

Abstract
Although the early outcome of acute coronary syndrome (ACS) has considerably improved in the last decade, cardiovascular diseases still represent the main cause of morbidity and mortality worldwide. This is mainly due to the fact that recurrence of ACS eventually leads to the pandemics of heart failure and sudden cardiac death, thus calling for a reappraisal of the mechanisms responsible for coronary instability. In this review, we will discuss recent advances in our understanding of how adaptive immunity contributes to the pathogenesis of ACS, as well as the clinical implications that arise from these new pathogenic concepts.
Keywords: outcome, pathogenesis, signaling, treatment
Introduction
In patients with acute coronary syndromes (ACS) and systemic evidence of inflammation, the higher frequency of activated T-cells compared with stable angina (1–4) suggests that the sudden changes leading to coronary instability might be related to mechanisms involving T-cell immunity. In this review, we will discuss recent advances in our understanding of how adaptive immunity contributes to the pathogenesis of ACS.
Helper T-cell dysregulation in ACS
Helper T-cells (CD4+ lymphocytes) are the key regulators of adaptive immunity. Following T-cell receptor (TCR) activation by antigen-presenting cells (APCs), T-cells differentiate into functionally polarized helper T-cells, classified according to cytokine production, surface markers, and the expression of lineage-specifying transcription factors. The cytokine environment controls the induction of different T-cell subsets by the amount of antigen and by TCR-mediated signal strength (5,6). The principal helper T-cell subsets are the Th1, Th2, Th17, and follicular helper (Tfh) CD4+ T-cells. Moreover, a subpopulation of Th1 cells, characterized by defective cell surface expression of the costimulatory molecule CD28, is expanded in different chronic inflammatory conditions (7). Emerging subsets of helper T-cells are Th22 (characterized by interleukin [IL]-22 production) and Th9 (producing IL-9) cells (6), but their role in ACS is still uncertain.
ACS patients have a skewed T-cell differentiation, oriented towards aggressive effector phenotypes and defective regulatory T-cells, the lymphocyte compartment able to suppress the excessive immune response (Table 1). Overall, such T-cell abnormalities characterize about half of ACS patients (8). In this subset of ACS patients, helper T-cell dysregulation might affect the biological outcome of the immune response and contribute to plaque destabilization through multiple damaging pathways (Figure 1).
Table 1.
Dysregulation of Helper T-cell Subsets in ACS Patients
| Helper T-cell Subpopulation |
Type of Dysregulation | Study Populations | Experimental Technique(s) |
References |
|---|---|---|---|---|
| Th1 | Increased frequency* | NSTEMI, STEMI, UA, SA, HC | Flow cytometry | 9–13 |
| Increased IFN-γ production* | NSTEMI, STEMI, UA, SA, HC | Flow cytometry, ELISA, real-time PCR | 9–13 | |
| Increased expression of STAT-4 and T-bet* | NSTEMI, STEMI, UA, SA, HC | Flow cytometry, western blot, real-time PCR | 9,11 | |
| CD28null | Increased frequency*, | NSTEMI, UA, SA, HC | Flow cytometry | 8,16–18 |
| Increased cytotoxicity*,y | UA, STEMI, NSTEMI, SA | Cytotoxicity assay, flow cytometry | 15,18 | |
| Increased resistance to apoptosis*,y | NSTEMI, STEMI, SA | Flow cytometry, proliferation and apoptosis assay | 19 | |
| Th17 | Increased frequency* | NSTEMI, AMI, UA, SA, HC | Flow cytometry | 8,22,23 |
| Increased IL-17 production* | NSTEMI, AMI, UA, SA, HC | Flow cytometry, ELISA | 22,23 | |
| Increased STAT-3 phosphorylation* | NSTEMI, AMI, UA, SA, HC | Flow cytometry, western blot | 23 | |
| Treg | Reduced frequency* | NSTEMI, STEMI, AMI, UA, SA, HC | Flow cytometry, western blot, real-time PCR, multiplex technology | 8,26–29 |
| Reduced immunosuppressive efficiency*,y | NSTEMI, STEMI, SA, HC | Functional suppression assays | 26 | |
| Increases susceptibility to apoptosis*,y | NSTEMI, SA, CPS | Apoptosis assay | 29 | |
| Reduced TCR-induced generation*,y | NSTEMI, SA, HC | Stimulation assay, flow cytometry | 36 |
All evidence comes from observational studies; some are ex vivo, others are in vitro.
Ex vivo observational study in cohorts
In vitro study
AMI = acute myocardial infarction; CPS = chest pain syndrome; ELISA = enzyme-linked immunosorbent assay; HC = healthy controls; IFN = interferon; NSTEMI = non-ST-segment elevation myocardial infarction; PCR = polymerase chain reaction; SA = stable angina; STEMI = ST-segment elevation myocardial infarction; TCR = T-cell receptor; UA= unstable angina
Figure 1. Helper T-cell Dysregulation in ACS.

Although no direct evidence exists for some of the pathways proposed, it could be hypothesized that the proinflammatory environment in ACS patients, due to the skewed CD4+ T-cell differentiation oriented towards an aggressive phenotype, may induce a positive feed-forward loop, causing inflammation and local T-cell recruitment that could damage atherosclerotic plaque through multiple pathways. ACS = acute coronary syndrome; APC = antigen-presenting cell; EC = endothelial cell; IFN-g = interferon-gamma; IL = interleukin; SMC = smooth muscle cell; TGF-b = transforming growth factor beta; TNF-α = tumor necrosis factor alpha; Treg = regulatory T-cell.
