

Abstract
Despite antiretroviral therapy (ART), respiratory infections increase mortality in individuals living with chronic human immunodeficiency virus (HIV) infection. In experimental and clinical studies of chronic HIV infection, alveolar macrophages (AMs) exhibit impaired phagocytosis and bacterial clearance. Peroxisome proliferator-activated receptor (PPAR)γ, NADPH oxidase (Nox) isoforms Nox1, Nox2, Nox4, and transforming growth factor-beta 1 (TGFβ1) are critical mediators of AM oxidative stress and phagocytic dysfunction. Therefore, we hypothesized that HIV alters AM expression of these targets, resulting in chronic lung oxidative stress and subsequent immune dysfunction. A cross-sectional study of HIV-infected (n = 22) and HIV-uninfected (n = 6) subjects was conducted. Bronchoalveolar lavage (BAL) was performed, and AMs were isolated. Lung H2O2 generation was determined by measuring H2O2 in the BAL fluid. In AMs, PPARγ, Nox1, Nox2, Nox4, and TGFβ1 mRNA (quantitative real-time polymerase chain reaction) and protein (fluorescent immunomicroscopy) levels were assessed. Compared with HIV-uninfected (control) subjects, HIV-infected subjects were relatively older and the majority were African American; ∼86% were on ART, and their median CD4 count was 445, with a median viral load of 0 log copies/ml. HIV infection was associated with increased H2O2 in the BAL, decreased AM mRNA and protein levels of PPARγ, and increased AM mRNA and protein levels of Nox1, Nox2, Nox4, and TGFβ1. PPARγ attenuation and increases in Nox1, Nox2, Nox4, and TGFβ1 contribute to AM oxidative stress and immune dysfunction in the AMs of otherwise healthy HIV-infected subjects. These findings provide novel insights into the molecular mechanisms by which HIV increases susceptibility to pulmonary infections.
Keywords: : HIV, immunology, molecular biology
Introduction
Antiretroviral therapy (ART) has led to a decrease in acquired immune deficiency syndrome (AIDS)-specific opportunistic infections in individuals living with chronic human immunodeficiency virus (HIV) infection.1,2 However, despite these advances, individuals living with HIV continue to be susceptible to serious bacterial and viral infections, such as pneumococcus and influenza,3–6 which lead to greater morbidity and mortality. Increased susceptibility of individuals living with HIV to pulmonary infections is at least partially due to the fact that HIV impairs alveolar macrophage (AM) immune functions.7–11 In fact, we have previously shown that the AM harbors HIV, even in otherwise healthy subjects with undetectable plasma viral loads, suggesting that the AM is a potential reservoir for the virus.12 HIV infection also causes derangements in multiple steps of an immune response, including abnormal activation,13–16 diminished oxidative burst,15 increased anti-inflammatory cytokine secretion,13 and impaired phagocytic capacity.8,9,12 Most notably, in our previous prospective cross-sectional study of healthy HIV-infected subjects with systemic viral suppression and on ART, AMs from HIV-infected subjects exhibited impaired phagocytic index compared with control subjects.12
Impaired phagocytosis by AMs in the HIV lung may be due to increased pulmonary oxidative stress. In preclinical7,17,18 and clinical studies,19,20 HIV infection causes systemic and pulmonary oxidative stress. Oxidative stress can be generated through several mechanisms, including the oxidation of extracellular thiols, such as glutathione (GSH) to glutathione disulfide (GSSG),21 but other underlying mechanisms may contribute to oxidative stress. Upregulation of peroxisome proliferator-activated receptor (PPAR)-γ is essential in protecting against pulmonary oxidant injury.22 Therefore, decreases in PPARγ impair the capacity of the lung to downregulate oxidative stress.23 In AMs, the expression of NADPH oxidases (Nox) 1 and 2 is required for Nox4 expression24 and increased Nox activity enhances oxidative stress and phagocytic dysfunction.24,25 Transforming growth factor β1 (TGFβ1) is a mediator of reactive oxygen species (ROS) and phagocytic dysfunction in the AM,26 and further, increased signaling through TGFβ1 has been shown to upregulate Nox4.27
Therefore, we hypothesize that individuals living with chronic HIV infection have an increased susceptibility to respiratory infections due to increased alveolar oxidative stress and impaired lung immunity. Specifically, chronic HIV infection is associated with AM oxidative stress through decreases in PPARγ expression and increases in the expression of Nox1, Nox2, Nox4, and TGFβ1. Increased oxidative stress is expected to subsequently impair the ability of the AM to phagocytize and clear infectious microbes from the alveolar space in individuals living with HIV, which may contribute to the increased risk for lung infections.
