tumorally with Flt3-ligand-derived DC or control injected with PBS 72 hr prior to intravenous injection of effector 2C T cells. 3 days post-T cell injection, the percent (B) and absolute number (C) of 2C T cells and overall T cells (D) in the tumor and the percentage of 2C T cell within CD8+ T cells in the TdLN (E) were assessed and are depicted as number per gram of tumor or percent in CD8+ T cells n = 4, 4, 3 or 6 mice per group (from left to right); data are pooled from two independent experiments). The tumor weight at time of analysis across all experiments was 1.3 g ± 0.1 g for BP-SIY + 2C, 1.7 g ± 0.22 g for BPC-SIY + 2C, 1.5 g ± 0.1 g for BPC-SIY + DC and 1.4 g ± 0.18 g for BPC-SIY + DC + 2C (mean ± SD). Box plots show median with 95th percentile, maximal deviation shown by error bars; significance was assumed with p ≤ 0.05, with * ≤ 0.05; ** ≤ 0.01. See also Figure S5.
To restore Batf3-lineage DC within the TME we prepared Flt3-ligand-derived bone marrow DC and activated them with polyI:C in vitro. Gene expression analysis indicated that these in vitro-derived DC produce high levels of CXCL9 and CXCL10 (Figure S5C). These were injected into BPC-SIY tumors 48 h before intravenous transfer of in vitro-activated effector 2C T cells. T cell accumulation at the tumor site was assessed 72h later and was significantly increased in mice receiving intra-tumoral DC compared to PBS-injected mice (Figure 7B–D). A single injection of Flt3-ligand DC was sufficient to enhance effector 2C T cell recruitment in BPC-SIY tumors to similar levels as observed in BP-SIY mice, while accumulation in the TdLN and spleen seemed to be unaffected (Spleen: 2C 0.8% ± 0.1 SEM; DC + 2C 1.1% ± 0.3 SEM; TdLN: 2C 1.2 % ± 0.34 SEM; DC + 2C 1.9% ± 0.5 SEM). Injection of Flt3-ligand derived DC mildly affected the recruitment of endogenous T cells into BPC-SIY tumors, while transfer of effector 2C T cell had no impact on the endogenous T cell response (Figure 7E). To further affirm that DC-derived CXCL10 is required for effector T cell recruitment, we generated bone marrow chimeras using WT or Cxcl10−/− bone marrow, engrafted into BP-SIY mice (Dufour et al., 2002). Following tumor induction with TAM we performed adoptive T cell transfer of 2C effector T cells and assessed recruitment of such into the TME. Indeed, we detected 2C T cells within the TME of BP-SIY tumors reconstituted with WT bone marrow but failed to detect infiltration in tumor of BP mice reconstituted with Cxcl10−/− bone marrow (Figure S5D). As a complementary approach, we generated bone marrow chimeras using CD11c-DTR donor cells in BP-SIY hosts. This allowed the TME to be established in a T cell-inflamed context and administration of diphtheria toxin resulted in depletion of any endogenous DC. Subsequently, Flt3-ligand-derived WT or Cxcl10−/− DC were injected into the tumor lesions followed by administration of 2C effector T cells. Consistent with the previous results, we observed lack of T cell infiltration into tumors injected with Cxcl10−/− DC while accumulation into WT DC-injected tumors appeared normal (Figure S5E). These data indicate that absence of CXCL10 produced by Batf3-lineage DC in melanomas is rate-limiting for effector T cell recruitment into the TME of β-catenin-expressing tumors, at least in this murine melanoma model.
Batf3-dependent dendritic cells and effector chemokine expression within the TME correlate with effector T cell markers in human melanoma samples
In order to assess whether Batf3-driven DC might also correlate with effector T cell recruitment in human melanomas, we analyzed gene expression data obtained from The Cancer Genome Atlas database (metastatic malignant melanoma, SKCM release date 10 October 2013). In order to assess the presence of Batf3-driven, CD141+ DC within the TME we generated a Batf3-DC score, comprising gene expression of BATF3, IRF8, THBD (encoding CD141), CLEC9A and XCR1, five key genes associated with this subset of DC. Using this score we observed a range of expression of Batf3-DC transcripts within the tumor samples (Figure 8). High expression of the Batf3-DC signature was strongly correlated with expression the CXCR3-binding chemokines CXCL9, CXCL10 and CXCL11 (Figure 8A–C). We further assessed whether the Batf3-DC score would correlate with the presence of effector T cells, assessed by a CD8+ effector T cell score (CD8A, CD8B, IFNG, PRF1). Indeed, a striking correlation was observed between the Batf3-DC score and the CD8+ effector T cell signature (Figure 8D), indicating that also in human melanoma tumor-residing Batf3-DC might be a dominant component in effector T cell recruitment into the TME.