Th1 cells
Th1 cells are characterized by the production of interferon-gamma (IFN-γ) and by the expression of the transcription factor T-bet. They contribute to tissue destruction and participate in the pathophysiology of autoimmune diseases, such as rheumatoid arthritis (RA) and inflammatory vascular diseases (6). The importance of Th1 and IFN-γ in atherosclerosis progression is well described in animal models (2), whereas their role in plaque destabilization is suggested by numerous studies, showing a marked increase in Th1 frequency and increased expression of Th1-related effectors, such as IFN-γ, signal transducer and activator of transcription (STAT)-4, and T-bet in patients with ACS (9–13). IFN-γ can contribute to plaque destabilization in several ways: by the recruitment and activation of macrophages in the atherosclerotic lesions; by reducing collagen synthesis; by increasing the production of extracellular matrix-degrading proteins; and by activating APCs (14). Furthermore, the increased IFN-γ expression induces a positive feed-forward loop of Th1 induction, keeping a proinflammatory state.
When activated in the intima, Th1 cells produce proinflammatory cytokines and enhance the expression of CD40 ligand. Ligation of CD40 on APCs by CD40 ligand induces release of extracellular matrix-degrading metalloproteinases and the expression of tissue factor, the key initiator of the coagulation cascade, which has been well documented in experimental models (14).
CD4+CD28null T-cells are distinct from classic helper T-cells in several respects. This terminally differentiated subpopulation shows an increased resistance to apoptosis and a wide range of proinflammatory properties (7). The proinflammatory functions of CD4+CD28null T-cells include the production of high levels of IFN-γ, tumor necrosis factor-alpha (TNF-α), and IL-2 (9) to direct cytotoxic ability (15). Indeed, they express killer immunoglobulin (Ig)-like receptors and directly kill endothelial cells in vitro by cytolytic enzymes, such as perforin, granzyme A, and granzyme B, that are usually present in killer T-cells and natural killer (NK) cells. Several studies showed the association of CD4+CD28null T-cells with infections and chronic inflammatory diseases, particularly rheumatoid arthritis. CD4+CD28null T-cells are present preferentially in unstable ruptured atherosclerotic plaques (4), and their frequency significantly increases the risk of ACS (8,16), particularly in patients with diabetes (17).
In ACS patients, CD4+CD28null T-cells circulating in the peripheral blood and infiltrating the unstable atherosclerotic plaque show high levels of the costimulatory receptors OX40 and 4-1BB, which mediate IFN-γ and TNF-α production, and modulate cell degranulation (18), as well as increased resistance to apoptosis (19). Overall, these molecular abnormalities could enhance the inflammatory and cytotoxic function of CD4+CD28null T-cells, and could contribute to atherosclerotic plaque destabilization. One study failed to replicate the differences in CD4+CD28null T-cells from ACS and controls in patients treated with high-dose statins (20).
Th17 cells
Th17 cells are characterized by the expression of retinoid-related orphan receptor (ROR)γt, the master regulator transcription factor responsible for IL-17 production. This T-cell subset, implicated in several autoimmune disorders, is important in the immune responses against fungi and extracellular bacteria (6). The precise role of IL-17 in atherosclerosis and ACS remains controversial, as discussed in detail in a recent editorial (21). On the one hand, experimental studies in mouse models have provided direct evidence that IL-17 is predominantly proatherogenic; on the other hand, these cells promote procollagen expression, which might reduce the risk of fibrous cap fissure (22).
Similarly, in the clinical setting, the Th17 subset is expanded in ACS patients (8,23), and the Th17/regulatory T-cell (Treg) balance might play a role in the development of inflammatory disorders, including atherosclerosis, and autoimmune diseases; however, in a cohort of ACS patients, lower serum levels of IL-17 were associated with a higher risk of major cardiovascular events (24). In conclusion, the issue of whether Th17 cells are protective or detrimental in ACS remains unsettled.
Tregs
Tregs are a subset of CD4+ T-cells able to suppress the immune response. These lymphocytes are characterized by expression of the transcription factor Foxp3, by high levels of the IL2 receptor CD25, and by down-regulation of the surface molecule CD127 (6). Tregs induce immunosuppression through multiple mechanisms, including production of the anti-inflammatory cytokines IL-10 and transforming growth factor-beta (TGF-β), direct cytolysis, and inhibition of dendritic cell (DC) maturation and function (6). In experimental atherosclerosis, the role of Tregs is well appreciated. Indeed, this T-cell subpopulation inhibits atherosclerosis development and progression by suppressing effector T-cell responses (25). Recently published data have consistently demonstrated a defective Treg compartment in the peripheral blood of ACS patients. ACS patients show low levels of circulating Tregs, a reduced suppressive efficiency of Tregs, and increased Treg susceptibility to apoptosis (26–29).
B cells
The contribution of B cells in autoimmunity is well recognized, and their function has been evaluated in various animal models of atherosclerosis (30). B1a cells and their secretion of IgM seem to mediate the protective effects of B cells, whereas B2 cells are proatherogenic through modulation of T-cell dependent mechanisms. Consistent with this hypothesis, the lack of B cell-activating factor receptor (BAFFR), which is critical for maintaining mature B2 cells, substantially mitigates atherosclerosis in LDL receptor-deficient (Ldlr−/−) mice by altering mature B2 cell-dependent cellular and humoral immune responses, while preserving the B1a cell subtype and production of natural IgM antibodies (31). The discovery of a new B cell subtype, the regulatory B cell (Breg), which secretes IL-10 and TGFβ, and affects Treg development, has revealed that inhibition of anti-inflammatory activity, is another mechanism by which B cells can influence autoimmunity and thereby atherosclerosis. Moreover, antibodies against several oxidation-specific epitopes of low-density lipoprotein (LDL) and other atherosclerosis-related antigens are found in animals and humans, both in the circulation and in atherosclerotic plaques, and are also found in ACS patients (32). Although the mechanisms by which these antibodies act remain unclear, many experimental atherosclerosis studies have shown that vaccination is protective, and that the protective effects of vaccination can be mediated by the induction of protective Treg responses and protective antibodies (33).