Materials and Methods
Study design and protocol
All participants signed the Emory University Institutional Review Board approved consent to participate in the study. Subjects were recruited and enrolled as previously described.12 In brief, we performed a cross-sectional study of HIV-infected (HIV, n = 22) and HIV-uninfected (hereafter referred to as control subjects) (Control, n = 6) individuals within the Grady Health System in Atlanta, GA. Exclusion criteria included active liver disease (known cirrhosis and/or total bilirubin >2.0 mg/dl), heart disease [ejection fraction <50%, history of acute myocardial infarction, New York Heart Association (NYHA) II–IV cardiac symptoms, severe valvular dysfunction], current renal disease (dialysis dependent or creatinine >2.0 mg/dl), current lung disease [spirometry revealing a forced vital capacity or forced expiratory volume in 1 s <80% of predicted], diabetes, current pregnancy, malnutrition (body mass index <17), current tobacco use, and age <25 years. All subjects completed a pre-enrollment evaluation, and subjects with exclusions were excluded from further participation.
After completing the pre-enrollment evaluation, subjects presented to the Grady Memorial Hospital Clinical Interaction Site, and demographic data were collected. Flexible fiberoptic bronchoscopy with standardized bronchoalveolar lavage (BAL) was performed by using standard conscious sedation techniques, by installing and withdrawing with low-pressure hand suction, 30 ml aliquots of 0.9% nonbacteriostatic normal saline solution until a total of 180 ml had been administered.
Human AMs cell culture
Human AMs were isolated from the BAL fluid24,25 and were determined to be ∼90% pure, as measured by Diff-Quik (Dade Behring) staining and cell counting.28 AMs were resuspended in RPMI-1640 medium containing 2% fetal bovine serum and 1% penicillin/streptomycin and cultured for 2 h, allowing AMs to attach to the plastic petri dishes, before experiments. Some AMs were treated with 10 μM pioglitazone (PIO, United States Biological, Salem, MA) or dimethyl sulfoxide vehicle (0.01%) for 24 h.
Measurement of H2O2 generation
H2O2 generation in collected BAL fluid was determined by using Amplex Red assay (Invitrogen). In brief, 50 μl of BAL fluid was incubated with 500 μl Amplex Red (20 μM) and horseradish peroxidase (0.1 U/ml) at 37°C for 30 min in the dark. Fluorescence of reaction mixtures was measured in duplicate (excitation 540 nm, emission 590 nm), and H2O2 concentrations were calculated by utilizing standard curves that were generated with reagent H2O2. BAL total protein concentrations were determined by using BCA assay. H2O2 concentrations were normalized to total protein concentrations and expressed as mean ± standard error of the mean (SEM), relative to average control values.
mRNA studies
Total RNA was extracted from AMs by using TRIzol reagent (Invitrogen, Carlsbad, CA). mRNA levels of PPARγ, Nox1, Nox2, Nox4, and TGFβ1 were determined and quantified by quantitative real-time polymerase chain reaction using methods previously described,23,24 and specific mRNA primers are shown in Table 1. Values for each target are expressed relative to mRNA levels of 9s in the same sample.
Table 1.
Human mRNA Primer Sequences to Measure mRNA Expression Using Quantitative Real-Time Polymerase Chain Reaction
| Primer | Forward sequence (5′-3′) | Reverse sequence (5′-3′) |
|---|---|---|
| PPARγ | GAGTTCATGCTTGTCAAGGATGC | CGATATCACTGGAGATCTCGCC |
| Nox1 | CGCTCCCAGCAGAAGGTCGTGATTACCAAG | GGAGTGACCCCAATCCCTGCCCCAACCA |
| Nox2 | GTCACACCCTTCGCATCCATTCTCAAGTCAGT | CTGAGACTCATCCCAGCCAGTGAGGTAG |
| Nox4 | CTGGAGGAGCTGGCTCGCCAACGAAG | GTGATCATGAGGAATAGCACCACCACCATGCAG |
| TGFβ1 | CAGAAATACAGCAACAATTCCTGG | TTGCAGTGTGTTATCCCTGCTGTC |
| 9s | GAGCTAGCCTCTGCCAGAGG | TCCAAGCCTCAAGACAGGAA |
PPARγ, peroxisome proliferator activated-receptor gamma; Nox, NADPH oxidase; TGFβ1, transforming growth factor beta-1.
Protein studies
Protein levels of PPARγ, Nox1, Nox2, Nox4, and TGFβ1 in AMs were assessed by fluorescent immunomicroscopy. Cultured AMs were fixed to chamber slides with 4% paraformaldehyde. Cells were then incubated with primary antibodies (Santa Cruz Biotechnology) for PPARγ (1:100), Nox1 (1:100), Nox2 (1:100), Nox4 (1:100), and TGFβ1 (1:100), followed by incubation with fluorescent TRITC-labeled secondary antibodies.24,29 After staining, fluorescent microscopy images were obtained with Fluoview analysis (Olympus, Melville, NY). Fluorescence units of TRITC-target stained cells were normalized to DAPI nuclear stain and are expressed as mean relative fluorescent units (RFU) per cell ± SEM, relative to control. We previously reported that quantitating protein targets with immunostaining methods correlated well with western blotting of the same targets.24,25 RFUs were measured in images that contain at least 10 cells per field for which there are 10 fields per experimental condition. Microscope settings, such as gain and gamma settings, were consistent for each field and experimental condition. The contrast of all provided representative images was increased by 50% to aid in visualization, without causing over-saturation of the fluorescent staining.
