

Abstract
Alzheimer’s disease (AD), the most common neurodegenerative disorder in the aged, is characterized by the cerebral deposition of fibrils formed by the amyloid β-protein (Aβ), a 40–42 amino acid peptide. The folding of Aβ into neurotoxic oligomeric, protofibrillar, and fibrillar assemblies is hypothesized to be the key pathologic event in AD. Aβ is formed through cleavage of the Aβ precursor protein by two endoproteinases, β-secretase and γ-secretase, that cleave the Aβ N-terminus and C-terminus, respectively. These facts support the relevance of therapeutic strategies targeting Aβ production, assembly, clearance, and neurotoxicity. Currently, no disease-modifying therapeutic agents are available for AD patients. Instead, existing therapeutics provide only modest symptomatic benefits for a limited time. We summarize here recent efforts to produce therapeutic drugs targeting Aβ assembly. A number of approaches are being used in these efforts, including immunological, nutraceutical, and more classical medicinal chemical (peptidic inhibitors, carbohydrate-containing compounds, polyamines, “drug-like” compounds, chaperones, metal chelators, and osmolytes), and many of these have progressed to phase III clinical trails. We also discuss briefly a number of less mature, but intriguing, strategies that have therapeutic potential. Although initial trials of some disease-modifying agents have failed, we argue that substantial cause for optimism exists.
INTRODUCTION
Alzheimer’s disease (AD) is the most common late-life neurodegenerative disorder [1], affecting an estimated 5.2 million in the U.S. and >27 million worldwide [2]. These case numbers are expected to triple or quadruple by 2050 [2]. If this should occur, the economic cost of AD patient care, now estimated at >$100 billion per year [3], will bankrupt the U.S. health care system [4]. Unfortunately, no disease-modifying therapies now exist. Coupled with the unquantifiable misery suffered by AD patients and their families around the world, the need for ameliorative and curative drugs is especially acute.
The most prominent current working hypothesis of AD pathogenesis posits that the amyloid β-protein (Aβ), an ~4,300–4,500 molecular weight peptide, is the proximate neurotoxic agent (for a recent review, see Roychaudhuri et al. [5]). Neurotoxicity is thought to result from the self-association of Aβ into oligomeric and higher order assemblies. Aβ itself is produced through the sequential action of two endoproteinases, β-secretase and γ-secretase, that cleave the Aβ N-terminus and C-terminus, respectively, from the larger Aβ precursor protein (APP) [1]. These facts support the relevance and attractiveness of two predominant strategies for the development of AD therapeutics: (1) blocking Aβ production; and (2) blocking Aβ self-assembly.
In this review, we focus on efforts to develop therapeutic agents targeting Aβ assembly (Table 1, Fig. 1). This process is surprisingly complex [5], which may explain why no one agent or class of agents yet has emerged as an obvious and favored choice for drug development. In fact, in addition to classical drug-like compounds, immunoglobulins, proteins, peptides, carbohydrate-containing compounds, lipids, nucleic acids, polyamines, osmolytes, chelators, polyphenols, vitamins, and other agents all are being studied. Such a broadly based search for efficacious compounds is especially valuable in light of initial, and well-publicized, failures of clinical trials representing diverse classes of therapeutic agents. In the sections that follow, we seek to provide the reader with a comprehensive, but necessarily brief, introduction to each of the better-developed approaches extant, as well as some insight into nascent but exciting new therapeutic strategies.
Table 1.
Current Aβ Assembly Therapeutics
| Section | Therapy | Mechanism | Example(s) | Clinical Trials |
|---|---|---|---|---|
| 1a | Active immunization | Aβ assembly, phagocytosis (antibody-dependent and independent), peripheral sequestration/elimination of cerebral Aβ, neutralizing antibodies | AN1792 ACC-001 CAD-106 V-950 |
Phase IIa Phase IIb Phase Ic Phase Id |
| 1b | Passive immunization | Bapineuzumab LY2062430 IVIg RN-1219 R-1450 |
Phase IIIe Phase IIf Phase IIg Phase Ih Phase Ii |
|
| 2 | Nutraceuticals | Aβ assembly, anti-oxidant | Curcumin Ginkgo biloba |
Phase IIj Phase IIIk |
| 3 | Peptidic inhibitors | Aβ assembly | β-sheet breakers N-methyl peptides |
None |
| 4 | Carbohydrate-containing compounds | Aβ assembly | GM1 ganglioside | None |
| 5 | Polyamines | Aβ assembly | Quinacrine | None |
| 6 | “Drug-like” compounds | Aβ assembly | Alzhemed™ Nicotine |
Phase IIIl None |
| 7 | Chaperones | Aβ assembly | Hsp20, Hsp27, Hsp70, Hsp90 | None |
| 8 | Chelation | Aβ assembly, metal sequestration | Clioquinol PBT2 |
Phase II/IIIm Phase IIn |
| 9 | Osmolytes | Aβ assembly | Cyclohexanehexol | None |
Fig. (1).
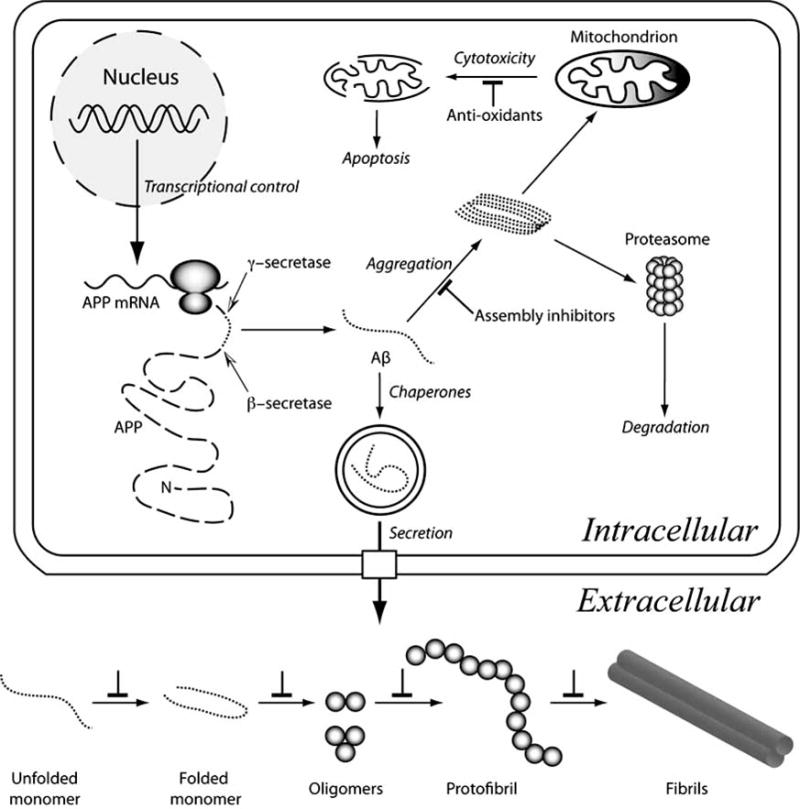
Aβ metabolism and assembly. Aβ (dotted lines) is produced by the sequential endoproteolytic cleavage of APP (dashed line). β-secretase cleavage (black-white arrowhead) produces the Aβ N-terminus, after which γ-secretase (black-white arrowhead) releases the Aβ C-terminus from APP. Transcriptional, translational, and endoproteolytic events all are targets for therapies to block Aβ production. The unstructured Aβ monomer may fold or aggregate intracellularly to produce toxic assemblies. One postulated cytotoxic mechanism is mitochondrial injury, which produces reactive oxygen species, mitochondrial injury, and apoptosis. Anti-oxidants could ameliorate redox effects directly. Assembly inhibitors would block this and other effects arising from formation of pathologic assemblies. Folding chaperones also would assist in this process. Aggregates may be eliminated through proteasomal digestion, but saturation of this system would result in cytotoxicity. Aβ secretion is a normal cellular process. Extracellular assembly of Aβ may occur in a variety of milieus. The micromolecular (pH, chemical composition) and macromolecular (proteins, lipids, carbohydrates) characteristics of these milieus differ, thus Aβ assembly pathways and kinetics also are likely to differ. Nevertheless, in vitro and in vivo experiments suggest that Aβ proceeds along a linear pathway comprising many populated monomer conformational states, a population of partially folded states (some of which facilitate peptide oligomerization), a more restricted distribution of oligomers (with distinct distributions for Aβ40 and Aβ42), protofibrils, and fibrils (of which multiple morphologies exist). Each of the inter-state transitions is a potential therapeutic target (⊥ symbol).
THERAPEUTIC APPROACHES
1. Immunotherapy
Immunotherapy involves the activation of cell-mediated or humoral (antibody) immune responses to eliminate noxious agents from the body. In AD, as discussed above, the “noxious agent” appears to be Aβ. Immunological approaches to AD therapy thus have focused on humoral immune responses specific for Aβ. Two classes of immunotherapeutic intervention have been explored: active and passive immunization. In the former class, various types of Aβ immunogens are used to elicit endogenous Aβ-specific antibody production. In the latter class, antibodies are produced exogenously, e.g., through monoclonal antibody methods, and then administered passively to the affected host.
Several mechanisms of action are thought to underlie the effects of Aβ-specific antibodies on Aβ assembly and amyloid plaques. One mechanism suggests that antibodies cross the blood-brain barrier (BBB), enter the brain, bind to Aβ in plaques, and target them for phagocytosis [6]. Active immunization trials in transgenic (Tg) mouse models of AD support this hypothesis. For example, Schenk et al. demonstrated that active immunization with Aβ42 prevented plaque formation in young mice and significantly reduced the plaque load in older mice [7]. The remaining Aβ plaques in these immunized mice exhibited significant amounts of bound antibodies. Major histocompatibility complex (MHC) II-expressing cells, thought to be activated microglia and monocytes, also were associated with these remaining plaques [7]. A second mechanism is direct antibody-mediated dissociation of Aβ fibrils and aggregates, which produces soluble forms of Aβ that can be eliminated more readily from the body. Such fibril disruption has been observed in vitro, where Aβ-specific antibodies can prevent or reverse fibril formation [8–10]. A third mechanism suggests that antibodies alter brain Aβ levels by affecting the equilibrium between brain and plasma Aβ. This “peripheral sink” theory posits that circulating Aβ-specific antibodies bind to and eliminate extra-cranial Aβ. To reestablish the intrinsic equilibrium between intra-and extra-cranial Aβ, insoluble Aβ in plaques is consumed and peripheral Aβ levels again rise. Antibodies thus need not cross the BBB to cause plaque elimination [11, 12]. Holtzmann et al. showed that peripheral administration of a monoclonal antibody (m266) specific for the central region of Aβ to PDAPP (platelet-derived growth factor promoter-expressing amyloid precursor protein) mice caused a 1,000-fold increase in plasma Aβ levels [11, 12]. Furthermore, the m266 antibody markedly reduced Aβ deposits in the mice, despite the inability of the antibodies to bind directly to these deposits.
Other mechanisms also may operate. For example, antibodies may neutralize neurotoxic Aβ assemblies by direct binding, a process independent of antigen clearance mechanisms. Antibody-independent mechanisms also may occur, as suggested by studies of innate immune responses mediated by macrophages and other phagocytic cells [13], responses that do not depend on the action of opsonizing antibodies [14].
a. Active Immunization
The first active immunization trial involved the administration of the immunogen AN1792, which is aggregated Aβ, along with a QS21 (Quillaja Saponaria 21) surface active saponin adjuvant. Immunization with AN1792 decreased plaque load in Tg mice that usually form plaques similar to those found in AD patients [7] and ameliorated behavioral impairments [15, 16]. Based on this evidence, phase I clinical trials were started on 80 patients with mild-to-moderate AD. The patients were immunized with AN1792 in adjuvant four times over a six-month period. Overall, the results from this trial showed promising results, including the induction of an Aβ-specific antibody response and delayed cognitive decline, based on the Disability Assessment Scale for Dementia (DAD). These results supported the initiation of a phase II trial [17]. However, during the phase II trial, one of the patients from the phase I trial developed subacute encephalitis and died from non-CNS related causes [18]. Subsequently, 18 of the 300 patients in the phase II trial developed subacute encephalitis, resulting in the early termination of the study [19]. Other Aβ vaccination protocols are under development. Elan/Wyeth started phase I testing of ACC-001, a vaccine that comprises the N-terminal seven amino acids of Aβ attached to a mutated diphtheria toxin carrier protein, CRM 197 (cross-reacting material 197). However, recently (April 2008), experimental testing of this vaccine was suspended after one patient developed skin lesions [20, 21]. It was unclear whether this patient was a member of the placebo or vaccine-treated group. Two other phase I vaccination trials are underway, CAD-106 from Novartis [22] and V-950 from Merck [23]. CAD-106 consists of Immunodrug™ carrier Qb coupled to an undisclosed fragment of Aβ. The nature of the V-950 immunogen is unknown.
b. Passive Immunization
i. Bapineuzumab (AAB-001)
Bapineuzumab, from Elan/Wyeth, is a humanized monoclonal antibody (half-human, half-mouse antibody produced in a mammalian cell culture system) specific for the N-terminus of Aβ. Pre-clinical assays showed that the mouse version of this antibody had affinity to both soluble and aggregated Aβ. Bapineuzumab currently is being tested in a phase III trial on patients with mild-to-moderate AD at 200 study sites in North America [24] and >150 study sites in 20 countries outside the U.S. [25, 26].