TCR signaling alterations in ACS
The first step in lymphocyte activation is TCR binding with specific peptides presented by APCs. TCR triggering initiates a cascade of phosphorylation events, culminating in the activation and nuclear translocation of transcription factors. A complex molecular coordination is required, including positive and negative feedback loops necessary to avoid lymphocyte hyper-reactivity and the breaking of immune tolerance. The proper tuning of TCR signaling is also critical for T-cell differentiation. Indeed, in addition to the cytokine environment, the induction of different T helper cell lineages is driven by TCR-mediated signal strength (5). In patients with ACS, several abnormalities of TCR signaling have been identified (Table 2 and Figure 2).
Table 2.
TCR Signaling Alteration in ACS Patients
| TCR molecular pathway |
Type of Alteration | Study Populatio ns |
Experimental Technique(s) |
References |
|---|---|---|---|---|
| CD3 | Increased accumulation in IS* | ACS, CAD, HC | Confocal microscopy | 34,35,41 |
| Intracellular [Ca2+] signals | Enhanced* | ACS, CAD, HC | Confocal microscopy | 34,35 |
| Phosphotyrosine levels | Enhanced* | ACS, SA, NSTEMI, CAD, HC | Pflow experiments, confocal microscopy | 35,36 |
| Zap70 | ACS, SA, NSTEMI, CAD, HC | Pflow experiments, confocal microscopy, ELISA | 34–36, 41 | |
| LCK | Insufficient deactivation* | ACS, CAD, HC | Pflow experiments, confocal microscopy | 35 |
| MAPKs | NSTEMI, SA, HC | Pflow experiments | 41 | |
| STATs | AMI, SA, HC | Pflow experiments | 23 | |
| CREB | NSTEMI, SA, HC | Pflow experiments, immunofluorescence microscopy, ChIP assay | 36 | |
| CD31 | Reduced expression and signaling during acute phase*,y | NSTEMI, SA, HC | Flow cytometry, Pflow experiments | 41 |
| PTPN22 | Increased expression*,y | NSTEMI, SA, HC | Flow cytometry, realtime PCR | 36 |
All evidence comes from observational studies, some in vitro, and others ex-vivo.
In vitro study
Ex vivo observational study in cohorts
ACS = acute coronary syndrome; CAD = coronary artery disease; ChIP = chromatin immunoprecipitation; CREB = cyclic adenosine monophosphate response element-binding protein; ERK = extracellular signal-related kinase; IS = immunological synapse; LCK = lymphocyte-specific protein tyrosine kinase; MAPK= mitogen-activated protein kinase; Pflow = phosphoflow cytometry; PTPN22 = protein tyrosine phosphatase nonreceptor type 22; STAT = signal transducer and activator of transcription; Zap70 = zeta-chain-associated protein kinase of 70 kD. Other abbreviations as in Table 1.
Figure 2. Schematic Outline of TCR Signaling Pathway Abnormalities Observed in ACS.

Proper tuning of TCR activation is critical for the functional regulation of helper T-cells, including cytokine production, lymphocyte differentiation, clonal expansion, and cytotoxic effector functions. The biochemical signal initiated by TCR triggering is dynamically regulated by protein tyrosine kinases and phosphatases. Fine molecular coordination is required to maintain the feedback loop necessary to avoid lymphocyte hyper-reactivity and the breaking of immune tolerance. ACS patients have excessive amplification of proximal TCR-mediated signals and unbalanced downstream molecular phosphorylation. CREB = cyclic adenosine monophosphate response element-binding protein; LCK= lymphocyte-specific protein tyrosine kinase; MAPK= mitogen-activated protein kinase; PTPN22 = protein tyrosine phosphatase nonreceptor type 22; STAT = signal transducer and activator of transcription; TCR = T-cell receptor; Zap70 = zeta-chain-associated protein kinase of 70 kD. Other abbreviations as in Figure 1.
CD4+ T-cells from ACS patients show an enhanced response to TCR stimulation (34–36) and have a lower setting of the T-cell activation threshold, attributable to enhanced amplification of proximal TCR-mediated signals. CD4+ T-cells show increased accumulation of CD3 complexes and zeta-chain-associated protein kinase of 70 kD (Zap70) in the immunological synapse, higher early tyrosine phosphorylation after TCR stimulation, and defective deactivation of the lymphocyte-specific protein tyrosine kinase (LCK) (34–36). These abnormalities involving the upstream TCR signaling pathway strongly contribute to the higher cytotoxic ability of helper T-cells in ACS patients; indeed, CD4+ T-cells from ACS patients are able to kill endothelial and vascular smooth muscle cells in vitro (15,34).
Recent studies suggest a defect in the protective signals necessary for appropriate T-cell activation. Indeed, CD4+ T-cells from ACS patients exhibit reduced phosphorylation of Zap70 at its inhibitory residue Tyr-292 (36). This residue is implicated in TCR/CD3 complex internalization, immunological synapse formation, and down-modulation of the proximal events in TCR signaling. ACS patients show also enhanced expression of the protein tyrosine phosphatase nonreceptor type 22 (PTPN22). This enzyme plays a key role in controlling the intensity of early TCR signal transduction acting on LCK and Zap70 (37). Although PTPN22 does not bind Tyr-292 of Zap70 directly, PTPN22 may be brought to phospho-Tyr-292 through binding of the SH2 domain of the c-Cbl ubiquitin ligase complex. Thus, the reduced Zap70 Tyr-292 phosphorylation might be indirectly due to increased expression of PTPN22 by a c-Cbl-mediated mechanism. Defective regulation of TCR signaling and increased PTPN22 expression persist in ACS patients 1 year after the index event, during a stable phase of the disease, suggesting persistent lymphocyte hyper-reactivity (36).