In vitro phagocytosis studies
In vitro phagocytic capacity was measured by using pHrodo Staphylococcus aureus BioParticles conjugate (Invitrogen).29 Briefly, cells were incubated with 1 × 106 particles of pH-sensitive pHrodo S. aureus BioParticles conjugate for 2 h and then fixed with 4% paraformaldehyde. After fixing, cells from 10 fields per experimental condition were assessed for phagocytosis, and fluorescent microscopy images were obtained with Fluoview analysis (Olympus, Melville, NY). Cells were considered positive for phagocytosis if any internalized bacteria were present. Phagocytosis was calculated as the percentage of cells that were positive for phagocytosis multiplied by the relative fluorescence units of S. aureus per cell and given as phagocytic index.
Statistical analyses
Univariate comparisons between HIV-infected subjects and control subjects were calculated and evaluated for a significance level of 0.05 by using a chi-squared test for categorical variables and a two-sample t-test for continuous variables. The data were log transformed or a Wilcoxon Rank-Sum Test was used when the data were not normally distributed. Data are presented as means ± SEM. In studies with more than two groups, statistical significance was calculated by using one-way ANOVA followed by Tukey-Kramer post hoc tests to detect differences between individual groups using GraphPad Prism version 5 (GraphPad, San Diego, CA). p < .05 was considered statistically significant.
Results
Baseline characteristics of HIV-uninfected and HIV-infected subjects
A total of 22 HIV-infected (HIV) subjects and 6 HIV-uninfected (control) subjects were enrolled (Table 2). The HIV-infected subjects were significantly older than control subjects. There were no significant differences in median gender, but the majority of subjects in both groups were African American. Overall, 86.4% of HIV-infected subjects were on ART and their median CD4 count was 445, with a median viral load of 0 log copies/ml.
Table 2.
Characteristics of HIV-Uninfected Subjects (Control, n = 6) and HIV-Infected Subjects (HIV, n = 22) Enrolled in the Study
| Variables | HIV uninfected (control) | HIV infected (HIV) | p |
|---|---|---|---|
| Number of subjects (N) | 6 | 22 | |
| Age (years)a | 38 (28.3–48.3) | 51 (46.8–55.5) | .009 |
| Gender (% male) | 66.7 | 59.1 | .74 |
| Race n (%) | |||
| White | 0 (0) | 1 (4.5) | .02 |
| Black | 4 (66.7) | 21 (95.5) | |
| Other | 2 (33.3) | ||
| BMIa | 28.5 (26.0–34.5) | 30 (26.8–33.5) | .65 |
| SMASTa | 0 (0–1.3) | 0 (0–4) | .33 |
| AUDITa | 0.5 (0–4.5) | 2 (0–3.3) | .71 |
| CD4 counta | 445 (317–557.3) | ||
| Viral load (log copies/ml)a | 0 (0–0) | ||
| Use of ART (%) | 86.4 | ||
Results are represented as medians with interquartile range.
BMI, body mass index; SMAST, Short Michigan Alcoholism Screening Test; AUDIT, Alcohol Use Disorders Identification Test; ART, antiretroviral therapy.
Chronic HIV infection is associated with H2O2 increases in BAL
In previous studies conducted in our laboratory, chronic HIV infection was associated with increased oxidative stress, as defined by decreased glutathione levels and altered glutathione redox balance in the alveolar space, as well as by increased H2O2 levels in lung tissue.17 Measurements of H2O2 were performed in the BAL cell-free fluid collected from control and HIV-infected subjects. Compared with controls, BAL from HIV-infected subjects showed a 2.3 ± 0.1-fold increase in H2O2 generation (Fig. 1). These data suggest that HIV infection is associated with increased H2O2 and oxidative stress in the alveolar space.
FIG. 1.

Chronic HIV infection is associated with increases in BAL oxidative stress. BAL fluid was collected from HIV-uninfected control subjects (Control, n = 6) and from HIV-infected subjects (HIV, n = 22). H2O2 generation was measured by Amplex Red assay (n = 28, in duplicate). Values were normalized to total protein and are expressed as mean ± SEM, relative to control. *p < .05 versus control. BAL, bronchoalveolar lavage; SEM, standard error of the mean.
