

Abstract
Neuronal accumulation of oligomeric amyloid-β (Aβ) is considered the proximal cause of neuronal demise in Alzheimer disease (AD) patients. Blood-borne macrophages might reduce Aβ stress to neurons by immigration into the brain and phagocytosis of Aβ. We tested migration and export across a blood-brain barrier model, and phagocytosis and clearance of Aβ by AD and normal subjects’ macrophages. Both AD and normal macrophages were inhibited in Aβ export across the blood-brain barrier due to adherence of Aβ-engorged macrophages to the endothelial layer. In comparison to normal subjects’ macrophages, AD macrophages ingested and cleared less Aβ, and underwent apoptosis upon exposure to soluble, protofibrillar, or fibrillar Aβ. Confocal microscopy of stained AD brain sections revealed oligomeric Aβ in neurons and apoptotic macrophages, which surrounded and infiltrated congophilic microvessels, and fibrillar Aβ in plaques and microvessel walls. After incubation with AD brain sections, normal subjects’ monocytes intruded into neurons and uploaded oligomeric Aβ. In conclusion, in patients with AD, macrophages appear to shuttle Aβ from neurons to vessels where their apoptosis may release fibrillar Aβ, contributing to cerebral amyloid angiopathy.
Keywords: Macrophages, Amyloid-beta, Apoptosis, Alzheimer, Congophillic, Angiopathy
Introduction
The amyloid hypothesis proposes that amyloid-β (Aβ) accumulation in the brain is the cause of Alzheimer disease [20]. Aβ is potentially generated from Aβ precursor protein (APP) wherever APP and the β- and γ-secretases are located, such as the endoplasmic reticulum and Golgi apparatus, but most is produced at the plasma surface or in the secretory pathway [25]. Aβ generation may also occur during autophagic turnover of APP-rich organelles [32]. Increasing molecular weight assemblies of Aβ accumulate both extra- and intra-neuronally in Alzheimer disease (AD) brain [25]; of these assemblies, at least the intraneuronal oligomeric Aβ has pathological consequences [43]. Aβ is cleared physiologically across the blood-brain barrier by low-density lipoprotein receptor-related protein-1 [35]. Despite this physiological clearance, Aβ is found to accumulate in neurons and extracellular deposits since the first year of life. The pathological consequences in AD patients are intraneuronal accumulation of oligomeric Aβ in multivesicular bodies [18] and neuronal death. Surprisingly, the total neuronal load of Aβ is not predictive of neurofibrillary degeneration [47].
Brain amyloidosis of AD patients is considered to be related to insufficient clearance rather than over-expression of Aβ [42]. Promising strategies for immune clearance of Aβ include Aβ vaccination [48], intravenous gamma-globulin [39], humanized anti-Aβ antibody (Bapineuzumab®), and transcriptional modulation of macrophages, such as by use of curcuminoids [15]. Studies of AD brain tissues [11] and, recently, of APP transgenic mice brain tissues [28, 30, 37] suggest that blood-derived macrophages and microglia, rather than resident brain microglia, have a key role in the clearance of Aβ.
Previous studies have identified basic immune mechanisms necessary for clearance of Aβ by macrophages. Aβ-induced adhesion molecules and chemokines CCL3 (MIP-1 α), CCL4 (MIP-1β), and CCL2 [9, 17, 38] act on macrophages, microglia, and astrocytes, promoting monocyte migration, differentiation [9], and survival [19]. In mouse macrophages and microglia, the class B scavenger receptor type 2 (CD36) is crucial for induction by Aβ of chemokines, cytokines, and reactive oxygen species [6, 34], and exposure to Aβ initiates the signaling cascade that links CD36 scavenger receptor to the actin cytoskeleton and diverse processes such as cellular migration, adhesion, and phagocytosis [41]. Fucoidan inhibits both class A and B scavenger receptor interactions [21], but, in our experience, does not inhibit Aβ phagocytosis by human macrophages (Fiala et al. unpublished data). Recent studies suggest a role for Toll-like receptors (TLR’s) in Aβ clearance: TLR’s are transcriptionally downregulated in AD patients’ monocytes [15], and TLR-2 signaling enhances Aβ uptake by microglia [4]. Importantly, mononuclear cells of most patients differ from those of normal subjects by transcriptional downregulation of β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucos-aminyltransferase (MGAT-3), an enzyme important in phagocytosis of Aβ [15].
Immunohistochemical studies brought new insights into the immune clearance of Aβ by macrophages in human brain. AD brain contains 11 times as many cyclooxygenase (COX-2)-positive macrophages as age-matched control brain [11], and a subset of these macrophages is inducible nitric oxide (NO) synthase (iNOS)-positive [10]. Monocyte immigration might be orchestrated by neuronal and microglial chemokine RANTES and cytokine interleukin-1β (IL-1P) [13, 31]. In CCR2-deficient APP transgenic mice, clearance of Aβ is depressed [7]. Despite the permeation of AD brain by macrophages, clearance of neuritic plaques is randomly incomplete, suggesting functional heterogeneity of AD macrophages [13].
AD is a human disease with specific immune and biochemical defects [15], which have not been engineered in APP transgenic mice. To clarify the pathophysiology of Aβ clearance in AD, we examined Aβ clearance using human brain tissues and human macrophages, and observed that the AD innate immune system is defective in proper disposal of Aβ in the brain.
Materials and methods
Antibodies and reagents
We stained macrophages using mouse anti-human CD68 (Dako, Carpinteria, CA) and goat anti-human CD68 (Santa Cruz Biotech, Santa Cruz, CA, USA). Neurons were stained with mouse anti-human neuronal nuclei (NeuN, Chemicon, Temecula, CA, USA); mouse anti-human microtubule associated protein 2 (MAP2, Sigma, St Louis, MO, USA); and rabbit anti-human neuron specific enolase (Immunostar, Hudson, WI, USA). To visualize Aβ in brain tissue and in macrophages, we utilized rabbit anti-Aβ 1–42 (COOH-terminal epitope) (Millipore, Billerica, MA, USA); mouse biotinylated anti-Aβ 1-42 (COOH-terminal epitope) (Signet); rabbit anti-oligomer A11 (Biosource, Carlsbad, CA, USA), which recognizes Aβ-42 and Aβ-40 pre-fibrillar oligomers [22]; and rabbit anti-fibrillar OC, which stains Aβ fibrils, as well as α-synuclein fibrils and islet amyloid polypeptide fibrils [23]. To stain apoptotic markers, we used anti-caspase-6, -7, and -8 antibodies, which were raised in rabbits using catalytic subunits of the relevant autoprocessed recombinant caspases as immunogens (Burnham Institute, La Jolla, CA, USA) [24]. Secondary antibodies were anti-mouse, anti-rabbit, and anti-goat IgG’s conjugated to Alexa Fluor 488, 555, and 647 (Invitrogen, Carlsbad, CA, USA). The reagents were monocyte chemotactic protein-1 (MCP-1) (PeproTech, Rocky Hill, NJ, USA); fluorescein isothiocyanate (FITC)-conjugated Aβ (Anaspec, San Jose, CA, USA); fibrillar FITC-Aβ and protofibrillar Aβ prepared by M. Inayathullah; and 14C-labeled Aβ from C. Glabe, UCI. Aβ was used at 2 μg/mL in most experiments.