CD31 is a member of the Ig superfamily of cell adhesion molecules expressed on most cells of the hematopoietic lineage, including platelets, monocytes, neutrophils, and a small population of T-cells. It plays an important role in the inflammatory response through the modulation of leukocyte activation, cytokine production, and the maintenance of vascular barrier integrity. CD31 is involved in TCR-signaling immunomodulation by reducing Zap70 phosphorylation through the action of protein tyrosine phosphatases (38). Accordingly, loss of Ig-like domains 1 to 5 of CD31 prevents homophilic binding interactions and consequent activation of the CD31 immunoreceptor tyrosine-based inhibition motif (ITIM)/SHP2 inhibitory pathway, finally leading to uncontrolled T-cell activation. CD31 has been implicated also in the development of atherosclerosis in animal models (39) and of its clinical complications in humans (40). ACS patients exhibit a transient reduction of CD31 expression and function on CD4+ T-cells (41). In 1 study, the expression and function of CD31 on CD4+ T-cell subsets was higher in stable angina (SA) patients compared with controls and in ACS patients during the acute phase of the disease. Of note, after 1 year of follow-up, CD31 expression and function recovered in ACS, becoming similar to that observed in SA patients and higher than that in healthy controls. A similar pattern has been observed in the CD14+CD16+ monocyte subset (42). Taken together, these data suggest an enhanced CD31-mediated protective mechanism operating in SA patients, which is transiently down-regulated in ACS patients during the acute phase of the disease. Thus, CD31 might have a role in containing the immune response in patients with SA. In contrast, in patients with ACS, the protective function of CD31 is reduced during the acute phase of the disease, thus leading to the uncontrolled lymphocyte activation typically observed in a subset of ACS patients.
A critical downstream molecule activated by TCR triggering is the cyclic adenosine monophosphate response element-binding protein (CREB), a transcription factor believed to be particularly important for the generation and maintenance of Tregs, and for IL-2 and IL-10 production. CD4+ T-cells of ACS patients show reduced activation of CREB during the acute phase of the disease compared with SA patients and controls, which improves during follow-up (36). The observed transient expression of CD31 during ACS might explain the reduced activation of CREB. Indeed, the CD31-ITIM–SH2 phosphatase signaling pathway elicits the activity of extracellular signal-related kinase (ERK), which is involved also in CREB activation. In summary, the transiently reduced expression of CD31 in the acute setting of ACS might contribute to reduced CREB activation, although CREB phosphorylation is regulated by several stimuli, in addition to TCR triggering.
Recently, the importance of the TCR signal strength in the differentiation of helper T-cell subsets has increasingly been defined (5). A lower setting of the T-cell activation threshold could affect the direction of T-cell differentiation, leading to the unbalanced expansion of the helper T-cell subset observed in ACS patients. Indeed, strong TCR signaling in the early stages of lymphocyte activation promotes Th1 and Th17 differentiation, and negatively regulates Treg induction (5).
Lessons from autoimmune diseases
An increasing body of evidence supports the notion that atherosclerosis shares surprising similarities with other inflammatory/autoimmune diseases. Chronic autoimmune disorders, such as systemic lupus erythematosus (SLE) and RA, characterized by chronic relapsing-remitting systemic inflammation, are recognized as risk factors for accelerated atherosclerosis. Therapies directed towards inflammatory process crucially reduce cardiovascular disease morbidity and mortality in SLE and RA patients (Figure 3). Furthermore, RA and other inflammatory diseases are associated with increased ACS incidence and poorer outcomes (43).
Figure 3. Lessons From Autoimmune Diseases.
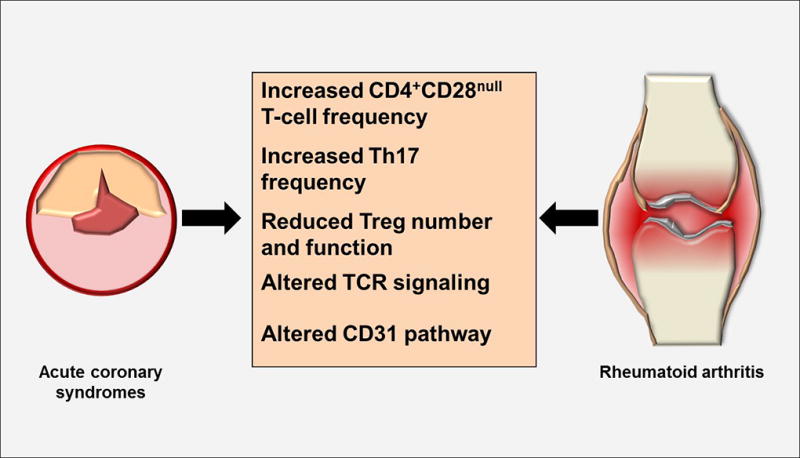
ACS shares surprising similarities with inflammatory/autoimmune diseases, such as rheumatoid arthritis (RA), characterized by chronic relapsing-remitting systemic inflammation. In particular, helper T-cell dysregulation and TCR signaling alterations characterize both ACS and RA. TCR = T cell receptor. Abbreviations as in Figures 1 and 2.
