

Abstract
Dipeptidylpeptidase 4 (DPP4/CD26) enzymatically cleaves select penultimate amino acids of proteins, including colony stimulating factors (CSFs), and has been implicated in cellular regulation. To better understand the role of DPP4 regulation of hematopoiesis, we analyzed the activity of DPP4 on the surface of immature blood cells and then comparatively assessed the interactions and functional effects of full-length (FL) and DPP4 truncated factors [(T)-GM-CSF and- IL-3] on both in vitro and in vivo models of normal and leukemic cells. T-GM-CSF and T-IL-3 had enhanced receptor binding, but decreased CSF activity, compared to their FL forms. Importantly, T-GM-CSF and T-IL-3 significantly, and reciprocally, blunted receptor binding and myeloid progenitor cell proliferation activity of both FL-GM-CSF and FL-IL-3 in vitro and in vivo. Similar effects were apparent in vitro using cluster forming cells from patients with Acute Myeloid Leukemia (AML) regardless of cytogenetic or molecular alterations and in vivo utilizing animal models of leukemia. This suggests that DPP4 T-molecules have modified binding and functions compared to their FL counterparts and may serve regulatory roles in normal and malignant hematopoiesis.
Introduction
Dipeptidylpeptidase 4 (DPP4/CD26), a serine protease found as membrane bound and soluble forms, enzymatically cleaves select penultimate amino acids of proteins that regulate multiple aspects of hematopoiesis.1,2 We reported that DPP4 truncated (T)-GM-CSF has diminished colony stimulating activity, and intracellular signaling compared to FL-GM-CSF, and blunted the in vitro colony forming effects of FL-GM-CSF.3 Although a large number of cytokines, chemokines, and growth factors (including GM-CSF and IL-3) have previously unrecognized putative DPP4 truncation sites1–3 their potential interactive roles in hematopoiesis (including modifying the function of other molecules and acting as regulatory molecules) are poorly understood. Our objective was to clarify the regulatory effects of DPP4 truncated GM-CSF and IL-3 on in vivo and in vitro modulation of steady and diseased state hematopoiesis.
Hematopoietic progenitor (HPC)/precursor cells from patients with Acute Myeloid Leukemia (AML) respond to cytokines such as GM-CSF and IL-3 for enhanced growth, survival and resistance to therapy.4–8 GM-CSF and IL-3 have additive to synergistic proliferative effects on normal and malignant cell growth,9–15 and patients with AML manifest increased serum GM-CSF and IL-3 before induction therapy compared to normal controls, and a decline of GM-CSF and IL-3 after successful remission.16 GM-CSF and IL-3 share a common receptor beta-chain, IL-3 competes for GM-CSF binding on AML blasts,10 and targeting the IL-3 receptor has been suggested as a useful therapeutic in AML.16, 17 Therefore, it is important to understand how T-GM-CSF and T-IL-3 may influence normal and malignant hematopoiesis. We now report that T-GM-CSF and T-IL-3 reciprocally blunt receptor binding, as well as functional interactions, of both FL-GM-CSF and FL-IL-3 in vitro in primary human cord blood (CB) HPC, primary AML HPC/precursors, and in the human growth factor dependent TF-1 cell line. Moreover, these blunting effects of T-GM-CSF and T-IL-3 were recapitulated in vivo in murine models of normal and malignant hematopoiesis, thus demonstrating an additional, and perhaps crucial, layer of cell regulation for normal and leukemic hematopoiesis.
Materials and Methods
Mice, mouse cells, human cord blood, TF-1 and primary patient AML cells
C57BL6/J mice were purchased from Jackson Laboratories (Bar Harbor, ME) and treated with Diprotin A (Peptides International; Louisville, KY) or PBS control as reported.3 dpp4−/−18, FLT3-ITD19, and PtpnE76K/+LysM-Cre+ (Ptpn11E76K)20 mice are all on a C57BL6/J mouse strain background, and have been previously described. Mouse bone marrow (mBM) and human cord blood [(CB) from the Eskenazi (formerly Wishard) Hospital, Indiana University School of Medicine, Indianapolis, Indiana, USA, and from Cord:Use Cord Blood Bank, Orlando, Florida, USA] were used as described.21 AML patient samples were obtained from peripheral blood samples of patients under an approved IRB (9402–10 & 9812–11).
The Indiana University Committee on Use and Care of Animals, and the Indiana University Institutional Review Board, respectively, approved mBM, human CB, and AML studies. CB received was from to be discarded material. TF-1 cells, originally obtained from T. Kitamura, Japan, are available from American Type Culture Collection (CRL-2003) and were used as described.3, 22, 23
HPC Colony Assay
C57BL6/J, dpp4−/−, FLT-3/ITD, and Ptpn11E76K mice were injected s.c. with combinations of 10ug of recombinant murine (rm) FL- or T- GM-CSF and/or -IL-3. After 24 hours mice were sacrificed, femurs flushed, and mBM cells plated at ~5×104 cells/ml in 1% methylcellulose culture medium in the presence of hemin 0.1 mM, 30% FBS (Hyclone, Utah) with the following growth factors, unless otherwise noted: 1 U/ ml recombinant human (rh) erythropoietin (Amgen Corporation, South San Francisco, CA), 50 ng/ml rmSCF (R&D Systems, Minneapolis, MN), and 5% vol/vol pokeweed mitogen mouse spleen cell–conditioned medium (PWMSCM).18, 21 Percent HPCs in S phase of the cell cycle was estimated by high-specific-activity tritiated thymidine kill technique. Colonies were scored after a 7 day incubation, and CFU-GM, BFU-E and CFU-GEMM progenitors distinguished.21 Human CB cells and primary AML patient samples were separated into a low-density fraction and plated at ~5×104 cells/ml, and TF-1 cells were plated at 250–1,000 cells/ml in 0.3% semisolid agar medium (Difco) with 10ng/ml (or concentrations listed) of rhFL- or T- GM-CSF or -IL-3. Colony formation for CB was counted on day 14, and colony formation for TF-1, or cluster formation of AML patient samples,24–26 at day 7–10. CD34+ CB cells (>95% CD34+) were purified.(27) Rm and rhGM-CSF, IL-3, and SCF were from R&D Systems. Cell cultures were incubated at lowered (5%) O2 tension in 5% CO2 in a humidified chamber.3, 21, 27
DPP4 Activity Assay
CB, AML patient samples, TF-1 or mBM were harvested and enriched (CD34+) or depleted (Lineage -) using Miltenyi (San Diego, CA) commercially available kits.3, 28 Approximately 1-5×105 cells (untreated or incubated with 5mM DiprotinA for 30 minutes) were plated/ well in duplicate (N= at least 3 individual samples or mice), and left intact in 50ul of PBS in a 96-well flat-bottom microtiter plate. The assay was carried out by combining cells with 50 μl of the DPP4 substrate Gly-Pro-aminoluciferin and assay buffer optimized for this assay (DPP4-Glo Protease Assay; Promega, Madison, WI, USA). Plates were incubated at 37 °C for 30 min, and surface DPP4 enzyme activity was measured using an LMAX luminometer (Molecular Devices, Sunnyvale, CA, USA).(3)
Flow Cytometry
Human TF-1 cells were fixed with 1% formaldehyde after stimulation (with 10ng/ml rhFL, T or FL:T IL-3), permeabilized using BD Phosflow Perm Buffer III (#558050) and stained in BD staining buffer with primary anti-P-Stat5 (BD) and anti-P-Jak2 antibodies (Cell Signaling) and secondary antibody (Cell signaling). Mouse bone marrow was washed in PBS and 2×106 cells were stained with the following phenotyping antibodies to determine HSC and progenitor populations29 prior to fixation [Lineage cocktail (Biolegend,), CD34, SCA, KIT, FLT3, Fcgamma receptor (BD)]. All samples were run on BD machines (LSR 4 or Fortessa) and analyzed using FlowJo Software.