Patients and controls
A total of ten patients [mean age 76.9 ± 5.8 years, mean Mini-Mental State Exam (MMSE) score of 21.7 ±5.1] with a diagnosis of probable AD established by the National Institute of Neurological and Communication Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association criteria [29] were recruited into the study since 2004 through the University of California, Los Angeles (UCLA), Alzheimer’s Disease Research Center under a UCLA Institutional Review Board-approved protocol. In addition, eight aged-matched control subjects (mean age 77.5 + 6.0 years) and three young control subjects (ages 20, 21, 44) were recruited from UCLA personnel and families of patients.
Isolation of PBMC’s
Peripheral blood mononuclear cells (PBMC’s) were isolated from venous blood of the AD patients and control subjects by Ficoll–Hypaque gradient centrifugation as previously described [13]. Monocytes were purified using RosetteSep Monocyte Enrichment Cocktail (StemCell Technologies, Vancouver, BC, Canada) from PBMC’s of normal subjects (“normal monocytes” or “normal macrophages”) and AD patients (“AD monocytes” or AD macrophages”).
Preparation of fibrils and protofibrils of Aβ (1–42)
Aβ fibrils were prepared by dissolving 1 mg of Aβ (1–42) peptide in 100 μL of 10 mM NaOH. The solution was diluted to a volume of 0.5 mL with milliQ water followed by addition of 0.5 mL of 10 mM (2×) phosphate buffer (pH 7.4). The resulting solution was then centrifuged at 16,000×g for 10 min. One-half milliliter of the resulting supernatant was transferred into a new tube and incubated at 37°C for 7 days. The fibrils were pelleted out by centrifugation for 10 min and the supernatant was transferred to another tube for protofibril isolation. The fibril pellet was washed thrice with MilliQ water and the resulting pellet thereafter resuspended in MilliQ water after each wash.
Protofibrils were isolated from the supernatant by filtering through a Centricon filter (molecular weight cutoff of 35 kDa) to remove any small oligomers and monomers and collecting the filtrate. The pellet was washed three times with MilliQ and then reconstituted and diluted with MilliQ water. A small aliquot of each sample was analyzed by amino acid analysis to determine the protein concentration. The samples were characterized by size exclusion chromatography and electron microscopy (Fig. 2). Fibrillar and protofibrillar Aβ also was prepared in smaller amounts from FITC-Aβ.
Fig. 2.
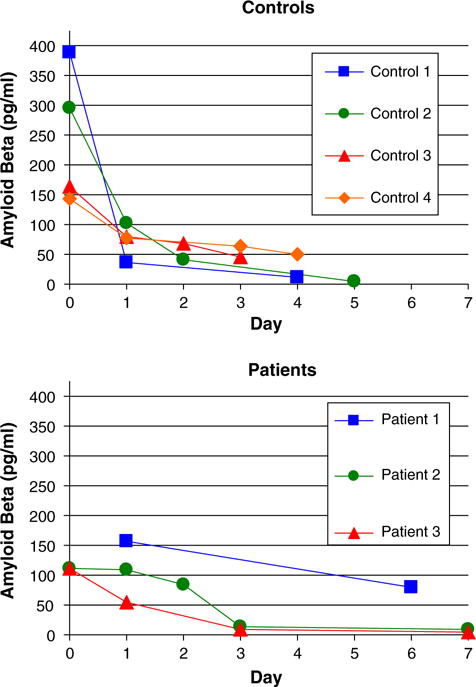
Uptake and removal of Aβ by normal monocytes are greater compared to AD monocytes. Monocytes were exposed to Aβ (2 μg/mL) for 2 h, washed, and incubated for the indicated number of days when Aβ was measured by the ELISA assay. Control monocytes (a) showed higher uptake of Aβ after 2-h exposure than AD monocytes (b) and similarly showed more rapid removal of Aβ in comparison to AD monocytes
Monocyte migration across a human blood-brain barrier model
A human blood-brain barrier model (BBB) model with primary human brain microvascular endothelial cells (BMVEC’s) was constructed in a 24-well plate as described previously [8, 12]. In the model, 50,000 BMVEC’s in passages 4–8 coated either the upper surface (“regular model”) or the lower surface of a porous membrane insert (Collaborative Biomedical Products, Bedford, MA, USA) (“reverse model”) which rests above a well. Both the well and the membrane insert contained RPMI medium with 10% fetal bovine serum or 10% autologous serum.
In migration experiments, 250,000 monocytes from two control subjects (ages 74 and 78) and two AD patients (ages 80 and 84 with MMSE scores 20 and 23, respectively) were allowed to migrate across the BBB for 17 h. The number of transmigrated cells was determined by triplicate cell counting in eight sections of a hemocytometer chamber. The inserts were washed gently with a buffer containing 0.1 M sodium cacodylate plus 0.2 M sucrose X2, then fixed with 3% glutaraldehyde in 0.1 M sodium cacodylate buffer for 1 h at 25°C [26]. The cells were stored in this buffer at 4°C, post-fixed with 1 % osmium tetroxide at 4°C, dehydrated in an ethanol series and embedded in plastic [27]. One-micron sections were stained with toluidine blue and examined by bright field and transmission electron microscopy.
Aβ phagocytosis and apoptosis by macrophages
Macrophages of four controls (ages 74, 74, 81, 90) and four AD patients (ages 70, 77, 82, 86 with MMSE scores 15, 27, 19, 27, respectively) were prepared in 8-well chamber slides as described [13]. The cultures were incubated with fibrillar or protofibrillar Aβ for 3 days. Macrophage apoptosis was determined using the FLICA VAD-FMK polycaspases assay kit (Immunohistochemistry Technologies, Bloomington, MN, USA). This assay utilizes a membrane permeant, sulforhodamine B (SR)-labeled inhibitor targeted to all active caspases to covalently label apoptotic cells. Macrophage cultures were incubated with the FLICA apoptosis detection probe for 1 h at 37°C and then washed to remove any non-covalently bound probe from non-apoptotic cells. Cells were examined with an Olympus Bmax fluorescence microscope with 100× objective. Fluorescence density was determined by Image-Pro Plus 4.1 (Media Cybernetics, Silver Spring, MD). We examined the middle strip of each well for 6–9 consecutive fields with macrophages, analyzing the integrated optical density (IOD) of Aβ (green) and FLICA (red) per macrophage in three experiments for each group.
In a set of related experiments, control and AD macrophages were incubated with soluble FITC-Aβ for 1 h, or 3, 5, or 7, and apoptosis was detected using the FLICA assay. In some experiments, macrophages were incubated with soluble unconjugated Aβ for 1 h, washed twice, then incubated for 2, 4, or 6 days. Apoptosis was determined by the FLICA assay, and Aβ fibrils and oligomers were detected by the indirect technique using OC and A11 antibodies, respectively.
ELISA assay of Aβ clearance from monocytes
Purified monocytes (300,000 per sample) were incubated with soluble Aβ (2 μg/mL) for 2 h, washed four times, re-incubated for the indicated number of days, and the amount of intracellular Aβ remaining at each time point was determined (0, 1, 2, 3, 5, and 7 days). To measure the amount of intracellular Aβ, the cells were harvested into an ELISA lysis buffer (Invitrogen), supplemented with a protease inhibitor cocktail (Sigma) and assayed using the Aβ 1–42 ELISA kit (Invitrogen) by spectrophotometry. Monocytes from four control subjects and three AD patients were tested.
Brain tissues
Frozen sections from the frontal lobe of two control subjects with no neuropathology (ages 23 and 37) and four AD patients were provided by the UCLA Brain Bank. The AD brain sections were from: (1) a 62-year-old patient with Binswanger encephalopathy and scattered senile plaques in the entorhinal cortex and hippocampus; (2) a 64-year-old Braak stage VI patient; (3) a 69-year-old Braak stage VI patient; (4) an 82-year-old Braak stage VI patient with Lewy body disease.