Helper T-cell dysregulation
It is tempting to speculate that CD4+CD28null T-cells are likely candidates for driving the accelerated atherosclerosis process observed in autoimmune disorders. Indeed, RA patients with high frequencies of CD4+CD28null T-cells have increased carotid artery intima-media thickness (IMT) and decreased flow-mediated vasodilatation (FMD) compared with RA patients in whom this T-cell subset does not expand. The mechanisms involved in CD4+CD28null T-cell expansion remain poorly understood. It has been suggested that CD28 down-regulation is antigen-driven (44). An alternative hypothesis implicates inflammatory cytokines in CD4+CD28null T-cell expansion, as CD4+CD28null T-cell clones from RA patients down-regulated CD28 transcription following TNF-α treatment in vitro (45).
A large body of evidence supports the concept that an imbalance between Treg and Th17 cells is a crucial aspect in the pathogenesis of RA. Although often contradictory, most studies agree that there is an overall depletion of Tregs and a parallel increase of Th17 cells in the peripheral blood and target organs in RA patients. In addition, intrinsic cell abnormalities involving genetic and epigenetic modifications may explain the defective suppressive activity of Tregs in RA.
TCR signaling alterations
Abnormalities in proximal TCR signaling have been implicated in autoimmunity, malignancy, and immunodeficiency. T-cell responsiveness in type 1 diabetes, SLE, and RA have been found to be abnormal, with a multitude of mechanisms involved, ranging from alterations in TCR/CD3 complex composition to excessive expression of costimulatory receptors (46).
A key role in controlling the intensity of TCR activation is played by protein phosphatases, signal transducing enzymes that dephosphorylate cellular phosphoproteins. PTPN22 is an interesting enzyme because genetic variants are associated with an increased risk for several chronic inflammatory diseases; it is therefore considered an important non-human leukocyte antigen (non-HLA) autoimmunity gene. Interestingly, the R620W polymorphic variant of PTPN22 has recently emerged as a major risk factor for the development of several autoimmune diseases, including type 1 diabetes mellitus, RA, and SLE, suggesting that aberrant activity of this phosphatase can perturb normal lymphocyte functions and homeostasis (37), although it has not been specifically investigated in ACS.
The Ig-like immunomodulatory molecule CD31 plays an important role in controlling the extent of T-cell-mediated inflammation in several experimental models of immune-mediated disease, such as experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA). The progression and the severity of these experimental diseases are increased in mice lacking CD31 (38). Moreover, CD31-deficent mice show accelerated allograft and tumor rejection. Furthermore, loss of CD31 expression is associated with reduced immunosuppressive activity of naturally occurring Tregs, although the molecular mechanism underlying this effect is unclear (38).
Knowledge gaps
The first knowledge gap concerns the association between adaptive immunity alterations in patients with ACS as compared with SA, healthy controls, and coronary thrombosis/myocardial ischemia-necrosis: are the former the cause of the latter or vice versa? Obviously we do not have an answer. It is tempting to speculate that alterations persisting after clinical stabilization, such as the enhanced expression of PTPN22 might have a causal role, whereas transient alterations, like the reduced expression of CD31, can be interpreted both ways. In this case, only Mendelian randomization or intervention trials can clarify the direction of causality.
The second knowledge gap concerns the causes of the adaptive immunity alterations associated with ACS: are they caused by genetic alterations leading to hyper-reactive inflammatory cells, by antigenic stimuli, such as oxidized LDL, heat shock proteins (HSPs), or infectious agents, or by a combination of both? The lessons learned from classical autoimmune diseases suggest that both mechanisms probably cooperate.
The third gap concerns the differences between ACS and classical autoimmune diseases. Which alterations of autoimmunity are specific to ACS? The answer to this critical question might allow the identification of new diagnostic techniques and treatments specific for ACS.
The fourth gap is the identification of the subset of patients in which adaptive immunity alterations are likely to play a causal role in ACS. It is indeed increasingly clear that the causes of ACS are multiple, and that systemic inflammation plays a role in a subset of patients, which is probably represented by those exhibiting systemic evidence of inflammation and plaque rupture (1).
The fifth gap is the correlation between alteration of adaptive immune markers in the plaque and in peripheral blood. Indeed, although at one extreme, CD4+CD28null T-cells are similarly expanded in the unstable plaque and in peripheral blood (4), this might not be the case for Treg and Th17 cells (47).
Clinical perspective
In addition to the underlying atherosclerotic process itself, which is addressed with cholesterol-lowering drugs and blood pressure control, coronary thrombosis is the final common pathway leading to ACS and our main current therapeutic target. More potent antithrombotic regimens have recently been found to increase the risk of major bleeding (48), whereas the rate of death, MI, and recurrent ACS at 1-year follow-up is still very high, approaching about 25% (49). Furthermore, recurrent ACS determines a progressive impairment of myocardial function, fueling the chronic heart failure pandemic.
Despite the knowledge gaps highlighted in the preceding section, the extensive studies reported in this review strongly support the notion that adaptive immunity should be seriously addressed in the management of ACS, as is the case in classical autoimmune diseases. A change of focus from soluble markers of inflammation to markers of adaptive immunity regulation, such as the molecules involved in TCR signaling, might prove to be more rewarding in the management of patients presenting with ACS, although this approach is currently limited by the lack of testing for these biomarkers available at the point of care (Central Illustration).
Central Illustration. Responses to Antigen Presentation Observed in Acute Coronary Syndrome Patients.

ACS patients show a unique expression pattern of signaling molecules involved in T-cell activation and differentiation. During the acute phase of the disease, ACS patients show reduced expression of CD31 and increased expression of PTPN22, two molecules that play a critical role in T-cell receptor signaling. ACS patients also have reduced activation of CREB, a transcription factor required for Treg generation and maintenance. After 1 year of follow-up, in a stable phase of the disease, CD4+ T-cells still show increased expression of PTPN22, which is partially counterbalanced by enhanced expression of CD31. Furthermore, at 1 year of follow-up, ACS patients have increased phosphorylation of CREB. Overall, these stable-phase signaling conditions may enhance the functionality of the Treg compartment, dampening excessive immune activation. ACS = acute coronary syndrome; CREB = cyclic adenosine monophosphate response element-binding protein; PTPN22 = the protein tyrosine phosphatase nonreceptor type 22; Treg = regulatory T-cell.