Generation of DPP4 truncated (T) -GM-CSF and -IL-3
Soluble human or porcine DPP4, prepared from human placental tissue or porcine kidney, were purchased from MP Biomedicals, LLC or Sigma Aldrich (#D7052), respectively, and used at approximately 0.25ug for every 1ug of full-length protein per digestion at 37°C for at least 18 hours.3
Equilibrium Receptor Binding
Receptor binding analysis of FL- and T- rh GM-CSF and IL-3 were done under equilibrating conditions, and data analyzed by Scatchard plotting. 3, 30, 31 using TF-1 cells, CD34+ CB cells, and AML primary patient samples. Carrier-free rhGM-CSF and IL-3 (R&D Systems; Minneapolis, MN) were radio-iodinated by chloramine-T method by Phoenix Pharmaceuticals, Inc. (Burlingame, CA). The [125I]-GM-CSF and IL-3 were re-purified to a concentration of approximately 0.67ug/ul and had a specific radioactivity of ~60 uCi/mmole. The chloramine-T method iodinates on tyrosine residues in proteins; therefore, truncation by DPP4 does not reduce the specific radioactivity of the protein by the enzyme’s removal of the N-terminal Ala-Pro. Nonspecific binding was assessed by competing rh[125I]-GM-CSF or IL-3 with a 1,000-fold excess of unlabeled rhGM-CSF or IL-3. Using TF-1 cells, this results in greater than 95% reduction in non-competed c.p.m. of bound ligand, which was near background radiation, so specific binding under these conditions was considered to be 100% of measured 125I, as determined by Beckman Coulter Gamma 5500B gamma counter (Brea, CA). Linear-regression coefficient of correlation (r2) was considered acceptable at a value of 0.90 or greater.
Statistical analyses
For all colony assays, three plates per experimental point were scored. Results of colony assays and animal studies were assessed by Two-tailed students T test or Analysis of Variance (ANOVA). Receptor binding studies were assessed by Student’s t-test. Numbers of experiments per group are noted in the figure legends, and all animal experiments analyzing effects in vivo contained at least 3 mice per group.
Results
A.) Interactions of FL- and T-GM-CSF and –IL-3 on Cellular Proliferation in vitro and in vivo
Ai.) DPP4 is active on the cell surface of immature cells
DPP4 has been shown to be present as an active, membrane bound form on specific cell types. We detected active DPP4 on the surface of the human growth factor-dependent TF-1 cell line, human CD34+ CB cells, primary AML patient samples, and lineage negative mBM and this activity was blunted by Diprotin A (DPA), an ILE-PRO-ILE DPP4 inhibitor (Figure 1A) on all samples evaluated.3, 18, 28
Figure 1. DPP4 is active on the cell surface of immature blood cells and DPP4 truncated molecules (T-GM CSF and T- IL3) block colony formation across molecules in vitro at less than a 1:1 ratio.
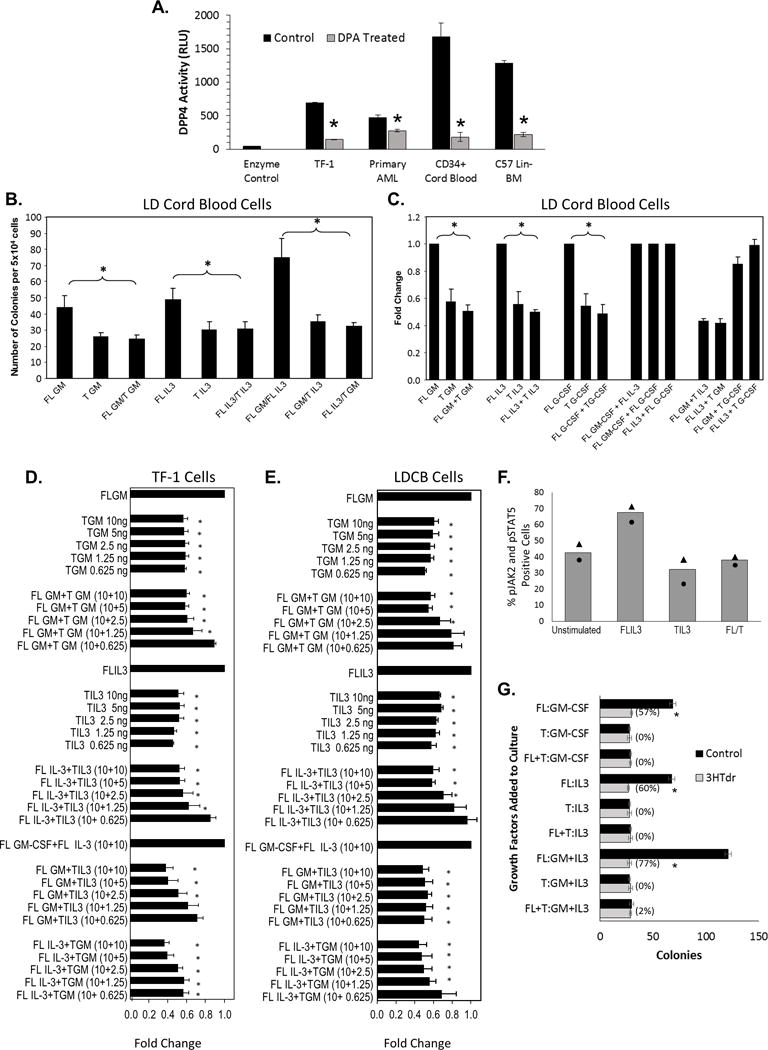
(A) 1–5×105 cells/well from TF-1, primary AML samples, CD34+ CB and lineage negative C57 BL6/J mBM were analyzed for baseline DPP4 activity. Diprotin A (DPA, a DPP4 inhibitor), was added as a control 30 minutes prior to the start of the assay (for some samples) to show assay specificity for DPP4 activity on the cell surface and ability to inhibit the DPP4 enzyme. N=3 samples per group done in triplicate for all except for the primary AML samples where N=5 and plating for data shown was lower than other samples (~1×105/well) due to sample availability. AML samples were from those shown in Table 1. Low density cord blood cells (B,C, and E) or TF-1 cells (D) were stimulated with 10ng/ml of full length (FL) or DPP4 truncated (T) GM-CSF, IL-3 or G-CSF unless otherwise listed. (F) TF-1 cells were factor starved and treated with 10ng/ml of FL, T or 1:1 ration of IL-3 for 5 minutes, fixed and assessed for induction of JAK/STAT signaling by flow cytometry (n=2 independent experiments). (G) Mouse BM was plated at 5×104 cells/ml in the presence of 10ng/ml rmFL, T or a mixture of FL/T GM-CSF and/or IL-3 and colony formation and cycling status of the progenitor cells were determined. All experiments contained 3 mice per group or were performed at least 3 times (unless noted otherwise) and ANOVA was used to determine statistical significance with the exception of 1A (students-T Test). * = p ≤ 0.05 compared to control (FL GM, IL-3 or FL GM/IL-3, FL G-CSF etc.) For the combined experiments shown as fold change the average number of control colonies were: FL GM= 45, IL-3= 55, FL GM/IL-3= 95 for LDCB and FL GM= 220, IL-3= 170, FL GM/IL-3= 310 for TF-1.
