

Abstract
The nervous system and immune system have broad and overlapping distributions in the body, and interactions of these ubiquitous systems are central to the field of neuroimmunology. Over the past two decades, there has been explosive growth in our understanding of neuroanatomical, cellular, and molecular mechanisms that mediate central modulation of immune functions through the autonomic nervous system. A major catalyst for growth in this field was the discovery that vagal nerve stimulation (VNS) caused a prominent attenuation of the systemic inflammatory response evoked by endotoxin in experimental animals. This effect was mediated by acetylcholine (ACh) stimulation of nicotinic receptors on splenic macrophages. Hence, the circuit was dubbed the “cholinergic anti-inflammatory pathway”. Subsequent work identified the α7 nicotinic ACh receptor (α7nAChR) as the crucial target for attenuation of pro-inflammatory cytokine release from macrophages and dendritic cells. Further investigation made the important discovery that cholinergic T cells within the spleen and not cholinergic nerve cells were the source of ACh that stimulated α7 receptors on splenic macrophages. Given the important role that inflammation plays in numerous disease processes, cholinergic anti-inflammatory mechanisms are under intensive investigation from a basic science perspective and in translational studies of animal models of diseases such as inflammatory bowel disease and rheumatoid arthritis. This basic work has already fostered several clinical trials examining the efficacy of VNS and cholinergic therapeutics in human inflammatory diseases. This review provides an overview of basic and translational aspects of the cholinergic anti-inflammatory response and relevant pharmacology of drugs acting at the α7nAChR.
Keywords: Vagal nerve stimulation, Cholinergic neurons, Cholinergic T cells, α7 Nicotinic ACh receptor, Macrophage, Microglial cell, Inflammation
1. Introduction
The nervous system and immune system are two so-called “supersystems” that play a vital role in achieving homeostatic adaptations to routine daily conditions and to challenging or adverse environmental conditions caused by injury, infections, and exposure to toxins (Elenkov et al., 2000). Given the ubiquitous distribution for both of these systems, it is not surprising that research over the past two decades has shown that the immune and nervous systems engage in an ongoing dialogue (Andersson and Tracey, 2012; Bellinger and Lorton, 2014; Elenkov et al., 2000; Olofsson et al., 2012). Crucial links between the brain and the immune system are provided by the autonomic nervous system, sensory nerves and chemical mediators released from neurons, and immune cells.
1.1. Afferent nerves and reflex responses
Sensory neurons have the vital function of continually sampling the peripheral environment and relaying this information to the central nervous system (CNS), where it is integrated and appropriate responses are made through activation of efferent neurons and endocrine mechanisms. This review will focus on efferent neuronal interactions with the immune system.
Sensory neurons comprise the afferent limb for reflex modulation of the immune system just as they serve as the sensors for cardiovascular reflexes and reflex control of other autonomic effector organs (Bonaz et al., 2016b). Baroreflex control of cardiac function serves as a model system for comparison. Vagal afferent nerve endings in the aortic arch contain mechanoreceptors that sense wall tension and are activated by increases in blood pressure. Activation of these receptors triggers action potentials in the vagal afferent nerve fibers, and this signal is transmitted to the brainstem by central projection fibers of the same sensory neurons whose cell bodies are located in the nodose ganglia (i.e., vagal sensory ganglia). This signal is processed centrally and causes a reflex decrease in sympathetic input to the heart and vasculature as well as an increase in vagal parasympathetic input to the heart. Analogous processes occur during reflex activation of the immune system, but the stimuli that activate afferent nerves during reflex modulation of the immune system are chemicals generated at sites of inflammation or injury (Bonaz et al., 2016b; Santoni et al., 2015). These include pathogen associated molecular patterns, danger associated molecular patterns, and cytokines/chemokines released in response to the former. Vagal afferents nerves and afferent nerves originating from the dorsal root ganglia can function in reflex modulation of the immune system. Circulating inflammatory mediators can also stimulate the CNS directly at specific sites that lack a blood-brain barrier (Bonaz et al., 2016b).
1.2. Sympathetic ganglia and nerves
Postganglionic sympathetic neurons and nerve fibers, which utilize norepinephrine (NE) as their primary neurotransmitter, are the most abundant conduit for efferent modulation of the immune system by the CNS (Elenkov et al., 2000; Jung et al., 2017; Nance and Sanders, 2007). These noradrenergic neurons provide innervation to all primary and secondary lymphoid tissues, including the bone marrow, spleen and lymph nodes (Jung et al., 2017; Nance and Sanders, 2007). Most immune cells express one or more type of adrenergic receptor (Table 1), but β2-adrenoceptors have the widest distribution and mediate most effects of sympathetic nerves on immune function (Bellinger and Lorton, 2014; Elenkov et al., 2000; Lorton and Bellinger, 2015; Padro and Sanders, 2014).
Table 1.
Adrenergic and cholinergic receptors associated with immune cells
| Receptor type Subtype | Leukocyte type | References | ||||
|---|---|---|---|---|---|---|
| Neutrophils | Monocytes | Macrophages/Dendritic cells | T cells | B cells | ||
| Adrenergic receptors | ||||||
| α1 | + | + | + | Bellinger and Lorton (2014) | ||
| β2 | + | + | + | + | + |
Elenkov et al.
(2000) Bellinger and Lorton (2014) |
| Cholinergic receptors | ||||||
| Nicotinic subunits | ||||||
| α7 | + | |||||
| α2, α5, α7 | + | + | + | Sato et. al. (1999) | ||
| β2 (variable) | ||||||
| α4(α5)β2 | + | Skok et al. (2007) | ||||
| α7(α5)β4 | + | |||||
| α2–α7, α9 | + | Safronova et al. (2016) | ||||
| β2–β4 | ||||||
| Muscarinic | ||||||
| M1–M3 (variable) | + | + | + | Sato et. al. (1999) | ||
| M4 and M5 | ||||||
Sympathetic nerves affect both innate and adaptive immune responses through stimulation of adrenergic receptors. Most evidence indicates that activation of β2-adrenoceptors has immunosuppressive effects on monocytes and macrophages (Lorton and Bellinger, 2015; Padro and Sanders, 2014). However, these effects are context dependent, and pro-inflammatory responses can occur under some circumstances. Effects on T cells, which contain only β2-adrenoceptors, have been studied most in CD4+ T helper cells. Depending on conditions, stimulation of β2-adrenoceptors on naive CD4+ T cells (Th0) cause differentiation into Th1 cells that enhance cellular immunity or into Th2 cells that decrease cellular immunity (Lorton and Bellinger, 2015; Padro and Sanders, 2014). Norepinephrine can also produce a β2-mediated anti-inflammatory effect via release of acetylcholine (ACh) from cholinergic splenocytes (see section 2. Vagal anti-inflammatory reflex). Lastly, there is evidence that NE and β2 agonists can augment adaptive immunity by increasing T cell-dependent antibody production by B cells (Bellinger and Lorton, 2014; Padro and Sanders, 2014).
1.3. Cholinergic neurons and non-neuronal cholinergic cells
Postganglionic parasympathetic and sympathetic nerve fibers provide dual innervation to most targets of the autonomic nervous system and have opposing effects on target function. However, most lymphoid organs lack cholinergic nerves and receive efferent input exclusively from sympathetic nerves (Bellinger and Lorton, 2014; Jung et al., 2017; Nance and Sanders, 2007). Recent neuroanatomical studies suggest that the bone marrow is an exception (Jung et al., 2017). Cholinergic nerve fibers have been identified in this tissue by immunostaining for choline acetyltransferase (ChAT) (Artico et al., 2002), the ACh synthetic enzyme, and for vesicular ACh transporter (VAChT) (Bajayo et al., 2012), the protein responsible for storage of ACh in secretory vesicles in nerve endings (Fig. 1). Transneuronal tracing experiments have identified corresponding preganglionic neurons in the sacral spinal cord (Bajayo et al., 2012), but localization of intervening parasympathetic ganglia has not been established. Furthermore, cholinergic innervation of the bone marrow appears to have a role in regulation of bone dynamics rather than hematopoiesis (Bajayo et al., 2012; Jung et al., 2017).
Figure 1.
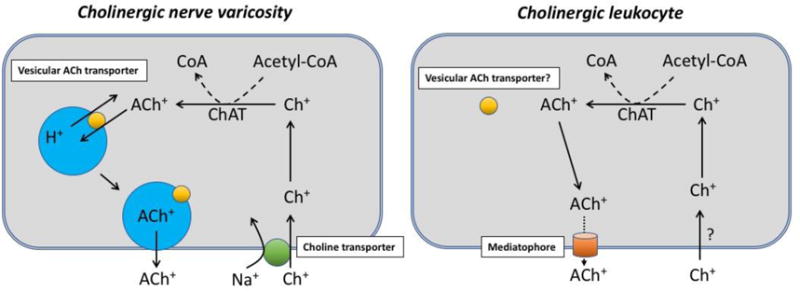
Diagrams depicting the cholinergic biochemistry of cholinergic nerve varicosities and cholinergic leukocytes. Choline (Ch) and acetylcholine (ACh) carry a positive charge at all pH values and require specialized mechanisms for crossing membranes. The cell membrane of cholinergic varicosities contains high affinity, sodium-dependent Ch+ transporters, which facilitate entry of Ch+ into the cytosol. It remains unclear how Ch+ enters leukocytes; this could involve a different transporter, breakdown of choline phospholipids, or both mechanisms. Both cell types contain choline acetyltransferase (ChAT), which catalyzes the synthesis of ACh+ from Ch+ and acetyl coenzyme A (acetyl-CoA). ACh+ in neurons is stored in vesicles and released by exocytosis. Depolarization of varicosities causes influx of Ca2+, which triggers docking of vesicles with the cell membrane. Storage of ACh+ in vesicles requires the vesicular ACh transporter (VAChT), which exchanges vesicular H+ for cytosolic ACh+. It is unclear if leukocytes contain VAChTs, but there is no ultrastructural evidence for vesicles in lymphocytes. Release of ACh+ from leukocytes may occur through a hemichannel named mediatophore (see text), but relatively little is known about this protein or ACh+ release dynamics in leukocytes.
The lack of cholinergic nerves in other lymphoid tissues and organs is surprising, since historically, ACh was first isolated from horse spleen almost 90 years ago (Dale and Dudley, 1929). An explanation for this discrepancy has been provided by recent evidence that many immune cells, particularly T and B cells can exhibit a cholinergic phenotype (Kawashima et al., 2012; Kawashima et al., 2015; Reardon et al., 2013; Rosas-Ballina et al., 2011). Identification of these cells in situ had been difficult since most available antibodies have a low efficacy for labeling ChAT in the periphery. This obstacle was overcome by using transgenic mice that express enhanced green fluorescent protein (eGFP) under control of the ChAT promotor (Tallini et al., 2006). Using these mice, it is possible to identify cholinergic immune cells in the spleen, mesenteric lymph nodes, and other tissues (Reardon et al., 2013; Rosas-Ballina et al., 2011) (Fig. 2).
Figure 2.
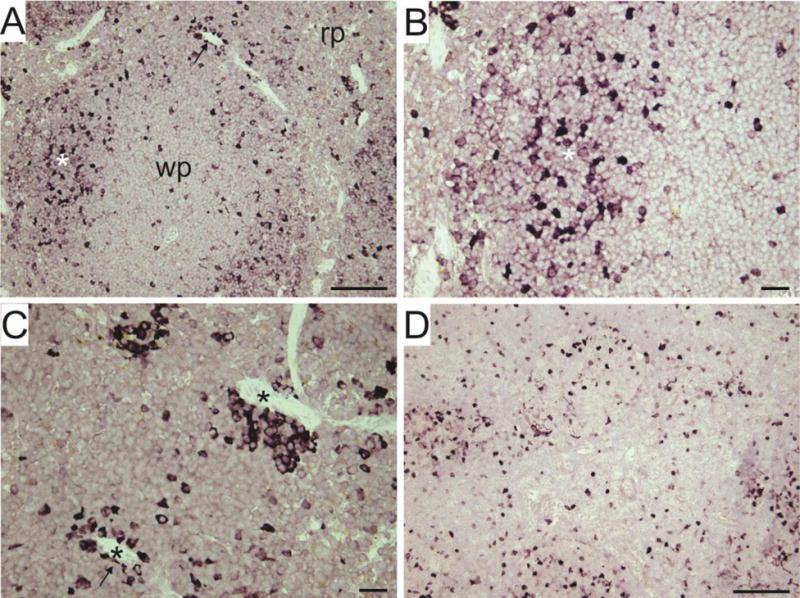
Immunohistochemical identification and localization of cholinergic leukocytes in 5 μm paraffin sections of mouse spleen (A–C) and mesenteric lymph node (D). Transgenic mice that express eGFP under control of the ChAT promoter were used in these experiments, and cholinergic cells were labeled using the standard ABC method for Immunohistochemical detection of GFP. The chromogen (Impact VIP peroxidase substrate, Vector Laboratories) yields an intense purple reaction product. A: Low magnification view of spleen showing white pulp (wp) and red pulp (rp). White asterisk and arrow indicates regions shown at higher magnification in B and C, respectively. C: Black asterisks indicate blood vessels with surrounding GFP+ leukocytes. Scale bar indicates 100 μm in A and D; 25 μm in B and C.
2. Vagal anti-inflammatory reflex
2.1. Studies contributing to the primary hypothesis
The concept of targeting the vagus nerve and cholinergic mechanisms to suppress inflammation took root when Tracey and colleagues reported that vagal nerve stimulation (VNS) attenuated the systemic inflammatory response (i.e., increased serum and hepatic tumor necrosis factor-α and hypotension) evoked by intravenous injection of lipopolysaccharide (LPS) in rats anesthetized with urethane (Borovikova et al., 2000). These beneficial effects were mediated by the efferent vagus nerve, since they occurred after bilateral vagotomy and stimulation of a distal cut end of the vagus nerve. Interestingly, LPS-evoked elevation of serum corticosterone levels, due to stimulation of vagal afferents by LPS and reflex activation of the hypothalamic-pituitary-adrenal axis, was also attenuated by vagotomy, but this effect was unaltered by efferent VNS. Since ACh is recognized as the neurotransmitter for pre- and postganglionic vagal efferent nerves and macrophages are a major source of tumor necrosis factor-α (TNF-α) and other pro-inflammatory cytokines, these investigators evaluated the effects of cholinergic agonists on LPS-evoked release of pro-inflammatory cytokines from human macrophages in culture. They found that ACh stimulated nicotinic receptors on the macrophages to cause a concentration-dependent inhibition of pro-inflammatory cytokine release. Based on these findings, Tracey and colleagues coined the term “cholinergic anti-inflammatory pathway” (Borovikova et al., 2000). Over the intervening years, many studies have built on this initial description of the cholinergic anti-inflammatory pathway to yield our current view, which casts the spleen as the major effector (Andersson and Tracey, 2012; Bonaz et al., 2016b; Pavlov and Tracey, 2015; Pereira and Leite, 2016).