Chronic HIV infection is associated with alterations in mRNA and protein levels of PPARγ, NADPH oxidases, and TGFβ1 in AMs
Since chronic HIV infection was associated with increased BAL H2O2 generation, we determined whether previously established markers of oxidative stress were altered in the AMs,29 particularly PPARγ, Nox1, Nox2, Nox4, and TGFβ1. Compared with controls, AMs from HIV-infected subjects demonstrated a 5.3 ± 0.1-fold decrease in PPARγ mRNA (Fig. 2A), a 2.1 ± 0.03-fold increase in Nox1 mRNA (Fig. 2B), a 2.44 ± 0.03-fold increase in Nox2 mRNA (Fig. 2C), a 2.5 ± 0.27-fold increase in Nox4 mRNA (Fig. 2D), and a 2.9 ± 0.21-fold increase in TGFβ1 mRNA (Fig. 2E). Although HIV status was not significantly associated with Nox mRNA after a multi-regression analysis with age as a confounder, HIV was still associated with PPARγ and TGFβ1 mRNA levels in AMs.
FIG. 2.

Chronic HIV infection is associated with alterations in mRNA levels of PPARγ, Nox1, Nox2, Nox4, and TGFβ1 in AMs. AMs were collected from HIV-uninfected control subjects (Control, n = 6) and from HIV-infected subjects (HIV, n = 22). mRNA levels of PPARγ (A), Nox1 (B), Nox2 (C), Nox4 (D), and TGFβ1 (E) were measured by quantitative real-time polymerase chain reaction (n = 28, in duplicate), normalized to 9s mRNA, and expressed as mean ± SEM, relative to control. *p < .05 versus control. AMs, alveolar macrophages; PPARγ, peroxisome proliferator-activated receptor gamma; TGFβ1, transforming growth factor beta-1.
Concomitant with HIV-induced alterations in mRNA levels in AMs, there was attenuation of PPARγ protein (Fig. 3A) and augmentation of protein levels of Nox1 (Fig. 3B), Nox2 (Fig. 3C), Nox4 (Fig. 3D), and TGFβ1 (Fig. 3E). These data indicate that HIV infection was associated with decreased mRNA and protein expression of PPARγ as well as with increased mRNA and protein expression of the Nox isoforms and TGFβ1, all of which are indicators of exaggerated oxidative stress in the AM.
FIG. 3.

Chronic HIV infection is associated with alterations in protein levels of PPARγ, Nox1, Nox2, Nox4, and TGFβ1 in AMs. AMs were collected from HIV-uninfected control subjects (Control, n = 6) and from HIV-infected subjects (HIV, n = 22). Protein levels of PPARγ (A), Nox1 (B), Nox2 (C), Nox4 (D), and TGFβ1 (E) were determined in AMs (n = 28, 10 fields per experimental condition) by using confocal fluorescence microscopy. Representative images are provided. Quantification of fluorescence values was normalized to DAPI nuclear stain and expressed as mean relative fluorescence units (RFU) per cell ± SEM, relative to control. *p < .05 versus control.
Ex vivo PIO treatment reverses chronic HIV infection-associated alterations in mRNA levels of PPARγ, NADPH oxidases, and TGFβ1 and improves phagocytosis in AMs
Since PPARγ ligands have been previously shown to downregulate established markers of oxidative stress in AMs,29 we determined whether PIO can reverse alterations in the mRNA levels of PPARγ, Nox1, Nox2, Nox4, and TGFβ1 that are associated with chronic HIV infection. Compared with AMs from HIV-infected subjects, PIO treatment ex vivo (n = 4) increased PPARγ mRNA levels (Fig. 4A) and decreased the mRNA levels of Nox1 (Fig. 4B), Nox2 (Fig. 4C), Nox4 (Fig. 4D), and TGFβ1 (Fig. 4E). There were no statistically significant differences in raw Ct values between the dimethyl sulfoxide vehicle and PIO treatment groups, indicating that 0.01% dimethyl sulfoxide is not responsible for the expression of the targeted genes. PIO treatment ex vivo also improved phagocytosis in AMs from HIV-infected individuals (Fig. 4F). These data suggest that the exaggerated AM oxidative stress response and impaired phagocytic function associated with chronic HIV infection can be improved with PIO treatment.
FIG. 4.

Ex vivo PIO treatment reverses chronic HIV infection-associated alterations in mRNA and protein levels of PPARγ, Nox1, Nox2, Nox4, and TGFβ1 and improves phagocytosis in AMs. AMs were collected from HIV-infected subjects and were treated with PIO (HIV + PIO) or dimethyl sulfoxide vehicle (HIV) (n = 4). mRNA levels of PPARγ (A), Nox1 (B), Nox2 (C), Nox4 (D), and TGFβ1 (E) were measured by qRT-PCR (n = 8, in duplicate), normalized to 9s mRNA, and expressed as mean ± SEM, relative to HIV. AM phagocytic ability (F) was assessed by phagocytosis assay and fluorescent microscopy imaging (10 fields per experimental condition). Phagocytic index was calculated as the percentage of phagocytic cells multiplied by the relative fluorescence units of S. aureus per cell. All values were expressed as mean ± SEM, relative to HIV. *p < .05 versus HIV. PIO, pioglitazone.