Aii.) T-GM-CSF and T-IL-3 blunt colony stimulating effects of both FL-GM-CSF or FL-IL-3 in vitro on human (CB and TF-1) and mouse cells
To delineate the ability of the FL- and T-GM-CSF and -IL-3 molecules to reciprocally modulate each other’s function, as well as to better understand the practical relevance of their functions, we analyzed the colony stimulating activity of these molecules using primary human low density CB HPCs (Figure 1B and 1C). This reciprocal interaction was specific for GM-CSF and IL-3, and likely involved the common beta chain they share in their receptors, as DPP4 T-G-CSF (whose receptor does not share the common beta-chain) was only able to blunt colony formation induced by FL-G-CSF, but not that of FL-GM-CSF or FL-IL-3 (Figure 1C). Importantly, the T-factors could be serially diluted to approximately 12% of the FL amount and still significantly blunt stimulation by either FL-GM-CSF or FL-IL-3 in both TF-1 (Figure 1D) and CB (Figure 1E) cells. Further, as previously demonstrated for GM-CSF,3 T-IL-3 induced less activation of the JAK/STAT pathway than FL-IL-3 and blunted the activation induced by FL-IL-3 (Figure 1F). T-GM-CSF and T-IL-3 alone or in combination blunted numbers of mBM colonies stimulated by their FL forms and manifested decreased percentages of HPCs in S-phase of the cell cycle (Figure 1G).
Aiii) Effects of T- and FL- CSFs on HPC in vivo mimic in vitro effects
To evaluate if the functional data above would be replicated in an in vivo situation, we studied effects of m FL- and T-GM-CSF and -IL-3 for effects on the absolute numbers and cycling status of mBM HPCs. We used dpp4−/− mice on a C57BL/6 strain background (Figure 2A), C57BL/6 mice that were pre-treated with DPA (Figure 2B), or C57BL/6 mice left untreated (Figure 2C). Mice were administered a single s.c. dose of rmFL- or T-GM-CSF or -IL-3, or various combinations of FL-, and T-, GM-CSF or -IL-3. FL-vs. T-CSFs were injected separately at different sites in the same mouse. In all 3 animal models, T-GM-CSF and T-IL-3 resulted in less in vivo stimulatory activity than their respective FL-CSFs with respect to HPC numbers (CFU-GM, CFU-GEMM, and BFU-E)/femur and their cell cycling, as assessed by percent of HPC in S-Phase of the cell cycle (Figure 2). Moreover, T-GM-CSF and T-IL-3 blunted effects of both FL-GM-CSF and FL-IL-3 similar to that seen in vitro. T-GM-CSF or T-IL-3 each resulted in in vivo suppression of the effects of both FL-GM-CSF and FL-IL-3 (Figure 2). Most importantly, and impressively, combinations of T-CSFs were able to blunt the increased additive potency of combinations of FL-GM-CSF plus FL-IL-3. This functional blunting, with respect to HPC numbers/femur and cycling in vivo, demonstrates that both T-GM-CSF and T-IL-3 functionally display down-modulating effects on HPC over both FL- factors.
Figure 2. Truncated GM-CSF (T-GM-CSF) and IL-3 (T-IL-3) block colony formation across molecules in vivo.
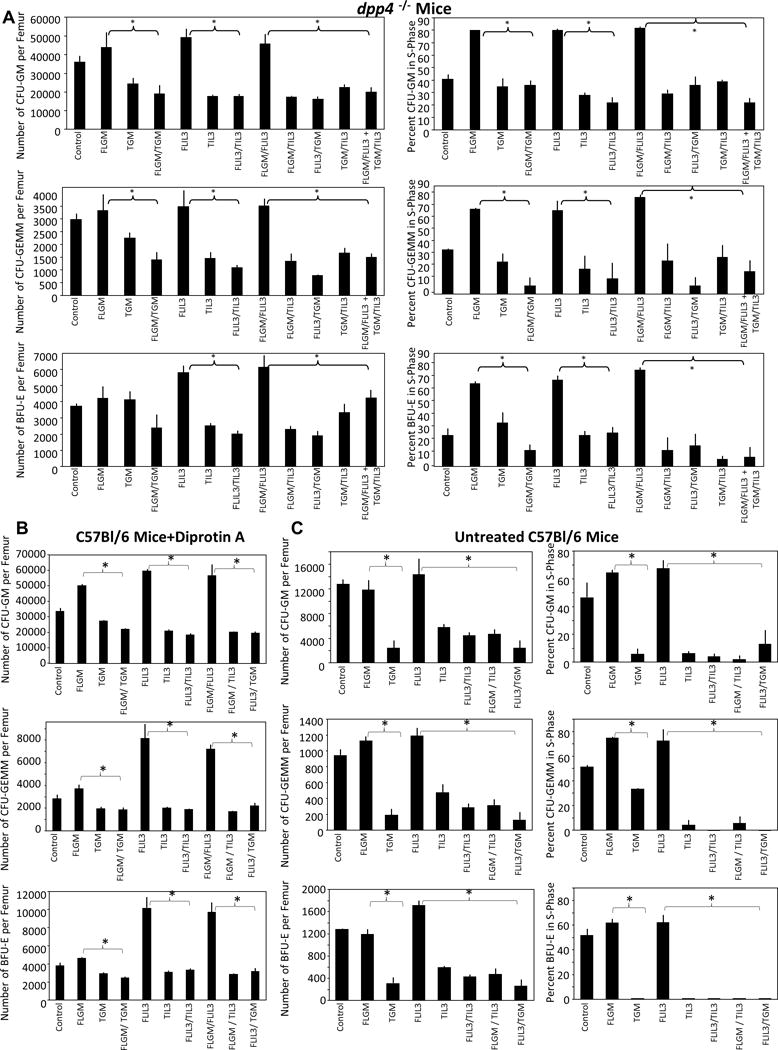
dpp4−/− mice (A), C57BL6/J mice treated with Diprotin A (B), or untreated C57BL6/J mice (C) were injected with 10ug of either rmFL, T or a combination of FL/T GM-CSF and/or IL-3 s.c. at different sites. Mice were sacrificed 24 hours later, femurs were flushed, and colony formation and cycling status of the progenitor cells were determined. *= p ≤ .05 or less by ANOVA. N=3 animals/group with each mouse individually accessed. 1 of 2 representative experiments shown for dpp4−/− mice.