Shortly after the identification of the cholinergic anti-inflammatory pathway, experiments with α7 nicotinic ACh receptor (α7nAChR) knockout mice established that this receptor was essential for vagal inhibition of the inflammatory response to LPS (Wang et al., 2003). Furthermore, treatment with LPS actually caused a larger increase in serum TNF-α in knockout mice compared to wild-type controls, suggesting that the cholinergic anti-inflammatory pathway might be activated reflexively during endotoxemia. The same study also demonstrated expression of α7nAChR mRNA by human macrophages and the presence of these receptors on the cell surface by using FITC-labeled α-bungarotoxin (α-bgt). Experiments with human macrophages and mouse peritoneal macrophages completed the story by establishing that ACh and nicotine inhibited LPS-evoked release of TNF-α from macrophages by stimulating α7 receptors (Wang et al., 2003).
The next crucial step in identifying the components of the cholinergic anti-inflammatory pathway was elucidation of the role the spleen played. The spleen is a major secondary lymphoid tissue that contains lymphocytes in the white pulp and myeloid lineage cells (e.g., monocytes, macrophages, and dendritic cells) in the red pulp. It serves as a major depot from which monocytes are recruited to sites of inflammation and infection (Swirski et al., 2009), and it is the major source of serum TNF-α during endotoxemia (Huston et al., 2006). Splenectomy or VNS in rats with intact spleens resulted in identical suppression of the LPS-evoked increase in serum TNF-α (Huston et al., 2006). Furthermore, VNS attenuated the LPS-evoked increase in TNF-α levels in the spleen in control rats. Like all lymphoid tissues, the spleen receives noradrenergic innervation from postganglionic sympathetic neurons. Neuronal tracer experiments identified the celiac/mesenteric ganglia as major sources of sympathetic input to the rat spleen (Bellinger et al., 1989). However, the link between efferent VNS and responses in the spleen is unclear. The initial hypothesis proposed that branches of the vagus nerve provide preganglionic cholinergic input to the sympathetic neurons that innervate the spleen (Andersson and Tracey, 2012). This hypothesis has substantial experimental support based on neuroanatomical tracer experiments (Berthoud and Powley, 1993; Cano et al., 2001) and selective nerve transection experiments (Huston et al., 2006), all performed using rats. Specifically, the latter study probed the circuitry responsible for attenuation of LPS-evoked increases in serum and splenic TNF-α by cutting branches of the vagus nerve below the diaphragm. Cutting the ventral branch had no effect, but cutting the common celiac branch eliminated the vagal anti-inflammatory response. Thus, it was proposed that efferent VNS activates sympathetic neurons in the celiac ganglia directly by means of preganglionic vagal cholinergic nerves that travel in the celiac branch of the vagus nerve (Huston et al., 2006). Subsequent experiments completed the wiring diagram by showing that postganglionic sympathetic nerve fibers travel to the spleen via the splenic nerve (Rosas-Ballina et al., 2008; Vida et al., 2011a). However, the spleen lacks cholinergic innervation, and the source of ACh that activates α7 nicotinic receptors on macrophages was unclear.
This question was answered by recent work showing that splenic T cells are the source of ACh and might also be the link between NE and the suppression of splenic macrophages (Rosas-Ballina et al., 2011). First, these investigators used microdialysis to show that VNS released ACh in spleens of LPS-treated mice. They also showed that NE can stimulate release of ACh from isolated spleen cells, albeit at high concentrations. However, this was a mixed population of immune cells. Two lines of evidence implicated T cells. First, they found that VNS suppressed serum TNF-α in LPS-treated BALB/c mice but not in nude mice that lack T cells. To identify putative ACh-producing T cells, they used transgenic mice that express eGFP under control of the promoter for ChAT, the ACh synthetic enzyme. Only ~3% of the total T cells isolated from the spleens of these mice expressed eGFP, but adoptive transfer of these cells to nude mice restored the anti-inflammatory response to VNS while adoptive transfer of ChAT-eGFP negative T cells did not. Activation of the ChAT-eGFP T cells increased the expression of eGFP suggesting that ChAT expression would likewise be increased under this condition. In fact, several other investigators had already established that ChAT mRNA, protein, and activity are upregulated when T cells are activated (Fujii et al., 1998; Fujii et al., 2008; Fujii et al., 2012b; Kawashima and Fujii, 2008). Subsequent studies by Ulloa and coworkers provided strong support for a T cell link between noradrenergic nerves and splenic macrophages, but their data implicated a different subgroup of helper T cells (Peña et al., 2011; Vida et al., 2011b). They also showed that β2-adrenergic receptors on these T cells are essential for the anti-inflammatory effects of VNS.
Collectively, previous work has made a strong case establishing the cellular components of the cholinergic anti-inflammatory pathway (Fig. 3). This pathway begins with cholinergic transmission at the celiac/superior mesenteric ganglion, and transgenic mouse experiments and other studies have provided definitive evidence that ganglionic transmission at this site is mediated at least in part by α7 nicotinic receptors (Downs et al., 2014; Vida et al., 2011a; Vida et al., 2011b). Action potentials generated in sympathetic neurons travel to the spleen via the splenic nerve and cause depolarization of sympathetic varicosities, which triggers release of NE. Next, NE stimulates β2-adrenoceptors on a small population of cholinergic T cells, which then release ACh. The actual mechanism for release of ACh remains unclear but probably differs significantly from the well characterized mechanism for release of ACh from varicosities of cholinergic nerves (Fig. 1). In fact, there is some evidence that ACh release from immune cells occurs by a non-vesicular mechanism involving a membrane hemichannel called mediatorphore (Fujii et al., 2012a; Fujii et al., 2012b). Lastly, T cell-derived ACh stimulates α7nAChRs on splenic macrophages to elicit the primary anti-inflammatory response.
Figure 3.
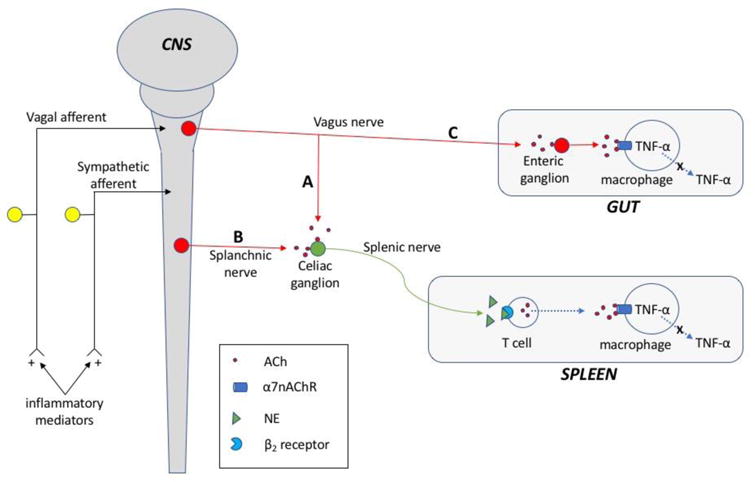
Diagram showing neuroimmune pathways mediating cholinergic anti-inflammatory responses. Afferent input that signals the presence of inflammation to the CNS is shown at the left. Efferent pathways are shown on the right. A: Vagal anti-inflammatory pathway to the celiac ganglion and spleen. B: Sympathetic anti-inflammatory pathway to the celiac ganglion and spleen. A and B pathways both have sympathetic input to β2-adrenergic receptors on cholinergic T cells causing the release of ACh. ACh then elicits an anti-inflammatory response by stimulation of α7nAChRs on splenic macrophages. C: Vagal anti-inflammatory pathway to the gut. Preganglionic cholinergic neurons project to cholinergic neurons in the enteric ganglia, and projections of postganglionic cholinergic neurons release ACh that stimulates α7nAChRs on resident macrophages in the muscle layers of the gut.
From a pharmacological perspective, this pathway is expected to have a high threshold for activation since 1) NE is not the preferred ligand for β2-adrenoceptors, having an affinity 100-fold less than that of epinephrine (Bylund et al., 1994) and 2) micromolar concentrations of ACh are required to stimulate homomeric α7nAChRs (Cuevas and Berg, 1998; McLaughlin et al., 2009). So why does this system work? Regarding the first point, there is definitive experimental evidence supporting a specific role for NE rather than epinephrine in the vagal anti-inflammatory response (Vida et al., 2011a). VNS increases plasma NE concentration without affecting plasma epinephrine. Furthermore, splenectomy eliminates the effect of VNS to increase plasma NE, supporting the conclusion that plasma NE is released from the spleen into the circulation during VNS. Additionally, adrenalectomy removes the major source of plasma epinephrine but does not affect the ability of VNS to inhibit the LPS-evoked increase in serum TNF-α (Vida et al., 2011a). Immunohistochemical studies have established that sympathetic nerves form close contacts with T cells and macrophages in the periarteriolar lymphatic sheath of the spleen (Felton et al., 1987). It is likely that this anatomical proximity produces local concentrations of NE that are high enough to activate β2 receptors on target immune cells. Likewise, proximity of cholinergic T cells to macrophages in the periarteriolar lymphatic sheath should facilitate cholinergic signaling between these immune cells. Since choline is a selective agonist at α7nAChRs (Albuquerque et al., 2009), it might also contribute to cholinergic signaling between T cells and macrophages. The absence of high affinity choline transporters in the spleen (Hoover et al., 2013) might amplify cholinergic signaling by prolonging the presence of extracellular choline generated from hydrolysis of ACh.
2.2. Alternative or additional mechanisms
While substantial evidence supports the primary hypothesis of the vagal anti-inflammatory pathway, there is some controversy regarding the role of efferent vagal connections to the celiac ganglia as a driving factor in stimulating noradrenergic input to the spleen (Bratton et al., 2012; Martelli et al., 2014a; Martelli et al., 2014b). Using sophisticated electrophysiological approaches, it was found that sympathetic neurons, with known projections to the spleen (i.e., projections traveling in splenic nerve), were unaffected by VNS but did fire action potentials in response to stimulation of preganglionic sympathetic axons in the splanchnic nerve. Based on these findings, it was proposed that noradrenergic input to the spleen is controlled by traditional preganglionic sympathetic input from the spinal cord rather than direct vagal efferent projections to the celiac ganglion (Fig. 3). This concept is certainly consistent with the well know reflex activation of the sympathetic nervous system during inflammation (Elenkov et al., 2000; Pongratz and Straub, 2014). However, these findings do not provide an explanation for the wealth of experimental data showing anti-inflammatory responses to selective stimulation of vagal efferent nerve fibers. This controversy is addressed in several recent reviews (Bonaz et al., 2016b; Martelli et al., 2014a; Pereira and Leite, 2016).
2.3. Translational targets
Regardless of the specific anatomical mechanism(s) contributing to the vagal anti-inflammatory response, there are two major approaches for targeting this system clinically. One of these is VNS and the other is pharmacologic stimulation of α7nAChRs on sympathetic ganglia and myeloid linage cells such as macrophages. With the former approach, the vagus nerves are intact, so both afferent and efferent nerve fibers will be activated. Stimulation of afferent vagal fibers would trigger reflex activation of preganglionic sympathetic (cholinergic) input to the celiac ganglia, carried by the splanchnic nerves. Stimulation of vagal efferent nerve fibers might also activate noradrenergic neurons in the celiac ganglia. As discussed later, vagal efferent projections to the gut can also have an anti-inflammatory effect mediated by ACh released from enteric cholinergic neurons (see section 7. Vagal anti-inflammatory pathway to the gut).
3. Properties of 7nAChRs
3.1. Structure of nicotinic receptors
Nicotinic ACh receptors comprise a large family of ligand-gated ion channels that have a broad distribution with localization to neurons and non-neuronal cells throughout the body (Albuquerque et al., 2009). These receptors are all pentamers formed by five, interacting subunits, which are classified as α, β, γ, δ, and ε based on structure. There are seven variants of the α subunit (1–7, 9 and 10), and most of the ligand binding site for nicotinic agonists is localized to this subunit. The remaining subunit repertoire includes four β subunits (β1 – β4) and once each of γ, δ, and ε subunits. Each subunit has a large NH2-terminal, extracellular domain, four transmembrane (TM) domains, a cytoplasmic loop between TM3 and TM4, and a small, extracellular COOH-terminal domain. Most of the variations in subunit structure occur at the cytoplasmic loop (Albuquerque et al., 2009). All functional nicotinic receptors contain at least two α subunits, and heteropentamers have various combinations of the other subunits, bringing the total to five. Homodimers can be formed by α7, α9, or α10 subunits, but only the 7 subunit is believed to form a native homodimer (Millar et al., 2014). Occupation of two ligand binding sites on the nAChR is required for optimum channel opening, hence the requirement for at least two α subunits. As noted previously, α7nAChRs on macrophages are essential for cholinergic anti-inflammatory responses, but other nicotinic receptors are present on immune cells (Table 1).