Co-incubation of PBMC’s with brain tissues
Frozen sections of post-mortem brain tissues of four AD patients and two controls were co-incubated with 500,000 PBMC’s (from seven normal donors, ages 20, 21, 72, 74, 74, 79, and 85 years old) for 1 to 6 days in Dulbecco’s Minimum Essential Medium (DMEM) with 10% fetal calf serum, washed with PBS, fixed with 4% paraformaldehyde, and processed for immunofluorescence.
In some experiments, PBMC’s were first pre-labeled with Qtracker 525, which distributes green Qdot nanocrystals in cytoplasmic vesicles, or CellTracker CMFDA (Invitrogen, Carlsbad, CA, USA), which undergoes an esterase reaction to produce a green fluorescent product in the cytoplasm. After isolation of PBMC’s from blood, cells were incubated with Qtracker or CellTracker according to the manufacturer’s recommendations, pelleted and washed twice with DMEM, and then incubated with tissues as above.
Immunofluorescence and confocal microscopy of brain tissues
Fixed tissues were permeabilized with 1% Triton X-100 and blocked with 1% bovine serum albumin (BSA) at 37°C for 30 min each. Brain sections were then incubated with various primary antibodies for 48 h at 4°C, washed with PBS, and incubated with appropriate secondary antibodies labeled with Alexa 488, Alexa 555, and Alexa 647 fluorophores for 1 h at 37°C. A mixture consisting of 0.2% Triton X-100 and 1% BSA was used as the diluent of both primary and secondary antibodies. As control staining, the sections were stained by secondary antibodies without primary antibodies. The preparations were examined using a Zeiss 510 Meta multiphoton confocal microscope or a Bio-Rad Laboratories MRC-1024 Es laser scanning confocal system attached to a Nikon E800 fluorescent microscope.
Statistical analysis
The data on monocyte migration and 14C-Aβ transport were analyzed by t test and Mann–Whitney and Kruskall–Wallis tests.
Results
Aβ uptake by monocytes inhibits monocyte emigration and Aβ export across BBB
In the human BBB model, we compared the rate of migration by normal monocytes in both directions using either a regular model (blood chamber above apical side of BMVEC’s) or a reverse model (brain chamber above abluminal side of BMVEC’s). The rate was greater in the direction from brain to blood (36.4 ± 0.7%) compared to that from blood to brain (13.6 ± 2%). In the course of inflammation, chemokines stimulate and lipoxins inhibit leukocyte migration; to confirm the value of this model in replicating in vivo mononuclear cell migration, we used either the chemokine MCP-1 (CCL2) alone or both MCP-1 and lipoxin A4 together in the lower chamber. MCP-1 (50 ng/mL) increased monocyte migration from 0.4 ± 0.8% to 6.5 ± 0.2%, and lipoxin A4 decreased this migration in a dose-responsive fashion from 6.5% with MCP-1 alone to 3.1 ± 0.1% [0.1 nM (lipoxin A4, plus 50 ng/mL MCP-1)], 1.6 ± 0.1% (1 nM), and 0.8 ± 0.1% (10 nM). We then investigated Aβ export using the reverse model with the chemokine MCP-1 in the lower chamber. As expected, the emigration of monocytes was highest (77,250 ± 9,032) in the presence of MCP-1 in the lower chamber and lowest without MCP-1 (5,375 ± 411). When 14C-Aβ (10,000 DPM or 1 μg/mL) was placed in the brain chamber, monocyte emigration was reduced by 62% (from 77,250 ± 9,032 to 29,600 ± 8,988) (Table 1). Furthermore, maximal 14C-Aβ export across the blood brain barrier was found in the absence of monocytes (mean ± SEM, 1,960 ± 198 CPM). The presence of monocytes in the upper chamber diminished export by 13% (to 1,712 ± 94 CPM), and monocytes in the upper chamber with MCP-1 in the lower chamber reduced the export by 26% (1,456 ± 168 CPM, P < 0.05) (Table 1). In two subsequent experiments with two different control/AD subject pairs, both AD and normal monocytes were inhibited in migration by Aβ-loading (SI Fig. 1).
Table 1.
Aβ uptake by monocytes in the brain chamber inhibits monocyte migration and Aβ export into the blood chamber
| Treatment | Brain Chamber
|
Blood Chamber
|
|||
|---|---|---|---|---|---|
| Monocytes | 14C-Aβ (CPM) | MCP-1 (nM) | Monocytes | 14C-Aβ (CPM) | |
| A | 250,000 | – | 20 | 77,250 ± 9,032 | – |
| B | – | 10,000 | – | – | 1,960 ± 198 |
| C | 250,000 | 10,000 | – | 13,750 ± 3,774 | 1,712 ± 94 |
| D | 250,000 | 10,000 | 20 | 29,600 ± 8,998 | 1,456 ± 168 |
| E | 250,000 | – | – | 5,375 ± 411 | – |
A total of 250,000 monocytes and/or 10,000 CPM of 14C-Aβ were placed into the upper (brain) chamber of a blood-brain barrier model with or without MCP-1 in the lower (blood) chamber. Monocytes and 14C-Aβ were measured in the lower chamber after 24 h
To visualize monocyte migration, the BBB models from the wells with Aβ [either fibrillar (Fig. 1a) or soluble (Fig. 1b)] or without Aβ (Fig. 1c, d) were fixed, sectioned, stained using toluidine blue, and examined by light and transmission electron microscopy. Large macrophages with vacuoles suggestive of uploaded Aβ were adherent to thin endothelial cells (Fig. 1a, b). In the wells without Aβ, monocytes freely migrated across the BBB model and, the endothelial cells displayed tall cytoplasm and abundant mitochondria (Fig. 1c, d). Thus macrophages loaded with Aβ adhere to endothelial cells and are inhibited in emigration across the BBB.
Fig. 1.

MM’s with Aβ adhere to brain endothelial cells, whereas MM’s without Aβ transmigrate. Monocytes, which were exposed to Aβ (2 μg/mL) [fibrillar (a) and soluble (b)] in the upper chamber and attracted for migration by MCP-1 (50 ng/mL) in the lower chamber, adhered to brain endothelial cells, which appeared flat. Monocytes, which were not exposed to Aβ, migrated freely across endothelial cells (c) (light microscopy, ×100) and the endothelial cells exposed to migrating monocytes became tall and disorganized with a monocyte migrating to the abluminal side (d) (transmission electron microscopy, ×22,000)
Normal monocytes bind and clear more Aβ in comparison to AD monocytes
When tested in bulk by Aβ ELISA, normal monocytes uploaded more Aβ and cleared it more rapidly than AD monocytes (Fig. 2). After the initial 2 h incubation, control monocytes bound an average of 256 pg/mL Aβ, compared to 112 pg/mL by AD monocytes. After thorough washing and re-incubation for 24 h, control monocytes had cleared on average 54% of the uploaded Aβ while AD monocytes had degraded only 28%.
Fibrillar, protofibrillar, and soluble Aβ induce apoptosis of AD macrophages
After 3-day exposure to fibrillar Aβ (Fig. 3a), AD macrophages displayed large fibrils and showed a robust apoptotic signal, whereas in normal macrophages Aβ was found in small fragments and the macrophages showed a low apoptotic signal (Fig. 4a). Similarly, after 3-day exposure to protofibrillar Aβ (Fig. 3b), AD macrophages displayed a robust apoptotic signal, whereas normal macrophages had almost no apoptotic signal although they showed a high uptake of protofibrils (Fig. 4b).