Another unmet need is a specific anti-inflammatory treatment (50). Expanding pathogenic concepts in ACS beyond the simplified view of chronic inflammation considerably widens the spectrum of therapeutic possibilities and enables a more tailored therapeutic approach that may avoid unwanted adverse effects of aggressive immunosuppression. Indeed, TCR activation signaling offers several targets for strongly immunosuppressive therapy, such as cyclosporin, tacrolimus, sirolimus, azathioprine, and imatinib mesylate. These drugs prevent T-cell activation, and reduce the hyper-reactivity of the adaptive immune system and the development of autoimmunity (51,52) (Table 3). Moreover, a synthetic CD31-derived peptide, able to engage a truncated extracellular CD31 fragment expressed by T cells that apparently lack CD31, was recently shown to have an immunosuppressive effect in vivo through restoration of the CD31 inhibitory pathway (53). Moreover, the development of a novel series of PTPN22 inhibitors might contribute to a better understanding its role in immune dysregulation in ACS patients (54).
Table 3.
Potential Therapeutic Targets Modulating TCR Signaling and Immune Response in ACS
| Drugs | Molecular Target and Mechanisms | Ref |
|---|---|---|
| Azathioprine | CD28 signaling and purine biosynthesis inhibition* | 51 |
| Cyclosporine and tacrolimus | NFAT and IL-2 inhibition, increases TGF-b expression* | 51 |
| Sirolimus | mTOR pathway inhibition* | 51 |
| Imatinib mesylate | Early TCR signaling inhibitiony | 52 |
| Anti-CD3 (OKT-3) | Atherosclerosis inhibition by enhancing Treg responsesz | 25 |
| CD31-binding peptides | CD31 signaling enhancementy,z | 53 |
| PTPN22 inhibitors | PTPN22 signaling inhibitionz | 54 |
| 3-hydroxyanthranilic acid | Atherosclerosis inhibition by regulating lipid metabolism and T-cell-dependent inflammation z | 56 |
Another intervention target might be restoring the balance between effector T-cells and Tregs. Isolation and ex vivo expansion of Tregs might restore the defective expression and function of this T-cell subset in ACS patients (55). Indeed, in mice, CD3 antibody treatment induced rapid regression of established atherosclerosis via reducing CD4+ T cells and increasing the proportion of Tregs. The tryptophan metabolite 3-hydroxyanthranilic acid was recently shown to have immune regulatory properties that can be used to decrease atherosclerosis in mice by regulating T-cell dependent inflammation and lowering plasma lipids (56).
Finally, the identification of antigens that trigger adaptive immunity may open the way for specific vaccinations. Of note, the demonstration that vaccination against influenza might be associated with a reduction in cardiovascular events is the first example of this approach (57).
Abbreviations and Acronyms
- ACS
acute coronary syndrome
- CREB
cyclic adenosine monophosphate response element-binding protein
- IFN
interferon
- IL
interleukin
- PTPN22
protein tyrosine phosphatase nonreceptor type 22
- RA
rheumatoid arthritis
- STAT
signal transducer and activator of transcription
- TCR
T-cell receptor
- Treg
regulatory T-cell
- Zap70
zeta-chain-associated protein kinase of 70 kD
Footnotes
Disclosures: The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
References
- 1.Crea F, Liuzzo G. Pathogenesis of acute coronary syndromes. J Am Coll Cardiol. 2013;61:1–11. doi: 10.1016/j.jacc.2012.07.064. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Liuzzo G, Kopecky SL, Frye RL, et al. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation. 1999;100:2135–9. doi: 10.1161/01.cir.100.21.2135. [DOI] [PubMed] [Google Scholar]
- 4.Liuzzo G, Goronzy JJ, Yang H, et al. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. 2000;101:2883–8. doi: 10.1161/01.cir.101.25.2883. [DOI] [PubMed] [Google Scholar]
- 5.Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4+ T cells toward distinct T-helper cell subsets. Immunol Rev. 2013;252:12–23. doi: 10.1111/imr.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geginat J, Paroni M, Facciotti F, et al. The CD4-centered universe of human T cell subsets. Semin Immunol. 2013;25:252–62. doi: 10.1016/j.smim.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Dumitriu IE, Araguás ET, Baboonian C, et al. CD4+ CD28null T cells in coronary artery disease: when helpers become killers. Cardiovasc Res. 2009;81:11–9. doi: 10.1093/cvr/cvn248. [DOI] [PubMed] [Google Scholar]
- 8.Liuzzo G, Montone RA, Gabriele M, et al. Identification of unique adaptive immune system signature in acute coronary syndromes. Int J Cardiol. 2013;168:564–7. doi: 10.1016/j.ijcard.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Liuzzo G, Vallejo AN, Kopecky SL, et al. Molecular fingerprint of interferon-gamma signaling in unstable angina. Circulation. 2001;103:1509–14. doi: 10.1161/01.cir.103.11.1509. [DOI] [PubMed] [Google Scholar]
- 10.Methe H, Brunner S, Wiegand D, et al. Enhanced T-helper-1 lymphocyte activation patterns in acute coronary syndromes. J Am Coll Cardiol. 2005;45:1939–45. doi: 10.1016/j.jacc.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 11.Rainer TH, Graham CA, Chan RWY, et al. Early time-dependent dynamic changes of TBET and GATA3 mRNA expressions in patients with acute coronary syndrome. Dis Markers. 2013;35:419–29. doi: 10.1155/2013/139895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Y, Li L, Yan H, et al. Endothelial microparticles exert differential effects on functions of Th1 in patients with acute coronary syndrome. Int J Cardiol. 2013;168:5396–404. doi: 10.1016/j.ijcard.2013.08.050. [DOI] [PubMed] [Google Scholar]