3.2. Function of nicotinic receptors
Nicotinic receptors are ligand gated cation channels that open when both ligand binding sites are occupied by ACh, nicotine, or another nicotinic agonist. Receptor occupancy triggers rapid and complex changes in protein structure that are transmitted to the TM2 domains causing rotations that form a hydrophilic channel of about 8 Å diameter that allows the influx of Na+ and Ca2+ (Albuquerque et al., 2009). The α7nAChRs expressed by neurons are unique in having much higher Ca2+ permeability than other nicotinic receptors and undergoing concentration-dependent desensitization more rapidly when exposed to agonists (Albuquerque et al., 2009). In this regard, it should be noted that choline, a selective agonist at α7nAChRs, dissociates from the receptor more rapidly than ACh and produces a desensitization that is less stable. The channel function of α7nAChRs expressed on macrophages has received far less attention, but stimulation with nicotinic ligands does caused small Ca2+ transients in these cells (Báez-Pagán et al., 2015) and increases [Ca2+]i (Matteoli et al., 2014). Desensitization properties of α7nAChRs on macrophages have not been reported.
3.3. Pharmacology of α7nAChRs
Acetylcholine and nicotine are non-selective, full agonists that are able to activate all nicotinic receptors including α7nAChRs. Structural differences in and around the ligand binding sites of different nicotinic receptors have enabled the development of agonists and antagonists that have a high degree of selectivity for the α7nAChR (Table 2). It is interesting to note that choline, a metabolite of ACh and substrate for ACh synthesis, functions as a full, selective agonist at α7nAChRs (Albuquerque et al., 2009), and it is sometimes used as a selective agonist for in vivo and in vitro experiments. The snake toxin, α-bgt, was instrumental in pioneering studies that isolated nicotinic receptors from the electric organ of Torpedo, and subsequent work established that α-bgt is a potent antagonist at α7nAChRs (Albuquerque et al., 2009). Since α-bgt is a polypeptide that blocks nicotinic receptors at the neuromuscular junction, use of this agent in neuroimmunology experiments is generally limited to blockade of α7 receptors in cell culture studies and localizing α7 receptors with fluorescent or radiolabeled α-bgt. Several α7-selective agonists, partial agonists, and antagonists are suitable for both in vivo and in vitro experiments (Albuquerque et al., 2009; Briggs et al., 1995; Meyers et al., 1998; Millar et al., 2014; Williams et al., 2011). GTS-21 and methyllycaconitine, respectively, are examples of small molecule agonists and antagonists used to study cholinergic anti-inflammatory mechanisms.
Table 2.
Drugs acting at α7 nicotinic acetylcholine receptors
| Drug class | Agent | Reference |
|---|---|---|
| Nonselective agonists | ACh Nicotine |
Albuquerque et al. (2009) |
| Selective α7 agonists | Choline A-582941 AR-R1779 PHA-543613 PNU-282987 |
Albuquerque et
al. (2009) Millar et al. (2014) Papke et al., (2004) |
| α7 Partial agonists | Encenicline GTS-21 |
Millar et al.
(2014) Meyer et al., (1998) Briggs et al., (1995) |
| Selective α7 antagonists | Methyllycaconitine α-Conotoxin |
Millar et al. (2014) |
| Positive allosteric modulators | ||
| Type 1 | Galantamine LY-20087101 Ivermectin |
Williams et
al. (2011) Millar et al. (2014) |
| Type 2 | PNU-120596 TQS A-867744 |
|
| Type 1/Type 2 intermediate | JNJ1930942 |
Nicotinic receptors also have modulatory allosteric binding sites that are distinct from the agonist binding sites (Williams et al., 2011). These sites function in a similar fashion to benzodiazepine binding sites on the GABAA receptor. Research has identified allosteric agonists for heteromeric nAChRs and some that are selective allosteric modulators of the α7nAChR (Williams et al., 2011). The latter are much more interesting from a therapeutics perspective, since α7nAChRs are viable targets in several CNS and inflammatory disorders (Uteshev, 2014). Binding of agonists to these modulatory allosteric sites does not elicit any current or functional responses but does facilitate responses to agonists, such as ACh and nicotine, which occupy the primary ligand binding site. In effect this shifts the concentration-response curve for ACh and nicotine to the left. Positive allosteric modulators of the α7nAChR have been placed into two major categories, type I and type II, based on specific ways that they produce an enhanced response to agonists (Grønlien et al., 2007; Williams et al., 2011). Some agents that fall between these categories in their properties are classified as type I/type II intermediate. Type II agents hold the greatest potential for therapeutic use since they are more selective, slow response decay time substantially, and have the ability to activate receptors that were desensitized by exposure to high concentrations of primary agonists. These features are particularly valuable for α7nAChRs since they have a low probability for opening and desensitize rapidly (Williams et al., 2011). PNU-120596, a type II agent used commonly in inflammation studies, is listed in Table 2 along with selected other positive allosteric modulators.
3.4. Localization of α7nAChRs and other cholinergic receptors to leukocytes
There is abundant evidence that α7nAChRs are expressed by macrophages, monocytes, and dendritic cells and mediate anti-inflammatory responses when activated on these cells. Neutrophils, T cells and B cells also express α7nAChR but they contain an array of other nicotinic and muscarinic receptors as well (Table 1). Accordingly, VNS and pharmacological administration of α7nAChR agonists have the potential to affect a wide range of leukocytes.
3.5. Human-specific, partially duplicated α7nAChR gene
The large family of nicotinic receptors evolved long ago from a primordial CHRNA7 gene that encodes α7nAChR (Changeux, 2012). More recently this gene has undergone a partial duplication to form a new gene termed CHRFAM7A, which is only present in humans (Gault et al., 1998; Sinkus et al., 2015). This gene is co-expressed with CHRNA7 in neuronal and non-neuronal cells, including immune cells, and undergoes translation at a low efficiency to form a peptide termed dupα7 (Gault et al., 1998; Sinkus et al., 2015). Studies with in vitro expression systems (i.e., oocytes) have shown that dupα7 and normal α7 subunits can form pentamers, which have reduced binding of [125I]-α-bgt and impaired gating function. Thus, dupα7 has been called a dominant-negative inhibitor (Araud et al., 2011). In this regard, it is interesting to note that treatment with PNU-120596, a positive allosteric modulator, appears to overcome the effect of dupα7 (Araud et al., 2011).
4. Cellular and molecular actions of ACh and nicotinic agonists on macrophages
4.1. Macrophage functions
Macrophages play a vital role in innate cellular immunity and the body’s response to infection (Zhang and Wang, 2014). Neutrophils and macrophages are the first immune cells recruited to sites of infection, and both cell types make important contributions to the elimination of pathogens (Hurst et al., 2001; Soehnlein et al., 2009). However, excessive activity of both cell types can contribute to pathology as occurs dramatically in sepsis (Iskander et al., 2013). The pro-inflammatory actions of specific macrophages can also contribute to the pathophysiology of many other diseases such as arthritis, atherosclerosis and type 2 diabetes (Alexandraki et al., 2006; Ouchi et al., 2003; Tobias and Curtiss, 2005). However, macrophages are a heterogeneous population of cells that have the ability to switch between M1 (pro-inflammatory) and M2 (anti-inflammatory) phenotypes (Chinetti-Gbaguidi et al., 2011; Zhang and Wang, 2014). M2 macrophages contribute importantly to the elimination of infection and repair of tissue damage (Gordon and Martinez, 2010). This review will focus on M1 macrophages since they are a primary target of cholinergic anti-inflammatory therapies.
Macrophages are activated to the M1 pro-inflammatory phenotype after stimulation by interferon-γ and toll-like receptor (TLR) ligands such as LPS, a bacterial cell membrane component that specifically stimulates TLR4 (Dey et al., 2015; Zhang and Wang, 2014). Activation of TLR4 by LPS triggers two parallel and interacting pathways in macrophages to increase the expression, synthesis and release of type I interferon and inflammatory cytokines, including TNF-α and IL-6. These substances play important roles in further activation of the innate immune system and combating infection, but dysregulated and excessive production of inflammatory cytokines can cause host tissue damage. In extreme cases, such as sepsis, tissue and organ damage can be life threatening. Macrophages also have the ability to migrate to sites of inflammation, phagocytize microbes and other substances, and to present antigens to lymphocytes (Nourshargh and Alon, 2014; Price and Vance, 2014). Most studies of cholinergic anti-inflammatory mechanisms have focused on the ability of nicotinic agonists to suppress the synthesis and release of inflammatory cytokines from activated macrophages.
4.2. Nicotinic receptor-mediated inhibition of the synthesis and release of pro-inflammatory cytokines
Several studies have demonstrated that nicotinic agonists can cause concentration-dependent inhibition of the synthesis and release of pro-inflammatory mediators such as IL-1β, TNF-α, IL-6, and HMGB1 from activated macrophages and macrophage cell lines but generally do not affect anti-inflammatory cytokines such as IL-10 (Borovikova et al., 2000; de Jonge et al., 2005; Kox et al., 2009; Pavlov et al., 2007; Sun et al., 2013b; Wang et al., 2004). The typical design for these experiments includes pretreatment with a nicotinic agonist followed by challenge with LPS or another TLR ligand to evoke a cytokine response. When evaluated at strategic times after LPS treatment, the nicotinic agonists (non-selective and α7 selective) attenuated the amount of cytokine released into the media. Such effects can be blocked by non-selective and α7 selective antagonists and are absent or reduced in cells from α7 knockout mice or wild type cells treated with siRNA to reduce levels of the α7nAChR (Khan et al., 2012; Wang et al., 2003; Wang et al., 2004).
4.3. Channel function of the macrophage α7 receptor
The ligand-gated ion channel properties of α7nAChRs expressed by neurons have been studied extensively and play a major role in signal transduction in the nervous system (Albuquerque et al., 2009). However, very little is known about the channel function of macrophage α7nAChRs. One group performed whole-cell recordings from human peripheral blood mononuclear cells and did not detect ACh- or nicotine-evoked currents, but these cells were not activated and lacked surface binding sites for α-bgt (Villiger et al., 2002). Recently, other investigators have studied single-channel currents in macrophages obtained by treatment of human monocytes with macrophage colony-stimulating factor. (Báez-Pagán et al., 2015). They could not detect ACh-evoked whole-cell currents either but did observe short duration, single-channel currents evoked by ACh and choline. The frequency and duration of agonist-evoked channel openings were increased by PNU-120596, an allosteric modulator of α7 receptors, and blockade of α7nAChRs with α-bgt inhibited current responses to choline. However, further work is needed to determine if these currents contribute to or are required for the intracellular signaling processes triggered by α7 agonists.
4.4. Intracellular signaling pathways mediating anti-inflammatory response to α7nAChR stimulation
Stimulation of α7nAChRs on macrophages and macrophage cell lines inhibits the synthesis and release of pro-inflammatory cytokines by activating several signaling pathways, but the actual link between the receptor and intracellular proteins is not known. Recent evidence suggests that α7 receptors may have dual signal transduction capabilities, functioning as 1) ligand-gated ion channels and 2) metabotropic receptors that signal through protein interactions with the cytosolic domain of the nicotinic receptor (Stokes et al., 2015). The latter mechanism has not received much attention, but one group has provided evidence for recruitment of Janus kinase 2 (Jak2) to the α7nAChR after treating peritoneal macrophages with nicotine (de Jonge et al., 2005). After metabotropic signaling is initiated, it seems likely that it and the cellular consequences of such signaling would persist even after desensitization of the α7nAChRs.
Stimulation of membrane localized TLRs (e.g., TLR4 by LPS) causes rapid nuclear translocation of nuclear factor-kappaB (NF-κB), and this transcription factor increases expression of pro-inflammatory cytokine genes. NF-κB is normally sequestered in the cytosol by inhibitor of kappaB binding (IκB), but TLR activation stimulates IκB kinase (IKK), which phosphorylates IκB, leading to its degradation and release of NF-κB. Early experiments with RAW264.7 macrophages showed that treatment with nicotine attenuated the effects of LPS and peptidoglycan to cause nuclear translocation and activation of NF-κB (Wang et al., 2004). Later work confirmed these findings using human monocytes and U937 cells, and this group further demonstrated that pretreatment with nicotine inhibited upstream phosphorylation of IκB by LPS (Yoshikawa et al., 2006). There is also evidence that α7nAChRs on peritoneal macrophages can inhibit LPS-evoked release of inflammatory cytokines by a mechanism that involves recruitment of Jak2 to the nicotinic receptor, phosphorylation of STAT3 and translocation of pSTAT3 to the nucleus where it binds to DNA and inhibits cytokine production (de Jonge et al., 2005). The latter finding appears at odds with one report, which suggests that anti-inflammatory effects of α7 receptors are mediated by unphosphorylated STAT3 (Peña et al., 2010). However, all of these studies agree in showing that α7nAChR activation can reduce the release of inflammatory cytokines from macrophages by inhibiting NF-κB promoter activity.
Given the complexity of mechanisms for regulation of cytokine synthesis and release, it is not surprising that recent studies have identified alternative or additional signaling pathways that can contribute to the anti-inflammatory effect of α7 agonists on macrophages. One extensive study has implicated IRAK-M (Maldifassi et al., 2014), which is a recognized negative regulator of TLR receptor evoked pro-inflammatory cytokine responses that acts early in the TLR receptor signaling cascade. Pretreatment of human and mouse macrophages with nicotine caused a concentration-dependent increase in IRAK-M mRNA and protein levels, which was mediated by α7 receptors. Furthermore, upregulation of IRAK-M by a lower concentration of nicotine was potentiated by the allosteric modulator, PNU-120596 (Maldifassi et al., 2014). Knockdown of IRAK-M reduced the ability of nicotine to attenuate LPS-evoked release of TNF-α, thereby implicating IRAK-M in the cholinergic anti-inflammatory effect.
Another study implicated microRNA-124 in α7 receptor mediated anti-inflammatory signaling in macrophages (Sun et al., 2013b). This work, which employed macrophage cell lines and thioglycollate-elicited peritoneal macrophages, showed that nicotine suppressed the release of IL-6 and TNF-α but only decreased levels of IL-6 mRNA. These effects were associated with an α7 receptor mediated increase in miRNA-124. Further experiments provided evidence that miRNA-124 reduced IL-6 mRNA by inhibition of the STAT3 pathway and reduced TNF-α protein levels by a post-translational mechanism involving inhibition of proTNF-α conversion to TNF-α (Sun et al., 2013b).