B) Primary progenitor/precursor cells from patients with AML respond in vitro to DPP4 T-rhGM-CSF and T-IL-3, in a manner similar to normal progenitors
GM-CSF and IL-3 are important in growth and maintenance of AML cells.6, 10, 32–35 Modifications in cytogenetic and molecular status, such as internal tandem duplication (ITD) mutations in the FMS-like tyrosine kinase 3 gene (FLT3/ITD), are common and can alter growth and aggressive progression of disease. Therefore, to understand if HPC/precursors from patients with AML, with various cytogenetic backgrounds and molecular mutations (Table 1), are sensitive to T- factors in a fashion similar to that of normal HPC, we assessed AML patient samples with varied cytogenetic and molecular alterations for responsiveness. Patients with newly diagnosed, or relapsed, AML have varied growth patterns compared to normal cells, and usually form clusters (3-40 cells) rather than colonies (>40 cells) or no colonies or clusters in semi-solid agar culture medium.24–26 In the cases shown (Table 1, Figure 3 A–D), cells from these patients with AML did not grow without CSFs, but formed clusters (~3-20 cells/clone) upon stimulation with either rhFL-GM-CSF or FL-IL-3, and T-GM-CSF and -IL-3 were each less effective stimulating factors on these cluster forming cells than their FL-forms. The FL-GM-CSF and FL-IL-3 stimulated cluster formation of the AML patient samples was reciprocally diminished by either T-GM-CSF or T-IL-3 in patient samples tested, regardless of cytogenetic or molecular status. Additionally, for two patient samples tested, and as seen in the CB and TF-1 in vitro models (Figure 1), less than a 1:1 ratio of T-CSF to FL-CSF down-modulated maximal stimulation noted with combinations of FL-GM-CSF and FL-IL-3 (Figure 3 C and D, patients 3135 and 3131). Further, cycling analysis showed an identical trend to that seen in the in vivo studies evaluating normal hematopoietic progenitors, where T- molecules induced less stimulatory activity than the FL-CSFs counterparts with respect to numbers of clusters formed and percent of cells in S-Phase of the cell cycle (Figure 3E). This dramatic suppressive response of AML patient samples in response to the T-factors, regardless of aggressive cytogenetic or molecular alterations, prompted us to compare the extent of the inhibition by fold change in primary CB vs primary AML patient samples. Figure 3F shows that, in general in this small sampling of patient cells, AML samples were significantly more sensitive to the T-factors than the CB cells, regardless of cytogenetic/molecular status, suggesting a possibility for consideration of future use of DPP4 T-factors to down-modulate growth of AML progenitors/precursors. We then assessed effects of T-CSFs in vivo in two mouse models of leukemia.
Table 1. Characterization of AML primary patient samples based on disease state, cytogenic/molecular phenotype and utilization.
AML patient samples were obtained and samples utilized were determined to be at least 90% tumor blasts. Yes or No (refers to growth in culture), “cycling” refers to being used in cycling assays
| Patient Number | De Novo or Relapse | Cytogenetic/Molecular Alterations | Experimental Use |
|---|---|---|---|
| 3011 | De Novo | inv 16 | Yes (cycling) |
| 3099 | De Novo | Flt3 ITD | N/A receptor binding only* |
| 3127 | De Novo | t(9;22)(q34;q11.2) | Yes* |
| 3130 | De Novo | normal karyotype, F1t3 TKD | Yes* |
| 3131 | De Novo | 7q- Flt3 ITD | Yes* (cycling) |
| 3135 | De Novo | inv 3(q21;q26) [EVI 1 activiation] | Yes* |
| 3136 | De Novo | normal karyotype, Flt3 TKD, Mnpml | No* |
| 3142 | Relapse | T(12;15)(Q13,Q11.2),Flt3ITD | N/A receptor binding only* |
| 3146 | De Novo | 46(X;X) [13 metaphases]; t(l;13) (p32;ql2) [ 8 metaphases] dmut CEBPA, and negative Flt3ITD/TKD, negative NPM1 | # |
| 3152 | Relapse | normal karyotype (46[X,Y]), NPM1 negative, CEBPA negative, FltlTD negative, Flt3 D835+ | # |
| 3155 | De Novo | complex, with monosomy 7, negative for Flt3ITD/TKD and negative for NPM1 | # |
| 3157 | De Novo | 46(X;X) negative Flt3ITD, neg NPM1, neg CEBPA. | # |
| 3159 | De Novo | normal karyotype; FLT3ITD+, NPM1−,CEBPA− | # |
| 3224 | Relapse | normal karyotype, [46(X,Y)], WT1 18% | Yes (cycling) # |
designates use for receptor binding studies
designates use in DPP4 activity assay.
Figure 3. DPP4 Truncation of GM-CSF and IL-3 inhibit AML cluster formation stimulated by FL-CSFs.