Fig. 3.
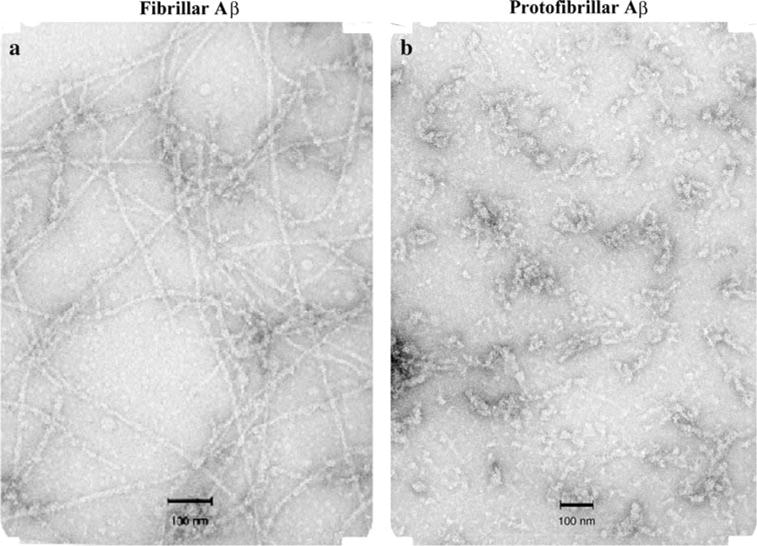
Fibrillar and protofibrillar Aβ (1–42). Five microlitre of samples were spotted on a glow-discharged, carbon-coated Formvar grid and incubated for 5 min, washed with distilled water, fixed with 2.5% glutaral-dehyde, stained with a 1% (w/v) aqueous uranyl acetate solution, and examined using a JEOL transmission electron microscope
Fig. 4.
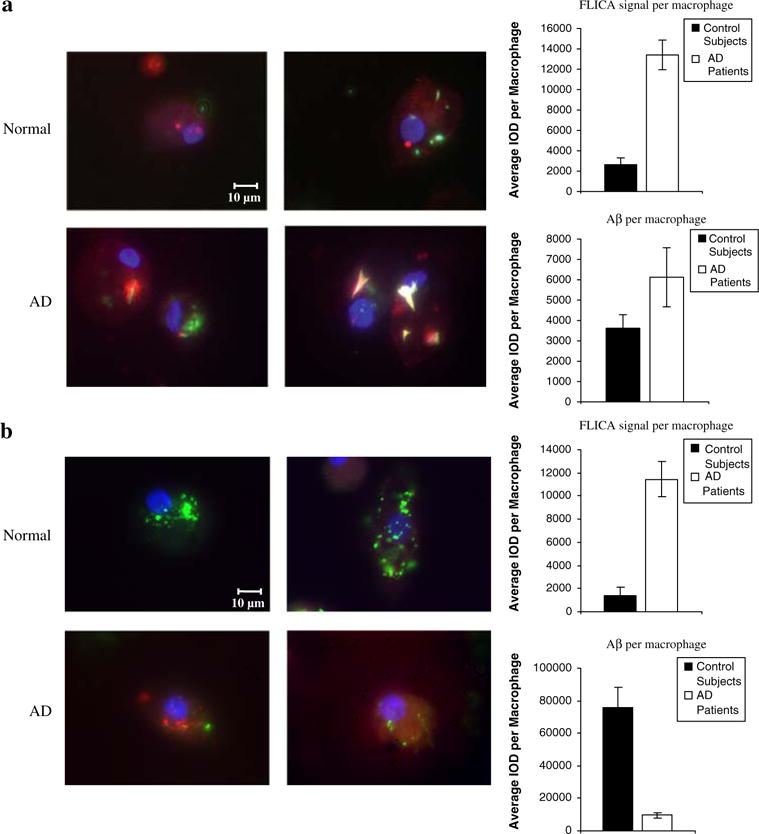
Fibrillar and protofibrillar Aβ induce apoptosis of AD macrophages. After overnight exposure to fibrillar (a) or proto-fibrillar Aβ (2 μg/mL) (b), normal macrophages displayed a low apoptotic FLICA signal (red) but a high uptake of proto-fibrillar Aβ (green), whereas AD macrophages showed a robust FLICA signal and a high uptake of large fibrils (yellow or white due to spectral bleed-through of FITC) but low uptake of protofibrils. FLICA signal (red), protofibrillar or fibrillar Aβ (green) (duplicate images, fluorescence microscopy, ×100). Charts show the mean IOD of FLICA and Aβ with error bars indicating standard error of mean
After only 1-h exposure to soluble FITC-Aβ, an apoptotic FLICA signal became visible in AD, but not control, macrophages; by 5 days AD macrophages were seen to be disrupted (Fig. 5a). In a follow-up experiment AD macrophages which were incubated with soluble Aβ for 1 h, washed, and re-incubated for 4 or 6 days demonstrated a strong apoptotic signal and lysed and heterogeneously contained and released oligomeric and fibrillar Aβ (Fig. 5b). In comparison, control macrophages showed a weak FLICA signal, intracellular Aβ decreased and the cells remained intact.
Fig. 5.
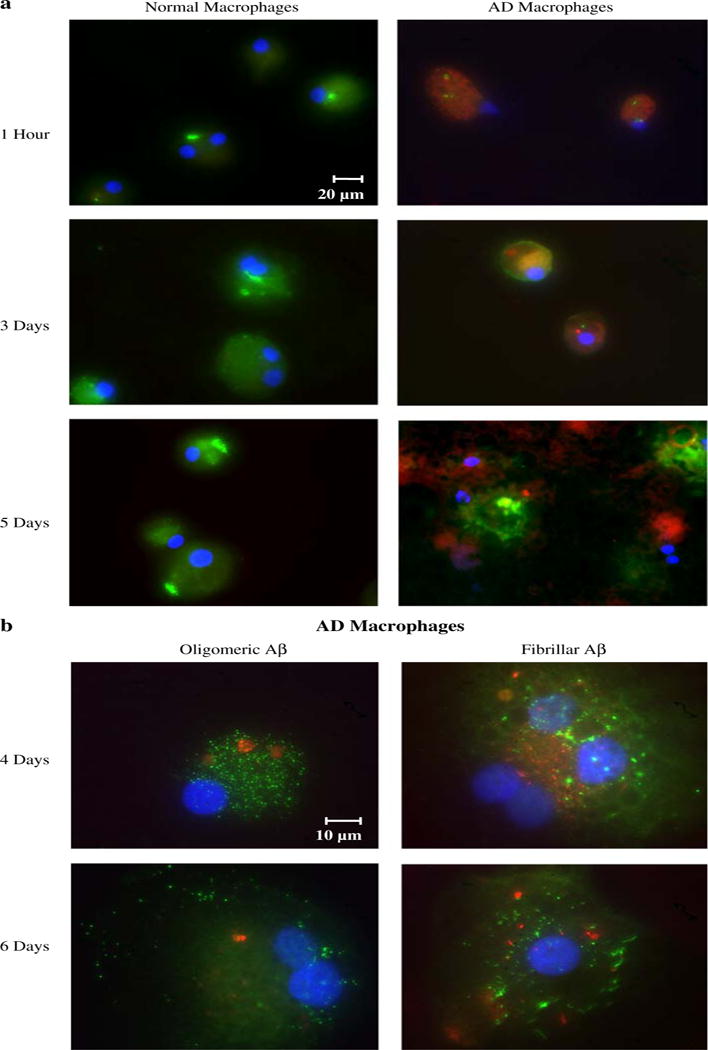
Soluble Aβ induces apoptosis of AD macrophages and release of oligomeric and fibrillar Aβ. a After incubation with FITC Aβ (2 μg/mL), AD macrophages displayed a significantly stronger apoptotic FLICA signal (red) compared to control macrophages. AD macrophages heterogeneously lysed by 5 days (fluorescence microscopy, ×40). b AD macrophages were incubated with soluble Aβ (2 μg/mL) for 1 h, washed with PBS, incubated for 4 or 6 days, and stained with FLICA (red) and anti-oligomeric or anti-fibrillar antibodies (green). After 4-day incubation, Aβ oligomers and fibrils are visible within the apoptotic macrophages, and by 6 days the cells appear disrupted. Aβ oligomers diffused from the lysed macrophages whereas the denser Aβ fibrils remained stuck in place on the glass slides (fluorescence microscopy, ×100)
To explore macrophage clearance of Aβ from AD brain, we first examined the distribution of Aβ assemblies in AD brain sections.