- 13.Guo M, Mao X, Ji Q, et al. miR-146a in PBMCs modulates Th1 function in patients with acute coronary syndrome. Immunol Cell Biol. 2010;88:555–64. doi: 10.1038/icb.2010.16. [DOI] [PubMed] [Google Scholar]
- 14.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–93. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakajima T, Schulte S, Warrington KJ, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002;105:570–5. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- 16.Liuzzo G, Biasucci LM, Trotta G, et al. Unusual CD4+CD28null T lymphocytes and recurrence of acute coronary events. J Am Coll Cardiol. 2007;50:1450–8. doi: 10.1016/j.jacc.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 17.Giubilato S, Liuzzo G, Brugaletta S, et al. Expansion of CD4+CD28null T-lymphocytes in diabetic patients: exploring new pathogenetic mechanisms of increased cardiovascular risk in diabetes mellitus. Eur Heart J. 2011;32:1214–26. doi: 10.1093/eurheartj/ehq499. [DOI] [PubMed] [Google Scholar]
- 18.Dumitriu IE, Baruah P, Finlayson CJ, et al. High levels of costimulatory receptors OX40 and 4-1BB characterize CD4+CD28null T cells in patients with acute coronary syndrome. Circ Res. 2012;110:857–69. doi: 10.1161/CIRCRESAHA.111.261933. [DOI] [PubMed] [Google Scholar]
- 19.Kovalcsik E, Antunes RF, Baruah P, et al. Proteasome-mediated reduction in proapoptotic molecule Bim renders CD4+CD28null T cells resistant to apoptosis in acute coronary syndrome. Circulation. 2015;131:709–20. doi: 10.1161/CIRCULATIONAHA.114.013710. [DOI] [PubMed] [Google Scholar]
- 20.Téo FH, de Oliveira RT, Mamoni RL, et al. Characterization of CD4+CD28null T cells in patients with coronary artery disease and individuals with risk factors for atherosclerosis. Cell Immunol. 2013;1:11–9. doi: 10.1016/j.cellimm.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Liuzzo G, Trotta F, Pedicino D. Interleukin-17 in atherosclerosis and cardiovascular disease: the good, the bad, and the unknown. Eur Heart J. 2013;34:556–9. doi: 10.1093/eurheartj/ehs399. [DOI] [PubMed] [Google Scholar]
- 22.Cirillo P, Golino P, Piscione F, et al. Transcoronary Th-17 lymphocytes and acute coronary syndromes: new evidence from the crime scene? Int J Cardiol. 2011;153:215–6. doi: 10.1016/j.ijcard.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 23.Zheng Y, Wang Z, Deng L, et al. Modulation of STAT3 and STAT5 activity rectifies the imbalance of Th17 and Treg cells in patients with acute coronary syndrome. Clin Immunol. 2015;157:65–77. doi: 10.1016/j.clim.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Simon T, Taleb S, Danchin N, et al. Circulating levels of interleukin-17 and cardiovascular outcomes in patients with acute myocardial infarction. Eur Heart J. 2013;34:570–7. doi: 10.1093/eurheartj/ehs263. [DOI] [PubMed] [Google Scholar]
- 25.Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. 2014;35:280–7. doi: 10.1161/ATVBAHA.114.303568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mor A, Luboshits G, Planer D, et al. Altered status of CD4+CD25+ regulatory T cells in patients with acute coronary syndromes. Eur Heart J. 2006;27:2530–7. doi: 10.1093/eurheartj/ehl222. [DOI] [PubMed] [Google Scholar]
- 27.Han SF, Liu P, Zhang W, et al. The opposite-direction modulation of CD4+CD25+ Tregs and T helper 1 cells in acute coronary syndromes. Clin Immunol. 2007;124:90–7. doi: 10.1016/j.clim.2007.03.546. [DOI] [PubMed] [Google Scholar]
- 28.Wigren M, Björkbacka H, Andersson L, et al. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler Thromb Vasc Biol. 2012;32:2000–4. doi: 10.1161/ATVBAHA.112.251579. [DOI] [PubMed] [Google Scholar]
- 29.Zhang WC, Wang J, Shu YW, et al. Impaired thymic export and increased apoptosis account for regulatory T cell defects in patients with non-ST segment elevation acute coronary syndrome. J Biol Chem. 2012;287:34157–66. doi: 10.1074/jbc.M112.382978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caligiuri G, Nicoletti A, Poirier B, et al. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–53. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sage P, Tsiantoulas D, Baker L, et al. BAFF receptor deficiency reduces the development of atherosclerosis in mice—brief report. Arterioscler Thromb Vasc Biol. 2012;32:1573–6. doi: 10.1161/ATVBAHA.111.244731. [DOI] [PubMed] [Google Scholar]
- 32.Biasucci LM, Liuzzo G, Ciervo A, et al. Antibody response to chlamydial heat shock protein 60 is strongly associated with acute coronary syndromes. Circulation. 2003;107:3015–7. doi: 10.1161/01.CIR.0000078632.76653.6C. [DOI] [PubMed] [Google Scholar]
- 33.Shah PK, Chyu K, Dimayuga PC, et al. Vaccine for atherosclerosis. J Am Coll Cardiol. 2014;64:2779–91. doi: 10.1016/j.jacc.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 34.Pryshchep S, Sato K, Goronzy JJ, et al. T cell recognition and killing of vascular smooth muscle cells in acute coronary syndrome. Circ Res. 2006;98:1168–76. doi: 10.1161/01.RES.0000220649.10013.5c. [DOI] [PubMed] [Google Scholar]