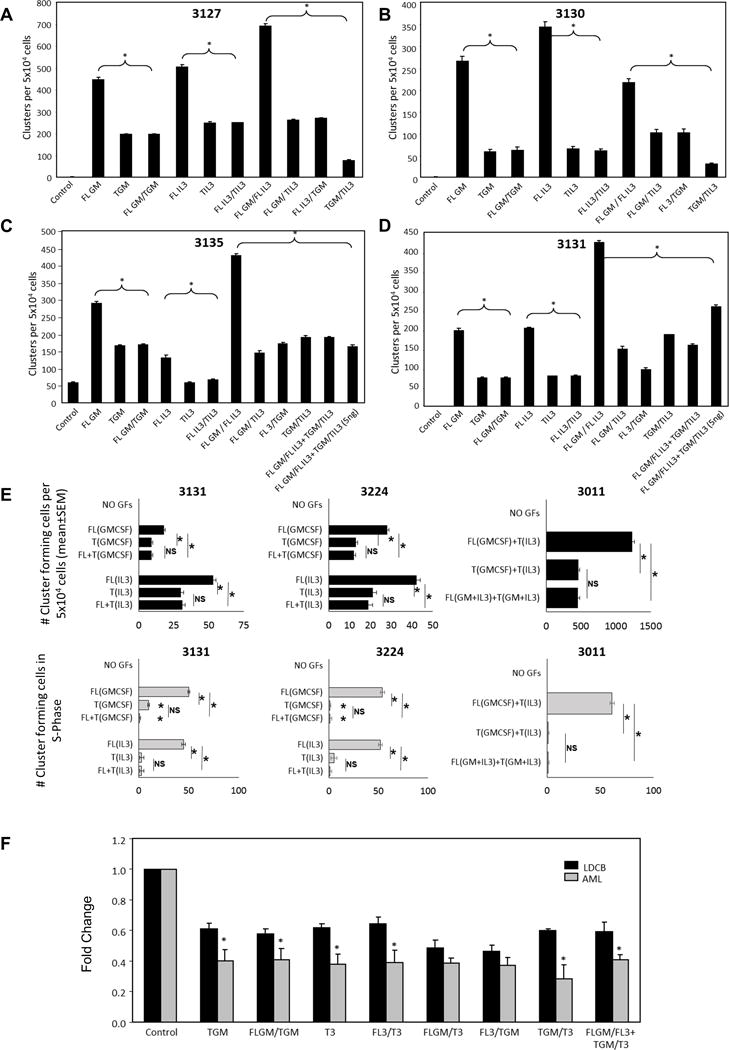
A-D) 5 × 104 cells from ficolled peripheral blood of AML patients with 90% or greater blast burden (Table 1) were plated in agar with 10ng/ml (unless otherwise noted) of rhFL- and/or T-GM-CSF and/or IL-3. Cells were plated in triplicate and incubated at 37°C/5% O2 for 7–10 days prior to enumeration of clusters formed. E) Cell cycling evaluation of AML patient samples (n=3, 5 × 104) treated with FL or T factors. F) Fold change comparison of CB response to DPP4 truncated factors compared to AML response. N=4 for AML samples and N=7 for LDCB * = p ≤ .05 or less by ANOVA compared to control (FLGM, FL3 or FLGM/FL3)
Bi) Effects of T- and FL-CSFs in vivo in mouse models of leukemia
AML primary patient samples were sensitive to the FL- and T-factors in vitro, regardless of cytogenetic or molecular status. Therefore, to begin to assess the potential clinical utility of T-GM-CSF and -IL-3 in models of malignant hematopoiesis in vivo, FLT-3/ITD mice were first utilized.19 Similarly, to in vivo experiments with normal mice (Figure 2), FLT-3/ITD mice were injected with rm FL- and T-GM-CSF and -IL-3, and effects on absolute numbers and cycling status of BM HPCs were analyzed. Mice showed a leukemia phenotype with significantly enhanced numbers of stem and progenitor cells in both the bone marrow and spleen (Supplemental Figure 1A and B, respectively) compared to control. In vivo treatment of FLT3-ITD mice with rmFL-factors (GM-CSF and IL-3 in combination to maximize stimulation) resulted in functional increases in numbers of progenitors (Figure 4A and C), and cycling status (Figure 4B and D), respectively in BM and spleen. T-factors (GM-CSF and IL-3 combination) not only reduced numbers and cycling status of HPCs compared to baseline levels of control mice but also significantly blunted the enhanced stimulation obtained with FL-CSFs in both the bone marrow (A and B) and spleen (C and D). Another model of murine leukemia containing an E76K activating mutation in protein tyrosine phosphatase, non-receptor type 11 (Ptpn11, Ptpn11E76K),20 commonly detected in childhood acute leukemia and myeloproliferative disorders, was also evaluated for response to rmFL- and T-GM-CSF and IL-3. These mice showed a leukemia phenotype with significantly enhanced numbers of progenitors in both the bone marrow and spleen (Supplemental Figure 2A and B, respectively) compared to control. Identical trends to those seen with the FLT-3/ITD mice were detected with respect to progenitor cell numbers and cycling in the spleen (Figure 4E and F). The BM of the Ptpn11E76K mice showed dramatic reductions in multiple phenotypically and functionally-defined cell populations, in response to both FL- and T- factors, such that there was >90% reduction in BM ST-HSCs, MPPs, CMPs and GMPs with few or no CFU-GM, BFU-E or CFU-GEMM detected in these mice after FL-, T-, or FL+T-GM-CSF plus -IL-3 administration (data not shown). These decreases in HPC numbers in BM likely reflect large mobilization effects suggesting that these mice may be ultra-sensitive to CSF-induced mobilization regardless of treatment with FL- or T- molecules. Data from these two leukemia mouse models suggest that T-CSFs are able to modify growth and cycling of leukemia cells with molecular alterations in vivo which may represent a heretofore unknown mechanism of leukemia cell regulation.
Figure 4. Effects of T- and FL- CSFs on in vivo model of leukemia.
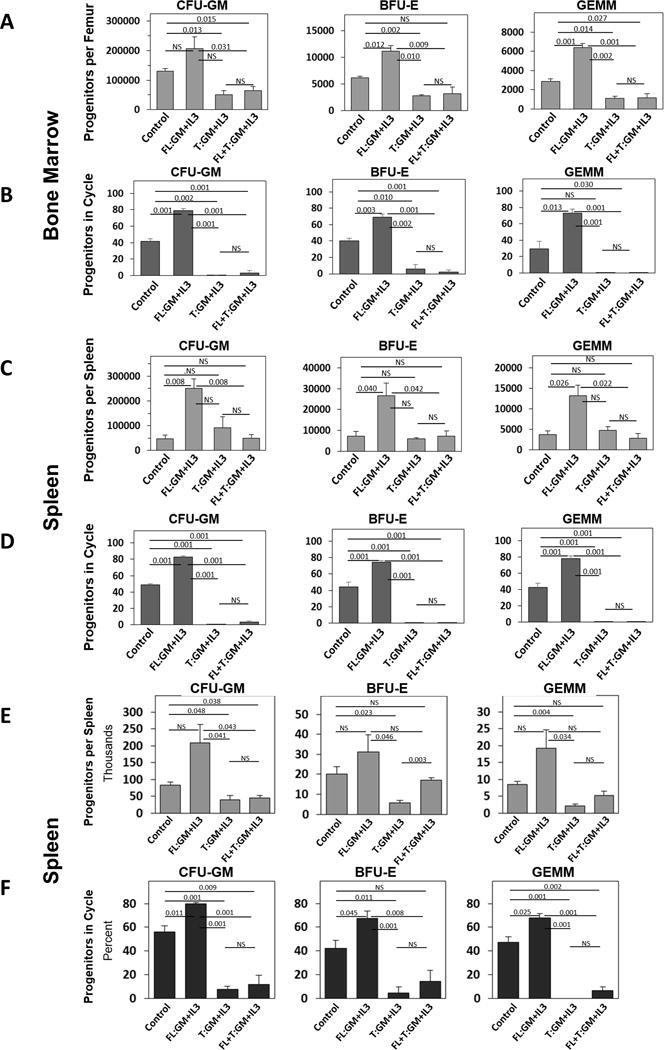
FLT3-ITD (A-D) and Ptpn11E76K ((F, G) mice were injected with 10ug of a combination of either rmFL-GM-CSF and FL-IL-3, T-GM-CSF and T-IL-3 or a mixture of both FL- and T- (GM-CSF and IL-3) s.c. at different sites. Mice were sacrificed 24 hours later, femurs flushed, spleens made into single cell suspensions and colony formation (A,C,E) and cycling status (B,D,F) of the progenitor cells were determined.