Soluble and oligomeric Aβ are present in neurons and macrophages, and fibrillar Aβ in plaques and congophilic microvessels
In AD brain tissues, Aβ was detected in neurons in a patchy but distinct fashion using immunofluorescence with anti-Aβ 1–42 (COOH terminal) (Fig. 6a) or anti-oligomer antibodies (Fig. 6b). Fibrillar Aβ was detected in neuritic plaques and brain vessels using the OC antibody; CD68-positive cells infiltrated plaques and vessels (Fig. 6c). Clusters of macrophages, which were loaded with oligomeric Aβ (Fig. 6d), abutted and infiltrated the wall of brain vessels and displayed activated caspases: initiator caspase-6, and effector caspases -7 and -8, indicating their apoptosis (Fig. 6e, f, g). The brain vessels displayed fibrillar (Fig. 6c) but not oligomeric Aβ (Fig. 6d). Unstained AD tissues, as well as tissues incubated with secondary but not primary antibodies, were inspected by fluorescence microscopy and their weak fluorescent signal due to autofluorescence was easily distinguished from the specific signal in preparations stained with both primary and secondary antibodies. In addition, control brain tissues did not disclose many infiltrating macrophages.
Fig. 6.
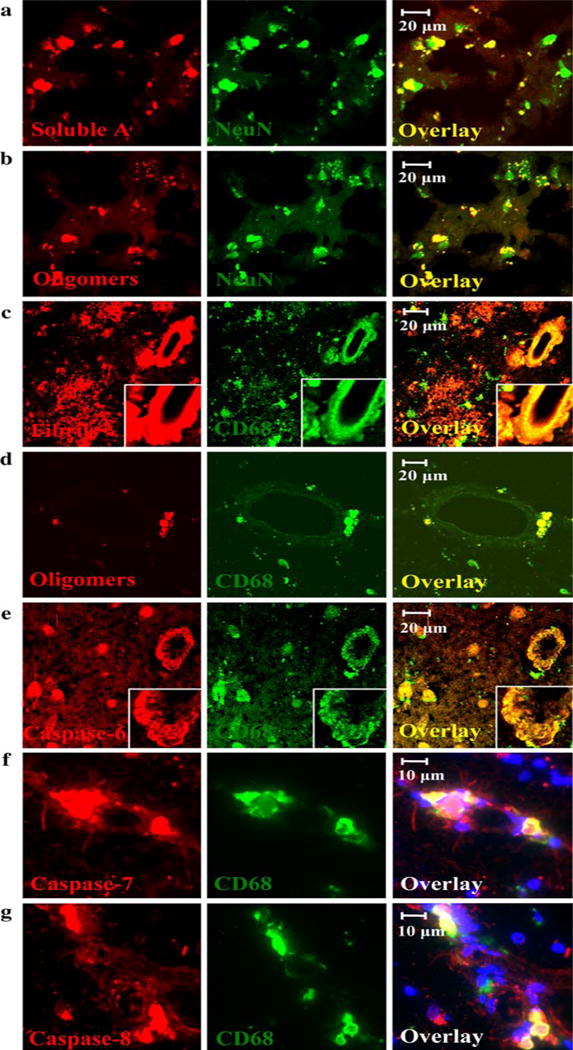
Aβ assemblies in AD brain: oligomeric and soluble Aβ is found in neurons and perivascular apoptotic macrophages, and fibrillar Aβ in neuritic plaques and vessels. AD brain tissues show monomeric (a) and oligomeric Aβ (b) in neurons in a patchy fashion, fibrillar Aβ in plaques and microvessel walls (c), and oligomeric Aβ in perivascular macrophages (d). Perivascular macrophages display activated caspases -6, -7, and -8 (e, f, and g). Red soluble Aβ, oligomeric Aβ, fibrillar Aβ, and activated caspases. Green neurons and macrophages. Blue nuclei. (a–c, e: ×40 objective with Zeiss 510 Meta confocal microscope; d: ×60 objective with Bio-Rad Laboratories MRC-1024 Es confocal system; f–g: ×40 objective with Olympus Bmax fluorescence microscope)
Monocytes upload soluble and oligomeric Aβ in neurons
To experimentally investigate the brain clearance of Aβ, we co-incubated AD brain sections with control PBMC’s for 3 days. Examination of these tissues by confocal microscopy showed monocytes (green) colocalizing in a patchy fashion with neurons (red) and uploading oligomeric Aβ (blue). The soma of the monocytes was found in higher z-sections compared to the soma of the neurons suggesting intrusion of these monocytes into neurons from above. In midsections, neurons, monocytes, and Aβ all colocalized (Fig. 7a). In addition, after 24 h incubation with AD brain sections, CellTracker and Qtracker pre-labeled monocytes were seen to co-localize with neurons, confirming intrusion of exogenous monocytes into neurons (Fig. 6b). We did not notice obvious differences in clearance of Aβ between the monocytes of younger and elderly control subjects but did not analyze these differences. We also stained AD brain tissues co-incubated without exogenous monocytes but did not find CD68 positive monocytes intruding into neurons.
Fig. 7.