Results from the phase II clinical trial reported recently at the International Conference on Alzheimer’s Disease (ICAD) were disappointing [27]. In the double-blind, randomized, placebo-controlled trial (DBRPCT), 234 patients received one of four doses of Bapineuzumab (0.15 mg/kg, 0.5 mg/kg, 1.0 mg/kg, 2.0 mg/kg) or placebo by intravenous (i.v.) administration every 13 weeks for 18 months. No significant differences were observed in the overall study population in the clinical efficacy measures Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) or DAD. Furthermore, Bapineuzumab did not exhibit a clear does-response profile typical of most pharmaceutical agents. Nevertheless, post hoc apolipoprotein ε4 (APOE4) haplotype analysis revealed promising statistical results. In non-APOE4 subjects, the Bapineuzumab-treated group showed significant improvement compared to the placebo group by several metrics, including ADAS-Cog (p=0.026), Neuropsychological Test Battery (NTB; p=0.006), Clinical Dementia Rating Sum of Boxes (CDR-SB; p=0.04), and decrease in brain volume as determined by the Brain Boundary Shift Integral (BBSI; p=0.004). Additionally, trends toward improved DAD and smaller increases in ventricular volume were noted in the Bapineuzumab-versus placebo-treated non-APOE4 subjects. In APOE4 subjects, in contrast, no statistically significant changes were observed in any of the cognitive or functional endpoints, although favorable trends were noted.
Bapineuzumab treatment exhibited a generally mild and transient side effect profile, except for the appearance of vasogenic edema that presented in 12 treatment patients receiving higher Bapineuzumab doses. The incidence of vasogenic edema, which correlated positively with APOE4 allele status, resolved after cessation of treatment.
Despite the lack of a clear dose-response, the potentially serious side effect of vasogenic edema in the APOE4 patients, and the difficulty in establishing efficacy in the overall population group, Bapineuzumab may have promise for non-APOE4 AD patients. It is hoped that results from the ongoing phase III trial of Bapineuzumab, in which a much larger number of participants exists than in the phase II trial, will provide an answer.
ii. LY2062430
LY2062430 is passive immunization therapy currently in phase II trials [28] that was developed by Eli Lilly and uses a human monoclonal antibody targeting the central domain of Aβ. Preclinical studies with the mouse analogue of this antibody, m266, demonstrated decreased plaque formation [11] and increased behavioral performance [29, 30]. Promising results from the phase II clinical trial recently were reported at ICAD. In this 12-week study, 52 patients with mild-to-moderate AD were given placebo or LY2062430 using dosing schedules of 100 mg or 400 mg every week or 100 mg or 400 mg every month. LY2062430 was well tolerated and no evidence was observed of inflammation, bleeding, or other cerebral side effects up to the tested 400 mg weekly infusions. Importantly, i.v.-administered LY2062430 increased plasma and cerebrospinal fluid (CSF) concentrations of Aβ, suggesting that the treatment facilitated plaque elimination.
iii. Intravenous Immunoglobulin (IVIg)
Intravenous immunoglobulin (IVIg) is an allogeneic polyclonal antibody treatment that putatively reduces Aβ levels in the brain and improves cognitive abilities in AD patients [31]. IVIg already was an FDA-approved therapy for several autoimmune and immunodeficiency diseases, yet its efficacy in AD has not been established. Human IVIg strongly inhibits fibril formation [32] and dissociates preformed Aβ fibrils in vitro [33]. IVIg also almost completely abrogates Aβ-induced neurotoxicity in rat hippocampal cells [32]. The mechanism of IVIg action may include increased cellular resistance to Aβ neurotoxicity and enhanced microglial phagocytosis of Aβ deposits [33].
A pilot clinical study of five AD patients suggested that IVIg administered over 6 months resulted in significant decreases in total Aβ levels in CSF and serum, with significant cognitive improvement as measured using the ADAS-Cog [34]. A follow-up open label study was conducted on 8 mildly affected AD patients treated with 2 g IVIg/kg for 6 months, followed by a 3-month washout period, and a final treatment regimen of 9 months [31]. CSF Aβ concentrations decreased during IVIg administration but returned to pre-treatment levels after washout. Interestingly, mini-mental state examination (MMSE) scores increased an average of 2.5 points after the initial treatment, but returned to baseline after washout and remained stable during the second phase of treatment [31]. No significant adverse events were noted in this study, suggesting that IVIg treatment is well tolerated. Currently, IVIg is in phase II testing in a 6-month long DBRPCT [35].
iv. Other Trails
Two other passive immunization trials currently are in phase I testing, RN-1219, a humanized Aβ-specific monoclonal antibody, and R-1450 [36], a monoclonal antibody developed from a fully synthetic human combinatorial antibody library (HuCAL) [37].
2. Nutraceuticals
Nutraceuticals are natural products or extracts therefrom that display medicinal or health benefits, independent of their nutritive properties. In the majority of cases, claims of health benefits from these compounds are unsupported by rigorous scientific studies. However, an increasing body of pre-clinical research, and certain clinical studies, suggest that some nutraceuticals may be of value as AD therapeutic agents.
a. Grape Seed Extracts
Recent epidemiological studies suggest that consumption of moderate amounts of red wine may protect against the development of AD [38–42]. Red wine is known to contain a broad range of polyphenols derived from the skin and seeds of grapes [43–47]. Several reports on the effects of these polyphenols suggest why consumption of red wine correlates with decreased AD risk. Wine-related polyphenols are potent anti-oxidants. Oxidation reactions have been postulated to be important pathologic processes in AD. Polyphenols thus may protect neurons from oxidative injury by scavenging reactive oxygen species, including hydroxyl or superoxide radicals [48, 49]. However, a number of reports show that wine-related polyphenols also inhibit Aβ fibril formation, fibril elongation, and dissociate pre-formed Aβ fibrils in vitro [50–52]. These polyphenols include tannic acid (TA), myricetin (Myr), morin (Mor), quercetin (Qur), kaempferol (Kmp), (+)-catechin (Cat), and (−)-epicatechin (epi-Cat). The rank order of polyphenol activity, from most to least active, was TA >> Myr = Mor = Qur > Kmp > Cat = epi-Cat [50, 51]. Finally, a recent study showed that a commercially available polyphenol-containing grape seed extract, MegaNatural-AZ, significantly attenuated AD-type cognitive deterioration and reduced cerebral Aβ concentrations in Tg2576 mice [52]. The effects of this preparation appear to come from its antioxidant activity and from its ability to block Aβ folding and assembly [53].
b. Curcumin and Rosmarinic Acid
Curcumin (Cur), a potent polyphenolic antioxidant and anti-inflammatory agent [54–56], is a main component of the Indian curry spice turmeric. Rosmarinic acid (RA), another polyphenol, is an ester of caffeic acid and 3,4-dihydroxyphenyllacetic acid [57]. RA comes from the mint family of plants and possesses several interesting biological activities, including antioxidative, anti-inflammatory, anti-mutagenic, anti-bacterial, and anti-viral [58].
Employing Thioflavin T (ThT) assays and electron microscopy (EM), Ono et al. demonstrated that Cur and RA inhibited Aβ fibril formation and elongation and destabilized preformed fibrils [59]. Interestingly, both Cur and RA showed similar anti-amyloidogenic activity. Cur and RA possess two 4-hydroxy-3-methoxyphenyl rings and two 3,4-dihydroxyphenyl rings, respectively, separated by a short symmetric hydrocarbon chain that confers symmetry and compactness [60]. This structural arrangement may be critical for specific binding to free Aβ and for subsequent inhibition of Aβ fibril formation [59, 60]. Alternatively, this structure might mediate binding and destabilization of the cross-β core of Aβ fibrils [59].
Several reports suggest that Cur could be a useful AD therapeutic. In addition to data showing that Cur can protect cells from Aβ-induced oxidative injury [61], studies in Tg2576 mice show that Cur decreases the amounts of soluble and insoluble Aβ as well as lessening plaque burden in many affected brain regions [62]. Recently, Yang et al. also reported that Cur inhibits formation of Aβ oligomers and fibrils, binds plaques, and reduces plaque burden in vivo [63]. Moreover, systemic administration of Cur for 7 days to APPswe/PS1ΔE9 mice reduced existing plaques and caused significant reversal of structural changes in dystrophic dendrites [64]. In humans, phase II clinical trials of curcumin C3 complex (Cur, demethoxycurcumin, and bisdemethoxycurcumin) in persons with mild-to-moderate AD are ongoing [65].
c. Ginkgo Biloba
Ginkgo biloba leaves have been used for centuries in traditional Chinese medical practice [66]. Ginkgo leaf extract, which on average contain ~24% flavonol glycosides, ~6% terpene trilactones (ginkgolides, bilobalide), ~7% proanthocyanidins, and other constituents, is prescribed for conditions that include cerebrovascular insufficiency, peripheral vascular insufficiency, and cognitive impairment associated with aging and AD [66]. Pre-clinical and clinical studies indicate that Ginkgo biloba extract counteracts neural and vascular damage, enhances cognition, improves blood flow, enhances tissue metabolism, and opposes the detrimental effects of ischemia [67, 68].
Ginkgo biloba extract has been reported to inhibit Aβ aggregation [69] and prevent Aβ-induced neurotoxicity in vitro [70, 71]. These effects may underlie results showing that chronic Ginkgo biloba treatment blocks age-dependent decline in spatial cognition in Tg2576 mice without altering Aβ levels or suppressing protein oxidation [72]. We note, however, that one study has shown that Ginkgo biloba treatment may inhibit Aβ production by lowering free cholesterol levels in aging rats [66].
Several clinical studies have suggested that Ginkgo biloba treatment may benefit patients with acquired cognitive impairment, including AD and vascular dementia, based on significant improvements in measures such as clinical global improvement (CGI), activities of daily living (ADL), and Neuropsychiatric Inventory (NPI) [73–75]. However, a recent DBRPCT involving 513 patients administered Ginkgo biloba did not show any treatment benefit [76]. It is possible that the lack of cognitive and functional decline in placebotreated patients in this study obscured any treatment difference resulting from the administration of Gingko biloba. Ongoing studies involving a broader spectrum of AD patients, from mild to severe, may help to determine whether Gingko biloba has therapeutic value [77].
d. Anti-Oxidants
Many studies indicate that oxidative injury is apparent in the brains of AD patients and that it may be involved in AD pathogenesis [78–80]. For this reason, a variety of antioxidants, including vitamin E (α-tocopherol) [81–85], vitamin C (ascorbic acid) [86, 87], and vitamin A [88, 89], have been suggested as potential therapeutic agents. In fact, administration of vitamins reportedly delays the progression of dementia [88, 90–94]. However, a recent study of vitamin E treatment of patients with mild cognitive impairment showed no benefits [95].
Several mechanisms have been postulated to explain the observed effects of vitamins, including protection of neurons from Aβ-induced neurotoxicity [81–87]. However, in addition to effects on redox chemistry, anti-oxidants also may affect Aβ assembly directly. To explore this possibility, Ono et al. [96] studied the effects of antioxidant vitamins, including vitamin A (all-trans-retinol, all-trans-retinal, and all-trans-retinoic acid), β-carotene, and vitamins B2, B6, C, and E on the formation, elongation, and destabilization of Aβ fibrils in vitro. Among these antioxidants, vitamin A and β-carotene inhibited the formation and elongation of Aβ fibrils and destabilized preformed fibrils. The rank order of inhibitory activity of the molecules examined was all-trans-retinol = all-trans-retinal > β-carotene > all-trans-retinoic acid. Consistent with the high potency of all-trans-retinol, Aβ40 fibrils pre-treated with all-trans-retinol have been found to be less toxic than untreated Aβ40 fibrils in MTT assays on HEK cells [96].