C) T-GM-CSF and -IL-3 have high receptor binding affinity and compete with FL-GM-CSF and -IL-3 for receptor binding in primary CB and AML cells
To further identify potential mechanistic interactions of T- vs. FL-GM-CSF and -IL-3 at a receptor level, receptor equilibrium binding studies were performed (Figure 5). We first utilized the TF-1 cell line stimulated with rhFL- and T-GM-CSF and -IL-3 that was I125 labeled or unlabeled. Both high and low affinity binding sites for FL- and T-GM-CSF and -IL-3 were detected by Scatchard analysis (Figure 5). By calculating dissociation constants (Kd), we determined that T-GM-CSF and T-IL-3 bind with higher affinity than their FL-counterparts, and both truncated forms are better competitors for receptor binding than FL-GM-CSF and -IL-3 (Figure 5A). We also noted, through binding competition studies using pooled CD34+ CB cells (Figure 5B) and multiple primary AML samples (Figure 5C), that the T-GM-CSF and T-IL-3 reciprocally competed with and blunted the receptor binding of both their own, as well as the other FL-CSFs.
Figure 5. T-IL-3 and T-GM-CSF bind with greater affinity than FL-IL-3 or FL-GM-CSF, and T-IL-3 and T-GM-CSF blunt receptor binding of both FL-IL-3 and FL-GM-CSF.
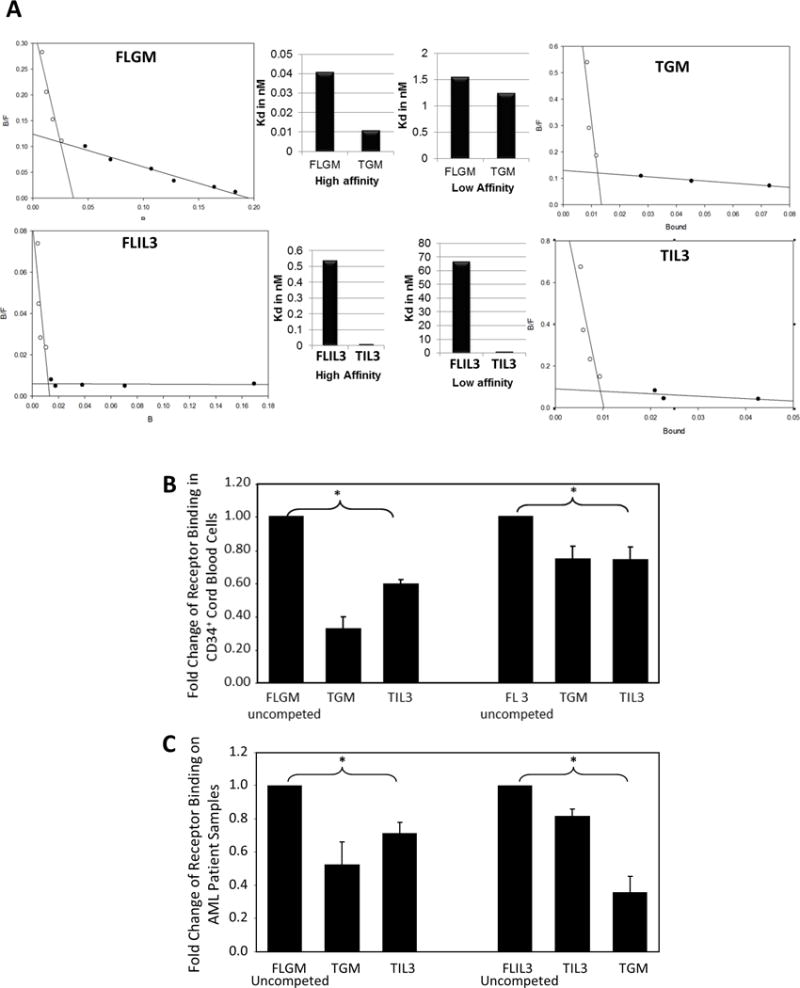
(A) Scatchard analysis and Kd determination of TF-1(n=2) was performed with I125 labeled IL-3. 1×106 TF-1 cells were plated per well, and iodinated full length or truncated IL-3 was added for one hour. Cells were harvested and levels of I125 labeled ligand bound were detected using a Beckman Coulter Gamma 5500B and low and high affinity binding sites, as well as the dissociation constant (Kd) were derived as previously published.(3) Pooled CD34+ CB (B) or primary AML patient samples (C) were incubated with I125 labeled FL-IL-3 or FL-GM-CSF alone or in addition to cold T-IL-3 and T-GM-CSF in excess for 1 hour. Cells were harvested and the CPM (or levels of I125 labeled ligand bound) were detected using a Beckman Coulter Gamma 5500B to analyze the ability of T-IL-3 and T-GM-CSF to compete with FL-IL-3 and FL-GM-CSF. * = p ≤ 0.05 AML patient samples n= 7 distinct samples; CB n= 5 independent experiments with CD34+ cells pulled from multiple cord bloods for each experiment.