5. Impact of cholinergic therapy and α7nAChRs in models of sepsis and burn injury
Endotoxemia, sepsis, burn injury, and trauma can all lead to a systemic inflammatory response due to excessive activation of the innate immune system (Binkowska et al., 2015; Iskander et al., 2013; Sherwood and Toliver-Kinsky, 2004). The spleen plays a central role in the pathophysiology of the hyperinflammatory state triggered by these threatening conditions, and splenic macrophages are a dominant source of pro-inflammatory cytokines. Accordingly, there has been much interest in defining the effect of cholinergic anti-inflammatory therapies in animal models of these conditions.
5.1. Endotoxemia and sepsis
Most experimental studies that examined the effects of cholinergic anti-inflammatory strategies in the context of systemic inflammation have utilized models of endotoxemia induced by intraperitoneal administration of LPS or sepsis induced by cecal ligation and puncture (CLP). Endotoxemia is a rapid onset model of inflammation generally produced by intraperitoneal injection of LPS. While it is sometimes referred to as “sterile” peritonitis, intestinal barrier function is disrupted and translocation of intestinal bacteria occurs (Wells et al., 1992). CLP produces polymicrobial peritonitis and bacteremia. Early studies in this field found that treatment with nicotine 30 min before injection of a lethal dose of LPS and for 3 days afterward caused a dose-dependent increase in 8-day survival (Wang et al., 2004). Furthermore, delayed treatment of septic mice with nicotine beginning 24 h after CLP increased 21-day survival compared to vehicle treated mice. The efficacy of delayed nicotine treatment was attributed to its ability to suppress the release of high mobility group box 1 (HMGB1) from macrophages (Wang et al., 2004). This effect is noteworthy since many inflammatory cytokines are already elevated before 24 h post-CLP and delayed initiation of cholinergic anti-inflammatory therapy is anticipated in a clinical setting. Analogous studies latter implicated α7nAChRs by showing that treatment with GTS-21, an α7 partial agonist, also inhibited release of HMGB1 into the circulation and increased survival in endotoxemia and CLP models (Pavlov et al., 2007). Further studies by the same group demonstrated that choline, an endogenous α7nAChRs agonist, can elicit the same beneficial effects on survival in endotoxemia and CLP models (Parrish et al., 2008). Choline was given by intraperitoneal injection in these experiments and was effective as a pretreatment in the endotoxemia model or as twice daily therapy for three days started 24 h after surgery in the CLP model. Parallel cell culture experiments showed that choline caused an α7nAChR-mediated, concentration-dependent inhibition LPS-evoked release of TNF-α from peritoneal macrophages. As expected, choline had a lower potency than ACh but had comparable efficacy. Since choline is well tolerated, these findings suggest a potential therapeutic role for this endogenous α7nAChR agonist in treating inflammatory diseases (Parrish et al., 2008).
Several studies have demonstrated that upstream activation of cholinergic anti-inflammatory mechanisms in the spleen is also able to reduce circulating levels of pro-inflammatory cytokines and to increase survival in mice with endotoxemia or CLP-induced sepsis. Stimulation of M1 muscarinic receptors in the CNS activates the cholinergic anti-inflammatory pathway, and treatment with xanomeline can increase survival in LPS-induced endotoxemia and CLP-induced sepsis by this mechanism (Rosas-Ballina et al., 2015). Increased survival in the sepsis model occurred after delayed treatment with xanomeline started at 20 h after CLP surgery but had no effect when the same doses were given 20 h before CLP. Administration of a higher dose of xanomeline 20 h before surgery actually decreased survival, suggesting possible over activation of the anti-inflammatory circuit. Pretreatment with galantamine, a reversible cholinesterase inhibitor, increased survival in LPS-treated mice and reduced acute lung injury (Li et al., 2016; Pavlov et al., 2009). The effects of galantamine to increase survival and reduce serum TNF in LPS-treated mice were attributed to central inhibition of acetylcholinesterase and activation of the vagal anti-inflammatory pathway via stimulation of central muscarinic receptors (Pavlov et al., 2009). This conclusion was supported by experiments showing that the beneficial effects of galantamine were blocked by atropine sulfate, which enters the CNS, but not by atropine methyl nitrate, which has poor access to the brain. Other work has shown that two classical cholinesterase inhibitors, physostigmine and neostigmine, increase survival in mice with endotoxemia or CLP-induced sepsis (Hofer et al., 2008). Physostigmine readily enters the CNS, so it inhibits both central and peripheral cholinesterase. Neostigmine, on the other hand, is a quaternary amine and usually inhibits only peripheral cholinesterase, since the blood-brain barrier (BBB) excludes it from the CNS. However, disruption of the BBB occurs in sepsis (Danielski et al., 2017), allowing neostigmine to enter and also inhibit cholinesterase in the CNS.
While direct stimulation of the vagus nerve is not a practical approach for activation of cholinergic anti-inflammatory mechanisms during sepsis and systemic hyperinflammatory states, there is evidence that the vagal pathway can be activated mechanically by transcutaneous nerve stimulation (Huston et al., 2007). This noninvasive approach increased survival, reduced clinical sickness, and attenuated serum HMGB1 levels in mice with CLP-induced sepsis (Huston et al., 2007).
5.2. Burn injury
Burn injury that is extensive can trigger a systemic inflammatory response and multiple organ dysfunction such as seen in sepsis (Sherwood and Toliver-Kinsky, 2004). Several approaches have been used to determine if the cholinergic anti-inflammatory pathway and mechanisms can be targeted to attenuate inflammation and tissue damage in models of burn injury.
Application of bilateral VNS for 12 min, when initiated 60 min after thermal burn injury, prevented burn-induced increases in serum levels of TNF-α, IL-1β, and IL-6 and reduced levels of the same cytokines in the heart and liver at 3 h post-injury (Niederbichler et al., 2010). Other investigators confirmed this work using unilateral stimulation of the left vagus nerve, and further demonstrated VNS reduced pulmonary damage following burn injury (Song et al., 2010). Beneficial effects of VNS were mediated by nicotinic receptors since they were blocked by hexamethonium but unaffected by atropine. Consistent with nicotinic receptor mediation of responses to VNS, transdermal administration of nicotine to rats at the time of burn injury attenuated or inhibited elevation of tissue pro-inflammatory cytokine levels, normalized blood pressure, and improved myocardial contractile function (Claassen et al., 2014; Claassen et al., 2015). While these were primarily acute studies, other investigators have demonstrated that treatment with GTS-21 increases 7-day survival in burned mice to 100% as compared to 25% in saline treated mice (Khan et al., 2012). Furthermore, in another study, GTS-21 treatment attenuated body weight loss and the decrease in skeletal muscle mass measured 3 days after burn injury (Kashiwagi et al., 2016).
6. Cholinergic anti-inflammatory therapy in experimental models of rheumatoid arthritis
There is a wealth of preclinical data that supports targeting of the cholinergic anti-inflammatory pathway in arthritis. Several rodent models of autoimmune arthritis are available that occur either spontaneously (i.e., mutations or transgenic mice) or are induced experimentally by injection of antigenic substance such as type II collagen (Moudgil et al., 2011). Collagen-induced arthritis has been used most commonly in cholinergic studies and has many characteristics similar to human disease including joint swelling, inflammation, pannus formation, destruction of cartilage, erosion of bone, and elevation of serum cytokines.
Shortly after discovery of the vagal anti-inflammatory pathway, clinical studies revealed and inverse relationship between vagal nerve activity, indexed by heart rate variability, and levels of inflammatory markers (e.g., HMGB1) in the serum of rheumatoid arthritis patients (Goldstein et al., 2007). A possible link between insufficient vagal activity and inflammatory disease in patients with arthritis was suggested. Recent work goes further providing evidence that reduced vagal function actually precedes the development of rheumatoid arthritis in patients determined to be at risk (Koopman et al., 2016). These observations have broad implications since vagal tone is decreased in a wide range of diseases and virtually all have an inflammatory component (Thayer et al., 2010; Thayer and Sternberg, 2006).
The first study to target cholinergic anti-inflammatory mechanisms in an animal model of arthritis evaluated the therapeutic response to nicotine and a selective α7 agonist (AR-R17779) in collagen-induced arthritis in mice (van Maanen et al., 2009). These investigators found that both drugs reduced clinical signs of arthritis, decreased joint swelling, reduced TNF-α expression in the synovium, decreased bone erosion and cartilage loss, and lowered serum levels of inflammatory cytokines (i.e., TNF-α and IL-6). Other studies confirmed these finding and demonstrated that the severity of arthritis was increased by vagotomy (Li et al., 2010) and deletion of the α7nAChR (van Maanen et al., 2010). Direct activation of the cholinergic anti-inflammatory pathway by VNS has also been tested using a rat model of collagen-induced arthritis (Levine et al., 2014). For this work, a custom electrode was implanted and the left vagus nerve was stimulated for 60 s once daily for 7 days. VNS reduced joint swelling and inflammation, cartilage damage, and bone resorption. Collectively, this work provides strong evidence that therapeutic responses can be evoked in experimental arthritis by direct stimulation of α7 receptors or by activating the vagal anti-inflammatory pathway. Potential targets within the inflamed joints include macrophages and fibroblast-like synoviocytes that express α7 receptors (Koopman et al., 2014).
7. Vagal anti-inflammatory pathway to the gut
7.1. Neuroanatomy
The gastrointestinal (GI) tract, which presents a huge area for exposure to microbes and foreign substances, is an important site for immune surveillance and a major target for vagal efferent nerve projections. These preganglionic vagal nerve fibers emanate from cholinergic neurons located in the brain stem in the dorsal motor nucleus and the nucleus ambiguus of the medulla, and they provide preganglionic cholinergic input to the ganglia of the enteric nervous system, where nicotinic receptors mediate ganglionic neurotransmission (Furness et al., 2014; Furness, 2012; Van Der Zanden et al., 2009). Postganglionic cholinergic neurons located in the myenteric and submucosal plexuses of the enteric nervous system send postganglionic nerve projects to effector tissues in the mucosal, submucosal and muscular layers of the gut. These cholinergic projections play well established roles in controlling motor and secretory functions of the GI tract, primarily through stimulation of muscarinic receptors on effector cells. The GI tract also contains abundant vagal afferent nerve fibers that can be activated to reflexly increase vagal efferent tone. Recent studies have shown that vagal input to the GI tract can also have important effects to modulate immune homeostasis (Matteoli and Boeckxstaens, 2013). Strong evidence for a separate cholinergic anti-inflammatory pathway to the gut has accrued from elegant studies of disease models in animals.
7.2. VNS prevents postoperative ileus
The first evidence for a separate vagal anti-inflammatory pathway to the GI tract was provided by studies showing that VNS prevented postoperative ileus in an animal model (Matteoli et al., 2014). This model entails controlled manipulation of the intestines, as might occur during surgery, and results in inflammation of the smooth muscle layers, which leads to impaired GI motor function with decreased propulsion. Surgical manipulation of the gut activates resident macrophages in the muscularis externa causing release of inflammatory cytokines, upregulation of adhesion molecules, and influx of neutrophils and other leukocytes. Application of unilateral VNS for 5 min before intestinal manipulation prevented this entire cascade of reactions and preserved GI motor function (Matteoli et al., 2014). Thus, resident macrophages play a crucial role in the process. Further experiments showed that the anti-inflammatory response was unaffected by splenectomy, so VNS prevented postoperative ileus by a direct pathway affecting the GI immune system. Furthermore, therapeutic effects of VNS persisted in Rag-1−/− mice, which lack mature T and B cells. Collectively, this evidence pointed to an effect of VNS on resident macrophages.
A series of further experiments provided definitive evidence that α7nAChRs on resident macrophages are the ultimate target for prevention of postoperative ileus by VNS (Matteoli et al., 2014). First it was shown that effects of VNS in this model were absent in 7nAChR knockout mice. Next, bone marrow chimera studies proved that the α7nAChRs were localized to immune cells rather than ganglia in the enteric nervous system, and resident macrophages were found to express much higher levels of α7nAChR mRNA than other immunes cells in the region. Finally, sophisticated immunohistochemical experiments showed that 1) postganglionic cholinergic nerve fibers form networks around resident macrophages, 2) resident macrophages bind FITC-labeled -bungarotoxin (an α7nAChR antagonist) and 3) both selective and non-selective α7 agonists increased [Ca2+]i of resident macrophages in situ (Matteoli et al., 2014). Thus, VNS acts directly through enteric cholinergic neurons to regulate immune function (homeostasis) in the gut (Fig. 3).
7.3. Influence of cholinergic anti-inflammatory mechanisms in inflammatory bowel disease
Inflammatory bowel diseases (IBD) include ulcerative colitis and Crohn’s disease, and the influence of cholinergic interventions has been investigated in both of these disorders. There are significant differences in the pathophysiology of these diseases, which may influence their susceptibility to cholinergic anti-inflammatory approaches. While both have an inflammatory root, inflammation is largely limited to the submucosa in ulcerative colitis but occurs across the bowel wall in Crohn’s disease (Matteoli and Boeckxstaens, 2013).
Evidence from preclinical studies and clinical practice suggests that cholinergic anti-inflammatory therapies could be beneficial in the treatment of colitis. Clinical experience has shown that smoking has beneficial effects in patients with ulcerative colitis whereas disease exacerbation can occur in patients with Crohn’s disease (Matteoli and Boeckxstaens, 2013). These effects are generally attributed to the nicotine content of tobacco. Animal models of colitis can be produced by placing 2,4,6-trinitrobenzene sulfonic acid (TNBS) into the lumen of the colon or by addition of dextran sulfate sodium (DSS) to the drinking water. Experiments using the TNBS model in rats have shown that VNS (3 h per day for 5 days) in freely moving animals or pretreatment with cholinesterase inhibitors (i.e., neostigmine and physostigmine) reduced the severity of colitis (Meregnani et al., 2011; Miceli and Jacobson, 2003). Using these models in mice, it was found that disease severity was increased after subdiaphragmatic bilateral vagotomy or removal of the splenic nerve to eliminate sympathetic nerves in the spleen (Munyaka et al., 2014; O’Mahony et al., 2009). Furthermore, activation of the vagal anti-inflammatory pathway to the spleen by injection of the M1 muscarinic receptor agonist, McN-A-343, into the cerebral ventricles reduced disease severity (Munyaka et al., 2014). Treatment with McN-A-343 commenced one days before inducing colitis and continued for the duration of study. Therapeutic effects achieved by central activation of the vagal nerves were eliminated by splenectomy. Thus, the beneficial effects of vagal stimulation in experimental colitis are mediated by the cholinergic anti-inflammatory pathway to the spleen (Munyaka et al., 2014) rather than the direct pathway to the gut (Matteoli et al., 2014).