- 35.Pryshchep S, Goronzy JJ, Parashar S, et al. Insufficient deactivation of the protein tyrosine kinase lck amplifies T-cell responsiveness in acute coronary syndrome. Circ Res. 2010;106:769–78. doi: 10.1161/CIRCRESAHA.109.206052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flego D, Severino A, Trotta F, et al. Increased PTPN22 expression and defective CREB activation impair regulatory T-cell differentiation in non-ST-segment elevation acute coronary syndromes. J Am Coll Cardiol. 2015;65:1175–86. doi: 10.1016/j.jacc.2015.01.027. [DOI] [PubMed] [Google Scholar]
- 37.Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol. 2014;10:602–11. doi: 10.1038/nrrheum.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marelli-Berg FM, Clement M, Mauro C, et al. An immunologist’s guide to CD31 function in T cells. J Cell Sci. 2013;126:2343–52. doi: 10.1242/jcs.124099. [DOI] [PubMed] [Google Scholar]
- 39.Caligiuri G, Groyer E, Khallou-Laschet J, et al. Reduced immunoregulatory CD31+ T cells in the blood of atherosclerotic mice with plaque thrombosis. Arterioscler Thromb Vasc Biol. 2005;25:1659–64. doi: 10.1161/01.ATV.0000172660.24580.b4. [DOI] [PubMed] [Google Scholar]
- 40.Caligiuri G, Rossignol P, Julia P, et al. Reduced immunoregulatory CD31+ T cells in patients with atherosclerotic abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2006;26:618–23. doi: 10.1161/01.ATV.0000200380.73876.d9. [DOI] [PubMed] [Google Scholar]
- 41.Flego D, Severino A, Trotta F, et al. Altered CD31 expression and activity in helper T cells of acute coronary syndrome patients. Basic Res Cardiol. 2014;109:448. doi: 10.1007/s00395-014-0448-3. [DOI] [PubMed] [Google Scholar]
- 42.Flego D, Severino A, Trotta F, et al. Reduced CD31 expression on CD14+CD16+ monocyte subset in acute coronary syndromes. Int J Cardiol. 2015;197:101–4. doi: 10.1016/j.ijcard.2015.06.039. [DOI] [PubMed] [Google Scholar]
- 43.Klingenberg R, Lüsher TF. Rheumatoid arthritis and coronary atherosclerosis: two cousins engaging in a dangerous liaison. Eur Heart J. 2015;36:3423–5. doi: 10.1093/eurheartj/ehv489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zal B, Kaski JC, Arno G, et al. Heat-shock protein 60-reactive CD4+CD28null T cells in patients with acute coronary syndromes. Circulation. 2004;10:1230–5. doi: 10.1161/01.CIR.0000118476.29352.2A. [DOI] [PubMed] [Google Scholar]
- 45.Bryl E, Vallejo AN, Matteson EL, et al. Modulation of CD28 expression with anti-tumor necrosis factor α therapy in rheumatoid arthritis. Arthritis Rheum. 2005;52:2996–3003. doi: 10.1002/art.21353. [DOI] [PubMed] [Google Scholar]
- 46.Singh K, Deshpande P, Pryshchep S, et al. ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J Immunol. 2009;183:8258–67. doi: 10.4049/jimmunol.0901784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Dijk RA, Duinisveld AJF, Schaapherder AF, et al. A change in inflammatory footprint precedes plaque instability: a systematic evaluation of cellular aspects of the adaptive immune response in human atherosclerosis. J Am Heart Assoc. 2015;4:e001403. doi: 10.1161/JAHA.114.001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexander JH, Lopes RD, James S, et al. APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365:699–708. doi: 10.1056/NEJMoa1105819. [DOI] [PubMed] [Google Scholar]
- 49.Roffi M, Patrono C, Collet JP, et al. ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2016;37:267–315. [PubMed] [Google Scholar]
- 50.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–25. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 51.Hartono C, Muthukumar T, Suthanthiran M. Immunosuppressive drug therapy. Cold Spring Harb Perspect Med. 2013;3:a015487. doi: 10.1101/cshperspect.a015487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seggewiss R, Loré K, Greiner E, et al. Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood. 2005;105:2473–9. doi: 10.1182/blood-2004-07-2527. [DOI] [PubMed] [Google Scholar]
- 53.Fornasa G, Clement M, Groyer E, et al. A CD31-derived peptide prevents angiotensin II-induced atherosclerosis progression and aneurysm formation. Cardiovasc Res. 2012;94:30–7. doi: 10.1093/cvr/cvs076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stanford SM, Krishnamurthy D, Falk MD, et al. Discovery of a novel series of inhibitors of lymphoid tyrosine phosphatase with activity in human T cells. J Med Chem. 2011;54:1640–54. doi: 10.1021/jm101202j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Putnam AL, Brusko TM, Lee MR, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes. 2009;58:652–62. doi: 10.2337/db08-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L, Ovchinnikova O, Jönsson A, et al. The tryptophan metabolite 3-hydroxyanthranilic acid lowers plasma lipids and decreases atherosclerosis in hypercholesterolaemic mice. Eur Heart J. 2012;33:2025–34. doi: 10.1093/eurheartj/ehs175. [DOI] [PubMed] [Google Scholar]
- 57.Phrommintikul A, Kuanprasert S, Wongcharoen W, et al. Influenza vaccination reduces cardiovascular events in patients with acute coronary syndrome. Eur Heart J. 2011;32:1730–5. doi: 10.1093/eurheartj/ehr004. [DOI] [PubMed] [Google Scholar]