Discussion
Our studies have now demonstrated that DPP4 exists in an active form on the cell surface of murine as well as immature normal and malignant human cells, and that DPP4 T-GM-CSF and -IL-3 have enhanced receptor binding as well as modified HPC functional regulatory activity compared to their FL counterparts both in vitro and in vivo for both normal and leukemia progenitors/precursor cells. These results allude to the importance of DPP4, via its regulation of normal and malignant cells by acting as a scaffolding molecule and truncating physiologically and pathologically relevant proteins, thus adding another layer of interactions and complexity to growth modulatory factors and their role in the BM microenvironment. Alterations in receptor binding and signaling after DPP4 truncation is likely not only important for the effects seen on normal and leukemic cells in the settings of GM-CSF and IL-3 that we have elucidated here, but also suggests the possibility for broader relevance for the many factors we have recently noted to have DPP4 putative or confirmed T sites1–3 and potentially for other proteins with DPP4 T-sites that have not yet been identified. This highlights the critical need to understand how DPP4 T alters receptor binding of individual proteins and how it may alter their ability to act as a negative or positive regulator on normal and malignant hematopoiesis, as well as other steady- and disease-states outside of hematopoiesis.1–3 Both T-GM-CSF and -IL-3 diminished receptor binding and function of both FL-GM-CSF and -IL-3, at less than a 1:1 ratio, likely through their shared common β chain receptor. Since DPP4 may play a more active role in hematopoiesis under stress conditions,3 a role for T- proteins in regulation of hematopoiesis may be even more apparent under stress.
DPP4 specific regulation of GM-CSF and IL-3 via its truncation, and in general as a modulator of CSF signaling and consequently alterations in hematopoietic function, may have potential clinical application. Patients with leukemia, may respond to T-factors regardless of molecular alterations or may have altered sensitivity to DPP4 T-factors based on leukemia type, similar to effects seen in our sampling of AML patient cells and the two in vivo models of leukemia assessed in our present studies. The knowledge we have gained in this research can possibly be used for investigation of the potential use of DPP4 T- proteins for future treatment of leukemia patients. This will require further mechanistic and therapeutic insight into the actions of T- vs FL- proteins in terms of what overlapping and distinct intracellular signals they elicit, and more in depth analysis of in vivo mouse models of leukemia to determine the effects on disease progression/relapse and animal survival.
Importantly, current assessment of protein levels, for the most part, do not distinguish between the FL- and DPP4 -T forms of these proteins and hence assessments of proteins by conventional methods such as ELISA, bioplex, and other methods that use antibodies that do not distinguish the FL- from the T- forms of the proteins therefore may not be fully telling in terms of the specific physiology or biological activities associated with these molecules. To that end, the possibility exists that functional outcomes that have been previously attributed to FL- molecules may, in fact, actually be due to T- molecules or a mixture of activities between the FL- and T- molecules. Thus, efforts to develop antibodies that can distinguish DPP4 T- from FL-proteins may be of practical, scientific, and potentially clinical value.
Supplementary Material
Key Points.
DPP4 truncated (T) GM-CSF & IL-3 reciprocally bind with higher affinity to both GM-CSF & IL-3 receptors compared to their full length (FL) forms.
DPP4 truncated (T) GM-CSF & IL-3 reciprocally blunt the activities of both full length (FL) GM-CSF & IL-3 in vitro and in vivo in primary AML patient samples and mouse models of normal/malignant hematopoiesis.
Acknowledgments
These studies were supported by (1) U.S. Public Health Service Grants from the National Institutes of Health: R01HL112669, R01HL67384, R01HL56416, P01DK90948, and U54DK106846 to H.E.B. and (2) Indiana University Bioinformatics Core Proposal Grant to H.A.O. H.A.O. and MC were supported by NIH T32DK07519 to H.E.B. We thank Giao Hangoc for help with some of the experiments and Heath D. Skinner for clinical insight.
Footnotes
Authorship Contributions: H.A.O. provided concepts, designed and performed experiments and wrote the manuscript. C.M., S.C., and M.C. performed experiments, H.S.B. provided clinical samples/knowledge, R.K., B.R., R.C., L.D., and C.K.Q. provided mouse models of leukemia and insight into their use. H.E.B. provided concepts, designed and performed experiments and helped in writing and editing of the manuscript. All authors read the manuscript and provided feedback for clarity and context.
Conflict of Interest Disclosure: HEB is a member of the Medical Scientific Advisory Board (MSAB) of Corduse, a cord blood banking company.
References
- 1.Ou X, O’Leary HA, Broxmeyer HE. Implications of DPP4 modification of proteins that regulate stem/progenitor and more mature cell types. Blood. 2013;122(2):161–9. doi: 10.1182/blood-2013-02-487470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Leary H, Ou X, Broxmeyer HE. The role of dipeptidyl peptidase 4 in hematopoiesis and transplantation. Current opinion in hematology. 2013;20(4):314–9. doi: 10.1097/MOH.0b013e32836125ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broxmeyer HE, Hoggatt J, O’Leary HA, Mantel C, Chitteti BR, Cooper S, et al. Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nature medicine. 2012;18(12):1786–96. doi: 10.1038/nm.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Favreau AJ, Sathyanarayana P. miR-590-5p, miR-219-5p, miR-15b and miR-628-5p are commonly regulated by IL-3, GM-CSF and G-CSF in acute myeloid leukemia. Leukemia research. 2012;36(3):334–41. doi: 10.1016/j.leukres.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller PH, Cheung AM, Beer PA, Knapp DJ, Dhillon K, Rabu G, et al. Enhanced normal short-term human myelopoiesis in mice engineered to express human-specific myeloid growth factors. Blood. 2013;121(5):e1–4. doi: 10.1182/blood-2012-09-456566. [DOI] [PubMed] [Google Scholar]
- 6.Riccioni R, Diverio D, Riti V, Buffolino S, Mariani G, Boe A, et al. Interleukin (IL)-3/granulocyte macrophage-colony stimulating factor/IL-5 receptor alpha and beta chains are preferentially expressed in acute myeloid leukaemias with mutated FMS-related tyrosine kinase 3 receptor. British journal of haematology. 2009;144(3):376–87. doi: 10.1111/j.1365-2141.2008.07491.x. [DOI] [PubMed] [Google Scholar]
- 7.Schiller GJ. High-risk acute myelogenous leukemia: treatment today … and tomorrow. Hematology/the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2013;2013:201–8. doi: 10.1182/asheducation-2013.1.201. [DOI] [PubMed] [Google Scholar]