Several lines of evidence have implicated nicotinic receptors as mediators of the vagal anti-inflammatory response in experimental colitis. Exacerbation of colitis after vagotomy was reversed by treatment with nicotine, and treatment of normal mice with the α7 agonist choline decreased the severity of DSS colitis (Ghia et al., 2006; Ghia et al., 2009). Severity of DSS colitis was increased in α7nAChR knockout mice (Ghia et al., 2009), further implicating this receptor in cholinergic anti-inflammatory mechanisms. However, other evidence suggests that selective α7 agonists may worsen or have no benefit in female mice with experimental colitis (Snoek et al., 2010). The latter finding might be explained, at least in part, by recent work demonstrating that α7 agonists are less efficacious in female mice with colitis (AlSharari et al., 2017).
Studies investigating the cellular and molecular mechanisms of spleen involvement in the cholinergic anti-inflammatory responses in experimental colitis have focused on dendritic cells and their ability to prime T cells (Munyaka et al., 2014) and on the presence of specific subtypes of T cells in the submucosa of the colon (Salaga et al., 2016). This work demonstrated that in vivo stimulation of α7nAChRs on dendritic cells via central vagal stimulation decreased release of Th1/Th17 pro-inflammatory cytokines from these cells in vitro and impaired their ability to prime T cells. It remains unclear how anti-inflammatory signals are transferred to the gut, but it most likely involves trafficking of cells from the spleen. Recent work has in fact demonstrated that treatment with encenicline, a partial agonist at the α7nAChR, reduced the infiltration of macrophages, neutrophils and B cells into the mucosa and submucosa of the colon in TNBS induced colitis in mice (Salaga et al., 2016). Furthermore, while the total number of T cells infiltrating the colon was unaffected by encenicline, the drug did reduce the abundance of FoxP3+ IL-17A+ T cells, which have a pro-inflammatory role in IBD.
8. Targeting α7nAChRs on microglia and macrophages in stroke and traumatic brain injury
8.1 Activated microglia express α7nAChRs and contribute to CNS pathology
Microglia are resident mononuclear phagocytes in the CNS and have a different origin from most peripheral macrophages (Dey et al., 2015). They are associated closely with neurons, and play important roles in development, maintenance, and homeostasis (Dey et al., 2015; Graeber et al., 2011). Nevertheless, activated microglia and macrophages have many properties in common, including the production and release of inflammatory cytokines that are known to be cytotoxic (Dey et al., 2015; Kalkman and Feuerbach, 2016; Shytle et al., 2004). Accordingly, it should not be surprising that a large body of literature suggests that activated microglia are key players in a wide range of CNS disorders (Graeber et al., 2011; Kalkman and Feuerbach, 2016). While some of these disorders do not involve inflammation in the classical sense (Graeber et al., 2011), there is the common thread that microglial activation to the M1 phenotype contributes to pathology (Graeber et al., 2011; Kalkman and Feuerbach, 2016). Furthermore, activated microglia are like macrophages in the expression of α7nAChRs that mediate inhibition of LPS-induced TNF-α release (Shytle et al., 2004). Thus, cholinergic anti-inflammatory mechanisms identified in the periphery may also operate in the CNS through stimulation of α7nAChRs on activated microglial cells and on macrophages that infiltrate when the blood brain barrier is disrupted. Although beyond the scope of this review, there is also evidence that neuroprotection can be achieved by stimulation α7nAChRs on neurons (Akaike et al., 2010). This review will focus on the role of α7nAChRs in immune responses to cerebral ischemia/stroke and traumatic brain injury.
8.2. Role of α7nAChRs is stroke
Cerebral ischemia that is produced by stroke triggers a robust inflammatory response in the brain (Guan et al., 2015) and a systemic inflammatory response (Mravec, 2010). Microglia, major resident immune cells in the brain, are activated, proliferate, and begin secreting large amounts of inflammatory cytokines that contribute to neuronal death (Guan et al., 2015). Studies using a rat model of transient forebrain ischemia showed that nicotine treatment, given 2–12 h after ischemia, decreased the death of hippocampal CA1 neurons when evaluated 7 days after injury (Guan et al., 2015). This neuroprotective effect of nicotine was associated with a marked attenuation of the ischemia induced increase of microglial cells and inflammatory cytokines (i.e., TNF-α and IL-1β) in the region. Further experiments showed that nicotine reduced proliferation of microglial cells in primary culture, and this effect was blocked by α-bgt, suggesting mediation by α7nAChRs. Other investigators have shown that treatment of mice with α7 agonists (i.e., PHA568478 or PNU-282987) immediately after permanent occlusion of the distal middle cerebral artery (Han et al., 2014) or one hour after induction of photothrombic stroke (Parada et al., 2013) reduced infarct size and functional deficits. In contrast, blockade of α7nAChRs with the selective antagonist, methyllycaconitine, increased infarct size, suggesting some degree of α7 stimulation by endogenous agonist (Han et al. 2014). Other investigators have suggested that endogenous choline that is released from injured brain tissue might fill this role (Sun et al., 2013a; Sun et al., 2017). These investigators found that treatment with PNU-120596, an allosteric modulator of α7nAChRs, decreased infarct size and improved performance on several neurological tests when given 6 h after transient occlusion of the middle cerebral artery in mice (Sun et al., 2013a). Subsequent work demonstrated that intranasal administration of PNU-120596 had similar therapeutic efficacy using the same stoke model in rats (Sun et al., 2017).
The relative contributions from stimulation of α7nAChRs on activated microglia cells and infiltrated macrophages are difficult to discern since they express similar markers. Nevertheless, therapeutic effects of the selective α7 agonist PHA568478 in the permanent middle cerebral artery occlusion model were associated with a decrease in the number of microglia/macrophages expressing the pro-inflammatory M1 phenotype in the peri-infarct zone at 3 and 14 days post-occlusion and an increase in number of cells with the anti-inflammatory M2 phenotype at 3 days post-occlusion (Han et al., 2014). These changes in cellular profiles were paralleled by corresponding changes in expression of M1 and M2 genes in the peri-infarct region and a gene expression profile favoring reduced oxidative stress. More direct evidence supporting a therapeutic role of α7 receptors on microglial cells comes from studies of oxygen and glucose deprivation followed by return of oxygen and glucose in organotypic hippocampal cultures (Parada et al., 2013). This model mimics ischemia/reperfusion in whole animal experiments. Treatment with PNU-282987 in this model reduced death of CA1 neurons by a mechanism that involved decreased TNF-α release, decreased production of reactive oxygen species, and increased expression of heme oxygenase-1 expression. Depletion of microglia from these preparations, by treatment with a microglial cell specific immunotoxin, increased neuronal death caused by oxygen and glucose deprivation and attenuated the neuroprotection and increased heme oxygenase-1 expression evoked by the α7 agonist.
Collectively, these observations are consistent in showing that treatment with an α7 agonist or positive allosteric modulator within a critical window after stroke in animal models can increase neuronal survival and neurological function. Several studies have provided evidence that activation by α7nAChRs on microglia/macrophages contributes to these therapeutic effects.
8.3. Role of α7nAChRs is traumatic brain injury
The pathophysiology of traumatic brain injury (TBI) has may similarities to stoke, and the rationale for targeting α7nAChRs is identical. TBI results in primary neuronal damage, which is unavoidable, and delayed secondary damage, which might be reduced by early therapeutic intervention (Gatson et al., 2015). In addition to causing primary neuronal damage, the initial insult to the brain activates microglial cells and astrocytes at the site of injury and triggers a systemic inflammatory response (Dash et al., 2016). Thus, damaging pro-inflammatory cytokines are produced by activated microglial cells at the site of injury and in the periphery as part of the systemic inflammatory response. Importantly, TBI disrupts the BBB and thereby allows infiltration of circulating inflammatory cells, inflammatory cytokines, and circulating fluids (Dash et al., 2016). Therapies directed at activating α7nAChRs might be beneficial through a direct neuroprotective effect on specific neuronal populations, by suppressing the pro-inflammatory phenotype in activated microglial cells, and by attenuating the systemic inflammatory response (Kelso and Oestreich, 2012).
Studies using nicotine treatment as a therapeutic approach in experimental TBI showed no benefit, possibly due to desensitization of α7 receptors (Gatson et al., 2015; Kelso and Oestreich, 2012). However, recent work using the positive allosteric modulator PNU-120596 have yielded encouraging results (Gatson et al., 2015). This agent works by enhancing the potency of the endogenous α7 receptor agonist, choline, which is elevated in the brain after TBI, while also decreasing receptor desensitization. Treatment of rats with this agent 5 min after TBI caused a marked decrease in neuronal death and astrocyte activation when evaluated 72 h later. Additional studies are needed to determine the cellular mechanism(s) underlying this effect (neuroprotection vs anti-inflammatory), to determine if neurological function is improved, the extent that treatment can be delayed while still getting therapeutic benefit, and the longevity of the therapeutic response. Another recent study provided evidence that intraperitoneal treatment with PNU-282987 (selective agonist) or PNU120596 (positive allosteric modulator) at 30 min, 12 h, and 20 h after TBI can reduce BBB permeability and the infiltration of neutrophils and macrophages by suppressing systemic inflammation (Dash et al., 2016). These effects required the activation of α7 receptors in the spleen, suggesting that they were mediated at least in part by peripheral cholinergic anti-inflammatory mechanisms.
9. Clinical studies and trials
Discovery of the vagal anti-inflammatory pathway has sparked intensive basic research to identify molecular, cellular, and systemic mechanisms that contribute to the anti-inflammatory response and to test the translational potential of these findings in animal models of diseases having a prominent inflammatory component. Although there is some disagreement in the field, it is widely accepted that anti-inflammatory responses can be elicited by VNS and by stimulation of α7nAChR on macrophages. Success in some of these preclinical studies has motivated movement into the clinical arena were recent and ongoing work is evaluating the safety and efficacy of cholinergic anti-inflammatory therapies in humans. Two approaches are being evaluated: bioelectrical stimulation and drug therapy.
9.1. Bioelectrical stimulation
Bioelectronic medicine is a growing field with VNS being a major therapeutic approach. Chronic VNS has been used clinically for over two decades, and is currently approved for treatment-refractory epilepsy and for chronic depression that is resistant to other therapies (Howland, 2014). VNS has a good record of safety and efficacy for these conditions, so it is not surprising that several clinical trials are evaluating VNS for other conditions such as heart failure and chronic inflammatory diseases such as IBD and rheumatoid arthritis. The current FDA-approved protocol requires general anesthesia for about 1 h of surgery needed to implant a pulse generator, run subcutaneous wires, and place the stimulation lead around either the left or right cervical vagus nerve. The left vagus nerve is stimulated for treatment of epilepsy and depression because it is thought to have less influence on cardiac rate than the right vagus nerve (Howland, 2014), but stimulation of either right or left vagus nerve appears to be safe and effective in patients with chronic heart failure (Premchand et al., 2014; Premchand et al., 2016). Stimulation of the vagus is phasic (e.g., 30 sec on and 5 min off) but continuous, and it is generally well tolerated. Therapeutic responses in epilepsy and depression are mediated by increased afferent input to the CNS. Given the need for surgery and chronic stimulation, this approach is most suitable for application in chronic inflammatory diseases such as IBD and rheumatoid arthritis.
Several clinical trials are in progress to evaluate VNS in patients with IBD, rheumatoid arthritis, and other chronic diseases (Table 3). While it is far too early to reach definitive conclusions, some encouraging results have been reported. The first evidence for a beneficial effect of VNS in Crohn’s disease was a case report documenting maintained remission in a patient with long-standing disease after continuous VNS for 12 months (Clarençon et al., 2014). More recently, the same group reported that 5 out of 7 patents with moderate to severe Crohn’s disease showed clinical, biological, and endoscopic improvement after 6 months of VNS at 10Hz and a duty cycle of 30 s ON and 5 min OFF (Bonaz et al., 2016a). Based on changes in the EEG and ECG, the authors concluded that VNS produced effects through stimulation of vagal afferent and efferent nerves (Clarençon et al., 2014). Recently, another group has reported that VNS attenuates production of inflammatory cytokines in patients with rheumatoid arthritis and decreases disease severity (Koopman et al., 2016). As a prelude to this study, the investigators first evaluated the effect of acute VNS on LPS-induced release of inflammatory cytokines from monocytes in whole blood. This experiment was done in epilepsy patients at the time when vagal stimulators were implanted. A single period of VNS for 30 s during surgery causes a significant reduction in the release of TNF, IL-6, and IL-1β from whole blood monocytes when evaluated 4 h later. Two cohorts of rheumatoid arthritis patients with active disease were evaluated in this study. The first cohort had active disease while taking methotrexate and either did not receive a TNF antagonist or could not tolerate it. The second cohort failed on traditional therapy and treatment with at least two unique biological agents. Both cohorts had significant improvement in the standard disease composite score after VNS for 5 weeks, regressed when stimulation was stopped for two weeks, and improved again when stimulation was resumed. It is noteworthy that stimulation was delivered 1–4 times daily for 60 s in this study (i.e., maximum of 240 s per day).
Table 3.