Discussion
Despite advances in HIV therapeutic strategies such as ART, individuals living with chronic HIV infection remain susceptible to serious bacterial and viral infections,3–6 leading to greater morbidity and mortality. Pulmonary infections, particularly bacterial pneumonia, are significantly more likely among individuals living with HIV.6 However, the mechanisms by which HIV increases the risk of developing respiratory infections remain poorly understood. For the first time, the results presented here describe that, in a healthy cohort of HIV-infected subjects who are nonsmokers and without lung disease, HIV infection is associated with: (a) increased H2O2 levels in BAL; (b) decreased PPARγ mRNA and protein levels in AMs; and (c) increased mRNA and protein levels of Nox1, Nox2, Nox4, and TGFβ1 in AMs. These results suggest that HIV infection is associated with the promotion of H2O2 levels in the alveolar space and the expression of oxidative stress markers in AMs, which may contribute to impaired lung immune functions and increased susceptibility to respiratory infections.
In the alveolar space, the AM is the first line of lung immune defense. HIV impairs AM immune functions7–11 through dysregulated activation,13–16 impaired oxidative burst,15 and enhanced anti-inflammatory cytokine secretion.13 In an experimental model using HIV transgenic rats, AM phagocytosis was significantly decreased in HIV-infected rats, compared with phagocytosis in AMs from wild-type rats.8,9 The HIV-infected subjects enrolled in our study were healthy individuals, as evidenced by their lack of underlying medical problems (exclusion criteria), nonsmoking status, and a majority of them being on ART where their median CD4 count was 445 and median viral load was 0 log copies/ml (Table 2). However, in our previous prospective cross-sectional study of otherwise healthy HIV-infected subjects on ART with systemic viral suppression, AMs from HIV-infected subjects exhibited a significantly lower median phagocytic index compared with control subjects.12 These observations suggest that individuals living with HIV have diminished immune responses in AMs, which may contribute to their increased susceptibility to respiratory infections.
Impaired AM phagocytic ability may be linked to increased oxidative stress within the AM. HIV has been shown to induce pulmonary oxidative stress in preclinical7,17,18 and clinical studies.19,20 Our data confirmed these studies and showed that HIV infection was associated with increased H2O2 levels in the BAL (Fig. 1). Lung oxidative stress can be generated through several mechanisms. In previous studies conducted by our laboratory, HIV infection was associated with altered alveolar redox state by decreasing levels of the primary alveolar antioxidant GSH and increasing the oxidized form GSSG.17 In addition, the loss of PPARγ in AMs has been shown to promote oxidative stress and phagocytic dysfunction.29 Upregulation of PPARγ has been shown to be essential in protecting against pulmonary oxidant injury via activation of NF-E2 related factor 2 (Nrf2) and binding to the antioxidant response element (ARE).22 Further, PPARγ activation by its ligand rosiglitazone decreased alcohol-induced lung oxidative stress through downregulation of eNOS23 and protected against AM phagocytic dysfunction by attenuating the expression of Nox proteins and TGFβ1.29 To our knowledge, this study is the first to show that AMs from a healthy cohort of HIV-infected subjects exhibit attenuated PPARγ mRNA (Fig. 2) and protein (Fig. 3) levels, compared with control subjects. These studies suggest a role for decreased PPARγ in HIV-associated AM oxidative stress and phagocytic dysfunction.
Previous studies also demonstrated that increased expression of Nox1, Nox2, and Nox424 and TGFβ126 promote AM oxidative stress and phagocytic dysfunction. In the AM, expression of Nox1 or Nox2 is required for expression of Nox4, a constitutively active Nox protein that is a primary source of H2O2.24 Increased Nox1, Nox2, and Nox4 expression in AMs is associated with increased oxidative stress and phagocytic dysfunction.24,25 TGFβ1, another mediator of alcohol-induced ROS and phagocytic dysfunction in the AM,26 has also been shown to upregulate Nox4 via activation of Smad2/327 and to suppress lung Nrf2-mediated activation of the ARE.30 For the first time, our data show that, in an otherwise healthy cohort of HIV-infected subjects, AMs exhibit increased expression of Nox1, Nox2, Nox4, and TGFβ1 mRNA (Fig. 2) and protein (Fig. 3). Since ex vivo treatments with PPARγ ligands reversed the expression of Nox1, Nox2, Nox4, and TGFβ1 and improved AM phagocytic ability in other model systems29 as well as in HIV-infected AMs (Fig. 4), we hypothesize that the immune dysfunction and risk of respiratory infections in HIV subjects is mediated by decreased PPARγ, resulting in increased Nox1, Nox2, Nox4, and TGFβ1 in the AM, and subsequent enhanced pulmonary oxidative stress (schema Fig. 5).