3. Peptidic Inhibitors
a. β-Sheet Breakers
Several reports have indicated that β-sheet-containing Aβ assemblies, including oligomers and fibrils, are neurotoxic [97–101]. It has been hypothesized that short peptides homologous to the central region of Aβ and with a low propensity to adopt a β-sheet conformation, “β-sheet breakers,” could inhibit Aβ oligomerization and fibril formation. In particular, Soto et al. [102] showed that an 11-residue β-sheet breaker peptide (iAβ11) binds to Aβ with high affinity and inhibits Aβ fibril formation in vitro. A 5-residue β-sheet breaker peptide (iAβ5), with the primary structure LPFFD, later was synthesized and found to inhibit Aβ fibrillation, disassemble preformed fibrils, and prevent fibril-induced neuronal death in cell culture [103, 104]. Treatment with iAβ5, which has good pharmacological properties and rapid BBB penetration, reduced significantly the amyloid burden, neurodegeneration, and brain inflammation normally observed in two different Tg AD mouse models [105]. Chacón et al. [106] have reported that treatment with iAβ5 significantly improved hippocampal-dependent spatial learning (Morris water maze and working memory analysis) in rats injected with Aβ fibrils. Finally, Austen et al. [107] created two peptide-based inhibitors comprising the AβKLVFF sequence and flanking G and R residues (to increase solubility). These peptides, OR1 (RGKLVFFGR) and OR2 (RGKLVFFGR-NH2), effectively inhibited Aβ fibril formation [107]. However, only the OR2 peptide also inhibited both Aβ oligomer formation and Aβ toxicity in human neuroblastoma SH-SY5Y cells [107].
b. N-Methyl Peptides
A fundamental aspect of Aβ fibril assembly is the formation of extended β-sheets. This suggests that disruption of interatomic interactions mediating β-sheet formation could have therapeutic benefits. One approach to β-sheet disruption has been the creation of N-methyl-peptide inhibitors. Methylation of backbone amides eliminates hydrogen atoms necessary for hydrogen bonding and prevents the close approach of other β-strands in a β-sheet through steric hindrance [108–112].
Incorporation of one N-methyl peptide as the edge strand of a β-sheet blocks subsequent addition of new strands [113]. Sciarretta et al. [114] have reported that incorporation of N-methyl amino acids into Aβ can disrupt both N- and C-terminal β-sheets and that this disruption renders Aβ “monomeric” and unable to form fibrils. In another study, using ThT fluorescence, EM, and MTT assays, Yan et al. [115] showed that an islet amyloid polypeptide (IAPP) analogue, [N-methyl-G24, N-methyl-I26]-IAPP (IAPP-GI), blocks formation of cytotoxic Aβ40 oligomeric and fibrillar assemblies. Decreases in the concentrations of these assemblies correlate with the formation of high-affinity Aβ40-IAPP-GI heteroassemblies [115].
4. Carbohydrate-Containing Compounds
a. GM1 Ganglioside
Ganglioside GM1, a glycosphingolipid localized to the outer leaf of the plasma membrane, exists at highest concentration in the brain. It has been suggested that GM1 and other gangliosides have potential as therapeutic agents in stroke and AD [116]. For example, GM1 administered intravenously to Tg PS/APP double transgenic mice reduces amyloid plaque load [117]. It is surprising then that GM1 ganglioside-containing vesicles have been found to associate avidly with Aβ and act as a template or “seed” that accelerates the rate of Aβ fibrillation in vitro [118–121]. The observed reductions in cerebral plaque load caused by GM1 thus may result from GM1 acting as a “peripheral sink” for Aβ, as discussed in section 1 regarding antibodies. This hypothesis is reasonable considering that GM1 likely cannot cross the BBB [122].
Clinical trials have assessed the utility of GM1 as an AD therapeutic. A DBRPCT trial of 12 AD patients injected daily (i.m.) with GM1 failed to show significant improvement in cognitive performance [123]. However, in another study, but one that was “open label” and lacked a placebo control, patients did exhibit improved motor performance, delayed deterioration of cognitive tasks, increased cerebral blood flow, and improved neuropsychological evaluation. In this study, five AD patients receiving intracerebroventricular injections of GM1 (20–30 mg/24h) were treated for one year [124]. Based solely on the results of these two small studies, it appears that peripheral administration of GM1 may be ineffective. However, the value of intracerebroventricular GM1 injection also is uncertain, pending follow-up studies that integrate rigorous controls and appropriate cognitive assessments of AD with a larger patient pool.
b. Glycosaminoglycans (GAGs)
Proteoglycans (PGs), glycosaminoglycan (GAG) chains attached to a protein core, are complexes found within plaques in AD patients [125]. PGs bind avidly to Aβ, accelerate fibril formation, stabilize fibrils, and protect Aβ against proteolysis [126–128]. It is believed that low molecular weight GAGs have the ability to antagonize the action of PGs [127, 129]. For example, sulphonate or sulphate-containing GAGs inhibit the aggregation and toxicity of Aβ, presumably through a mechanism involving competition for Aβ binding sites with structurally analogous GAG side chains on PGs [130–132]. These activities suggest that small GAGs may be useful therapeutic agents. However, no clinical studies of this question have been reported yet.
5. Polyamines
Polyamines are low molecular weight aliphatic amines. The naturally-occurring polyamine spermine (H2N-(CH2)3-NH-(CH2)4-NH-(CH2)3-NH2) has been reported to scavenge free radicals generated by Aβ that cause oxidative stress. However, treatment of cultured neurons with spermine and freshly prepared Aβ42 resulted in more toxicity than either spermine or Aβ alone [133]. Another polyamine with multiple medical applications, quinacrine (also known as Atabrine™), is a more promising potential inhibitor of Aβ assembly. Quinacrine has been investigated extensively for the treatment of prion diseases and has been shown to inhibit prion replication in vitro [134]. Recently, a multimeric quinacrine conjugate showed direct inhibition of Aβ self-assembly [135]. However, monomeric quinacrine showed no inhibitory effect on Aβ fibril formation [135]. Multimeric quinacrine compounds, and other polyamines, thus may comprise a new class of Aβ assembly inhibitors.
6. “Drug-Like” Compounds
“Drug-like” compounds are small molecules with physical and biological properties similar to those of drugs currently in use, which makes them attractive candidates for full-scale development efforts. These compounds contain functional groups or have physical properties that produce good absorption, distribution, metabolism, excretion, and toxicity profiles. A number of metrics are used to evaluate attractiveness of candidate drugs. One is Lipinski’s “rule of five” [136], which states that an orally active drug should not violate more than one of the following criteria: (1) no more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms); (2) no more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms); (3) a molecular weight < 500; and (4) a partition coefficient (log P) < 5. Other metrics include the number of rotatable bonds (≤8) and rings (≤4) [137]. Ghose et al. [138] have suggested that ideal ranges for the partition coefficient and molecular weight are −0.4–5.6 and 160–480, respectively. Ghose et al. also suggest a molar refractivity range of 40–130 m3 mol−1. “Drug-likeness” is a quality that is used frequently in the context of high-throughput screening (HTS) of combinatorial compound libraries [139].
a. “Drug-Like” Compounds Targeting A
A diversity of organic compounds inhibit Aβ fibrillogenesis in vitro, including nicotine [140], β-cyclodextrin [141], hemin and related porphyrins [142], the anthracycline 4′-iodo-4′-deoxydoxorubicin [143], hexadecyl-N-methylpiperidinium bromide [144], rifampicin [145], (−)-5,8-dihydroxy-3R-methyl-2R-(dipropylamino)-1,2,3,4-tetrahydronaphthalene [146], and melatonin [147]. Salvianolic acid B inhibits Aβ fibril formation, disaggregates preformed fibrils, and protects against Aβ-induced cytotoxicity [148].
In one study, Bartolini et al. [149] induced Aβ aggregation using human recombinant acetylcholinesterase (AChE) and then small molecules were tested for their ability to inhibit this process. Molecules including propidium iodide (a peripheral anionic-site ligand of human recombinant AChE), decamethonium bromide, donepezil, and physostigmine were active [149]. Propidium iodide, decamethonium bromide, physostigmine, and donepezil all are AChE inhibitors and four of the five FDA-approved therapeutic agents for AD belong to this drug class. Unfortunately, a relatively long history of use of these compounds in AD patients shows that they are not effective disease-modifying agents.
Non-steroidal anti-inflammatory drugs (NSAIDs) also have been reported to inhibit human aluminum-induced Aβ and IAPP aggregation in vitro [150, 151]. Kim and Lee found that 1,2-(dimethoxymethano)fullerene specifically binds to the 16–20 region of Aβ with high affinity, thus putatively inhibiting the early stages of amyloid formation [152]. The lack of structural similarity amongst the aforementioned drug-like compounds is striking, suggesting that these inhibitors bind to different sites on the Aβ monomer or to different structures formed by Aβ oligomerization or polymerization. This phenomenon contrasts with the actions of most other drugs, which depend on binding of one or more members of a class to a single target site. The diversity of Aβ structures to which the “drug-like” compounds bind complicates the execution of rational drug design strategies.
b. Tramiprosate
Tramiprosate (3-amino-1-propanesulfonic acid; Alzhemed™) has been shown to bind preferentially to pre-fibrillar (monomeric and oligomeric) forms of Aβ, preventing their conversion into higher-order aggregates and fibrils [153, 154]. Studies in primary neuronal cultures from rat brain showed that tramiprosate decreased Aβ42-induced cell death by 38% [153]. Administration of tramiprosate to Tg mice that develop cerebral amyloidosis resulted in significant reductions in plasma Aβ (60%), soluble Aβ40 (30%), insoluble Aβ40 (31%), soluble Aβ42 (25%), and insoluble Aβ42 (22%) [153]. Importantly, tramiprosate crosses the BBB and causes minimal toxicity in mice [153]. For these reasons, clinical trials of tramiprosate were undertaken. Unfortunately, the latest results from a phase III trial in patients with mild-to-moderate AD showed no significant benefits relative to placebo-treated controls [155]. Neurochem, the maker of Alzhemed™, has abandoned the study and apparently now is marketing this product as a “dietary supplement” [156].
c. Nicotine
An inverse correlation has been reported between the amount of cigarette smoking and the risk of AD [157]. Although tobacco is linked with cancer and many other health risks, an active ingredient in tobacco, nicotine, may be a beneficial pharmaceutical agent [158]. Nicotine, injected or introduced into the body using a skin patch, was shown to improve significantly learning and memory in animals [159, 160] and in patients with AD [161–163]. These nicotine-related positive effects were ascribed mainly to the neuroprotection conferred against Aβ-induced toxicity by upregulation of nicotinic receptors deficient in the AD brain [164, 165]. However, nicotine also has been found to inhibit Aβ fibril formation and destabilize preformed fibrils [140, 166]. In mice, as would be predicted from these earlier studies, Nordberg et al. [167] showed that chronic nicotine treatment caused a significant diminution in Aβ42-containing plaques in Tg2576 mice.
The anti-amyloidogenic properties of nicotine may originate from the β-sheet disrupting character of the N-methylpyrrolidine substituent. This notion is supported by the facts that N-methylpyrrolidine has similar anti-amyloidogenic properties to nicotine [166] and the N-methyl and 5′-methylene pyrrolidine moieties appear to bind to the His residues of Aβ(1–28) [140]. If this supposition is valid, then nicotine derivatives containing N-methylpyrrolidine moieties can be developed as agents that disrupt Aβ assembly, possibly avoiding the vasoconstrictive and thrombogenic properties of nicotine.
7. Chaperones
Cellular chaperone proteins prevent the misfolding of nascent polypeptides. Chaperones selectively recognize and bind to exposed hydrophobic surfaces of non-native proteins via non-covalent interactions, thereby inhibiting aggregation of those proteins in vivo [168] and in vitro [169]. In addition to the prevention of protein aggregation, chaperones facilitate correct protein folding [168]. Consistent with these activities, numerous studies have suggested that heat shock proteins (HSPs) are critical for the prevention of amyloid formation [170–175].
Molecular chaperones comprise several distinct classes of sequence-conserved proteins, most of which are stress-inducible HSPs. Major classes of these HSPs are Hsp100, Hsp90, Hsp70, Hsp60, and the small HSPs. Data from cell-free in vitro studies indicate that Hsp70 and Hsp90 inhibit early stages of Aβ42 aggregation [169] and that Hsp90 or Hsp70, with its co-chaperone Hsp40, dissociate preformed Aβ oligomers but not Aβ fibrils. The small heat shock proteins Hsp20, Hsp27, and αB-crystallin bind Aβ40 and inhibit Aβ aggregation and toxicity [176]. Other reports show that Hsp20 [177] and Hsp104 [178] also limit amyloid formation in vitro in neuronal cells and yeast, respectively. In vivo, chaperones have been shown to block formation of Aβ aggregates in Tg C. elegans [179].