Clinical trial
| Intervention | Condition | ClinicalTrials.gov Identifier |
|---|---|---|
| VNS vs prucalopride | Postoperative ileus | NCT02425774 |
| VNS | Crohn’s disease |
NCT02311660 NCT02951650 NCT01569503 |
| Transcutaneous VNS | Irritable bowel syndrome | NCT02420158 |
| VNS | Rheumatoid arthritis |
NCT01552941 NCT01552538 |
| Galantamine | Metabolic syndrome | NCT02283242 |
| Transcutaneous VNS | Systemic Lupus Musculoskeletal Pain |
NCT02822989 |
| Neostigmine methylsulfate Raceanisodamine HCl | Prevent/treat SIRS* | NCT02279147 |
SIRS, systemic inflammatory response syndrome
Other approaches to VNS, which are less invasive or non-invasive are also under investigation in clinical trials. Transvenous vagus stimulation is a less invasive approach that utilizes a catheter electrode inserted into the left jugular vein to stimulate the adjacent vagus nerve (Kox et al., 2015). Using this approach, researchers found the VNS for 30 min at 20 Hz did not affect the innate immune response triggered by intravenous injection of LPS in normal volunteers (Kox et al., 2015). Other non-invasive systems are designed to provide transcutaneous VNS (Akdemir and Benditt, 2016; Bonaz et al., 2016b, c; Howland, 2014). The most common of these non-invasive systems is inserted in the outer ear and works by stimulation of the auricular branch of the vagus nerve. Using this device, investigators have demonstrated efficacy at reducing sympathetic nerve activity in normal volunteers (Clancy et al., 2014). Another device, GammaCore (electroCore LLC, Basking Ridge, NJ) is hand-held and uses two disc electrodes for transcutaneous stimulation of the cervical vagus nerve (Bonaz et al., 2016c). Use of such non-invasive devices in treating chronic inflammatory diseases will need to consider patient compliance, which is also a concern with drug therapy (Bonaz et al., 2016c). The clinical efficacy of these approaches is currently under evaluation.
9.2. Pharmacologic activation of cholinergic anti-inflammatory pathway
Based on the preclinical evidence presented, there are several potential ways to activate cholinergic anti-inflammatory mechanisms pharmacologically. The most direct of these would use nicotine, selective α7nAChR agonists, or positive allosteric modulators of the α7nAChR. All of these would target α7nAChRs on macrophages and dendritic cells, which are a major source of pro-inflammatory cytokine release. Although, there is epidemiological data showing that nicotine (i.e., via smoking) is beneficial in some cases of IBD, use of nicotine therapeutically would not be viable due to adverse consequence from non-selective activation of all nicotinic receptors. Selective α7 agonists and positive allosteric modulators are a better option since they are expected to have fewer off-target actions. In addition to acting on immune effector cells in the spleen and other tissues (e.g., resident macrophages), these agents also have the potential to stimulate sympathetic neurons in the cholinergic anti-inflammatory pathway and thereby increase noradrenergic stimulation of cholinergic T cells in the spleen. The efficacy of positive allosteric modulators would be dependent on endogenous ACh or choline provided by reflex activation of cholinergic anti-inflammatory pathways. Another theoretical approach would be to combine lower doses of an α7nAChR agonist or partial agonist with a positive allosteric modulator. Lastly, it may be possible to activate cholinergic pathways by using centrally active drugs such as a M1 muscarinic receptor agonist, galantamine, or other cholinesterase inhibitors. However, the anti-inflammatory efficacy of cholinesterase inhibitors that act exclusively in the periphery has not been demonstrated. Theoretically, these agents might augment cholinergic anti-inflammatory responses in the periphery by enhancing ACh stability at ganglia, cholinergic T cell-macrophage interactions in the spleen, and cholinergic nerve-macrophage interactions in the gut. So far, only a few of these approaches have been tested clinically.
Published clinical studies specifically designed to evaluate cholinergic anti-inflammatory drug actions have been limited to the evaluation of effects of nicotine and GTS-21 on the innate immune response to LPS in normal volunteers (Kox et al., 2011; Wittebole et al., 2007). Both of these studies were unable to detect an effect of either agonist on plasma levels of pro-inflammatory cytokines during endotoxemia. However, there was an inverse correlation between the plasma levels (area under the curve) of GTS-21 and pro-inflammatory cytokines, supporting the suggestion that GTS-21 was having an effect at the individual level (Kox et al., 2011). Low, possibly sub-therapeutic, concentrations of agonist in the circulation as well as design issues, could have been factors in both of these studies (Kox et al., 2011).
10. Conclusions and future directions
The past two decades have seen tremendous advances in our understanding of neuroimmune mechanisms for maintaining and restoring homeostasis during normal and pathophysiologic conditions. Basic reflex mechanisms for sensing adverse conditions and responding to them through efferent vagal and sympathetic circuits have been identified, and many direct effects of neurotransmitters on leukocytes have been clarified with important clinical implications. Three major discoveries have been 1) the identification of anti-inflammatory responses to VNS, 2) the integration of cholinergic T cells into the efferent neuroimmune pathway within the spleen and 3) identification of α7nAChRs on macrophages, dendritic cells, and microglial cells as targets for suppression of inflammation by α7 selective drugs or by ACh derived from immune cells or neurons. These discoveries and a wealth of supporting basic science data have resulted in many translational studies in animal models of inflammation and several ongoing clinical trials. Furthermore, it is now recognized that vagal tone is reduced in a wide range of diseases that have an inflammatory component (Thayer and Sternberg, 2006; Bonaz et al., 2016) and that reduced vagal tone could be a risk factor associated with pathogenesis (Bonaz, 2016; Koopman et al., 2016). Therefore, assessing and normalizing vagal tone could be a major therapeutic strategy. Continued basic and pharmaceutical research into the function and pharmacology of α7nAChRs, including positive allosteric modulators and the human-specific dupα7 subunit, has the promise of providing novel therapeutics for CNS and inflammatory disorders. However, much remains to be learned in this field. For example, the roles of cholinergic B and T cells, which are abundant mobile sources of ACh, need to be determined as do specific target cells and their cholinergic receptors. Overall, it is likely we have just touched the surface in revealing the complexities and potential therapeutic targets presented by neuroimmune interactions.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (NIH R15GM107949) and NIH C06RR0306551
Abbreviations
- Ach
acetylcholine
- α-bgt
α-bungarotoxin
- BBB
blood-brain barrier
- CNS
central nervous system
- ChAT
choline acetyltransferase
- DSS
dextran sulfate sodium
- eGFP
enhanced green fluorescent protein
- GI
gastrointestinal
- HMGB1
high mobility group box 1
- IBD
inflammatory bowel disease
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- IL-10
interleukin-10
- IκB
inhibitor of kappaB binding
- IKK
IκB kinase
- IRAK-M
interleukin-1 receptor-associated kinase-M
- Jak2
Janus kinase 2
- LPS
lipopolysaccharide
- NE
norepinephrine
- α7nAChR
α7 nicotinic ACh receptor
- NF-κB
nuclear factor-kappaB
- STAT3
signal transducer and activator of transcription 3
- TBI
traumatic brain injury
- TLR
Toll-like receptor
- TM
transmembrane
- TNBS
2,4,6-trinitrobenzene sulfonic acid
- TNF-α
tumor necrosis factor-α
- VNS
vagal nerve stimulation
- VAChT
vesicular ACh transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The author declares that there are no conflicts of interest.
References
- Akaike A, Takada-Takatori Y, Kume T, Izumi Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of α4 and α7 receptors in neuroprotection. J Mol Neurosci. 2010;40:211–216. doi: 10.1007/s12031-009-9236-1. [DOI] [PubMed] [Google Scholar]
- Akdemir B, Benditt DG. Vagus nerve stimulation: An evolving adjunctive treatment for cardiac disease. Anatol J Cardiol. 2016;16:804–810. doi: 10.14744/AnatolJCardiol.2016.7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandraki K, Piperi C, Kalofoutis C, Singh J, Alaveras A, Kalofoutis A. Inflammatory process in type 2 diabetes: The role of cytokines. Ann N Y Acad Sci. 2006;1084:89–117. doi: 10.1196/annals.1372.039. [DOI] [PubMed] [Google Scholar]
- AlSharari DS, Bagdas D, Akbarali HI, Lichtman PA, Raborn ES, Cabral GA, Carroll FI, McGee EA, Damaj MI. Sex differences and drug dose influence the role of the α7 nicotinic acetylcholine receptor in the mouse dextran sodium sulfate-induced colitis model. Nicotine Tob Res. 2017;19:460–468. doi: 10.1093/ntr/ntw245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med. 2012;209:1057–1068. doi: 10.1084/jem.20120571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araud T, Graw S, Berger R, Lee M, Neveu E, Bertrand D, Leonard S. The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of α7*nAChR function. Biochem Pharmacol. 2011;82:904–914. doi: 10.1016/j.bcp.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artico M, Bosco S, Cavallotti C, Agostinelli E, Giuliani-Piccari G, Sciorio S, Cocco L, Vitale M. Noradrenergic and cholinergic innervation of the bone marrow. Int J Mol Med. 2002;10:77–80. [PubMed] [Google Scholar]
- Báez-Pagán AC, Delgado-Vélez M, Lasalde-Dominicci JA. Activation of the macrophage α7 nicotinic acetylcholine receptor and control of inflammation. J Neuroimmune Pharmacol. 2015;10:468–476. doi: 10.1007/s11481-015-9601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajayo A, Bar A, Denes A, Bachar M, Kram V, Attar-Namdar M, Zallone A, Kovács KJ, Yirmiya R, Bab I. Skeletal parasympathetic innervation communicates central IL-1 signals regulating bone mass accrual. Proc Natl Acad Sci U S A. 2012;109:15455–15460. doi: 10.1073/pnas.1206061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger LD, Felten SY, Lorton D, Felten DL. Origin of noradrenergic innervation of the spleen in rats. Brain Behav Immun. 1989;3:291–311. doi: 10.1016/0889-1591(89)90029-9. [DOI] [PubMed] [Google Scholar]
- Bellinger LD, Lorton D. Autonomic regulation of cellular immune function. Auton Neurosci. 2014;182:15–41. doi: 10.1016/j.autneu.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Berthoud RH, Powley TL. Characterization of vagal innervation to the rat celiac, suprarenal and mesenteric ganglia. J Auton Nerv Syst. 1993;42:153–169. doi: 10.1016/0165-1838(93)90046-w. [DOI] [PubMed] [Google Scholar]
- Binkowska MA, Michalak G, Słotwiński R. Current views on the mechanisms of immune responses to trauma and infection. Cent Eur J Immunol. 2015;40:206–216. doi: 10.5114/ceji.2015.52835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaz B. Autonomic dysfunction: A predictive factor of risk to develop rheumatoid arthritis. EBioMedicine. 2016;6:20–21. doi: 10.1016/j.ebiom.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaz B, Sinniger V, Hoffmann D, Clarençon D, Mathieu N, Dantzer C, Vercueil L, Picq C, Trocmé C, Faure P, Cracowski JL, Pellissier S. Chronic vagus nerve stimulation in Crohn’s disease: a 6-month follow-up pilot study. Neurogastroenterol Motil. 2016a;28:948–953. doi: 10.1111/nmo.12792. [DOI] [PubMed] [Google Scholar]
- Bonaz B, Sinniger V, Pellissier S. Anti-inflammatory properties of the vagus nerve: potential therapeutic implications of vagus nerve stimulation. J Physiol. 2016b;594:5781–5790. doi: 10.1113/JP271539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaz B, Sinniger V, Pellissier S. Vagal tone: Effects on sensitivity motility, and inflammation. Neurogastroenterol Motil. 2016c;28:455–462. doi: 10.1111/nmo.12817. [DOI] [PubMed] [Google Scholar]
- Borovikova VL, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- Bratton OB, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol. 2012;97:1180–1185. doi: 10.1113/expphysiol.2011.061531. [DOI] [PubMed] [Google Scholar]
- Briggs AC, McKenna DG, Piattoni-Kaplan M. Human α7 nicotinic acetylcholine receptor responses to novel ligands. Neuropharmacology. 1995;34:583–590. doi: 10.1016/0028-3908(95)00028-5. [DOI] [PubMed] [Google Scholar]
- Bylund BD, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, Molinoff PB, Ruffolo RR, Trendelenburg U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- Cano G, Sved AF, Rinaman L, Rabin BS, Card JP. Characterization of the central nervous system innervation of the rat spleen using viral transneuronal tracing. J Comp Neurol. 2001;439:1–18. doi: 10.1002/cne.1331. [DOI] [PubMed] [Google Scholar]
- Changeux JP. The nicotinic acetylcholine receptor: the founding father of the pentameric ligand-gated ion channel superfamily. J Biol Chem. 2012;287:40207–40215. doi: 10.1074/jbc.R112.407668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinetti-Gbaguidi G, Staels B. Macrophage polarization in metabolic disorders: functions and regulation. Curr Opin Lipidol. 2011;22:365–72. doi: 10.1097/MOL.0b013e32834a77b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen L, Papst S, Reimers K, Stukenborg-Colsman C, Steinstraesser L, Vogt PM, Kraft T, Niederbichler AD. Inflammatory response to burn trauma: nicotine attenuates proinflammatory cytokine levels. Eplasty. 2014;14:e46. [PMC free article] [PubMed] [Google Scholar]
- Claassen L, Papst S, Reimers K, Stukenborg-Colsman C, Steinstraesser L, Vogt PM, Kraft T, Niederbichler AD. Transdermal nicotine application attenuates cardiac dysfunction after severe thermal injury. Biomed Res Int. 2015;2015:292076. doi: 10.1155/2015/292076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy AJ, Mary DA, Witte KK, Greenwood JP, Deuchars SA, Deuchars J. Non-invasive vagus nerve stimulation in healthy humans reduces sympathetic nerve activity. Brain Stimul. 2014;7:871–877. doi: 10.1016/j.brs.2014.07.031. [DOI] [PubMed] [Google Scholar]