- 8.Thomas D, Powell JA, Green BD, Barry EF, Ma Y, Woodcock J, et al. Protein kinase activity of phosphoinositide 3-kinase regulates cytokine-dependent cell survival. PLoS biology. 2013;11(3):e1001515. doi: 10.1371/journal.pbio.1001515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broughton SE, Dhagat U, Hercus TR, Nero TL, Grimbaldeston MA, Bonder CS, et al. The GM-CSF/IL-3/IL-5 cytokine receptor family: from ligand recognition to initiation of signaling. Immunological reviews. 2012;250(1):277–302. doi: 10.1111/j.1600-065X.2012.01164.x. [DOI] [PubMed] [Google Scholar]
- 10.Onetto-Pothier N, Aumont N, Haman A, Park L, Clark SC, De Lean A, et al. IL-3 inhibits the binding of GM-CSF to AML blasts, but the two cytokines act synergistically in supporting blast proliferation. Leukemia. 1990;4(5):329–36. [PubMed] [Google Scholar]
- 11.Taketazu F, Chiba S, Shibuya K, Kuwaki T, Tsumura H, Miyazono K, et al. IL-3 specifically inhibits GM-CSF binding to the higher affinity receptor. Journal of cellular physiology. 1991;146(2):251–7. doi: 10.1002/jcp.1041460209. [DOI] [PubMed] [Google Scholar]
- 12.Broxmeyer HE, Williams DE, Cooper S, Shadduck RK, Gillis S, Waheed A, et al. Comparative effects in vivo of recombinant murine interleukin 3, natural murine colony-stimulating factor-1, and recombinant murine granulocyte-macrophage colony-stimulating factor on myelopoiesis in mice. The Journal of clinical investigation. 1987;79(3):721–30. doi: 10.1172/JCI112877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broxmeyer HE, Williams DE, Hangoc G, Cooper S, Gillis S, Shadduck RK, et al. Synergistic myelopoietic actions in vivo after administration to mice of combinations of purified natural murine colony-stimulating factor 1, recombinant murine interleukin 3, and recombinant murine granulocyte/macrophage colony-stimulating factor. Proc Natl Acad Sci U S A. 1987;84(11):3871–5. doi: 10.1073/pnas.84.11.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams DE, Straneva JE, Cooper S, Shadduck RK, Waheed A, Gillis S, et al. Interactions between purified murine colony-stimulating factors (natural CSF-1, recombinant GM-CSF, and recombinant IL-3) on the in vitro proliferation of purified murine granulocyte-macrophage progenitor cells. Experimental hematology. 1987;15(10):1007–12. [PubMed] [Google Scholar]
- 15.Williams DE, Straneva JE, Shen RN, Broxmeyer HE. Purification of murine bone-marrow-derived granulocyte-macrophage colony-forming cells. Experimental hematology. 1987;15(3):243–50. [PubMed] [Google Scholar]
- 16.Elbaz O, Shaltout A. Implication of Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) and Interleukin-3 (IL-3) in Children with Acute Myeloid Leukaemia (AML); Malignancy. Hematology. 2001;5(5):383–8. [PubMed] [Google Scholar]
- 17.Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–84. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 18.Christopherson KW, 2nd, Hangoc G, Mantel CR, Broxmeyer HE. Modulation of hematopoietic stem cell homing and engraftment by CD26. Science. 2004;305(5686):1000–3. doi: 10.1126/science.1097071. [DOI] [PubMed] [Google Scholar]
- 19.Lee BH, Tothova Z, Levine RL, Anderson K, Buza-Vidas N, Cullen DE, et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. 2007;12(4):367–80. doi: 10.1016/j.ccr.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. The Journal of experimental medicine. 2011;208(10):1977–88. doi: 10.1084/jem.20110450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. The Journal of experimental medicine. 2005;201(8):1307–18. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gotoh A, Takahira H, Mantel C, Litz-Jackson S, Boswell HS, Broxmeyer HE. Steel factor induces serine phosphorylation of Stat3 in human growth factor-dependent myeloid cell lines. Blood. 1996;88(1):138–45. [PubMed] [Google Scholar]
- 23.Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, et al. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. Journal of cellular physiology. 1989;140(2):323–34. doi: 10.1002/jcp.1041400219. [DOI] [PubMed] [Google Scholar]
- 24.Broxmeyer HE, Grossbard E, Jacobsen N, Moore MA. Evidence for a proliferative advantage of human leukemia colony-forming cells in vitro. J Natl Cancer Inst. 1978;60(3):513–21. doi: 10.1093/jnci/60.3.513. [DOI] [PubMed] [Google Scholar]
- 25.Broxmeyer HE, Grossbard E, Jacobsen N, Moore MA. Persistence of inhibitory activity against normal bone-marrow cells during remission of acute leukemia. N Engl J Med. 1979;301(7):346–51. doi: 10.1056/NEJM197908163010702. [DOI] [PubMed] [Google Scholar]
- 26.Moore MA, Spitzer G, Williams N, Metcalf D, Buckley J. Agar culture studies in 127 cases of untreated acute leukemia: the prognostic value of reclassification of leukemia according to in vitro growth characteristics. Blood. 1974;44(1):1–18. [PubMed] [Google Scholar]
- 27.Broxmeyer HE, Lee MR, Hangoc G, Cooper S, Prasain N, Kim YJ, et al. Hematopoietic stem/progenitor cells, generation of induced pluripotent stem cells, and isolation of endothelial progenitors from 21- to 23.5-year cryopreserved cord blood. Blood. 2011;117(18):4773–7. doi: 10.1182/blood-2011-01-330514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christopherson KW, 2nd, Hangoc G, Broxmeyer HE. Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34+ progenitor cells. Journal of immunology. 2002;169(12):7000–8. doi: 10.4049/jimmunol.169.12.7000. [DOI] [PubMed] [Google Scholar]
- 29.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–36. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Niu L, Golde DW, Vera JC, Heaney ML. Kinetic resolution of two mechanisms for high-affinity granulocyte-macrophage colony-stimulating factor binding to its receptor. Blood. 1999;94(11):3748–53. [PubMed] [Google Scholar]
- 31.Woodcock JM, Bagley CJ, Zacharakis B, Lopez AF. A single tyrosine residue in the membrane-proximal domain of the granulocyte-macrophage colony-stimulating factor, interleukin (IL)-3, and IL-5 receptor common beta-chain is necessary and sufficient for high affinity binding and signaling by all three ligands. The Journal of biological chemistry. 1996;271(42):25999–6006. doi: 10.1074/jbc.271.42.25999. [DOI] [PubMed] [Google Scholar]
- 32.Lemoli RM, Gulati SC, Strife A, Lambek C, Perez A, Clarkson BD. Proliferative response of human acute myeloid leukemia cells and normal marrow enriched progenitor cells to human recombinant growth factors IL-3, GM-CSF and G-CSF alone and in combination. Leukemia. 1991;5(5):386–91. [PubMed] [Google Scholar]
- 33.Feuring-Buske M, Gerhard B, Cashman J, Humphries RK, Eaves CJ, Hogge DE. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia. 2003;17(4):760–3. doi: 10.1038/sj.leu.2402882. [DOI] [PubMed] [Google Scholar]
- 34.Vellenga E, Ostapovicz D, O’Rourke B, Griffin JD. Effects of recombinant IL-3, GM-CSF, and G-CSF on proliferation of leukemic clonogenic cells in short-term and long-term cultures. Leukemia. 1987;1(8):584–9. [PubMed] [Google Scholar]
- 35.Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24(10):1785–8. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.