Figure 8. Batf3-driven DC gene signature correlates with CXCL9/10/11 expression and effector T cell gene signature in human melanoma patients.
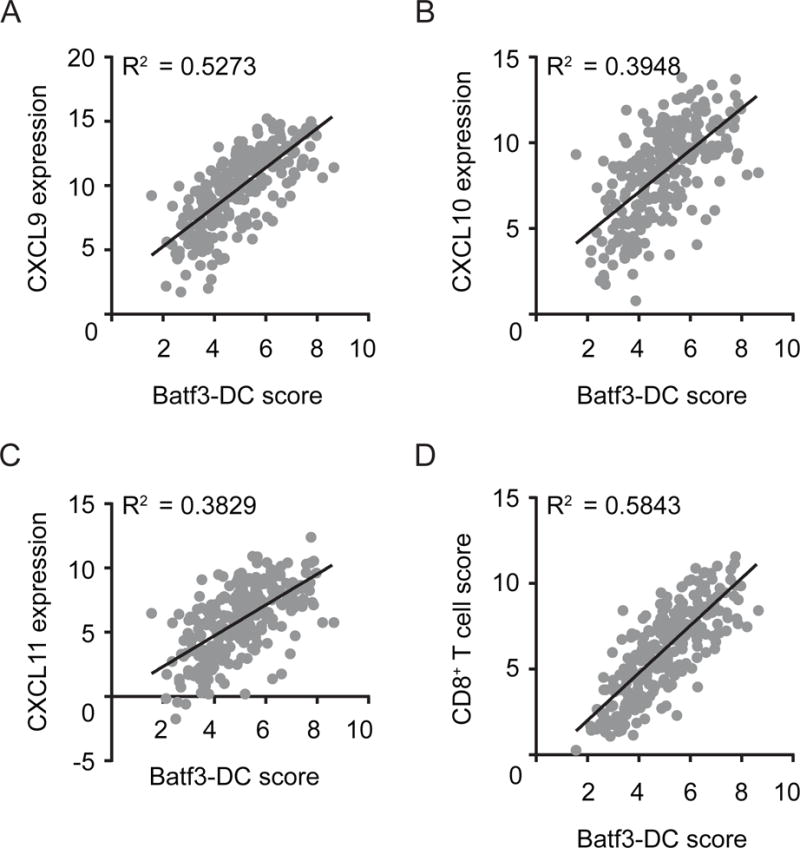
(A–C) Gene expression (normalized and log2 transformed) of CXCL9 (A) CXCL10 (B) and CXCL11 (C) from 266 human metastatic melanoma samples were plotted against a Batf3-DC score. (D) Correlation between the CD8+ effector T cell score and the Batf3-DC score. Correlation is shown using R2 and significance was determined using a Spearman correlation (p< 0.0001 for all correlations).
Discussion
Developing therapeutic strategies that help overcome a lack of T cell infiltration into the TME will be critical in order to increase the fraction of patients responding to immunotherapy. Our current work provides evidence that tumor-residing CD103+ DC are required for the recruitment of effector T cells into the TME and are a major component of the establishment of the T cell-inflamed tumor phenotype. Tumor-residing CD103+ DC were necessary for CD8+ effector T cell recruitment and their absence in β-catenin-expressing tumors resulted in failed T cell recruitment, and reconstitution of activated Batf3-DC restored effector T cell migration into the TME. In addition, a Batf3-DC gene signature was associated with an effector T cell phenotype in human melanoma metastases. Together, these data suggest that strategies to restore Batf3-DC recruitment and activation within the tumor microenvironment could have therapeutic utility in restoring effector T cell accumulation and clinical activity of checkpoint blockade and other immunotherapies in non-T cell-inflamed tumors.