Graef et al. [180, 181] have integrated the chaperone function of the FK506-binding protein (FKBP), a large soluble molecule that binds to the immunosuppressant FK506 and inhibits the enzyme calcineurin, with the amyloid binding activity of Congo red (CR) by linking the two through a synthetic ligand for FKBP (SLF). In this complex, CR putatively caps amyloid fibrils while FKBP sterically blocks the large contact surface of fibrils, thereby preventing their interaction and further assembly. SLF-CR/FKBP and SLF-benzidine-CR/FKBP complexes both blocked Aβ aggregation and inhibited the neurotoxic effects of such aggregates in primary cultures of rat hippocampal neurons [180].
It has been suggested that an imbalance between the capacity of chaperones to mediate proper protein folding and the number of nascent misfolded proteins is the cause of cytotoxicity [173]. Increased chaperone expression thus might suppress Aβ aggregate-induced neurotoxicity. Clinical studies of such treatments have not been done. Until they are, techniques can be developed that enhance chaperone activity or increase chaperone expression. It is possible that these approaches, in addition to ameliorating Aβ-specific pathologic aggregation, might also have beneficial effects on the folding of other proteins, including those involved in other neurodegenerative diseases.
8. Metal Chelation
Alterations in brain metal ion homeostasis, particularly of Cu2+ and Zn2+, have been implicated in Aβ-associated pathology [182]. Cu2+ and Zn2+ levels are elevated markedly in amyloid plaques, where the abundant Aβ present can coordinate these metal ions [183, 184]. Iron also has been found in Aβ deposits, and in vitro experiments indicate that all three metals, when coordinated by Aβ, may participate in redox chemistry leading to the production hydrogen peroxide and pathologic sequelae [185]. A logical therapeutic approach that has evolved from these findings is the sequestration of metals by chelation agents.
Bush et al. [186] have shown that chelators inhibit hydrogen peroxide production by Aβ in vitro. These compounds also can reverse the aggregation of Aβ in vitro and in vivo (based on analysis of post-mortem human brain specimens) [186]. A particularly important chelator is clioquinol (CQ), an analogue of 8-hydroxy-quinoline. CQ has moderate affinity for Cu2+ and Zn2+ and has been found to inhibit metal-induced Aβ aggregation and reactive oxygen species generation in vitro [187]. Oral administration of CQ to Tg2576 mice reduced brain Aβ burden by ~50% after 9 weeks [187].
In a pilot phase IIa trial of CQ (PBT1), AD patients showed evidence of delayed cognitive deterioration and significantly lowered plasma Aβ levels [188]. However, phase II/III trials were halted due to impurities in the formulation [189]. Adlard et al. [190] characterized a second-generation 8-hydroxy-quinoline analogue, PBT2, which also targets metal-induced Aβ aggregation and has greater BBB permeability. Compared to CQ, PBT2 significantly decreased soluble interstitial brain Aβ levels in Tg mice within hours and improved cognitive performance to levels that were equal to or better than those of non-transgenic littermates. Non-transgenic mice were unaffected by PBT2 administration.
A recent 12-week phase IIa DBRPCT of PBT2 was conducted on patients with mild-to-moderate AD [191, 192]. In this trial, participants received oral PBT2 (50 mg or 250 mg) or placebo. Patients treated with 250 mg PBT2 showed significant diminution in frontal lobe functional deficits, as measured by the Category Fluency Test and Trail Making Part B, and significant decreases in CSF Aβ42 levels. Importantly, no serious side-effects were noted in patients that received the PBT2 formulation. Future trials on a larger AD patient pool will be necessary to determine the clinical utility of PBT2.
9. Osmolytes
Osmolytes are small organic solutes produced by cells to prevent the denaturation of intracellular proteins by environmental stressors such as heat, dehydration, high salt or urea concentration [193, 194]. Osmolytes are chemically diverse and include chemical classes such as sugars, polyols, amino acids, and methylamine compounds [193, 195]. At high concentrations, which generally are toxic in humans, osmolytes facilitate correct protein folding and therefore function as “chemical chaperones.” However, compounds that bind specifically to target proteins may function like osmolytes and do so at low concentrations. These “pharmacological chaperones” could be attractive agents for prevention of pathologic Aβ folding and assembly [196, 197].
Some studies of osmolytic effects on Aβ aggregation have been done. Trehalose, a simple α-linked disaccharide, is an effective inhibitor of Aβ aggregation and reduces Aβ-induced cytotoxicity [198]. Sucrose delays Aβ fibril growth and also hinders the racemization of D-aspartic acid [199], which contributes to the production of Aβ deposits [200].
The best studied osmolyte is cyclohexanehexol (inositol), a naturally-occurring cyclic polyol compound that is found in foods such as nuts, beans, and fruits. McLaurin et al. [201, 202] showed that three cyclohexanehexol stereoisomers, scyllo-cyclohexanehexol, epi-cyclohexanehexol, and myocyclohexanehexol, inhibit Aβ fibril assembly, disassembled preformed fibrils, and blocked oligomer-induced toxicity in primary neuronal cultures. Although myo-cyclohexanehexol is the most abundant isomer found in the brain, scyllo-cyclohexanehexol and epi-cyclohexanehexol were shown to be better inhibitors of Aβ aggregation and toxicity [202]. Furthermore, orally administered scyllo-and epi-cyclohexanehexol stereoisomers block the deposition and neurotoxic effects of Aβ in the TgCRND8 mouse model of AD [203]. These isomers also inhibited the formation of soluble Aβ oligomers and insoluble Aβ aggregates in a dose-dependent manner and decreased AD-like behavioral deficits, AD-like pathology, and mortality in the TgCRND8 mice [203]. Scyllo-cyclohexanehexol inhibited the growth of plaques in mice with advanced stages of AD-like pathology and brain concentrations of the compound could be achieved that were an order of magnitude higher than that of the endogenous compound [204]. Inosose stereoisomers, which differ from inositols by the replacement of one hydroxyl group by a ketone, also can inhibit Aβ aggregation [205].
It has been hypothesized that the mechanism of inositol and inosose stereoisomer action is osmolyte modulation of Aβ folding [206] and competition for Aβ binding sites. Inositol and inosose stereoisomers can compete for Aβ binding sites [203] with phosphatidylinositol lipids that facilitate Aβ oligomerization and fibril formation [201, 207–209]. However, in addition to these direct effects on Aβ aggregation, osmolytes may increase expression of HSPs and other intracellular chaperones [210] and increase the efficiency of protein folding during cellular stress [211, 212]. In conclusion, cyclohexanehexols are attractive therapeutic agents as they readily cross the BBB, are found endogenously in the brain, and can diminish Aβ accumulation and neurotoxic effects before, during, and even after the onset of AD-like pathological events in mice.
10. Nascent Approaches
Thus far, we have reviewed therapeutic approaches for controlling Aβ assembly for which substantial pre-clinical or clinical data exist. We now turn to three nascent, but intriguing, approaches that also may have promise.
a. γ-secretase Modulators (GSMs)
As discussed in the introduction, a major therapeutic strategy for AD is blocking Aβ production. To do so, inhibitors are being sought that target the enzymes that produce Aβ, particularly β-secretase and γ-secretase, which cleave the N-terminus and C-terminus of Aβ, respectively, from APP. Until recently, small-molecule γ-secretase modulators (GSMs), such as tarenflurbil (also known as R-flurbiprofen or Flurizan™), have been hypothesized to target γ-secretase, although no clear mechanism of action has been delineated [213–215]. However, it recently has been demonstrated that enzyme inhibitors appear to bind APP directly [216]. Furthermore, the GSM interaction with APP was isolated to the 28–36 region of Aβ. Experimental evidence suggests that the N-terminal portion of this region is a critical mediator of Aβ folding and aggregation [217] and that this region would be a particularly important therapeutic target [218]. An unanticipated application for GSMs thus may be as inhibitors of Aβ folding and aggregation. The recognition of this novel use of GSMs is particularly fortuitous in light of recently presented results of phase III clinical trials of Flurizan™ that failed to show any benefit in AD patients [219].
b. Aptamers
Aptamers are short single-stranded DNA or RNA molecules that bind to various substrates, including nucleic acids and proteins, with high specificity and avidity. For example, an RNA aptamer specific for murine PrPSc possessed a dissociation constant of ~2.1 nM [220] and many other protein-specific aptamers have affinities in the nM range [221–223] to pM range [224]. Aptamers are produced from screens of large combinatorial libraries in a process called Systematic Evolution of Ligands by Exponential Enrichment (SELEX) that identifies and amplifies structures with high target affinity [225]. Aptamers may prove to be exceptionally useful agents for preventing or reversing the peptide aggregation and fibril formation associated with AD. Aptamers have some advantages over antibodies. Antibodies may be difficult to produce if the antigens being targeted are toxic or weakly immunogenic [224]. In addition, murine antibodies, which are the most common source of monoclonals, necessitate “humanization” for therapeutic use [224]. Aptamer synthesis, in contrast, does not have these difficulties. Aptamer synthesis is straightforward, cost-efficient, and amenable to automation [224]. In the AD field, RNA aptamers recently have been developed that specifically modulate protein factors that interfere with the recruitment of proteins that typically associate with BACE1 (β-secretase) [222].
c. Platinum-Based A Inhibitors
Aβ amino acid residues His6, His13, and His14 form coordination complexes with certain metals that accelerate Aβ assembly and are involved in redox chemistry [226, 227]. As discussed above, metal-targeted strategies, e.g., the use of chelators, is one approach to blocking the coordination process and its sequelae. However, rather than target the metal itself, an alternative approach recently discussed by Barnham et al. [228] is the use of various 1,10-phenanthroline-PtCl2 complexes to bind to the high affinity metal coordination sites on Aβ. In an impressive set of experiments, Barnham et al. demonstrated that this binding alters Aβ structure, inhibiting fibril formation, production of reactive oxygen species, perturbation of cellular redox activity, and synaptotoxicity.
CAVEATS AND CONCLUSIONS
An unprecedented number of clinical trials are being conducted to identify AD drugs. The diversity of potential therapeutic agents (Table 1) reflects the complexity of the disease mechanism(s) (Fig. 1). Initial trails have been disappointing and thus the long awaited first disease-modifying therapeutic agent remains elusive. Such disappointments are not unusual, but they should stimulate discussion about current therapeutic strategies. Two topics are especially important: (1) is the amyloid cascade hypothesis correct; and (2) if it is, are the therapeutic targets extant the correct ones?
The answer to the first question is unknown. However, the innovative and complementary therapeutic strategies currently being executed may provide one. If efficacious drugs are discovered, support for the amyloid cascade hypothesis will have been strengthened and impetus for increased efforts to develop Aβ-specific therapeutics will exist. More importantly, patients finally would have ameliorative or curative therapeutic options.
If efficacious drugs are not discovered, both questions will remain. In that case, continued efforts to develop Aβ-specific drugs also are warranted to ensure that all reasonable targets are studied. These efforts must be conducted thoughtfully and rigorously, and if they are, the resultant information either will lead the field in more promising directions or provide compelling reasons for a shift to new working hypotheses of disease causation.
Acknowledgments
G.Y. was supported by grants from the UCLA Graduate Research Mentorship Program and the UCLA Chemistry-Biology Interface (CBI) Training Program. K.O. was supported by the Japan Human Science Foundation, a Pergolide Fellowship from Eli Lilly Japan, and the Mochida Memorial Foundation for Medical and Pharmaceutical Research. Support from the State of California Alzheimer’s Disease Research Fund (#07-65806), the Jim Easton Consortium for Alzheimer’s Drug Discovery and Biomarkers at UCLA, and NIH grants AG027818 and AT004511 also is gratefully acknowledged.
ABBREVIATIONS
- Aβ
Amyloid β-protein
- AChE
Acetylcholinesterase
- AD
Alzheimer’s disease
- ADAS-Cog
AD Assessment Scale-Cognitive Subscale
- APP
Amyloid β-protein precursor
- BBB
Blood-brain barrier
- Cat
(+)-Catechin
- CQ
Clioquinol (iodochlorhydroxyquin or 5-chloro-7-iodo-8-hydroxyquinoline)
- CR
Congo red
- Cur
Curcumin
- DAD
Disability Assessment Scale for Dementia
- DBRPCT
Double-blind, randomized, placebo-controlled trial
- EM
Electron microscopy
- Epi-Cat
(−)-Epicatechin
- FKBP
FK506-Binding protein
- GSMs
γ-Secretase modulators
- HEK
Human embryonic kidney
- HSP/Hsp
Heat shock protein
- iAβ
β-Sheet breaker peptide
- ICAD
International Conference on Alzheimer’s Disease
- i.m.