FIG. 5.
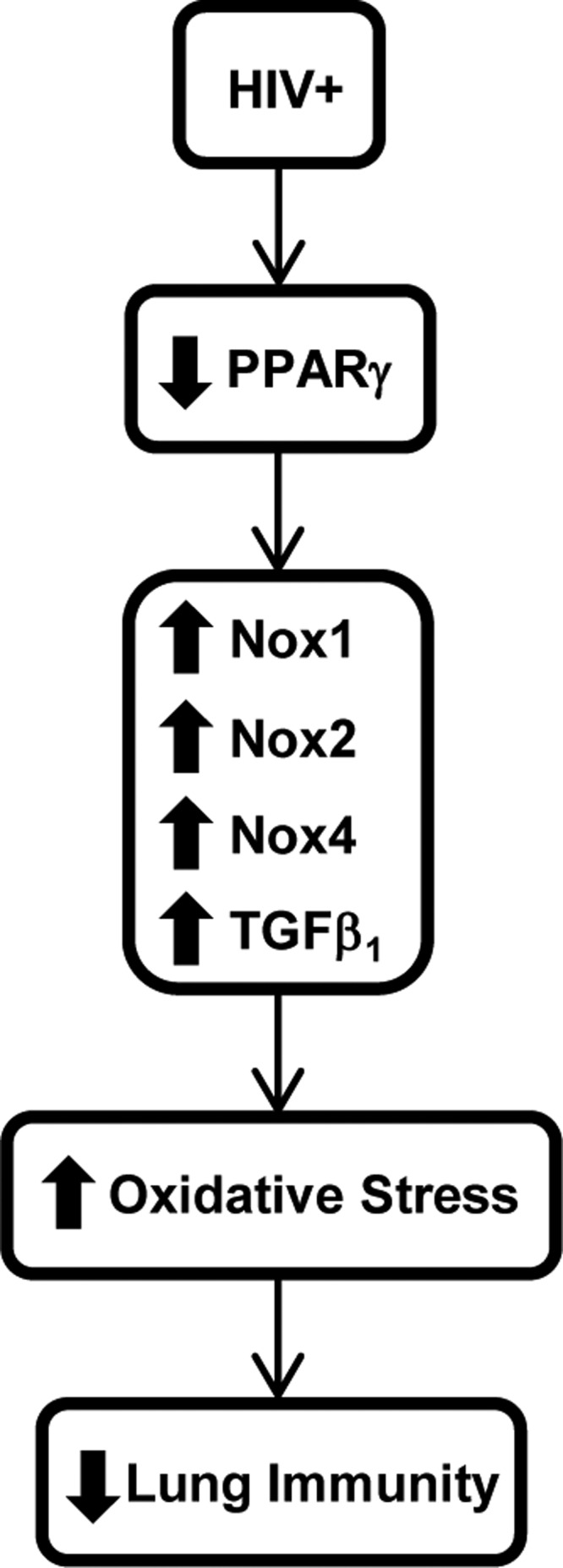
Hypothetical schema of the molecular mechanisms involved in HIV-associated lung immune dysfunction. Chronic HIV infection is associated with impaired lung immunity through decreases in AMs PPARγ expression. Attenuation of PPARγ increases AMs expression of Nox isoforms, and TGFβ1, which, in turn, enhances lung oxidative stress. This increase in lung oxidative stress causes the lung immune dysfunction observed in HIV-infected individuals.
Studies have shown impaired phagocytic capacity in small AMs of HIV-infected individuals31 and impaired AM phagosomal proteolysis function in HIV-infected individuals.31,32 These studies focused on whether the change in AM phenotype correlates with the HIV infection status of the AM or the volunteer, whereas our study focuses on elucidating the molecular mechanisms by which chronic HIV infection is associated with increased oxidative stress in the alveolar space and AM, whereby the lungs of HIV-infected individuals are potentially primed for an increased risk of developing respiratory infections. NADPH oxidase expression, specifically Nox2, has been shown to negatively regulate phagosomal proteolysis in macrophages,33 and our data show that chronic HIV infection is associated with increased NADPH oxidase expression in AM (Fig. 2B–D and Fig. 3B–D). Further studies are warranted to explore whether NADPH oxidases impair phagosomal proteolysis in AM in the context of chronic HIV infection.