T cell infiltration into the TME has been observed to correlate with efficacy of immunotherapeutic interventions including checkpoint blockade (Spranger et al., 2014; Tumeh et al., 2014). Productive infiltration includes CD8+ effector T cells and IFN-γ-producing CD4+ Th1 cells (Nishimura et al., 2000; Yu et al., 2005). Recruitment of these effector cell subsets has been associated with the expression of the chemokine receptor CXCR3 (Mikucki et al., 2015). Analysis of human tumors by gene expression profiling also has revealed a correlation between the presence of CD8+ T cells and expression of the CXCR3 ligands CXCL9 and CXCL10 (Harlin et al., 2009; Salerno et al., 2014). In our current work using autochthonous melanoma models, T cells infiltrating tumors were also found to express CXCR3. However, the critical cellular source of the corresponding chemokines CXCL9 and CXCL10 had not been known, and we found that the predominant source of CXCR3-ligands in the T cell-inflamed TME was the CD103+ DC population. These results are similar to what has recently been reported in the context of Listeria monocytogenes infection, where Batf3-lineage DC were found not only to recruit effector memory T cells but also to reactivate them at the site of infection (Alexandre et al., 2016). Our bone marrow chimera experiments with Batf3−/− hematopoietic cells clearly indicated that effector CD8+ T cells failed to migrate into tumors in the absence of CD103+ DC. Similarly, transfer of Ccr5−/− bone marrow, which eliminated migration of CD103+ DC via CCL4 produced by tumor cells (Spranger et al., 2015), also resulted in failed CD8+ effector T cell trafficking. Consistent with these results, deletion of Cxcl10 from the hematopoietic cell compartment or only the DC compartment resulted in a failed effector T cell recruitment into the tumor. Although previous work has suggested redundancy for CXCL9 and CXCL10 within the tumor microenvironment, we observed a requirement of CXCL10 derived from hematopoietic cells for effector T cell recruitment (Mikucki et al., 2015). This work is consistent with the original characterization of immune responses in Cxcl10−/− mice, which indicate significantly reduced inflammatory T cell response in peripheral tissues (Dufour et al., 2002). These data establish that the primary source of chemokines for effector T cell trafficking into the TME is the Batf3-lineage DC subset.
Additionally, the intra-vital imaging technique provided evidence that the presence of CD103+ DC might be associated with a highly motile behavior of effector T cells within the TME. A decrease in T cell velocity has been described for tissue-residing memory CD8+ T cells, a subset of T cells migrating and residing within peripheral tissues (Bottcher et al., 2015). Due to the small number of cells this might be a conceivable explanation for the non-migratory phenotype observed in β-catenin-expressing tumors, in particular since migration of tissue residing memory T cells appears to be independent of effector T cell chemokines. Additional experiments will be needed to fully understand the dynamic interaction of DC and T cells in DC-rich compared to DC-deprived tumor microenvironments.
It was noteworthy that the secondary expansion of CD8+ memory T cells failed to occur in the context of induced β-catenin-expressing tumors in our model. Since those tumors lack Batf3-lineage DC, these results argue that antigen presentation during the secondary challenge is also mediated by that DC subset. The fact that secondary expansion was seen both in the tumor-draining lymph node and in the second tumor site suggest that the site of memory expansion may be the lymph node, although direct expansion within the tumor itself cannot be excluded (Joshi et al., 2015). Our previous work had demonstrated that initial CD8+ T cell priming in response to a primary tumor also is dependent upon Batf3-lineage DC, with the initial site of CD8+ T cell proliferation being observed in the draining lymph node (Spranger et al., 2015). Together with our current work, these observations suggest that a subset of Batf3-lineage DC migrates to the draining lymph node to mediate T cell cross-priming, while another subset remains in the tumor site to produce CXCR3 ligands to recruit CD8+ effector T cells back to the target tissue. This notion is supported by a recent study combining systemic Flt3-ligand injection with intra-tumoral polyI:C in the BPC mouse model (Salmon et al., 2016). This therapeutic regimen not only enhanced CD103+ DC within the TME but also allowed effector T cell infiltration into the tumor.