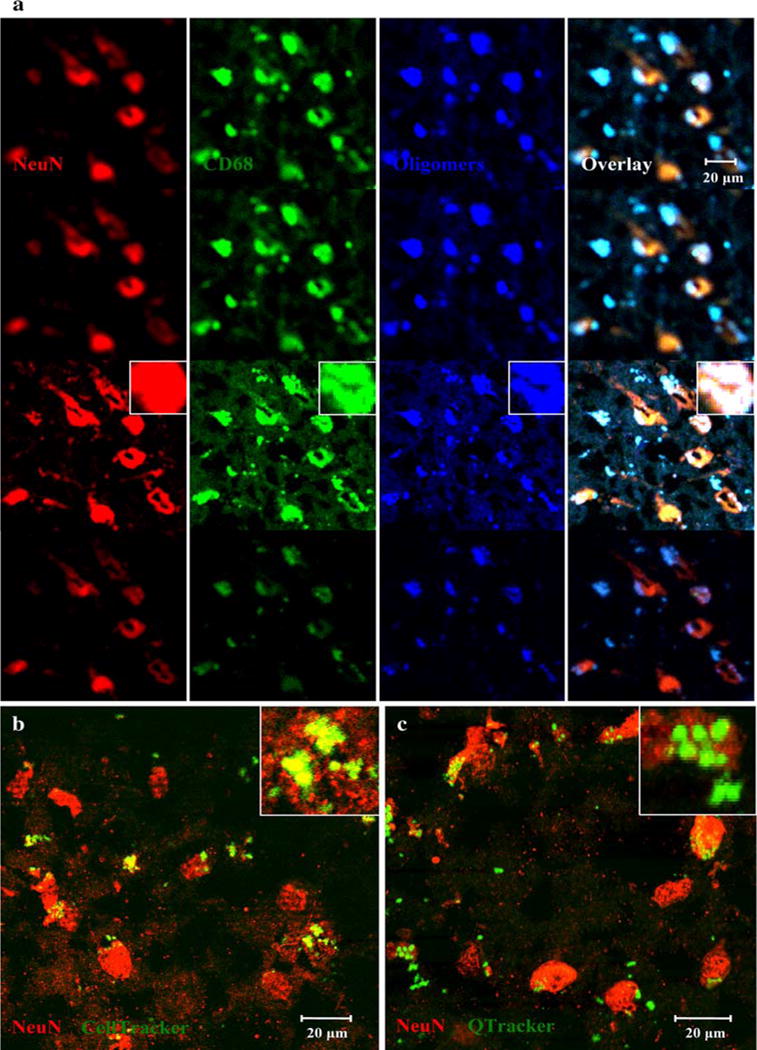
In vitro clearance of Aβ from AD brain: normal monocytes intrude and upload oligomeric Aβ from neurons. a Brain tissue was co-incubated with 500,000 normal PBMC’s for 3 days, stained and examined by a Zeiss 510 Meta confocal microscope (×40). The soma of macrophages is visible in the top sections co-localized with oligomeric Aβ (blue-green). Consecutively lower sections show higher densities of neurons (red) with less CD68 (green) present. The yellow and white patches within neurons show intrusion of macrophages within neurons and phagocytosis of Aβ. Macrophages stained with anti-CD68 (green), oligomeric Aβ with anti-oligomer A11 (blue), and neurons with NeuN (red). (b and c) Brain tissue was co-incubated for 24 h with control PBMC’s prestained with either CellTracker CMFDA (b) or Qtracker 525 (c), stained to visualize neurons, and examined with a Bio-Rad Laboratories MRC-1024 Es confocal system (×60). Round monocytes (green) decorate neurons (red). Insets in b and c show details of decoration of neurons (red) by monocytes (green)
Discussion
In transgenic animal models of AD, Aβ-induced pathology seems to be upstream of tau and is considered the primary mechanism [33]. We have attempted to glean the mechanisms responsible for clearance of Aβ from human brain by comparing the results of morphologic and experimental investigations. Although emigration of macrophages across the BBB has been infrequently investigated, macrophage immigration is well known in HIV-1 encephalitis and is becoming accepted in AD [11]. Our results in the model BBB exclude the emigration of Aβ-engorged macrophages as a mechanism of Aβ clearance since these adhered to the endothelium and were inhibited in emigration. Thus the fate of Aβ in aging brain seems to depend upon its handling and degradation inside the BBB.
Our observations suggest that oligomeric Aβ in perivascular macrophages may contribute to the fibrillar Aβ in congophilic vessels. AD macrophages are defective in uptake of Aβ (Fig. 3), yet are particularly susceptible to apoptosis from all assembly states of Aβ (Figs. 4, 5), contrasting with the ability of control macrophages to phagocytize Aβ [15] and to resist apoptosis (Figs. 4, 5). The phagocytic propensity of AD macrophages is compounded by their low clearance of Aβ (Fig. 2). These deficits might be related to downregulation of transcription of certain genes, including MGAT-3 and Toll-like receptors [15].
In AD brain, macrophages are apoptotic through activation of caspases −6, −7, and −8 (Fig. 6e, f, g), and they abut and infiltrate congophilic microvessels from the outside [11] (Fig. 6d), in agreement with the assembly of Aβ fibrils outside of the basal lamina [52]. Immunohistochemical studies of cerebral amyloid angiopathy (CAA) revealed its association with a significant increase and activation of macrophages in leptomeningeal and cortical vessels—implicating a central role for macrophages in amyloid angiopathy [53]. Furthermore, the BBB in AD brain is impaired [11] by immigrating macrophages (Fig. 1d), thus allowing leakage of plasma proteins into the brain and further aggravation of angiopathy. Taken together, AD macrophages loaded with oligomeric Aβ seem to suffer apoptosis, disintegrate and release oligomeric and fibrillar Aβ into the wall of congophilic vessels. Still, it is important to recognize that although phagocytosis of Aβ is generally greater in control monocytes compared to AD monocytes, this is a multifactorial and heterogeneous effect, and monocytes of some aged control subjects occasionally demonstrate inferior innate immunity [15] and phagocytosis of Aβ (SI Fig. 1).
Proposed mechanisms for the pathogenesis of CAA have included production of Aβ by myocytes in vessel walls [51], derivation of Aβ from blood or cerebrospinal fluid, and, more recently, deposition of Aβ from interstitial fluid being drained from the central nervous system [49]. In APP-transgenic mice, amyloid angiopathy developed specifically in areas of the brain that over-produce Aβ, suggesting that brain, as opposed to blood, is the major source of Aβ [44]. In mice, tracers were injected intracerebrally and found to be distributed, similarly to Aβ, within the basement membranes of intracerebral capillaries and arteries, and in leptomeningeal arteries [3]. Interestingly, within 24 h of injection the tracers were located in perivascular macrophages and these cells remained in brain microvessels, experiencing limited migration. Thus both drainage of the interstitial fluid and shuttling of Aβ by macrophages seem to lead to entrapment of Aβ in perivascular macrophages.
The results of the experimental co-culture of normal subjects’ monocytes with AD brain tissues show intrusion by these monocyte/macrophages into neurons and uploading of oligomeric Aβ, suggesting a protective neuronal mechanism by the normal innate immune system. However, AD monocyte/macrophages seem to be defective in such protection of neurons [15]. Although we have not investigated this mechanism by electron microscopy of AD brain tissues, we have observed that macrophages and monocytes exposed to Aβ in vitro develop prolific microvilli and larger dendrites, which could reach into neurons and upload Aβ [15].
In animal models of amyloidosis, microglia were first noted for their proinflammatory role [1]. Microglia of APOE4 transgenic mice produced more nitric oxide compared to APOE3 transgenic mice [5]. Subsequently, microglia were found to provide clearance of Aβ and were assisted in clearing Aβ deposits by antibodies [2] and by complement opsonization [16, 45, 46]. Clearance of Aβ was recognized to include two phases: the first mediated by antibodies and the second by microglia and antibodies [50]. However, in triple transgenic APP-thymidine kinase mice, bone marrow-derived rather than resident microglia restricted amyloid deposits as shown by ganciclovir destruction of microglia [36].
However, human brain disorders involve both microglial activation and macrophage recruitment [40]. In AD brain, the initial inflammatory signal is induced by microglia [1] and neuronal chemokine RANTES. These stimuli attract large macrophages (clearly not resembling ramified microglia) to migrate across brain microvessels and invade neuritic plaques in AD brain [11]. Enzyme studies mitigate against the role of microglia in Aβ clearance since, in comparison to macrophages, degradation of Aβ by microglia is poor due to defective lysosomal enzymes [28].