Similar to the aforementioned studies, our data also implicate HIV-associated AM dysregulation in the potential susceptibility of individuals living with HIV to developing respiratory infections; however, there are several limitations to our study. Subjects enrolled in the study represent a cross-sectional cohort, and the sample size of subjects enrolled (n = 28) and ex vivo treatment with PIO (n = 4) were relatively small. Further, HIV-infected subjects in this study are older; although oxidative stress increases with age,34 the effects of aging on the lung and lung immune function have not been well characterized. Multi-regression analysis with age as a confounder showed that HIV was not significantly associated with Nox mRNA, but HIV status was still significantly associated with PPARγ and TGFβ1 mRNA levels in AMs (Fig. 2). Since both age and HIV may have contributed to the Nox-mediated increase in oxidative stress in the lung, this is an important area for future investigations. Age-matched controls and HIV-infected subjects will be included in future studies.
In spite of these limitations, our results show that even individuals living with HIV, without any evidence of other medical problems, may be at risk for lung infections due to impaired immune responses of their AMs. In a longitudinal prospective study, the relative expression of these mediators could potentially be correlated with risk and severity of respiratory infections, morbidity, and mortality. Also, in a larger study, subjects could be further stratified into different groups according to age, smoking status, chronic obstructive pulmonary disease status, etc. This will allow for further clarification on how these AM mediators could be used in the future. Our data demonstrate the critical importance in evaluating BAL H2O2 levels and AM expression of PPARγ, Nox1, Nox2, Nox4, and TGFβ1 as potential biomarkers that might be predictive of poor outcomes and increased susceptibility to respiratory infections in the future of individuals living with chronic HIV infection.
Acknowledgments
The authors would like to thank the following people for contributing to this article: (1) Joel Andrews, RN, BSN, for all his help with the recruitment and enrollment of study subjects and IRB regulations; (2) Annette Esper, MD, MSc, and Greg Martin, MD, MSc, for their help with bronchoscopies. In addition, the corresponding author, Dr. Yeligar, would like to thank her co-authors for the following contributions: (1) study design and recruitment (L.B., D.G., S.C.); (2) analysis and interpretation of data (J.W., F.H., L.B., D.G., S.C.); and (3) article preparation (J.W., F.H., L.B., D.G., S.C.).
Sources of Support
National Institute on Alcohol Abuse and Alcoholism National Research Service Award 1F32AA020724-01; American Heart Association National Scientist Development Grant No. 13SDG13930003; National Institute on Alcohol Abuse and Alcoholism 1K99AA021803-01A1 and 4R00AA021803-03; National Heart, Lung and Blood Institute 1R01 HL125042 and 5R34 HL117351; and National Center for Advancing Translational Sciences of the National Institutes of Health KL2 TR000454 and UL1 TR000454.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Li TS, Tubiana R, Katlama C, Calvez V, Ait Mohand H, Autran B: Long-lasting recovery in CD4 T-cell function and viral-load reduction after highly active antiretroviral therapy in advanced HIV-1 disease. Lancet 1998;351:1682–1686 [DOI] [PubMed] [Google Scholar]
- 2.Mellors JW, Munoz A, Giorgi JV, Margolick JB, Tassoni CJ, Gupta P, et al. : Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV-1 infection. Ann Intern Med 1997;126:946–954 [DOI] [PubMed] [Google Scholar]
- 3.Gordin FM, Roediger MP, Girard PM, Lundgren JD, Miro JM, Palfreeman A, et al. : Pneumonia in HIV-infected persons: Increased risk with cigarette smoking and treatment interruption. Am J Respir Crit Care Med 2008;178:630–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordano Q, Falco V, Almirante B, Planes AM, del Valle O, Ribera E, et al. : Invasive pneumococcal disease in patients infected with HIV: Still a threat in the era of highly active antiretroviral therapy. Clin Infect Dis 2004;38:1623–1628 [DOI] [PubMed] [Google Scholar]
- 5.Strategies for Management of Antiretroviral Therapy Study Group; El-Sadr WM, Lundgren J, Neaton JD, Gordin F, Abrams D, et al. : CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006;355:2283–2296 [DOI] [PubMed] [Google Scholar]
- 6.Crothers K, Huang L, Goulet JL, Goetz MB, Brown ST, Rodriguez-Barradas MC, et al. : HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med 2011;183:388–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacob BA, Porter KM, Elms SC, Cheng PY, Jones DP, Sutliff RL: HIV-1-induced pulmonary oxidative and nitrosative stress: Exacerbated response to endotoxin administration in HIV-1 transgenic mouse model. Am J Physiol Lung Cell Mol Physiol 2006;291:L811–L819 [DOI] [PubMed] [Google Scholar]