Our results have several important implications for the clinic. First, they suggest that immune editing could provide evolutionary pressure on tumors not only to yield antigen-loss or class I MHC-negative variants but also to select for the outgrowth of tumor cells expressing an immune-evasive oncogene pathway such as β-catenin. Other ancillary oncogene pathways likely contribute to immune resistance in other tumor subsets, such as PI3K-pathway activation (Peng et al., 2015), and it will be of interest to determine whether Batf3-DC are also poorly recruited into those human tumors. Our current results do not exclude other potential mechanisms of immune evasion. Second, the fact that CD103+ DC-negative tumors are resistant to effector and memory CD8+ T cell responses suggests that adoptive T cell therapy also may be ineffective against tumors with oncogene-mediated DC/T cell exclusion, such as β-catenin pathway activation. This is a hypothesis that could be evaluated through tumor biopsies and genomic analysis in the setting of adoptive T cell-based clinical trials. Third, our data suggest that a critical rate-limiting step for initiating endogenous T cell responses against cancer may be the recruitment and activation of Batf3-lineage DC. Analysis of the melanoma TCGA data set has revealed a positive correlation between Batf3-driven DC subset, effector T cell chemokine production and a T effector gene signature, suggesting that tumors lacking this key DC subset may fail to prime and recruit CD8+ T cells. In our mouse BP melanoma model, the initial recruitment of CD103+ DC is dependent upon CCL4 produced by tumor cells, a chemokine which also shows a positive correlation with CD8 transcripts in human melanomas (Spranger et al., 2015). But in addition to their recruitment into the tumor site, those CD103+ DC require activation in order to mediate T cell priming and likely effector T cell recruitment. In our mouse BP melanoma model, tumor-infiltrating DC produce IFN-β, which indicates activation via an innate signaling pathway (Spranger et al., 2015). Our previous work combined with results presented here suggests the STING/TBK1/IRF3 pathway as critical for IFN-β production and DC cross-presentation (Woo et al., 2014). Taken together, these collective observations argue that strategies to restore both the recruitment and the activation of Batf3-lineage DC within non-T cell-inflamed tumors should be evaluated as an approach to expand the fraction of patients that respond to immunotherapy.
Star Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Thomas F. Gajewski (tgajewsk@medicine.bsd.uchicago.edu). Please note, that additional Material Transfer agreements with third parties will be needed to obtain BP, BPC, or any related mouse strain.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The following mouse strains were kind gifts from collaborators and were used to generate the models used in this study: Tyr:Cre-ER (gifted by Lynda Chin), loxP-BrafV600E (kindly provided by Martin MacMahon), loxP-Pten (provided by Tak Mak), loxP-CAT-STA (provided by Fotini Gounari), loxP-rosa-SIY and loxP-rosa-YFP (Jackson Laboratories, strain 006148) (Bosenberg et al., 2006; Dankort et al., 2009; DuPage et al., 2012; Gounari et al., 2002; Sirma Ekmekci et al., 2012; Spranger et al., 2015). Genotyping was performed as described previously(Spranger et al., 2015), mice were used at 6–12 weeks of age and gender matched and reandomized into treatment groups. 2C TCR-Tg mice were maintained as a source of SIY-specific CD8+ T cells (Manning et al., 1997) and RAGN12-F were obtained from Taconic Farms as hosts for generating tumor cell lines. Ccr5−/−, CD11c-DTR, Cxcl10−/− and Batf3−/− mice were obtained from Jackson laboratories, mice were used at 6–12 weeks of age. All mice were house under standard special pathogen free conditions. All animal procedures were approved by the IACUC Committee of the University of Chicago.
Cell lines
MC57-SIY cells were a gift from Dr. Hans Schreiber and were maintained as described previously (Spiotto et al., 2002). For primary tumor rejection as a vaccination, 1×106 MC57-SIY cells were injected subcutaneously on the flank of the GEMs (at 6–10 weeks of age).