- Clarençon D, Pellissier S, Sinniger V, Kibleur A, Hoffman D, Vercueil L, David O, Bonaz B. Long term effects of low frequency (10 hz) vagus nerve stimulation on EEG and heart rate variability in Crohn’s disease: a case report. Brain Stimul. 2014;7:914–916. doi: 10.1016/j.brs.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Cuevas J, Berg DK. Mammalian nicotinic receptors with α7 subunits that slowly desensitize and rapidly recover from α-bungarotoxin blockade. J Neurosci. 1998;18:10335–10344. doi: 10.1523/JNEUROSCI.18-24-10335.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale HH, Dudley HW. The presence of histamine and acetylcholine in the spleen of the ox and the horse. J Physiol. 1929;68:97–123. doi: 10.1113/jphysiol.1929.sp002598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielski GL, Giustina AD, Badawy M, Barichello T, Quevedo J, Dal-Pizzol F, Petronilho F. Brain barrier breakdown as a cause and consequence of neuroinflammation in sepsis. Mol Neurobiol. 2017 doi: 10.1007/s12035-016-0356-7. [DOI] [PubMed] [Google Scholar]
- Dash KP, Zhao J, Kobori N, Redell JB, Hylin MJ, Hood KN, Moore AN. Activation of alpha 7 cholinergic nicotinic receptors reduce blood-brain barrier permeability following experimental traumatic brain injury. J Neurosci. 2016;36:2809–2818. doi: 10.1523/JNEUROSCI.3197-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol. 2015;5:1–15. doi: 10.3389/fimmu.2014.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs MA, Bond CE, Hoover DB. Localization of α7 nicotinic acetylcholine receptor mRNA and protein within the cholinergic anti-inflammatory pathway. Neuroscience. 2014;266:178–185. doi: 10.1016/j.neuroscience.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Elenkov JI, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- Felton LD, Ackerman KD, Wiegand SJ, Felton SY. Noradrenergic sympathetic innervation of the spleen: I. nerve fibers associate with lymphocytes and macrophages in specific compartments of the splenic white pulp. J Neurosci Res. 1987;18:28–36. doi: 10.1002/jnr.490180107. [DOI] [PubMed] [Google Scholar]
- Fujii T, Takada-Takatori Y, Horiguchi K, Kawashima K. Mediatophore regulates acetylcholine release from T cells. J Neuroimmunol. 2012a;244:16–22. doi: 10.1016/j.jneuroim.2011.12.022. [DOI] [PubMed] [Google Scholar]
- Fujii T, Takada-Takatori Y, Kawashima K. Basic and clinical aspects of non-neuronal acetylcholine: expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J Pharmacol Sci. 2008;106:186–192. doi: 10.1254/jphs.fm0070109. [DOI] [PubMed] [Google Scholar]
- Fujii T, Takada-Takatori Y, Kawashima K. Regulatory mechanisms of acetylcholine synthesis and release by T cells. Life Sci. 2012b;91:981–985. doi: 10.1016/j.lfs.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Fujii T, Yamada S, Watanabe Y, Misawa H, Tajima S, Fujimoto K, Kasahara T, Kawashima K. Induction of choline acetyltransferase mRNA in human mononuclear leukocytes stimulated by phytohemagglutinin, a T-cell activator. J Neuroimmunol. 1998;82:101–107. doi: 10.1016/S0165-5728(97)00195-1. [DOI] [PubMed] [Google Scholar]
- Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9:286–294. doi: 10.1038/nrgastro.2012.32. [DOI] [PubMed] [Google Scholar]
- Furness BJ, Callaghan BP, Rivera LR, Cho HJ. The enteric nervous system and gastrointestinal innervation: integrated local and central control. Adv Exp Med Biol. 2014;817:39–71. doi: 10.1007/978-1-4939-0897-4_3. [DOI] [PubMed] [Google Scholar]
- Gatson JW, Simpkins JW, Uteshev VV. High therapeutic potential of positive allosteric modulation of α7 nAChRs in a rat model of traumatic brain injury: proof-of-concept. Brain Res Bull. 2015;112:35–41. doi: 10.1016/j.brainresbull.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, Moore T, Jacobs S, Meriwether J, Choi MJ, Kim EJ, Walton K, Buiting K, Davis A, Breese C, Freedman R, Leonard S. Genomic organization and partial duplication of the human α7 neuronal nicotinic acetylcholine receptor gene (CHRNA7) Genomics. 1998;52:173–185. doi: 10.1006/geno.1998.5363. [DOI] [PubMed] [Google Scholar]
- Ghia EJ, Blennerhassett P, Deng Y, Verdu EF, Khan WI, Collins SM. Reactivation of inflammatory bowel disease in a mouse model of depression. Gastroenterology. 2009;136:2280–2288. doi: 10.1053/j.gastro.2009.02.069. [DOI] [PubMed] [Google Scholar]
- Ghia EJ, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology. 2006;131:1122–1130. doi: 10.1053/j.gastro.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Goldstein SR, Bruchfeld A, Yang L, Qureshi AR, Gallowitsch-Puerta M, Patel NB, Huston BJ, Chavan S, Rosas-Ballina M, Gregersen PK, Czura CJ, Sloan RP, Sama AE, Tracey KJ. Cholinergic anti-inflammatory pathway activity and High Mobility Group Box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol Med. 2007;13:210–215. doi: 10.2119/2006-00108.Goldstein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Graeber BM, Li W, Rodriguez ML. Role of microglia in CNS inflammation. FEBS Lett. 2011;585:3798–3805. doi: 10.1016/j.febslet.2011.08.033. [DOI] [PubMed] [Google Scholar]
- Grønlien HJ, Håkerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, Malysz J. Distinct profiles of α7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. 2007;72:715–724. doi: 10.1124/mol.107.035410. [DOI] [PubMed] [Google Scholar]
- Guan Y-Z, Jin X-D, Guan L-X, Yan H-C, Wang P, Gong Z, Li S-J, Cao X, Xing Y-L, Gao T-M. Ncotine inhibits microglial proliferation and is neuroprotective in global ischemia rats. Mol Neurobiol. 2015;51:1480–1488. doi: 10.1007/s12035-014-8825-3. [DOI] [PubMed] [Google Scholar]
- Han Z, Shen F, He Y, Degos V, Camus M, Maze M, Young WL, Su H. Activation of α-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro-inflammatory macrophages and oxidative stress. PLoS ONE. 2014;9(8):e105711. doi: 10.1371/journal.pone.0105711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer S, Eisenbach C, Lukic IK, Schneider L, Bode K, Brueckmann M, Mautner S, Wente MN, Encke J, Werner J, Dalpke AH, Stremmel W, Nawroth PP, Martin E, Krammer PH, Bierhaus A, Weigand MA. Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Crit Care Med. 2008;36:404–408. doi: 10.1097/01.CCM.0B013E31816208B3. [DOI] [PubMed] [Google Scholar]
- Hoover DB, Bond CE, Ozment TR, McDonald D, Williams DL. Molecular evidence for upregulation of the cholinergic anti-inflammatory phenotype during sepsis. 36th Annual Conference on Shock, June 1–4, San Diego, CA. Shock. 2013;39(Suppl. 2):18. [Google Scholar]
- Howland RH. Vagus Nerve Stimulation. Curr Behav Neurosci Rep. 2014;1:64–73. doi: 10.1007/s40473-014-0010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst MS, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, Rose-John S, Fuller GM, Topley N, Jones SA. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705–714. doi: 10.1016/s1074-7613(01)00151-0. [DOI] [PubMed] [Google Scholar]
- Huston MJ, Gallowitsch-Puerta M, Ochani M, Ochani K, Yuan R, Rosas-Ballina M, Ashok M, Goldstein RS, Chavan S, Pavlov VA, Metz CN, Yang H, Czura CJ, Wang H, Tracey KJ. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med. 2007;35:2762–2768. doi: 10.1097/01.CCM.0000288102.15975.BA. [DOI] [PubMed] [Google Scholar]
- Huston MJ, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, Tracey KJ, Ulloa L. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203:1623–1628. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskander NK, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, Remick DG. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev. 2013;93:1247–1288. doi: 10.1152/physrev.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CW, Levesque JP, Ruitenberg MJ. It takes nerve to fight back: The significance of neural innervation of the bone marrow and spleen for immune function. Semin Cell Dev Biol. 2017;61:60–70. doi: 10.1016/j.semcdb.2016.08.010. [DOI] [PubMed] [Google Scholar]
- Kalkman H, Feuerbach OD. Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell Mol Life Sci. 2016;73:2511–2530. doi: 10.1007/s00018-016-2175-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi S, Khan MA, Yasuhara S, Goto T, Tompkins RG, Kaneki M, Martyn JA. Prevention of Burn-Induced Inflammatory Responses and Muscle Wasting By GTS-21, A Specific Agonist for α7 Nicotinic Acetylcholine Receptors. Shock. 2016;47:61–69. doi: 10.1097/SHK.0000000000000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima K, Fujii T. Basic and clinical aspects of non-neuronal acetylcholine: overview of non-neuronal cholinergic systems and their biological significance. J Pharmacol Sci. 2008;106:167–173. doi: 10.1254/jphs.fm0070073. [DOI] [PubMed] [Google Scholar]
- Kawashima K, Fujii T, Moriwaki Y, Misawa H, Horiguchi K. Reconciling neuronally and nonneuronally derived acetylcholine in the regulation of immune function. Ann N Y Acad Sci. 2012;1261:7–17. doi: 10.1111/j.1749-6632.2012.06516.x. [DOI] [PubMed] [Google Scholar]
- Kawashima K, Fujii T, Moriwaki Y, Misawa H, Horiguchi K. Non-neuronal cholinergic system in regulation of immune function with a focus on α7 nAChRs. Int Immunopharmacol. 2015;29:127–134. doi: 10.1016/j.intimp.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Kelso ML, Oestreich JH. Traumatic brain injury: central and peripheral role of α7 nicotinic acetylcholine receptors. Curr Drug Targets. 2012;13:631–636. doi: 10.2174/138945012800398964. [DOI] [PubMed] [Google Scholar]
- Khan AM, Farkhondeh M, Crombie J, Jacobson L, Kaneki M, Martyn JA. Lipopolysaccharide upregulates α7 acetylcholine receptors: stimulation with GTS-21 mitigates growth arrest of macrophages and improves survival in burned mice. Shock. 2012;38:213–219. doi: 10.1097/SHK.0b013e31825d628c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman AF, Schuurman PR, Vervoordeldonk MJ, Tak PP. Vagus nerve stimulation: a new bioelectronics approach to treat rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2014;28:625–635. doi: 10.1016/j.berh.2014.10.015. [DOI] [PubMed] [Google Scholar]
- Koopman AF, Tang MW, Vermeij J, De Hair MJ, Choi IY, Vervoordeldonk MJ, Gerlag DM, Karemaker JM, Tak PP. Autonomic dysfunction precedes development of rheumatoid arthritis: A prospective cohort study. EBioMedicine. 2016;6:231–237. doi: 10.1016/j.ebiom.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kox M, van Velzen JF, Pompe JC, Hoedemaeker CW, van der Hoeven JG, Pickkers P. GTS-21 inhibits pro-inflammatory cytokine release independent of the Toll-like receptor stimulated via a transcriptional mechanism involving JAK2 activation. Biochem Pharmacol. 2009;78:863–872. doi: 10.1016/j.bcp.2009.06.096. [DOI] [PubMed] [Google Scholar]
- Kox M, Pompe JC, Gordinou de Gouberville MC, van der Hoeven JG, Hoedemaekers CW, Pickkers P. Effects of the α7 nicotinic acetylcholine receptor agonist GTS-21 on the innate immune response in humans. Shock. 2011;36:5–11. doi: 10.1097/SHK.0b013e3182168d56. [DOI] [PubMed] [Google Scholar]
- Kox M, van Eijk LT, Verhaak T, Frenzel T, Kiers HD, Gerretsen J, van der Hoeven JG, Kornet L, Scheiner A, Pickkers P. Transvenous vagus nerve stimulation does not modulate the innate immune response during experimental human endotoxemia: a randomized controlled study. Arthritis Res Ther. 2015;17:150. doi: 10.1186/s13075-015-0667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AY, Koopman FA, Faltys M, Caravaca A, Bendele A, Zitnik R, Vervoordeldonk MJ, Tak PP. Neurostimulation of the cholinergic anti-inflammatory pathway ameliorates disease in rat collagen-induced arthritis. PLoS One. 2014;9:e104530. doi: 10.1371/journal.pone.0104530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Zhou CL, Zhou QS, Zou HD. Galantamine protects against lipopolysaccharide-induced acute lung injury in rats. Braz J Med Biol Res. 2016;49:e5008. doi: 10.1590/1414-431X20155008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Zuo X, Zhou Y, Wang Y, Zhuang H, Zhang L, Zhang H, Xiao X. The vagus nerve and nicotinic receptors involve inhibition of HMGB1 release and early pro-inflammatory cytokines function in collagen-induced arthritis. J Clin Immunol. 2010;30:213–220. doi: 10.1007/s10875-009-9346-0. [DOI] [PubMed] [Google Scholar]
- Lorton D, Bellinger DL. Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int J Mol Sci. 2015;16:5635–5665. doi: 10.3390/ijms16035635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldifassi CM, Atienza G, Arnalich F, López-Collazo E, Cedillo JL, Martín-Sánchez C, Bordas A, Renart J, Montiel C. A new IRAK-M-mediated mechanism implicated in the anti-inflammatory effect of nicotine via α7 nicotinic receptors in human macrophages. PLoS One. 2014;9:e108397. doi: 10.1371/journal.pone.0108397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci. 2014a;182:65–69. doi: 10.1016/j.autneu.2013.12.007. [DOI] [PubMed] [Google Scholar]
- Martelli D, Yao ST, McKinley MJ, McAllen RM. Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol. 2014b;592:1677–1686. doi: 10.1113/jphysiol.2013.268573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut. 2013;62:1214–1222. doi: 10.1136/gutjnl-2012-302550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoli G, Gomez-Pinilla PJ, Nemethova A, Di Giovangiulio M, Cailotto C, van Bree SH, Michel K, Tracey KJ, Schemann M, Boesmans W, Vanden Berghe P, Boeckxstaens GE. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut. 2014;63:938–948. doi: 10.1136/gutjnl-2013-304676. [DOI] [PubMed] [Google Scholar]
- McLaughlin TJ, Barron SC, See JA, Rosenberg RL. Conformational changes in α7 acetylcholine receptors underlying allosteric modulation by divalent cations. BMC Pharmacol. 2009;9:1–13. doi: 10.1186/1471-2210-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregnani J, Clarençon D, Vivier M, Peinnequin A, Mouret C, Sinniger V, Picq C, Job A, Canini F, Jacquier-Sarlin M, Bonaz B. Anti-inflammatory effect of vagus nerve stimulation in a rat model of inflammatory bowel disease. Auton Neurosci. 2011;160:82–89. doi: 10.1016/j.autneu.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Meyer ME, Kuryatov A, Gerzanich V, Lindstrom J, Papke RL. Analysis of 3-(4-hydroxy, 2-methoxybenzlidene) anabaseine selectivity and activity at human and rat alpha-7 nicotinic receptors. J Pharmacol Exp Ther. 1998;287:918–925. [PubMed] [Google Scholar]
- Miceli CP, Jacobson K. Cholinergic pathways modulate experimental dinitrobenzene sulfonic acid colitis in rats. Auton Neurosci. 2003;105:16–24. doi: 10.1016/S1566-0702(03)00023-7. [DOI] [PubMed] [Google Scholar]
- Millar N, Gotti SC, Marks MJ, Wonnacott S. Nicotinic acetylcholine receptors. IUPHAR/BPS Guide to PHARMACOLOGY. 2014 http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76.