Intramuscular
- i.v.
Intravenous
- Kmp
Kaempferol
- MHC
Major histocompatibility complex
- Mor
Morin
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Myr
Myricetin
- OR1
Peptide with the sequence RGKLVFFGR
- OR2
Peptide with the sequence RGKLVFFGR-NH2
- PC12
Pheochromocytoma 12
- PrPSc
Scrapie form of the prion protein
- Qur
Quercetin
- RA
Rosmarinic acid
- SLF
Synthetic ligand for FKBP
- TA
Tannic acid
- Tg
Transgenic
- Tg2576
Transgenic mouse that over-expresses human APP with the Swedish (K670N, M671L) mutation
- TgCRND8
Transgenic mouse that over-expresses human APP with Swedish (K670N, M671L) and Indiana (V717F) mutations
- ThT
Thioflavin T
References
References 229-231 are related articles recently published.
- 1.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–81. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s & Dementia. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 3.Meek PD, McKeithan K, Schumock GT. Economic considerations in Alzheimer’s disease. Pharmacotherapy. 1998;18:68–73. discussion 79–82. [PubMed] [Google Scholar]
- 4.Voelker R. Guideline: dementia drugs’ benefits uncertain. JAMA. 2008;299:1763. doi: 10.1001/jama.299.15.1763. [DOI] [PubMed] [Google Scholar]
- 5.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-protein assembly and Alzheimer’s disease. J Biol Chem. 2008 doi: 10.1074/jbc.R800036200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 7.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 8.Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer β-amyloid peptide. Proc Natl Acad Sci USA. 1996;93:452–5. doi: 10.1073/pnas.93.1.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc Natl Acad Sci U S A. 1997;94:4109–12. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Legleiter J, Czilli DL, Gitter B, DeMattos RB, Holtzman DM, Kowalewski T. Effect of different anti-Aβ antibodies on Aβ fibrillogenesis as assessed by atomic force microscopy. J Mol Biol. 2004;335:997–1006. doi: 10.1016/j.jmb.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 11.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science. 2002;295:2264–7. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- 13.Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid-β and inflammation: two faces of innate immunity in Alzheimer’s disease. J Alzheimers Dis. 2007;11:457–63. doi: 10.3233/jad-2007-11406. [DOI] [PubMed] [Google Scholar]
- 14.Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–93. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, et al. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–82. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 16.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–5. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 17.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, et al. Evaluation of the safety and immunogenicity of synthetic Aβ42 (AN1792) in patients with AD. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 18.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 19.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 20.Study evaluating safety, tolerability, and immunogenicity of ACC-001 in subjects with Alzheimer’s disease. http://www.clinicaltrials.gov/ct2/show/NCT00479557.
- 21.Study evaluating ACC-001 in mild to moderate Alzheimers disease subjects. http://www.clinicaltrials.gov/ct2/show/NCT00498602.
- 22.Safety and tolerability study in patients with mild to moderate Alzheimer’s disease (AD) http://www.clinicaltrials.gov/ct2/show/NCT00411580.
- 23.A study of V950 in people with Alzheimer disease. http://www.clinicaltrials.gov/ct2/show/NCT00464334.
- 24.Bapineuzumab in patients with mild to moderate Alzheimer’s disease. http://www.clinicaltrials.gov/ct2/show/NCT00575055, http://www.clinicaltrials.gov/ct2/show/NCT00574132.
- 25.Study evaluating the safety and efficacy of Bapineuzumab in Alzheimer disease patients. http://www.clinicaltrials.gov/ct2/show/NCT00676143.
- 26.Study evaluating the efficacy and safety of Bapineuzumab in Alzheimer disease patients. http://www.clinicaltrials.gov/ct2/show/NCT00667810.
- 27.Elan and Wyeth present encouraging results from phase 2 clinical trial of Bapineuzumab at International Conference on Alzheimer’s Disease. http://www.elan.com/News/full.asp?ID=1180940.
- 28.Effects of LY2062430 in subjects with mild-to-moderate Alzheimer’s disease and in healthy volunteers. http://www.clinicaltrials.gov/ct2/show/NCT00329082.
- 29.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nat Neurosci. 2002;5:452–7. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 30.Bales KR, Tzavara ET, Wu S, Wade MR, Bymaster FP, Paul SM, et al. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-Aβ antibody. J Clin Invest. 2006;116:825–32. doi: 10.1172/JCI27120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, et al. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.021. in press. [DOI] [PubMed] [Google Scholar]
- 32.Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, et al. Human anti-β-amyloid antibodies block β-amyloid fibril formation and prevent β-amyloid-induced neurotoxicity. Brain. 2003;126:1935–9. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- 33.Istrin G, Bosis E, Solomon B. Intravenous immunoglobulin enhances the clearance of fibrillar amyloid-β peptide. J Neurosci Res. 2006;84:434–43. doi: 10.1002/jnr.20886. [DOI] [PubMed] [Google Scholar]
- 34.Dodel RC, Du Y, Depboylu C, Hampel H, Frolich L, Haag A, et al. Intravenous immunoglobulins containing antibodies against β-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–4. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phase II study of intravenous immunoglobulin (IVIg) for Alzheimer’s disease. http://www.clinicaltrials.gov/ct2/show/NCT00299988.
- 36.A multiple ascending dose study of R1450 in patients with Alzheimer disease. http://www.clinicaltrials.gov/ct2/show/NCT00531804.
- 37.Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, et al. Fully synthetic human combinatorial antibody libraries (Hu-CAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000;296:57–86. doi: 10.1006/jmbi.1999.3444. [DOI] [PubMed] [Google Scholar]
- 38.Dartigues JF, Orgogozo JM, Letenneur L, Barberger-Gateau P. French epidemiological bases for the treatment of dementia syndromes and cognitive impairment in the elderly. Therapie. 1993;48:185–7. [PubMed] [Google Scholar]
- 39.Dorozynski A. Wine may prevent dementia. BMJ. 1997;314:997. [PMC free article] [PubMed] [Google Scholar]
- 40.Luchsinger JA, Tang MX, Siddiqui M, Shea S, Mayeux R. Alcohol intake and risk of dementia. J Am Geriatr Soc. 2004;52:540–6. doi: 10.1111/j.1532-5415.2004.52159.x. [DOI] [PubMed] [Google Scholar]
- 41.Orgogozo JM, Dartigues JF, Lafont S, Letenneur L, Commenges D, Salamon R, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris) 1997;153:185–92. [PubMed] [Google Scholar]
- 42.Truelsen T, Thudium D, Gronbaek M. Amount and type of alcohol and risk of dementia: the Copenhagen City Heart Study. Neurology. 2002;59:1313–9. doi: 10.1212/01.wnl.0000031421.50369.e7. [DOI] [PubMed] [Google Scholar]
- 43.Soleas GJ, Diamandis EP, Goldberg DM. Resveratrol: a molecule whose time has come? And gone? Clin Biochem. 1997;30:91–113. doi: 10.1016/s0009-9120(96)00155-5. [DOI] [PubMed] [Google Scholar]
- 44.Hertog MG, Hollman PC, Katan MB, Kromhout D. Intake of potentially anticarcinogenic flavonoids and their determinants in adults in The Netherlands. Nutr Cancer. 1993;20:21–9. doi: 10.1080/01635589309514267. [DOI] [PubMed] [Google Scholar]
- 45.Celotti E, Ferrarini R, Zironi R, Conte LS. Resveratrol content of some wines obtained from dried Valpolicella grapes: recioto and amarone. J Chromatogr A. 1996;730:47–52. doi: 10.1016/0021-9673(95)00962-0. [DOI] [PubMed] [Google Scholar]
- 46.Goldberg DM, Tsang E, Karumanchiri A, Diamandis E, Soleas G, Ng E. Method to assay the concentrations of phenolic constituents of biological interest in wines. Anal Chem. 1996;68:1688–94. doi: 10.1021/ac951083i. [DOI] [PubMed] [Google Scholar]
- 47.Sato M, Suzuki Y, Okuda T, Yokotsuka K. Contents of resveratrol, piceid, and their isomers in commercially available wines made from grapes cultivated in Japan. Biosci Biotechnol Biochem. 1997;61:1800–5. doi: 10.1271/bbb.61.1800. [DOI] [PubMed] [Google Scholar]
- 48.Virgili M, Contestabile A. Partial neuroprotection of in vivo excitotoxic brain damage by chronic administration of the red wine antioxidant agent, trans-resveratrol in rats. Neurosci Lett. 2000;281:123–6. doi: 10.1016/s0304-3940(00)00820-x. [DOI] [PubMed] [Google Scholar]
- 49.Inanami O, Watanabe Y, Syuto B, Nakano M, Tsuji M, Kuwabara M. Oral administration of (−)catechin protects against ischemia-reperfusion-induced neuronal death in the gerbil. Free Radic Res. 1998;29:359–65. doi: 10.1080/10715769800300401. [DOI] [PubMed] [Google Scholar]
- 50.Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzheimer’s disease. J Neurochem. 2003;87:172–81. doi: 10.1046/j.1471-4159.2003.01976.x. [DOI] [PubMed] [Google Scholar]
- 51.Ono K, Hasegawa K, Naiki H, Yamada M. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer’s β-amyloid fibrils in vitro. Biochim Biophys Acta. 2004;1690:193–202. doi: 10.1016/j.bbadis.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Ho L, Zhao W, Ono K, Rosensweig C, Chen L, et al. Grape-derived polyphenolics prevent Aβ oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J Neurosci. 2008;28:6388–92. doi: 10.1523/JNEUROSCI.0364-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ono K, Condron MM, Ho L, Wang J, Zhao W, Pasinetti GM, et al. Effects of grape seed-derived polyphenols on amyloid β-protein self-assembly and cytotoxicity. J Biol Chem. 2008 doi: 10.1074/jbc.M806154200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao BL, Li XJ, He RG, Cheng SJ, Xin WJ. Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys. 1989;14:175–85. doi: 10.1007/BF02797132. [DOI] [PubMed] [Google Scholar]
- 55.Sreejayan Rao MN. Nitric oxide scavenging by curcuminoids. J Pharm Pharmacol. 1997;49:105–7. doi: 10.1111/j.2042-7158.1997.tb06761.x. [DOI] [PubMed] [Google Scholar]
- 56.Xu YX, Pindolia KR, Janakiraman N, Chapman RA, Gautam SC. Curcumin inhibits IL1 alpha and TNF-alpha induction of AP-1 and NF-kB DNA-binding activity in bone marrow stromal cells. Hematopathol Mol Hematol. 1997;11:49–62. [PubMed] [Google Scholar]
- 57.Petersen M, Simmonds MS. Rosmarinic acid. Phytochemistry. 2003;62:121–5. doi: 10.1016/s0031-9422(02)00513-7. [DOI] [PubMed] [Google Scholar]
- 58.Parnham MJ, Kesselring K. Rosmarinic acid. Drugs of the Future. 1985;10:756–7. [Google Scholar]
- 59.Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J Neurosci Res. 2004;75:742–50. doi: 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- 60.Ono K, Naiki H, Yamada M. The development of preventives and therapeutics for Alzheimer’s disease that inhibit the formation of β-amyloid fibrils (fAβ), as well as destabilize preformed fAβ. Curr Pharm Des. 2006;12:4357–75. doi: 10.2174/138161206778793010. [DOI] [PubMed] [Google Scholar]
- 61.Kim DS, Park SY, Kim JK. Curcuminoids from Curcuma longa L. (Zingiberaceae) that protect PC12 rat pheochromocytoma and normal human umbilical vein endothelial cells from βA(1-42) insult. Neurosci Lett. 2001;303:57–61. doi: 10.1016/s0304-3940(01)01677-9. [DOI] [PubMed] [Google Scholar]
- 62.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–7. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 64.Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem. 2007;102:1095–104. doi: 10.1111/j.1471-4159.2007.04613.x. [DOI] [PubMed] [Google Scholar]
- 65.Ringman JM, Frautschy SA, Cole GM, Masterman DL, Cummings JL. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr Alzheimer Res. 2005;2:131–6. doi: 10.2174/1567205053585882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao ZX, Han Z, Drieu K, Papadopoulos V. Ginkgo biloba extract (Egb 761) inhibits β-amyloid production by lowering free cholesterol levels. J Nutr Biochem. 2004;15:749–56. doi: 10.1016/j.jnutbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 67.DeFeudis FV, Drieu K. Ginkgo biloba extract (EGb 761) and CNS functions: basic studies and clinical applications. Curr Drug Targets. 2000;1:25–58. doi: 10.2174/1389450003349380. [DOI] [PubMed] [Google Scholar]
- 68.Maclennan KM, Darlington CL, Smith PF. The CNS effects of Ginkgo biloba extracts and ginkgolide B. Prog Neurobiol. 2002;67:235–57. doi: 10.1016/s0301-0082(02)00015-1. [DOI] [PubMed] [Google Scholar]
- 69.Luo Y, Smith JV, Paramasivam V, Burdick A, Curry KJ, Buford JP, et al. Inhibition of amyloid-β aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc Natl Acad Sci U S A. 2002;99:12197–202. doi: 10.1073/pnas.182425199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bastianetto S, Ramassamy C, Dore S, Christen Y, Poirier J, Quirion R. The ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by β-amyloid. Eur J Neurosci. 2000;12:1882–90. doi: 10.1046/j.1460-9568.2000.00069.x. [DOI] [PubMed] [Google Scholar]
- 71.Yao Z, Drieu K, Papadopoulos V. The Ginkgo biloba extract EGb 761 rescues the PC12 neuronal cells from β-amyloid-induced cell death by inhibiting the formation of β-amyloid-derived diffusible neurotoxic ligands. Brain Res. 2001;889:181–90. doi: 10.1016/s0006-8993(00)03131-0. [DOI] [PubMed] [Google Scholar]
- 72.Stackman RW, Eckenstein F, Frei B, Kulhanek D, Nowlin J, Quinn JF. Prevention of age-related spatial memory deficits in a transgenic mouse model of Alzheimer’s disease by chronic Ginkgo biloba treatment. Exp Neurol. 2003;184:510–20. doi: 10.1016/s0014-4886(03)00399-6. [DOI] [PubMed] [Google Scholar]
- 73.Birks J, Grimley EV, Van Dongen M. Ginkgo biloba for cognitive impairment and dementia. Cochrane Database Syst Rev. 2002:CD003120. doi: 10.1002/14651858.CD003120. [DOI] [PubMed] [Google Scholar]
- 74.Napryeyenko O, Borzenko I. Ginkgo biloba special extract in dementia with neuropsychiatric features. A randomised, placebo-controlled, double-blind clinical trial. Arzneimittelforschung. 2007;57:4–11. doi: 10.1055/s-0031-1296579. [DOI] [PubMed] [Google Scholar]
- 75.Mazza M, Capuano A, Bria P, Mazza S. Ginkgo biloba and donepezil: a comparison in the treatment of Alzheimer’s dementia in a randomized placebo-controlled double-blind study. Eur J Neurol. 2006;13:981–5. doi: 10.1111/j.1468-1331.2006.01409.x. [DOI] [PubMed] [Google Scholar]
- 76.Schneider LS, DeKosky ST, Farlow MR, Tariot PN, Hoerr R, Kieser M. A randomized, double-blind, placebo-controlled trial of two doses of Ginkgo biloba extract in dementia of the Alzheimer’s type. Curr Alzheimer Res. 2005;2:541–51. doi: 10.2174/156720505774932287. [DOI] [PubMed] [Google Scholar]
- 77.Ginkgo biloba prevention trial in older individuals. http://www.clinicaltrials.gov/ct2/show/NCT00010803.