METHOD DETAILS
Autochthonous tumor induction, tissue harvest and generation of cell lines
For induction of the autochthonous tumors, mice were shaved on the back and 5 μl of 4-OH-Tamoxifen (Sigma) at a concentration of 10 μg/ml (dissolved in acetone) was applied. Mice were screened weekly for tumor induction. Tumor masses were measured by assessing length, width and height of major tumor mass using a digital caliper. Tumor volume was calculated using the formula TV= TL*TW*TH, since the tumor shape was rectangular and flat rather than spherical. The maximum tumor size was reached when the tumor mass reached approximately 10% of the body weight. At the indicated experimental endpoint, tumor tissue was harvested and single cell a suspension was prepared as described previously (Spranger et al., 2015). For tumor cell line generation, a single cell suspension of the tumor tissue was generated and used in its entirety for subcutaneous injection into Rag2-KO mice (RAGN12-F; Taconic). Following tumor outgrowth, the tumor tissue was harvested and reinjection into Rag2-KO mice and subsequently adapted to cell culture using DMEM (Gibco) with 10% FCS (Atlanta Biologics), 1x NEAA (Gibco) and 1x MOPS (Sigma). Prior to FACS staining for MHC-class I, cells were cultured for 24 h in the presence of 100μg/ml IFN-γ (Biolegend).
Adoptive T cell transfer
Tumor development was induced and intravenous transfer of 1×106 or 10×106 T cells was performed when tumors reached 600 – 1000 mm3 (5–6 weeks after induction). Transferred T cells were isolated from gender-matched 2C donor mice using the Miltenyi CD8+ enrichment Kit II for untouched CD8+ T cell isolation. Cells were activated for three days through plate-bound anti-CD3 (0.2 μg/ml; 145-2C11 clone; Biolegend) and anti-CD28 (0.5 μg/ml; 37.51 clone; BD) antibodies. Following activation, T cells were stained with 1 μM CFSE-solution (eBioscience) for 8 min at 37°C before intravenous injection. Tumor tissue, tumor-draining LNs, and spleen were harvested 3 days following adoptive transfer and used for flow cytometric analysis. This short time frame was chosen to avoid the reported leakiness of the SIY-transgene that has been associated with partial T cell activation within the spleen and to assure no additional proliferation would take place. For tumor tissues, the entirety of each sample was acquired and total number of CD3+CD8+ T cells and transferred 2C cells was assessed. The percentage 2C cells was calculated as [(100/CD3+/CD8+ T cells)*2C] and also the number of 2C cells per gram tumor. For therapeutic adoptive transfer experiments, T cell medium was supplemented with huIL-2, muIL-7 and muIL-15 at 5 ng/ml final concentration (Peprotech) and cells were cultured for 7 days prior to transfer. For adoptive transfer prior to intravital imaging, no IL-7 or IL-15 was included, and cells were labeled with CellTracker Deep Red (ThermoFisher) prior to adoptive transfer.
IFN-γ ELISpot
Splenocytes from naïve, tumor-challenged non-treated or treated mice were harvested on day 7 or day 14 after tumor inoculation. Single cell suspensions were prepared and 1×106 splenocytes were assayed per well. Cells were either left un-stimulated or stimulated with 160nM SIY-peptide (SIYRYYGL) or PMA 100ng/ml and Ionomycin 1μg/ml as positive control. After a 24 hr culture period, detection of INF-γ-producing T cells was performed according to manufacturer’s instructions using Millipore ELISpot plates.