The alternative emphasis on treating brain amyloidosis or inflammation has led to two different therapeutic strategies in AD: Aβ vaccination or anti-inflammatory drugs. In AD patients, a delicate balance between macrophage phagocytosis and microglial inflammation appears to be disturbed, leading to decreased phagocytosis and degradation, and increased inflammation. We propose that the innate immune clearance of Aβ in AD brain involves shuttling of oligomeric Aβ by AD macrophages from neurons to perivascular locations, apoptotic death of these macrophages, Aβ spillage into the vessel wall, and seeding of congophilic angiopathy (Fig. 8). We also speculate that brain amyloidosis may be cleared by stimulating AD macrophages using curcuminoids which increase phagocytosis [14, 15, 54], and other approaches which enhance resistance to apoptosis and decrease inflammation.
Fig. 8.
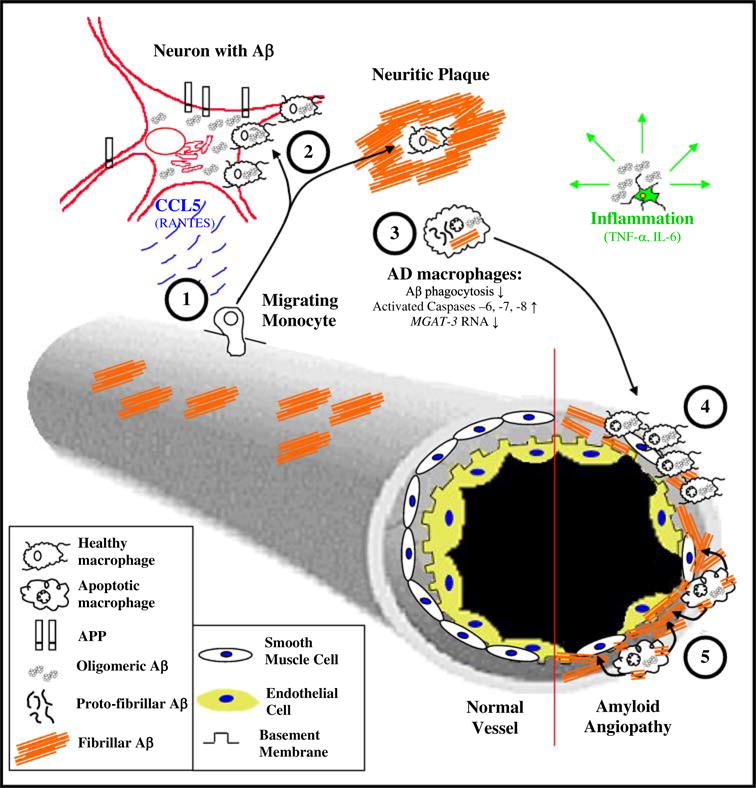
Congophilic angiopathy immunopathogenesis. Aβ is produced in neurons by cleavage of the amyloid-precursor protein (APP) by the β- and γ-secretases, releasing Aβ, which is re-uptaken into neurons and becomes oligomeric. Blood-borne monocytes (1) are attracted to neurons by chemokines produced by neurons (e.g. RANTES), intrude into neurons and neuritic plaques (2), and upload oligomeric Aβ from neurons and fibrillar Aβ in neuritic plaques. However, AD monocyte/macrophages (3) are heterogeneously deficient in Aβ clearance (uptake and degradation), and have deficient MGAT-III transcription and increased activation of caspases leading to their apoptosis. Some AD macrophages loaded with Aβ migrate to vessels (4) but are unable emigrate due to their engorgement with Aβ and consequently undergo apoptosis. Simultaneously, soluble and oligomeric Aβ have undergone peptide assembly into fibrillar Aβ, which is discharged into the vessel wall (5). Furthermore, congophilic angiopathy induces oxidative stress to neurons, and inflammatory cytokines produced by microglia potentiate neuronal damage
Supplementary Material
Acknowledgments
We thank J. Sayre for statistical analysis, S. Krajewski (Burnham Institute) for antibodies to activated caspases, B. Carter for nursing assistance with collection of blood specimens from patients and control subjects, B. Magrys for cutting sections of the blood-brain barrier model, C. Glabe for providing 14C-Aβ and A11 and OC antibodies, and C. Serhan for lipoxin A4. The UCLA Brain Bank provided the brain tissues for immunostaining.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-008-0481-0) contains supplementary material, which is available to authorized users.
Conflict of interest statement This work was supported by the Alzheimer’s Association, MPBio Ltd (MF) and NIH grant AG027818 (DBT).
Contributor Information
Justin Zaghi, Department of Orthopaedic Surgery, UCLA-Orthopaedic Hospital Research Center, 615 Charles E. Young Drive South, Room 410, Los Angeles, CA 90095-7358, USA.
Ben Goldenson, Department of Orthopaedic Surgery, UCLA-Orthopaedic Hospital Research Center, 615 Charles E. Young Drive South, Room 410, Los Angeles, CA 90095-7358, USA.
Mohammed Inayathullah, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA; Molecular Biology Institute, University of California, Los Angeles, CA 90095, USA; Brain Research Institute, University of California, Los Angeles, CA 90095, USA.
Albert S. Lossinsky, New Jersey Neuroscience Institute, JFK Medical Center, Edison, NJ 08818, USA
Ava Masoumi, Department of Orthopaedic Surgery, UCLA-Orthopaedic Hospital Research Center, 615 Charles E. Young Drive South, Room 410, Los Angeles, CA 90095-7358, USA.
Hripsime Avagyan, Department of Orthopaedic Surgery, UCLA-Orthopaedic Hospital Research Center, 615 Charles E. Young Drive South, Room 410, Los Angeles, CA 90095-7358, USA.
Michelle Mahanian, Department of Orthopaedic Surgery, UCLA-Orthopaedic Hospital Research Center, 615 Charles E. Young Drive South, Room 410, Los Angeles, CA 90095-7358, USA.
Michael Bernas, Department of Surgery, University of Arizona, Tucson, AZ, USA.
Martin Weinand, Department of Surgery, University of Arizona, Tucson, AZ, USA.
Mark J. Rosenthal, Department of Medicine, Greater LA VA Medical Center and UCLA School of Medicine, Los Angeles, CA 90095, USA
Araceli Espinosa-Jeffrey, Department of Neurobiology, David Geffen School of Medicine, Los Angeles, CA 90095, USA.
Jean de Vellis, Department of Neurobiology, David Geffen School of Medicine, Los Angeles, CA 90095, USA.
David B. Teplow, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA Molecular Biology Institute, University of California, Los Angeles, CA 90095, USA; Brain Research Institute, University of California, Los Angeles, CA 90095, USA.
Milan Fiala, Department of Orthopaedic Surgery, UCLA-Orthopaedic Hospital Research Center, 615 Charles E. Young Drive South, Room 410, Los Angeles, CA 90095-7358, USA.