- 78.Praticò D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer’s disease. Am J Med. 2000;109:577–85. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- 79.Rottkamp CA, Nunomura A, Raina AK, Sayre LM, Perry G, Smith MA. Oxidative stress, antioxidants, and Alzheimer disease. Alzheimer Dis Assoc Disord. 2000;14(Suppl 1):S62–6. doi: 10.1097/00002093-200000001-00010. [DOI] [PubMed] [Google Scholar]
- 80.Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:139–44. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 81.Behl C, Davis J, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid β protein toxicity. Biochem Biophys Res Commun. 1992;186:944–50. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 82.Pereira C, Santos MS, Oliveira C. Involvement of oxidative stress on the impairment of energy metabolism induced by Aβ peptides on PC12 cells: protection by antioxidants. Neurobiol Dis. 1999;6:209–19. doi: 10.1006/nbdi.1999.0241. [DOI] [PubMed] [Google Scholar]
- 83.Subramaniam R, Koppal T, Green M, Yatin S, Jordan B, Drake J, et al. The free radical antioxidant vitamin E protects cortical synaptosomal membranes from amyloid β-peptide(25–35) toxicity but not from hydroxynonenal toxicity: relevance to the free radical hypothesis of Alzheimer’s disease. Neurochem Res. 1998;23:1403–10. doi: 10.1023/a:1020754807671. [DOI] [PubMed] [Google Scholar]
- 84.Yatin SM, Yatin M, Aulick T, Ain KB, Butterfield DA. Alzheimer’s amyloid β-peptide associated free radicals increase rat embryonic neuronal polyamine uptake and ornithine decarboxylase activity: protective effect of vitamin E. Neurosci Lett. 1999;263:17–20. doi: 10.1016/s0304-3940(99)00101-9. [DOI] [PubMed] [Google Scholar]
- 85.Zhou Y, Gopalakrishnan V, Richardson JS. Actions of neurotoxic β-amyloid on calcium homeostasis and viability of PC12 cells are blocked by antioxidants but not by calcium channel antagonists. J Neurochem. 1996;67:1419–25. doi: 10.1046/j.1471-4159.1996.67041419.x. [DOI] [PubMed] [Google Scholar]
- 86.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–27. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 87.Yallampalli S, Micci MA, Taglialatela G. Ascorbic acid prevents β-amyloid-induced intracellular calcium increase and cell death in PC12 cells. Neurosci Lett. 1998;251:105–8. doi: 10.1016/s0304-3940(98)00515-1. [DOI] [PubMed] [Google Scholar]
- 88.Jama JW, Launer LJ, Witteman JC, den Breeijen JH, Breteler MM, Grobbee DE, et al. Dietary antioxidants and cognitive function in a population-based sample of older persons. The Rotterdam study Am J Epidemiol. 1996;144:275–80. doi: 10.1093/oxfordjournals.aje.a008922. [DOI] [PubMed] [Google Scholar]
- 89.Perrig WJ, Perrig P, Stahelin HB. The relation between antioxidants and memory performance in the old and very old. J Am Geriatr Soc. 1997;45:718–24. doi: 10.1111/j.1532-5415.1997.tb01476.x. [DOI] [PubMed] [Google Scholar]
- 90.Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, et al. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223–9. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 91.Morris MC, Beckett LA, Scherr PA, Hebert LE, Bennett DA, Field TS, et al. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:121–6. doi: 10.1097/00002093-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 92.Morris MC, Evans DA, Bienias JL, Tangney CC, Wilson RS. Vitamin E and cognitive decline in older persons. Arch Neurol. 2002;59:1125–32. doi: 10.1001/archneur.59.7.1125. [DOI] [PubMed] [Google Scholar]
- 93.Paleologos M, Cumming RG, Lazarus R. Cohort study of vitamin C intake and cognitive impairment. Am J Epidemiol. 1998;148:45–50. doi: 10.1093/oxfordjournals.aje.a009559. [DOI] [PubMed] [Google Scholar]
- 94.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s disease cooperative study. N Engl J Med. 1997;336:1216–22. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 95.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–88. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 96.Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M. Vitamin A exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp Neurol. 2004;189:380–92. doi: 10.1016/j.expneurol.2004.05.035. [DOI] [PubMed] [Google Scholar]
- 97.Lorenzo A, Yankner BA. β-Amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci U S A. 1994;91:12243–7. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–87. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–32. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 100.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 101.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 102.Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem Biophys Res Commun. 1996;226:672–80. doi: 10.1006/bbrc.1996.1413. [DOI] [PubMed] [Google Scholar]
- 103.Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer’s therapy. Nat Med. 1998;4:822–6. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- 104.Ono K, Hasegawa K, Yoshiike Y, Takashima A, Yamada M, Naiki H. Nordihydroguaiaretic acid potently breaks down pre-formed Alzheimer’s β-amyloid fibrils in vitro. J Neurochem. 2002;81:434–40. doi: 10.1046/j.1471-4159.2002.00904.x. [DOI] [PubMed] [Google Scholar]
- 105.Permanne B, Adessi C, Saborio GP, Fraga S, Frossard MJ, Van Dorpe J, et al. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer’s disease by treatment with a β-sheet breaker peptide. FASEB J. 2002;16:860–2. doi: 10.1096/fj.01-0841fje. [DOI] [PubMed] [Google Scholar]
- 106.Chacón MA, Barría MI, Soto C, Inestrosa NC. β-sheet breaker peptide prevents Aβ-induced spatial memory impairments with partial reduction of amyloid deposits. Mol Psychiatry. 2004;9:953–61. doi: 10.1038/sj.mp.4001516. [DOI] [PubMed] [Google Scholar]
- 107.Austen BM, Paleologou KE, Ali SA, Qureshi MM, Allsop D, El-Agnaf OM. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s β-amyloid peptide. Biochemistry. 2008;47:1984–92. doi: 10.1021/bi701415b. [DOI] [PubMed] [Google Scholar]
- 108.Tonelli AE. Conformational characteristics of L-proline oligomers. J Am Chem Soc. 1970;92:6187–90. doi: 10.1021/ja00724a015. [DOI] [PubMed] [Google Scholar]
- 109.Tonelli AE. On the stability of cis and trans amide bond conformations in polypeptides. J Am Chem Soc. 1971;93:7153–5. doi: 10.1021/ja00755a007. [DOI] [PubMed] [Google Scholar]
- 110.Tonelli AE. Conformational characteristics of polypeptides containing isolated L-proline residues with cis peptide bonds. J Mol Biol. 1974;86:627–35. doi: 10.1016/0022-2836(74)90185-5. [DOI] [PubMed] [Google Scholar]
- 111.Kumar NG, Izumiya N, Miyoshi M, Sugano H, Urry DW. Conformational and spectral analysis of the polypeptide antibiotic N-methylleucine gramicidin S dihydrochloride by nuclear magnetic resonance. Biochemistry. 1975;14:2197–207. doi: 10.1021/bi00681a024. [DOI] [PubMed] [Google Scholar]
- 112.Patel DJ, Tonelli AE. N-methylleucine gramicidin-S and (di-N-methylleucine) gramicidin-S conformations with cis L-Orn-L-N-MeLeu peptide bonds. Biopolymers. 1976;15:1623–35. doi: 10.1002/bip.1976.360150815. [DOI] [PubMed] [Google Scholar]
- 113.Gordon DJ, Sciarretta KL, Meredith SC. Inhibition of β-amyloid(40) fibrillogenesis and disassembly of β-amyloid(40) fibrils by short β-amyloid congeners containing N-methyl amino acids at alternate residues. Biochemistry. 2001;40:8237–45. doi: 10.1021/bi002416v. [DOI] [PubMed] [Google Scholar]
- 114.Sciarretta KL, Boire A, Gordon DJ, Meredith SC. Spatial separation of β-sheet domains of β-amyloid: disruption of each β-sheet by N-methyl amino acids. Biochemistry. 2006;45:9485–95. doi: 10.1021/bi0605585. [DOI] [PubMed] [Google Scholar]
- 115.Yan LM, Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A. IAPP mimic blocks Aβ cytotoxic self-assembly: cross-suppression of amyloid toxicity of Aβ and IAPP suggests a molecular link between Alzheimer’s disease and type II diabetes. Angew Chem Int Ed Engl. 2007;46:1246–52. doi: 10.1002/anie.200604056. [DOI] [PubMed] [Google Scholar]
- 116.Svennerholm L. Gangliosides: a new therapeutic agent against stroke and Alzheimer’s disease. Life Sci. 1994;55:2125–34. doi: 10.1016/0024-3205(94)00393-9. [DOI] [PubMed] [Google Scholar]