- 8.Joshi PC, Guidot DM: HIV-1 transgene expression in rats induces differential expression of tumor necrosis factor alpha and zinc transporters in the liver and the lung. AIDS Res Ther 2011;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi PC, Raynor R, Fan X, Guidot DM: HIV-1-transgene expression in rats decreases alveolar macrophage zinc levels and phagocytosis. Am J Respir Cell Mol Biol 2008;39:218–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pugliese A, Vidotto V, Beltramo T, Torre D: Phagocytic activity in human immunodeficiency virus type 1 infection. Clin Diagn Lab Immunol 2005;12:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stebbing J, Gazzard B, Douek DC: Where does HIV live? N Engl J Med 2004;350:1872–1880 [DOI] [PubMed] [Google Scholar]
- 12.Cribbs SK, Lennox J, Caliendo AM, Brown LA, Guidot DM: Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res Hum Retroviruses 2015;31:64–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denis M, Ghadirian E: Alveolar macrophages from subjects infected with HIV-1 express macrophage inflammatory protein-1 alpha (MIP-1 alpha): Contribution to the CD8+ alveolitis. Clin Exp Immunol 1994;96:187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans MR, Wansbrough-Jones MH: Alveolar macrophage activation in HIV infection. J Infect 1996;33:91–94 [DOI] [PubMed] [Google Scholar]
- 15.Koziel H, O'Riordan D, Warner A, Rose RM: Alveolar macrophage interaction with Pneumocystis carinii. Immunol Ser 1994;60:417–436 [PubMed] [Google Scholar]
- 16.Lipman MC, Johnson MA, Bray DH, Poulter LH: Changes to alveolar macrophage phenotype in HIV infected individuals with normal CD4 counts and no respiratory disease. Thorax 1995;50:777–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lassiter C, Fan X, Joshi PC, Jacob BA, Sutliff RL, Jones DP, et al. : HIV-1 transgene expression in rats causes oxidant stress and alveolar epithelial barrier dysfunction. AIDS Res Ther 2009;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louboutin JP, Agrawal L, Reyes BA, Van Bockstaele EJ, Strayer DS: HIV-1 gp120-induced injury to the blood-brain barrier: Role of metalloproteinases 2 and 9 and relationship to oxidative stress. J Neuropathol Exp Neurol 2010;69:801–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bilbis LS, Idowu DB, Saidu Y, Lawal M, Njoku CH: Serum levels of antioxidant vitamins and mineral elements of human immunodeficiency virus positive subjects in Sokoto, Nigeria. Ann Afr Med 2010;9:235–239 [DOI] [PubMed] [Google Scholar]
- 20.Coaccioli S, Crapa G, Fantera M, Del Giorno R, Lavagna A, Standoli ML, et al. : Oxidant/antioxidant status in patients with chronic HIV infection. Clin Ter 2010;161:55–58 [PubMed] [Google Scholar]
- 21.Jones DP: Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 2008;295:C849–C868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, et al. : Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med 2010;182:170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner MC, Yeligar SM, Brown LA, Michael Hart C: PPARgamma ligands regulate NADPH oxidase, eNOS, and barrier function in the lung following chronic alcohol ingestion. Alcohol Clin Exp Res 2012;36:197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeligar SM, Harris FL, Hart CM, Brown LA: Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol 2012;188:3648–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeligar SM, Harris FL, Hart CM, Brown LA: Glutathione attenuates ethanol-induced alveolar macrophage oxidative stress and dysfunction by down-regulating NADPH oxidases. Am J Physiol Lung Cell Mol Physiol 2014;306:L429–L441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown SD, Brown LA: Ethanol induced TGF-β1 and ROS production are necessary for ethanol induced alveolar macrophage dysfunction and induction of alternative activation Alcohol Clin Exp Res 2012;36:1952–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. : NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 2005;97:900–907 [DOI] [PubMed] [Google Scholar]
- 28.Brown SD, Gauthier TW, Brown LA: Impaired terminal differentiation of pulmonary macrophages in a Guinea pig model of chronic ethanol ingestion. Alcohol Clin Exp Res 2009;33:1782–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeligar SM, Mehta AJ, Harris FL, Brown LA, Hart CM: Peroxisome proliferator-activated receptor gamma regulates chronic alcohol-induced alveolar macrophage dysfunction. Am J Respir Cell Mol Biol 2016;55:35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sueblinvong V, Tseng V, Smith T, Saghafi R, Mills ST, Neujahr DC, et al. : TGFbeta1 mediates alcohol-induced Nrf2 suppression in lung fibroblasts. Alcohol Clin Exp Res 2014;38:2731–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jambo KC, Banda DH, Kankwatira AM, Sukumar N, Allain TJ, Heyderman RS, et al. : Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal Immunol 2014;7:1116–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jambo KC, Banda DH, Afran L, Kankwatira AM, Malamba RD, Allain TJ, et al. : Asymptomatic HIV-infected individuals on antiretroviral therapy exhibit impaired lung CD4(+) T-cell responses to mycobacteria. Am J Respir Crit Care Med 2014;190:938–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rybicka JM, Balce DR, Khan MF, Krohn RM, Yates RM: NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci U S A 2010;107:10496–10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finkel T, Holbrook NJ: Oxidants, oxidative stress and the biology of ageing. Nature 2000;408:239–247 [DOI] [PubMed] [Google Scholar]