Flow cytometry
For flow cytometric analysis, washed cells were resuspended in staining buffer (PBS with 10% FCS and 0.5 M EDTA (Ambion). Cells were incubated with live/dead staining dye (Invitrogen, wavelength 450) and Fc Block (clone 93; Biolegend) for 20 min on ice. Subsequently, specific antibodies were added (see resource table) and staining was continued for 40 min on ice. After a washing step, cells were either analyzed directly or fixed with 4% PFA (BD) solution for 30 min and stored in a 1 % PFA solution until analysis. Staining of SIY-specific cells was performed using the SIYRYYGL-pentamer (Proimmune), conjugated with Phycoerythrin (PE), or as a non-specific control with the SIINFEKL-pentamer. For staining, pentamers were diluted 1:50 in PBS + 10% FCS and incubated for 20 min at room temperature (RT). Following a washing step, cells were stained with specific antibodies for 30 min on ice prior to fixation in 4% PFA. For staining of 2C TCR Tg T cells a TCR-specific biotinylated mAb (1B2 clone) was obtained from the University of Chicago Monoclonal Core Facility. Subsequent to live/dead staining, TCR-specific mAb was added for 15 min on ice at a 1:100 dilution alone with surface Abs targeting other antigens added in for an additional 25 min thereafter. After a washing step, a 1:500 dilution of Streptavidin APC was added and incubated on ice for 20 min before cells were fixed in 4% PFA and stored in 1% PFA solution. For intracellular chemokine staining, Brefeldin A (Sigma) at 10 μg/ml was injected subcutaneously beneath the tumor 5 min before euthanasia of the animals and was included in all buffers used starting with the initial digestion buffer. Intracellular staining was performed using the FoxP3 staining kit (BD) according to manufacturer’s instructions. Flow cytometry sample acquisition was performed on a LSR2B (BD), and analysis was performed using FlowJo software (TreeStar). For cell sorting, staining protocols were carried out similarly under sterile conditions.
Intra-vital imaging and analysis
We adapted an imaging method previously described for transplantable tumors (Schietinger et al., 2013). Tumors were induced with TAM on the flank of the mice. Twenty-one days following tumor induction, 2C TCR Tg effector T cells labeled with cell tracker deep red (Life technologies) were adoptively transferred by intravenous injection. 24 hr post-adoptive transfer we implanted window chambers onto of the induced tumor moles imaged 24 hr later. For imaging, mice were anesthetized using isoflurane inhalation. The window was fastened to the stage of the microscope using a custom-made holder (Schietinger et al., 2013). A motorized microscope XY scanning stage and Leica LAS-AF software allowed recording individual 3-dimensional positions per field-of-view and returning to them later with high precision (stated accuracy +/− 3 μm; reproducibility < 1.0 μm). Confocal images were captured with a Leica SP5 II TCS Tandem scanner spectral confocal with 4 × and 25 × /0.45 LWD IR objectives from Olympus. The optical penetration of tissue ranged between 90–150 μm with the average of 120–150 μm. We used the 488 nm and 633 nm excitation laser lines. All images and movies were acquired with a 4.12 sec exposure time. Analysis was done using ImageJ (NIH) and MtrackJ plug-in software (Meijering et al., 2012). MtrackJ software enabled assessment of velocity as well as displacement for each analyzed T cell. Spider plots were obtained from single time point analysis. Net-displacement was calculated as √[(Xend)2+(Yend)2]- √[(Xstart)2+(Ystart)2]. Distance between T cell (center) and tumor cell (nearest edge) was assessed using ImageJ ROI manager.
Functional antigen-detection assay
CD8+ T cells from 2C donor mice were isolated using the Miltenyi CD8+ enrichment Kit II for untouched CD8+ T cell isolation. After isolation, cells were stained with 1 μM CFSE-solution (eBioscience) for 8 min at 37°C. Cell lines established from BP-SIY and BPC-SIY mice were co-cultured with CFSE-labeled 2C TCR-transgenic T cells for 72 hr at a ratio of 1 tumor cell (4000 cells) to 10 T cells (40000 cells). As a positive control, tumor cells were pulsed with 100mM SIY peptide for 1 hr prior to co-culture. Additionally, BP and BPC tumor cells derived from BP and BPC mice, respectively, were transduced with a retroviral vector containing a SIY-GFP fusion protein facilitating expression of the antigen as previously described (Kline et al.). Following co-culture, cells were harvested, stained with a live/dead dye as well as for CD3 and CD8 and subsequently analyzed for dilution of CFSE.
Molecular antigen detection assay
To assay for the presence of the SIY antigen within tumor cells, RNA was isolated and cDNA generated using Qiagen RNA Isolation Kit and Applied Biosystems Reverse Transcriptase Kit (according to manufactures instructions). PCR was performed using primers specific for the luciferase gene in case of BP-SIY and BPC-SIY derived cell lines or GFP for in vitro transduced cell lines with amplicons of 183 base pairs and 182 base pairs, respectively. 18S was used as a reference control.