- 117.Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, et al. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. J Neurosci. 2003;23:29–33. doi: 10.1523/JNEUROSCI.23-01-00029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Choo-Smith LP, Garzon-Rodriguez W, Glabe CG, Surewicz WK. Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J Biol Chem. 1997;272:22987–90. doi: 10.1074/jbc.272.37.22987. [DOI] [PubMed] [Google Scholar]
- 119.Yanagisawa K. GM1 ganglioside and the seeding of amyloid in Alzheimer’s disease: endogenous seed for Alzheimer amyloid. Neuroscientist. 2005;11:250–60. doi: 10.1177/1073858405275177. [DOI] [PubMed] [Google Scholar]
- 120.Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem. 2001;276:24985–90. doi: 10.1074/jbc.M100252200. [DOI] [PubMed] [Google Scholar]
- 121.Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M, et al. A seed for Alzheimer amyloid in the brain. J Neurosci. 2004;24:4894–902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wells JM, Ventura RF, Eisenhauer PB, McKenna DC, Fine RE, Ullman MD. Transport of GM1 and GM1 inner ester across an in vitro model of the blood-brain barrier. Neurosci Lett. 1996;217:121–4. [PubMed] [Google Scholar]
- 123.Flicker C, Ferris SH, Kalkstein D, Serby M. A double-blind, placebo-controlled crossover study of ganglioside GM1 treatment for Alzheimer’s disease. Am J Psychiatry. 1994;151:126–9. doi: 10.1176/ajp.151.1.126. [DOI] [PubMed] [Google Scholar]
- 124.Svennerholm L, Bråne G, Karlsson I, Lekman A, Ramström I, Wikkelsö C. Alzheimer disease - effect of continuous intracerebroventricular treatment with GM1 ganglioside and a systematic activation programme. Dement Geriatr Cogn Disord. 2002;14:128–36. doi: 10.1159/000063604. [DOI] [PubMed] [Google Scholar]
- 125.Snow AD, Sekiguchi RT, Nochlin D, Kalaria RN, Kimata K. Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer’s disease brain. Am J Pathol. 1994;144:337–47. [PMC free article] [PubMed] [Google Scholar]
- 126.Castillo GM, Ngo C, Cummings J, Wight TN, Snow AD. Perlecan binds to the β-amyloid proteins (Aβ) of Alzheimer’s disease, accelerates Aβ fibril formation, and maintains Aβ fibril stability. J Neurochem. 1997;69:2452–65. doi: 10.1046/j.1471-4159.1997.69062452.x. [DOI] [PubMed] [Google Scholar]
- 127.Gupta-Bansal R, Frederickson RC, Brunden KR. Proteoglycan-mediated inhibition of Aβ proteolysis. A potential cause of senile plaque accumulation. J Biol Chem. 1995;270:18666–71. doi: 10.1074/jbc.270.31.18666. [DOI] [PubMed] [Google Scholar]
- 128.Shaffer LM, Dority MD, Gupta-Bansal R, Frederickson RC, Younkin SG, Brunden KR. Amyloid β protein (Aβ) removal by neuroglial cells in culture. Neurobiol Aging. 1995;16:737–45. doi: 10.1016/0197-4580(95)00055-j. [DOI] [PubMed] [Google Scholar]
- 129.Leveugle B, Scanameo A, Ding W, Fillit H. Binding of heparan sulfate glycosaminoglycan to β-amyloid peptide: inhibition by potentially therapeutic polysulfated compounds. Neuroreport. 1994;5:1389–92. [PubMed] [Google Scholar]
- 130.Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG, Szarek WA. Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer’s disease. Nat Med. 1995;1:143–8. doi: 10.1038/nm0295-143. [DOI] [PubMed] [Google Scholar]
- 131.Pollack SJ, Sadler II, Hawtin SR, Tailor VJ, Shearman MS. Sulfonated dyes attenuate the toxic effects of β-amyloid in a structure-specific fashion. Neurosci Lett. 1995;197:211–4. doi: 10.1016/0304-3940(95)11939-t. [DOI] [PubMed] [Google Scholar]
- 132.Pollack SJ, Sadler II, Hawtin SR, Tailor VJ, Shearman MS. Sulfated glycosaminoglycans and dyes attenuate the neurotoxic effects of β-amyloid in rat PC12 cells. Neurosci Lett. 1995;184:113–6. doi: 10.1016/0304-3940(94)11182-i. [DOI] [PubMed] [Google Scholar]
- 133.Yatin SM, Yatin M, Varadarajan S, Ain KB, Butterfield DA. Role of spermine in amyloid β-peptide-associated free radical-induced neurotoxicity. J Neurosci Res. 2001;63:395–401. doi: 10.1002/1097-4547(20010301)63:5<395::AID-JNR1034>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 134.Macleod MA. Potential treatments and treatment strategies in Creutzfeldt-Jakob disease. IDrugs. 2003;6:345–50. [PubMed] [Google Scholar]
- 135.Dolphin GT, Chierici S, Ouberai M, Dumy P, Garcia J. A multimeric quinacrine conjugate as a potential inhibitor of Alzheimer’s-amyloid fibril formation. Chembiochem. 2008;9:952–63. doi: 10.1002/cbic.200700602. [DOI] [PubMed] [Google Scholar]
- 136.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 137.Oprea TI. Property distribution of drug-related chemical databases. J Comput Aided Mol Des. 2000;14:251–64. doi: 10.1023/a:1008130001697. [DOI] [PubMed] [Google Scholar]
- 138.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 139.Martin EJ, Critchlow RE. Beyond mere diversity: tailoring combinatorial libraries for drug discovery. J Comb Chem. 1999;1:32–45. doi: 10.1021/cc9800024. [DOI] [PubMed] [Google Scholar]
- 140.Salomon AR, Marcinowski KJ, Friedland RP, Zagorski MG. Nicotine inhibits amyloid formation by the β-peptide. Biochemistry. 1996;35:13568–78. doi: 10.1021/bi9617264. [DOI] [PubMed] [Google Scholar]
- 141.Camilleri P, Haskins NJ, Howlett DR. β-cyclodextrin interacts with the Alzheimer amyloid β-A4 peptide. FEBS Lett. 1994;341:256–8. doi: 10.1016/0014-5793(94)80467-2. [DOI] [PubMed] [Google Scholar]
- 142.Howlett D, Cutler P, Heales S, Camilleri P. Hemin and related porphyrins inhibit β-amyloid aggregation. FEBS Lett. 1997;417:249–51. doi: 10.1016/s0014-5793(97)01290-8. [DOI] [PubMed] [Google Scholar]
- 143.Merlini G, Ascari E, Amboldi N, Bellotti V, Arbustini E, Perfetti V, et al. Interaction of the anthracycline 4′-iodo-4′-deoxydoxorubicin with amyloid fibrils: inhibition of amyloidogenesis. Proc Natl Acad Sci U S A. 1995;92:2959–63. doi: 10.1073/pnas.92.7.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wood SJ, MacKenzie L, Maleeff B, Hurle MR, Wetzel R. Selective inhibition of Aβ fibril formation. J Biol Chem. 1996;271:4086–92. doi: 10.1074/jbc.271.8.4086. [DOI] [PubMed] [Google Scholar]
- 145.Tomiyama T, Shoji A, Kataoka K, Suwa Y, Asano S, Kaneko H, et al. Inhibition of amyloid β protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. J Biol Chem. 1996;271:6839–44. doi: 10.1074/jbc.271.12.6839. [DOI] [PubMed] [Google Scholar]
- 146.Parker MH, Chen R, Conway KA, Lee DH, Luo C, Boyd RE, et al. Synthesis of (−)-5,8-dihydroxy-3R-methyl-2R-(dipropylamino)-1,2,3,4-tetrahydronaphthalene: an inhibitor of β-amyloid(1-42) aggregation. Bioorg Med Chem. 2002;10:3565–9. doi: 10.1016/s0968-0896(02)00251-1. [DOI] [PubMed] [Google Scholar]
- 147.Pappolla M, Bozner P, Soto C, Shao H, Robakis NK, Zagorski M, et al. Inhibition of Alzheimer β-fibrillogenesis by melatonin. J Biol Chem. 1998;273:7185–8. doi: 10.1074/jbc.273.13.7185. [DOI] [PubMed] [Google Scholar]
- 148.Durairajan SS, Yuan Q, Xie L, Chan WS, Kum WF, Koo I, et al. Salvianolic acid B inhibits Aβ fibril formation and disaggregates preformed fibrils and protects against Aβ-induced cytotoxicty. Neurochem Int. 2008;52:741–50. doi: 10.1016/j.neuint.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 149.Bartolini M, Bertucci C, Cavrini V, Andrisano V. β-amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem Pharmacol. 2003;65:407–16. doi: 10.1016/s0006-2952(02)01514-9. [DOI] [PubMed] [Google Scholar]
- 150.Thomas T, Nadackal GT, Thomas K. Aspirin and diabetes: inhibition of amylin aggregation by nonsteroidal anti-inflammatory drugs. Exp Clin Endocrinol Diabetes. 2003;111:8–11. doi: 10.1055/s-2003-37493. [DOI] [PubMed] [Google Scholar]
- 151.Hirohata M, Ono K, Naiki H, Yamada M. Non-steroidal antiinflammatory drugs have anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. Neuropharmacology. 2005;49:1088–99. doi: 10.1016/j.neuropharm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 152.Kim JE, Lee M. Fullerene inhibits β-amyloid peptide aggregation. Biochem Biophys Res Commun. 2003;303:576–9. doi: 10.1016/s0006-291x(03)00393-0. [DOI] [PubMed] [Google Scholar]
- 153.Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28:537–47. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 154.Wright TM. Tramiprosate. Drugs Today (Barc) 2006;42:291–8. doi: 10.1358/dot.2006.42.5.973584. [DOI] [PubMed] [Google Scholar]
- 155.Neurochem announces results from Tramiprosate (ALZHEMED (TM)) North American phase III clinical trial. http://www.bellushealth.com/en/news/2007-7.php.
- 156.Neurochem announces important initiatives to provide medical and health benefits to patients. http://www.bellushealth.com/en/news/2007-4.php.