References
- 1.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 3.Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–144. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 4.Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 5.Colton CA, Brown CM, Cook D, Needham LK, Xu Q, Czapiga M, Saunders AM, Schmechel DE, Rasheed K, Vitek MP. APOE and the regulation of microglial nitric oxide production: a link between genetic risk and oxidative stress. Neurobiol Aging. 2002;23:777–785. doi: 10.1016/S0197-4580(02)00016-7. [DOI] [PubMed] [Google Scholar]
- 6.El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 8.Fiala M, Looney DJ, Stins M, Way DD, Zhang L, Gan X, Chiappelli F, Schweitzer ES, Shapshak P, Weinand M, Graves MC, Witte M, Kim KS. TNF-alpha opens a paracellular route for HIV-1 invasion across the blood-brain barrier. Mol Med. 1997;3:553–564. [PMC free article] [PubMed] [Google Scholar]
- 9.Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med. 1998;4:480–489. [PMC free article] [PubMed] [Google Scholar]
- 10.Fiala M, Liu NQ, Reddy S, Graves M. Macrophages infiltrate the brain and express COX-2 and iNOS in Alzheimer’s disease and AIDS. Alzheimers Rep. 2001;4:1–7. [Google Scholar]
- 11.Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32:360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- 12.Fiala M, Eshleman AJ, Cashman J, Lin J, Losssinsky AS, Suarez V, Yang W, Zhang J, Popik W, Singer EJ, Chiappelli F, Carro E, Weinand M, Witte M, Arthos J. Cocaine increases human immunodeficiency virus type 1 neuroinvasion through remodeling brain microvascular endothelial cells. J Neurovirol. 2005;11:1–11. doi: 10.1080/13550280590952835. [DOI] [PubMed] [Google Scholar]
- 13.Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. 2005;7:221–232. doi: 10.3233/jad-2005-7304. discussion 255–262. [DOI] [PubMed] [Google Scholar]
- 14.Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid-beta and inflammation: two faces of innate immunity in Alzheimer’s disease. J Alzheimers Dis. 2007;11:457–463. doi: 10.3233/jad-2007-11406. [DOI] [PubMed] [Google Scholar]
- 15.Fiala M, Liu PT, Espinosa-Jeffrey A, Rosenthal MJ, Bernard G, Ringman JM, Sayre J, Zhang L, Zaghi J, Dejbakhsh S, Chiang B, Hui J, Mahanian M, Baghaee A, Hong P, Cashman J. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci USA. 2007;104:12849–12854. doi: 10.1073/pnas.0701267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. see comments. [PMC free article] [PubMed] [Google Scholar]
- 17.Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, Stern D, Kim KS, Zlokovic B, Kalra VK. Beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol. 2000;279:C1772–C1781. doi: 10.1152/ajpcell.2000.279.6.C1772. [DOI] [PubMed] [Google Scholar]
- 18.Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton JA, Whitty G, White AR, Jobling MF, Thompson A, Barrow CJ, Cappai R, Beyreuther K, Masters CL. Alzheimer’s disease amyloid beta and prion protein amyloidogenic peptides promote macrophage survival, DNA synthesis and enhanced proliferative response to CSF-1 (M-CSF) Brain Res. 2002;940:49–54. doi: 10.1016/S0006-8993(02)02589-1. [DOI] [PubMed] [Google Scholar]
- 20.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 21.Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002;40:195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- 22.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 23.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krajewska M, Rosenthal RE, Mikolajczyk J, Stennicke HR, Wiesenthal T, Mai J, Naito M, Salvesen GS, Reed JC, Fiskum G, Krajewski S. Early processing of Bid and caspase-6, -8, -10, -14 in the canine brain during cardiac arrest and resuscitation. Exp Neurol. 2004;189:261–279. doi: 10.1016/j.expneurol.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 25.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 26.Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, Roberts J, Pushkarsky T, Bukrinsky M, Witte M, Weinand M, Fiala M. Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol. 2002;76:6689–6700. doi: 10.1128/JVI.76.13.6689-6700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lossinsky AS, Shivers RR. Studies of cerebral endothelium by scanning and high-voltage electron microscopy. In: Nag S, editor. The blood-brain barrier: biology and research protocols. Vol. 89. Humana Press; Totawa: 2003. pp. 67–82. [DOI] [PubMed] [Google Scholar]
- 28.Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, Maxfield FR. Degradation of fibrillar forms of Alzheimer’s amyloid beta-peptide by macrophages. Neurobiol Aging. 2008;29:707–715. doi: 10.1016/j.neurobiolaging.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 30.Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6C(hi)CCR2(+) monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 31.Mrak RE, Griffin WS. Potential inflammatory biomarkers in Alzheimer’s disease. J Alzheimers Dis. 2005;8:369–375. doi: 10.3233/jad-2005-8406. [DOI] [PubMed] [Google Scholar]
- 32.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 33.Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- 34.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/S0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 35.Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simard AR, Rivest S. Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer’s disease. Mol Psychiatry. 2006;11:327–335. doi: 10.1038/sj.mp.4001809. [DOI] [PubMed] [Google Scholar]
- 37.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 38.Smits HA, Rijsmus A, van Loon JH, Wat JW, Verhoef J, Boven LA, Nottet HS. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. 2002;127:160–168. doi: 10.1016/S0165-5728(02)00112-1. [DOI] [PubMed] [Google Scholar]
- 39.Solomon B. Intravenous immunoglobulin and Alzheimer’s disease immunotherapy. Curr Opin Mol Ther. 2007;9:79–85. [PubMed] [Google Scholar]
- 40.Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–247. doi: 10.1016/S0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- 41.Stuart LM, Bell SA, Stewart CR, Silver JM, Richard J, Goss JL, Tseng AA, Zhang A, El Khoury JB, Moore KJ. CD36 signals to the actin cytoskeleton and regulates microglial migration via a p130Cas complex. J Biol Chem. 2007;282:27392–27401. doi: 10.1074/jbc.M702887200. [DOI] [PubMed] [Google Scholar]
- 42.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 43.Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van Den Haute C, Mercken M, Moechars D, Laenen I, Kuiperi C, Bruynseels K, Tesseur I, Loos R, Vanderstichele H, Checler F, Sciot R, Van Leuven F. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am J Pathol. 2000;157:1283–1298. doi: 10.1016/S0002-9440(10)64644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol. 2000;161:127–138. doi: 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- 46.Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Antibody-mediated phagocytosis of the amyloid beta-peptide in microglia is differentially modulated by C1q. J Immunol. 2001;166:7496–7503. doi: 10.4049/jimmunol.166.12.7496. [DOI] [PubMed] [Google Scholar]
- 47.Wegiel J, Kuchna I, Nowicki K, Frackowiak J, Mazur-Kolecka B, Imaki H, Mehta PD, Silverman WP, Reisberg B, Deleon M, Wisniewski T, Pirttilla T, Frey H, Lehtimaki T, Kivimaki T, Visser FE, Kamphorst W, Potempska A, Bolton D, Currie JR, Miller DL. Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol. 2007;113:389–402. doi: 10.1007/s00401-006-0191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6:404–416. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 49.Weller RO, Nicoll JA. Cerebral amyloid angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res. 2003;25:611–616. doi: 10.1179/016164103101202057. [DOI] [PubMed] [Google Scholar]
- 50.Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wisniewski HM, Wegiel J. Beta-amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol. 1994;87:233–241. doi: 10.1007/BF00296738. [DOI] [PubMed] [Google Scholar]
- 52.Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 1992;84:117–127. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- 53.Yamada M, Itoh Y, Shintaku M, Kawamura J, Jensson O, Thorsteinsson L, Suematsu N, Matsushita M, Otomo E. Immune reactions associated with cerebral amyloid angiopathy. Stroke. 1996;27:1155–1162. doi: 10.1161/01.str.27.7.1155. [DOI] [PubMed] [Google Scholar]
- 54.Zhang L, Fiala M, Cashman J, Sayre J, Espinosa-Jeffrey A, Mahanian M, Zaghi J, Badmaev V, Graves M, Bernard G, Rosenthal M. Curcuminoids enhance amyloid-beta uptake by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. 2006;10:1–7. doi: 10.3233/jad-2006-10101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.