Gene expression analysis
To assess the level of gene expression, RNA was isolated using the Qiagen mRNA kit or the micro mRNA kit, dependent on the cell number. Isolated RNA was revere transcribed in cDNA using the Applied Biosystems Reverse Transcriptase Kit (according to manufactures instructions) and subsequently used for quantitative PCR analysis. We used 1 μl of cDNA per reaction in a final volume of 20 μl. Primer were used at 10 pM final concentration and are listed in Table 2. Analysis was conducted as described in (Schmittgen and Livak, 2008) and only samples were analyzed for which the house keeping gene (18S) was at least detected at a CT of 15 or smaller.
Bone marrow chimeras and dendritic cell depletion
To generate bone marrow chimeras, BP or BP-SIY mice were irradiated with 500 rad followed by a second dose of 550 rad 3 hr apart. To obtain donor bone marrow, femurs and tibiae were harvested and the bone marrow flushed out. After washing, total bone marrow cells were resuspended in PBS for transfer into mice 24 hr after irradiation. For Ccr5−/− bone marrow chimeras, 5×106 cells were injected per mouse; for CD11c-DTR/WT and CD11c-DTR/Batf3−/− mixed chimeras, 5×106 cells of a 1:1 mixture were injected and allowed to engraft for 8 weeks before we initiated the experiments. For depletion of DC, diphtheria toxin (500ng/dose/mouse) was administered i.p. 21 days post-tumor induction for 7 days (given every other day), and transfer of in vitro activated 2C T cell was performed on day 27.
Human tumor gene expression profiling
The gene expression of 266 malignant melanoma samples were downloaded, processed as described previously (Spranger et al., 2015). For gene expression correlation analysis, the expression level of each gene of interest was used following normalization and log2-transformation. To generate the Batf3-DC score and CD8+ effector T cell score, gene expression of BATF3, IRF8, THBD, CLEC9A and XRC1 or CD8A, CD8B, IFNG and PRF1, respectfully, were averaged. Statistical assessment was performed using a Spearman correlation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
All statistical analyses were performed using GraphPad Prism (GraphPad). Unless otherwise noted in the figure legend, all data are shown as mean +/− SEM combined with a two-tailed Mann-Whitney U test. Box plots show median with 95th percentile, maximal deviation shown by error bars. Significance was assumed with p ≤ 0.05, with * ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001, **** ≤ 0.0001, ***** ≤ 0.00001. All experiments shown were repeated at least in two independent experiments.
Supplementary Material
Significance.
Immunotherapeutic interventions, such as checkpoint blockade therapy and adoptive T cell transfer, are providing a therapeutic benefit for many cancer patients, including patients with malignant melanoma. Nonetheless, not all patients response equally well to those interventions and both primary and secondary resistance is being observed clinically. In the current study we show that failed accumulation of CD103+ dendritic cells, a cell type which is the major source of the T cell-recruiting chemokines CXCL9/10, in non-inflamed tumors mediates deficient entry of therapeutically activated T cells and immunotherapy resistance. Therefore, absence of CD103+ DC from the tumor microenvironment may be a dominant mechanism of resistance to multiple immunotherapies.
Acknowledgments
The authors would like to thank Tamara Cox and Michael Leung for support with mouse breeding and genotyping, the light microscopy core facility at the University of Chicago for excellent support, and Ainhoa Arina for advice regarding the establishment of the intra-vital imaging technology.
Funding. This study was supported by a Team Science Award from the Melanoma Research Foundation. SS was recipient of the Irvington Postdoctoral fellowship awarded by the Cancer Research Institute (2014–2016) and is currently supported by NCI (K99CA204595).