- 157.Fratiglioni L, Wang HX. Smoking and Parkinson’s and Alzheimer’s disease: review of the epidemiological studies. Behav Brain Res. 2000;113:117–20. doi: 10.1016/s0166-4328(00)00206-0. [DOI] [PubMed] [Google Scholar]
- 158.Jarvick ME. Beneficial effects of nicotine. Br J Addict. 1991;86:571–5. doi: 10.1111/j.1360-0443.1991.tb01810.x. [DOI] [PubMed] [Google Scholar]
- 159.Levin ED, Simon BB. Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl) 1998;138:217–30. doi: 10.1007/s002130050667. [DOI] [PubMed] [Google Scholar]
- 160.Kenney JW, Gould TJ. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol Neurobiol. 2008;38:101–21. doi: 10.1007/s12035-008-8037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Emilien G, Beyreuther K, Masters CL, Maloteaux JM. Prospects for pharmacological intervention in Alzheimer disease. Arch Neurol. 2000;57:454–9. doi: 10.1001/archneur.57.4.454. [DOI] [PubMed] [Google Scholar]
- 162.White HK, Levin ED. Four-week nicotine skin patch treatment effects on cognitive performance in Alzheimer’s disease. Psychopharmacology (Berl) 1999;143:158–65. doi: 10.1007/s002130050931. [DOI] [PubMed] [Google Scholar]
- 163.Wilson AL, Langley LK, Monley J, Bauer T, Rottunda S, McFalls E, et al. Nicotine patches in Alzheimer’s disease: pilot study on learning, memory, and safety. Pharmacol Biochem Behav. 1995;51:509–14. doi: 10.1016/0091-3057(95)00043-v. [DOI] [PubMed] [Google Scholar]
- 164.Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, et al. Nicotinic receptor stimulation protects neurons against β-amyloid toxicity. Ann Neurol. 1997;42:159–63. doi: 10.1002/ana.410420205. [DOI] [PubMed] [Google Scholar]
- 165.Zamani MR, Allen YS, Owen GP, Gray JA. Nicotine modulates the neurotoxic effect of β-amyloid protein(25-35) in hippocampal cultures. Neuroreport. 1997;8:513–7. doi: 10.1097/00001756-199701200-00027. [DOI] [PubMed] [Google Scholar]
- 166.Ono K, Hasegawa K, Yamada M, Naiki H. Nicotine breaks down preformed Alzheimer’s β-amyloid fibrils in vitro. Biol Psychiatry. 2002;52:880–6. doi: 10.1016/s0006-3223(02)01417-8. [DOI] [PubMed] [Google Scholar]
- 167.Nordberg A, Hellstrom-Lindahl E, Lee M, Johnson M, Mousavi M, Hall R, et al. Chronic nicotine treatment reduces β-amyloidosis in the brain of a mouse model of Alzheimer’s disease (APPsw) J Neurochem. 2002;81:655–8. doi: 10.1046/j.1471-4159.2002.00874.x. [DOI] [PubMed] [Google Scholar]
- 168.Welch WJ. Role of quality control pathways in human diseases involving protein misfolding. Semin Cell Dev Biol. 2004;15:31–8. doi: 10.1016/j.semcdb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 169.Evans CG, Wisén S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J Biol Chem. 2006;281:33182–91. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- 170.Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280:33097–100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- 171.Diamant S, Ben-Zvi AP, Bukau B, Goloubinoff P. Size-dependent disaggregation of stable protein aggregates by the DnaK chaperone machinery. J Biol Chem. 2000;275:21107–13. doi: 10.1074/jbc.M001293200. [DOI] [PubMed] [Google Scholar]
- 172.Dul JL, Davis DP, Williamson EK, Stevens FJ, Argon Y. Hsp70 and antifibrillogenic peptides promote degradation and inhibit intracellular aggregation of amyloidogenic light chains. J Cell Biol. 2001;152:705–16. doi: 10.1083/jcb.152.4.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Barral JM, Broadley SA, Schaffar G, Hartl FU. Roles of molecular chaperones in protein misfolding diseases. Semin Cell Dev Biol. 2004;15:17–29. doi: 10.1016/j.semcdb.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 174.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A. 2000;97:7841–6. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Raman B, Ban T, Sakai M, Pasta SY, Ramakrishna T, Naiki H, et al. αB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid β-peptide and β2-microglobulin. Biochem J. 2005;392:573–81. doi: 10.1042/BJ20050339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wilhelmus MM, Boelens WC, Otte-Holler I, Kamps B, de Waal RM, Verbeek MM. Small heat shock proteins inhibit amyloid-β protein aggregation and cerebrovascular amyloid-β protein toxicity. Brain Res. 2006;1089:67–78. doi: 10.1016/j.brainres.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 177.Lee S, Carson K, Rice-Ficht A, Good T. Hsp20, a novel α-crystallin, prevents Aβ fibril formation and toxicity. Protein Sci. 2005;14:593–601. doi: 10.1110/ps.041020705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–7. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 179.Fonte V, Kapulkin V, Taft A, Fluet A, Friedman D, Link CD. Interaction of intracellular β amyloid peptide with chaperone proteins. Proc Natl Acad Sci U S A. 2002;99:9439–44. doi: 10.1073/pnas.152313999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gestwicki JE, Crabtree GR, Graef IA. Harnessing chaperones to generate small-molecule inhibitors of amyloid β aggregation. Science. 2004;306:865–9. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]
- 181.Bose M, Gestwicki JE, Devasthali V, Crabtree GR, Graef IA. ‘Nature-inspired’ drug-protein complexes as inhibitors of Aβ aggregation. Biochem Soc Trans. 2005;33:543–7. doi: 10.1042/BST0330543. [DOI] [PubMed] [Google Scholar]
- 182.Adlard PA, Bush AI. Metals and Alzheimer’s disease. J Alzheimers Dis. 2006;10:145–63. doi: 10.3233/jad-2006-102-303. [DOI] [PubMed] [Google Scholar]
- 183.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, et al. Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003;42:2768–73. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 184.Opazo C, Huang X, Cherny RA, Moir RD, Roher AE, White AR, et al. Metalloenzyme-like activity of Alzheimer’s disease β-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J Biol Chem. 2002;277:40302–8. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 185.Varadarajan S, Yatin S, Butterfield DA. Alzheimer’s amyloid β peptide (1–42) fibrils are not always neurotoxic. Alzheimers Rep. 2000;3:71–76. [Google Scholar]
- 186.Bush AI. Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol Aging. 2002;23:1031–8. doi: 10.1016/s0197-4580(02)00120-3. [DOI] [PubMed] [Google Scholar]
- 187.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665–76. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 188.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, et al. Metal-protein attenuation with iodochlorhydroxyquin (Clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–91. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 189.Tschäpe JA, Hartmann T. Therapeutic perspectives in Alzheimer’s disease. Recent Patents CNS Drug Discov. 2006;1:119–27. doi: 10.2174/157488906775245354. [DOI] [PubMed] [Google Scholar]
- 190.Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 191.Study evaluating the safety, tolerability and efficacy of PBT2 in patients with early Alzheimer’s disease. http://www.clinicaltrials.gov/ct2/show/NCT00471211.
- 192.Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 193.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–22. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 194.Plaza del Pino IM, Sanchez-Ruiz JM. An osmolyte effect on the heat capacity change for protein folding. Biochemistry. 1995;34:8621–30. doi: 10.1021/bi00027a011. [DOI] [PubMed] [Google Scholar]
- 195.Yancey PH. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J Exp Biol. 2005;208:2819–30. doi: 10.1242/jeb.01730. [DOI] [PubMed] [Google Scholar]
- 196.Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: from thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006;1764:1677–87. doi: 10.1016/j.bbapap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 197.Morello JP, Petäjä-Repo UE, Bichet DG, Bouvier M. Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol Sci. 2000;21:466–469. doi: 10.1016/s0165-6147(00)01575-3. [DOI] [PubMed] [Google Scholar]
- 198.Liu R, Barkhordarian H, Emadi S, Park CB, Sierks MR. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol Dis. 2005;20:74–81. doi: 10.1016/j.nbd.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 199.Ueda T, Nagata M, Monji A, Yoshida I, Tashiro N, Imoto T. Effect of sucrose on formation of the β-amyloid fibrils and D-aspartic acids in Aβ 1-42. Biol Pharm Bull. 2002;25:375–378. doi: 10.1248/bpb.25.375. [DOI] [PubMed] [Google Scholar]
- 200.Tomiyama T, Asano S, Furiya Y, Shirasawa T, Endo N, Mori H. Racemization of Asp(23) residue affects the aggregation properties of Alzheimer amyloid β protein analogues. J Biol Chem. 1994;269:10205–10208. [PubMed] [Google Scholar]
- 201.McLaurin J, Franklin T, Chakrabartty A, Fraser PE. Phosphatidylinositol and inositol involvement in Alzheimer amyloid-β fibril growth and arrest. J Mol Biol. 1998;278:183–94. doi: 10.1006/jmbi.1998.1677. [DOI] [PubMed] [Google Scholar]
- 202.McLaurin J, Golomb R, Jurewicz A, Antel JP, Fraser PE. Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid β peptide and inhibit Aβ-induced toxicity. J Biol Chem. 2000;275:18495–502. doi: 10.1074/jbc.M906994199. [DOI] [PubMed] [Google Scholar]
- 203.McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MH, Phinney AL, et al. Cyclohexanehexol inhibitors of Aβ aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med. 2006;12:801–8. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- 204.Fenili D, Brown M, Rappaport R, McLaurin J. Properties of scyllo-inositol as a therapeutic treatment of AD-like pathology. J Mol Med. 2007;85:603–11. doi: 10.1007/s00109-007-0156-7. [DOI] [PubMed] [Google Scholar]
- 205.Nitz M, Fenili D, Darabie AA, Wu L, Cousins JE, McLaurin J. Modulation of amyloid-β aggregation and toxicity by inosose stereoisomers. FEBS J. 2008;275:1663–74. doi: 10.1111/j.1742-4658.2008.06321.x. [DOI] [PubMed] [Google Scholar]
- 206.Auton M, Bolen DW. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc Natl Acad Sci U S A. 2005;102:15065–8. doi: 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.McLaurin J, Chakrabartty A. Membrane disruption by Alzheimer β-amyloid peptides mediated through specific binding to either phospholipids or gangliosides. Implications for neurotoxicity. J Biol Chem. 1996;271:26482–9. doi: 10.1074/jbc.271.43.26482. [DOI] [PubMed] [Google Scholar]
- 208.McLaurin J, Chakrabartty A. Characterization of the interactions of Alzheimer β-amyloid peptides with phospholipid membranes. Eur J Biochem. 1997;245:355–63. doi: 10.1111/j.1432-1033.1997.t01-2-00355.x. [DOI] [PubMed] [Google Scholar]
- 209.Koppaka V, Axelsen PH. Accelerated accumulation of amyloid β proteins on oxidatively damaged lipid membranes. Biochemistry. 2000;39:10011–6. doi: 10.1021/bi000619d. [DOI] [PubMed] [Google Scholar]
- 210.Barth S, Huhn M, Matthey B, Klimka A, Galinski EA, Engert A. Compatible-solute-supported periplasmic expression of functional recombinant proteins under stress conditions. Appl Environ Microbiol. 2000;66:1572–9. doi: 10.1128/aem.66.4.1572-1579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kregel KC. Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol. 2002;92:2177–86. doi: 10.1152/japplphysiol.01267.2001. [DOI] [PubMed] [Google Scholar]
- 212.Sun Y, MacRae TH. The small heat shock proteins and their role in human disease. FEBS J. 2005;272:2613–27. doi: 10.1111/j.1742-4658.2005.04708.x. [DOI] [PubMed] [Google Scholar]
- 213.Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, et al. NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo. J Clin Invest. 2003;112:440–9. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, et al. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid β 42 production by direct modulation of γ-secretase activity. J Biol Chem. 2003;278:31831–7. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]
- 215.Leuchtenberger S, Kummer MP, Kukar T, Czirr E, Teusch N, Sagi SA, et al. Inhibitors of Rho-kinase modulate amyloid-β (Aβ) secretion but lack selectivity for Aβ42. J Neurochem. 2006;96:355–65. doi: 10.1111/j.1471-4159.2005.03553.x. [DOI] [PubMed] [Google Scholar]
- 216.Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, et al. Substrate-targeting-secretase modulators. Nature. 2008;453:925–9. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14:1581–96. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, et al. Familial Alzheimer’s disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc Natl Acad Sci U S A. 2007;104:16522–7. doi: 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Green RC, Schneider LS, Zavitz KH, Amato DH, Beelen AP, Swabb EA, et al. Safety and Efficacy of tarenflurbil in subjects with mild alzheimer’s disease: results from an 18-month multicenter phase 3 trial. ICAD Meeting. 2008 [Google Scholar]
- 220.Sekiya S, Nishikawa F, Noda K, Kumar PK, Yokoyama T, Nishikawa S. In vitro selection of RNA aptamers against cellular and abnormal isoform of mouse prion protein. Nucleic Acids Symp Ser (Oxf) 2005:361–2. doi: 10.1093/nass/49.1.361. [DOI] [PubMed] [Google Scholar]
- 221.Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002;62:4029–33. [PubMed] [Google Scholar]
- 222.Rentmeister A, Bill A, Wahle T, Walter J, Famulok M. RNA aptamers selectively modulate protein recruitment to the cytoplasmic domain of β-secretase BACE1 in vitro. RNA. 2006;12:1650–60. doi: 10.1261/rna.126306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yu J, Jiang Y, Ma X, Lin Y, Fang X. Energy landscape of aptamer/protein complexes studied by single-molecule force spectroscopy. Chem Asian J. 2007;2:284–9. doi: 10.1002/asia.200600230. [DOI] [PubMed] [Google Scholar]
- 224.Ulrich H, Trujillo CA, Nery AA, Alves JM, Majumder P, Resende RR, et al. DNA and RNA aptamers: from tools for basic research towards therapeutic applications. Comb Chem High Throughput Screen. 2006;9:619–32. doi: 10.2174/138620706778249695. [DOI] [PubMed] [Google Scholar]
- 225.Yang Y, Yang D, Schluesener HJ, Zhang Z. Advances in SELEX and application of aptamers in the central nervous system. Biomol Eng. 2007;24:583–92. doi: 10.1016/j.bioeng.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 226.Tickler AK, Smith DG, Ciccotosto GD, Tew DJ, Curtain CC, Carrington D, et al. Methylation of the imidazole side chains of the Alzheimer Disease amyloid β-peptide results in abolition of super-oxide dismutase-like structures and inhibition of neurotoxicity. J Biol Chem. 2005;280:13355–63. doi: 10.1074/jbc.M414178200. [DOI] [PubMed] [Google Scholar]
- 227.Smith DP, Smith DG, Curtain CC, Boas JF, Pilbrow JR, Ciccotosto GD, et al. Copper-mediated amyloid-β toxicity is associated with an intermolecular histidine bridge. J Biol Chem. 2006;281:15145–54. doi: 10.1074/jbc.M600417200. [DOI] [PubMed] [Google Scholar]
- 228.Barnham KJ, Kenche VB, Ciccotosto GD, Smith DP, Tew DJ, Liu X, et al. Platinum-based inhibitors of amyloid-β as therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2008;105:6813–8. doi: 10.1073/pnas.0800712105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Deane R, Sagare A, Zlokovic BV. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr Pharm Des. 2008;14(16):1601–5. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hoozemans JJ, Rozemuller JM, van Haastert ES, Veerhuis R, Eikelenboom P. Cyclooxygenase-1 and -2 in the different stages of Alzheimer’s disease pathology. Curr Pharm Des. 2008;14(14):1419–27. doi: 10.2174/138161208784480171. [DOI] [PubMed] [Google Scholar]
- 231.Potterat O, Hamburger M. Drug discovery and development with plant-derived compounds. Prog Drug Res. 2008;65(45):47–118. doi: 10.1007/978-3-7643-8117-2_2. [DOI] [PubMed] [Google Scholar]