- Moudgil DK, Kim P, Brahn E. Advances in rheumatoid arthritis animal models. Curr Rheumatol Rep. 2011;13:456–463. doi: 10.1007/s11926-011-0200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mravec B. The role of the vagus nerve in stroke. Auton Neurosci. 2010;158:8–12. doi: 10.1016/j.autneu.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Munyaka P, Rabbi MF, Pavlov VA, Tracey KJ, Khafipour E, Ghia JE. Central muscarinic cholinergic activation alters interaction between splenic dendritic cell and CD4+CD25− T cells in experimental colitis. PLoS One. 2014;9:e109272. doi: 10.1371/journal.pone.0109272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance MD, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007) Brain Behav Immun. 2007;21:736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederbichler DA, Papst S, Claassen L, Jokuszies A, Ipaktchi K, Reimers K, Hirsch T, Steinstraesser L, Kraft T, Vogt PM. Burn-induced organ dysfunction: vagus nerve stimulation improves cardiac function. Eplasty. 2010;10:e45. [PMC free article] [PubMed] [Google Scholar]
- Nourshargh S, Alon R. Leukocyte migration into inflamed tissue. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- O’Mahony C, van der Kleij H, Bienenstock J, Shanahan F, O’Mahony L. Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1118–26. doi: 10.1152/ajpregu.90904.2008. [DOI] [PubMed] [Google Scholar]
- Olofsson SP, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248:188–204. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K. Obesity, adiponectin and vascular inflammatory disease. Cur Opin Lipidol. 2003;14:561–566. doi: 10.1097/00041433-200312000-00003. [DOI] [PubMed] [Google Scholar]
- Padro JC, Sanders VM. Neuroendocrine regulation of inflammation. Semin Immunol. 2014;26:357–368. doi: 10.1016/j.smim.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RLJ, Papke KP, Rose GM. Activity of α7-selective agonists at nicotinic and serotonin 5HT3 receptors expressed in Xenopus oocytes. Bioorg Med Chem Lett. 2004;14:1849–1853. doi: 10.1016/j.bmcl.2003.09.104. [DOI] [PubMed] [Google Scholar]
- Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, Soares MP, López MG. The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal. 2013;19:1135–1148. doi: 10.1089/ars.2012.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish RW, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang L-H, et al. Modulation of TNF release by choline requires α7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med. 2008;14:567–574. doi: 10.2119/2008-00079.Parrish. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov AV, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, Levi J, Parrish WR, Rosas-Ballina M, Czura CJ, Larosa GJ, Miller EJ, Tracey KJ, Al-Abed Y. Selective α7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med. 2007;35:1139–1144. doi: 10.1097/01.CCM.0000259381.56526.96. [DOI] [PubMed] [Google Scholar]
- Pavlov AV, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, Chavan S, Al-Abed Y, Tracey KJ. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun. 2009;23:41–45. doi: 10.1016/j.bbi.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov AV, Tracey KJ. Neural circuitry and immunity. Immunol Res. 2015;63:38–57. doi: 10.1007/s12026-015-8718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña G, Cai B, Deitch EA, Ulloa L. JAK2 inhibition prevents innate immune responses and rescues animals from sepsis. J Mol Med (Berl) 2010;88:851–859. doi: 10.1007/s00109-010-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña G, Cai B, Ramos L, Vida G, Deitch EA, Ulloa L. Cholinergic regulatory lymphocytes re-establish neuromodulation of innate immune responses in sepsis. J Immunol. 2011;187:718–725. doi: 10.4049/jimmunol.1100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RM, Leite PE. The Involvement of Parasympathetic and Sympathetic Nerve in the Inflammatory Reflex. J Cell Physiol. 2016;231:1862–1869. doi: 10.1002/jcp.25307. [DOI] [PubMed] [Google Scholar]
- Pongratz G, Straub RH. The sympathetic nervous response in inflammation. Arthritis Res Ther. 2014;16:504. doi: 10.1186/s13075-014-0504-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premchand KR, Sharma K, Mittal S, Monteiro R, Dixit S, Libbus I, DiCarlo LA, Ardell JL, Rector TS, Amurthur B, KenKnight BH, Anand IS. Autonomic regulation therapy via left or right cervical vagus nerve stimulation in patients with chronic heart failure: results of the ANTHEM-HF trial. J Card Fail. 2014;20:808–816. doi: 10.1016/j.cardfail.2014.08.009. [DOI] [PubMed] [Google Scholar]
- Premchand KR, Sharma K, Mittal S, Monteiro R, Dixit S, Libbus I, DiCarlo LA, Ardell JL, Rector TS, Amurthur B, KenKnight BH, Anand IS. Extended follow-up of patients with heart failure receiving autonomic regulation therapy in the ANTHEM-HF study. J Card Fail. 2016;22:639–642. doi: 10.1016/j.cardfail.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Price JV, Vance RE. The macrophage paradox. Immunity. 2014;41:685–693. doi: 10.1016/j.immuni.2014.10.015. [DOI] [PubMed] [Google Scholar]
- Reardon C, Duncan GS, Brüstle A, Brenner D, Tusche MW, Olofsson PS, Olofsson P, Rosas-Ballina M, Tracey KJ, Mak TW. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc Natl Acad Sci U S A. 2013;110:1410–1415. doi: 10.1073/pnas.1221655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A. 2008;105:11008–11013. doi: 10.1073/pnas.0803237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Valdés-Ferrer SI, Dancho ME, Ochani M, Katz D, Cheng KF, Olofsson PS, Chavan SS, Al-Abed Y, Tracey KJ, Pavlov VA. Xanomeline suppresses excessive pro-inflammatory cytokine responses through neural signal-mediated pathways and improves survival in lethal inflammation. Brain Behav Immun. 2015;44:19–27. doi: 10.1016/j.bbi.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safronova GV, Vulfius CA, Shelukhina IV, Mal’tseva VN, Berezhnov AV, Fedotova EI, Miftahova RG, Kryukova EV, Grinevich AA, Tsetlin VI. Nicotinic receptor involvement in regulation of functions of mouse neutrophils from inflammatory site. Immunobiology. 2016;221:761–772. doi: 10.1016/j.imbio.2016.01.016. [DOI] [PubMed] [Google Scholar]
- Salaga M, Blomster LV, Piechota-Polańczyk A, Zielińska M, Jacenik D, Cygankiewicz AI, Krajewska WM, Mikkelsen JD, Fichna J. Encenicline, an α7 nicotinic acetylcholine receptor partial agonist, reduces immune cell infiltration in the colon and improves experimental colitis in mice. J Pharmacol Exp Ther. 2016;356:157–169. doi: 10.1124/jpet.115.228205. [DOI] [PubMed] [Google Scholar]
- Santoni G, Cardinali C, Morelli MB, Santoni M, Nabissi M, Amantini C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J J Neuroinflammation. 2015;3(12):21. doi: 10.1186/s12974-015-0239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato ZK, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett. 1999;266:17–20. doi: 10.1016/s0304-3940(99)00259-1. [DOI] [PubMed] [Google Scholar]
- Sherwood RE, Toliver-Kinsky T. Mechanisms of the inflammatory response. Best Pract Res Clin Anaesthesiol. 2004;18:385–405. doi: 10.1016/j.bpa.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Shytle DR, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR. Cholinergic modulation of microglial activation by α7 nicotinic receptors. J Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- Sinkus LM, Graw S, Freedman R, Ross RG, Lester HA, Leonard S. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation, and function. Neuropharmacology. 2015;96:274–288. doi: 10.1016/j.neuropharm.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skok VM, Grailhe R, Agenes F, Changeux JP. The role of nicotinic receptors in B-lymphocyte development and activation. Life Sci. 2007;80:2334–2336. doi: 10.1016/j.lfs.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Snoek AS, Verstege MI, van der Zanden EP, Deeks N, Bulmer DC, Skynner M, Lee K, Te Velde AA, Boeckxstaens GE, de Jonge WJ. Selective α7 nicotinic acetylcholine receptor agonists worsen disease in experimental colitis. Br J Pharmacol. 2010;160:322–333. doi: 10.1111/j.1476-5381.2010.00699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehnlein O, Lindbom L, Weber C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood. 2009;114:4613–4623. doi: 10.1182/blood-2009-06-221630. [DOI] [PubMed] [Google Scholar]
- Song MX, Li JG, Wang YL, Liang H, Huang Y, Yuan X, Zhou Q, Zhang ZZ. Effect of vagus nerve stimulation on thermal injury in rats. Burns. 2010;36:75–81. doi: 10.1016/j.burns.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Stokes C, Treinin M, Papke RL. Looking below the surface of nicotinic acetylcholine receptors. Trends Pharmacol Sci. 2015;36:514–523. doi: 10.1016/j.tips.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Jin K, Uteshev VV. A type-II positive allosteric modulator of α7 nAChRs reduces brain injury and improves neurological function after focal cerebral ischemia in rats. PLoS ONE. 2013a;8(8):e73581. doi: 10.1371/journal.pone.0073581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Johnson SR, Jin K, Uteshev VV. Boosting endogenous resistance of brain to ischemia. Mol Neurobiol. 2017;54:2045–2059. doi: 10.1007/s12035-016-9796-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Li Q, Gui H, Xu DP, Yang YL, Su DF, Liu X. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 2013b;23:1270–1283. doi: 10.1038/cr.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski KF, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallini NY, Shui B, Greene KS, Deng KY, Doran R, Fisher PJ, Zipfel W, Kotlikoff MI. BAC transgenic mice express enhanced green fluorescent protein in central and peripheral cholinergic neurons. Physiol Genomics. 2006;27:391–397. doi: 10.1152/physiolgenomics.00092.2006. [DOI] [PubMed] [Google Scholar]
- Thayer FJ, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361–372. doi: 10.1196/annals.1366.014. [DOI] [PubMed] [Google Scholar]
- Thayer FJ, Yamamoto SS, Brosschot JF. The relationship of autonomic imbalance, heart rate variability and cardiovascular disease risk factors. Int J Cardiol. 2010;141:122–131. doi: 10.1016/j.ijcard.2009.09.543. [DOI] [PubMed] [Google Scholar]
- Tobias P, Curtiss LK. Thematic review series: The immune system and atherogenesis. Paying the price for pathogen protection: toll receptors in atherogenesis. J Lipid Res. 2005;46:404–411. doi: 10.1194/jlr.R400015-JLR200. [DOI] [PubMed] [Google Scholar]
- Uteshev VV. The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur J Pharmacol. 2014;727:181–185. doi: 10.1016/j.ejphar.2014.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Zanden EP, Boeckxstaens GE, de Jonge WJ. The vagus nerve as a modulator of intestinal inflammation. Neurogastroenterol Motil. 2009;21:6–17. doi: 10.1111/j.1365-2982.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- van Maanen MA, Lebre MC, Poll T van der, LaRosa GJ, Elbaum D, Vervoordeldonk MJ, Tak PP. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009;60:114–122. doi: 10.1002/art.24177. [DOI] [PubMed] [Google Scholar]
- van Maanen MA, Stoof SP, Larosa GJ, Vervoordeldonk MJ, Tak PP. Role of the cholinergic nervous system in rheumatoid arthritis: aggravation of arthritis in nicotinic acetylcholine receptor α7 subunit gene knockout mice. Ann Rheum Dis. 2010;69:1717–1723. doi: 10.1136/ard.2009.118554. [DOI] [PubMed] [Google Scholar]
- Vida G, Peña G, Deitch EA, Ulloa L. α7-cholinergic receptor mediates vagal induction of splenic norepinephrine. J Immunol. 2011a;186:4340–4346. doi: 10.4049/jimmunol.1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida G, Peña G, Kanashiro A, Thompson-Bonilla MR, Palange D, Deitch EA, Ulloa L. β2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J. 2011b;25:4476–4485. doi: 10.1096/fj.11-191007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, Bertrand D, Romand JA. Expression of an α7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol. 2002;126:86–98. doi: 10.1016/s0165-5728(02)00057-7. [DOI] [PubMed] [Google Scholar]
- Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Wells CL, Barton RG, Wavatne CS, Dunn DL, Cerra FB. Intestinal bacterial flora, intestinal pathology, and lipopolysaccharide-induced translocation of intestinal bacteria. Circ Shock. 1992;37:117–123. [PubMed] [Google Scholar]
- Williams KD, Wang J, Papke RL. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem Pharmacol. 2011;82:915–930. doi: 10.1016/j.bcp.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittebole X, Hahm S, Coyle SM, Kumar A, Calvano SE, Lowry SF. Nicotine exposure alters in vivo human responses to endotoxin. Clin Exp Immunol. 2007;147:28–34. doi: 10.1111/j.1365-2249.2006.03248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa H, Kurokawa M, Ozaki N, Nara K, Atou K, Takada E, Kamochi H, Suzuki N. Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-κB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor α7. Clin Exp Immunol. 2006;146:116–123. doi: 10.1111/j.1365-2249.2006.03169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang CC. Inflammatory response of macrophages in infection. Hepatobiliary Pancreat Dis Int. 2014;13:138–152. doi: 10.1016/s1499-3872(14)60024-2. [DOI] [PubMed] [Google Scholar]