Footnotes
Authors’ contributions
SS designed and performed experiments, DD assisted with bone marrow chimera experiments, BH assisted with establishment of the imaging technology, TG and SS designed the overall study and wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
- Alexandre YO, Ghilas S, Sanchez C, Le Bon A, Crozat K, Dalod M. XCR1+ dendritic cells promote memory CD8+ T cell recall upon secondary infections with Listeria monocytogenes or certain viruses. The Journal of experimental medicine. 2016;213:75–92. doi: 10.1084/jem.20142350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosenberg M, Muthusamy V, Curley DP, Wang Z, Hobbs C, Nelson B, Nogueira C, Horner JW, 2nd, Depinho R, Chin L. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis. 2006;44:262–267. doi: 10.1002/dvg.20205. [DOI] [PubMed] [Google Scholar]
- Bottcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Hochst B, Eickhoff S, Rieckmann JC, Russo C, Bauer T, et al. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nature communications. 2015;6:8306. doi: 10.1038/ncomms9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer cell. 2014;26:638–652. doi: 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, Goodnow CC. Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. The Journal of experimental medicine. 1995;182:581–586. doi: 10.1084/jem.182.2.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature genetics. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. Journal of immunology. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, Jacks T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer cell. 2011;19:72–85. doi: 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. The Journal of experimental medicine. 2010;207:823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, Krummel MF. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer cell. 2012;21:402–417. doi: 10.1016/j.ccr.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. The Journal of experimental medicine. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends in immunology. 2013;34:67–73. doi: 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounari F, Signoretti S, Bronson R, Klein L, Sellers WR, Kum J, Siermann A, Taketo MM, von Boehmer H, Khazaie K. Stabilization of beta-catenin induces lesions reminiscent of prostatic intraepithelial neoplasia, but terminal squamous transdifferentiation of other secretory epithelia. Oncogene. 2002;21:4099–4107. doi: 10.1038/sj.onc.1205562. [DOI] [PubMed] [Google Scholar]
- Groom JR, Luster AD. CXCR3 in T cell function. Experimental cell research. 2011;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle S, Keyser KA, Stahl FR, Busche A, Marquardt A, Zheng X, Galla M, Heissmeyer V, Heller K, Boelter J, et al. In Vivo Killing Capacity of Cytotoxic T Cells Is Limited and Involves Dynamic Interactions and T Cell Cooperativity. Immunity. 2016;44:233–245. doi: 10.1016/j.immuni.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer research. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity. 2015;43:579–590. doi: 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunological reviews. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine. 2015 doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning TC, Rund LA, Gruber MM, Fallarino F, Gajewski TF, Kranz DM. Antigen recognition and allogeneic tumor rejection in CD8+ TCR transgenic/RAG(−/−) mice. Journal of immunology. 1997;159:4665–4675. [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods in enzymology. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nature communications. 2015;6:7458. doi: 10.1038/ncomms8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Nakui M, Sato M, Iwakabe K, Kitamura H, Sekimoto M, Ohta A, Koda T, Nishimura S. The critical role of Th1-dominant immunity in tumor immunology. Cancer chemotherapy and pharmacology. 2000;46(Suppl):S52–61. doi: 10.1007/pl00014051. [DOI] [PubMed] [Google Scholar]
- Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. Journal of leukocyte biology. 2002;71:669–676. [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer discovery. 2015 doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF. Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer cell. 2016 doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno EP, Olson WC, McSkimming C, Shea S, Slingluff CL., Jr T cells in the human metastatic melanoma microenvironment express site-specific homing receptors and retention integrins. International journal of cancer Journal international du cancer. 2014;134:563–574. doi: 10.1002/ijc.28391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44:924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Arina A, Liu RB, Wells S, Huang J, Engels B, Bindokas V, Bartkowiak T, Lee D, Herrmann A, et al. Longitudinal confocal microscopy imaging of solid tumor destruction following adoptive T cell transfer. Oncoimmunology. 2013;2:e26677. doi: 10.4161/onci.26677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Sirma Ekmekci S, C GE, Kandilci A, Gulec C, Akbiyik M, Emrence Z, Abaci N, Karakas Z, Agaoglu L, Unuvar A, et al. SET oncogene is upregulated in pediatric acute lymphoblastic leukemia. Tumori. 2012;98:252–256. doi: 10.1177/030089161209800212. [DOI] [PubMed] [Google Scholar]
- Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, Fu YX, Schreiber H. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17:737–747. doi: 10.1016/s1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. Journal for immunotherapy of cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature reviews Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, Fu YX. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. The Journal of experimental medicine. 2005;201:779–791. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.