

Abstract
We previously constructed a collection of fission yeast strains that express various mammalian cyclic nucleotide phosphodiesterases (PDEs) and developed a cell-based high throughput screen (HTS) for small molecule PDE inhibitors. Here we describe a compound, BC54, that is a selective inhibitor of enzymes from the cAMP-specific PDE4 and PDE7 families. Consistent with the biological effect of other PDE4 and PDE7 inhibitors, BC54 displays potent anti-inflammatory properties and is superior to a combination of rolipram (a PDE4 inhibitor) and BRL50481 (a PDE7A inhibitor) for inducing apoptosis in chronic lymphocytic leukemia (CLL) cells. We further exploited PKA-regulated growth phenotypes in fission yeast to isolate two mutant alleles of the human PDE4B2 gene that encode enzymes possessing single amino acid changes that confer partial resistance to BC54. We confirm this resistance to both BC54 and rolipram via yeast-based assays and, for PDE4B2T407A, in vitro enzyme assays. Thus, we are able to use this system for both chemical screens to identify biologically-active PDE inhibitors and molecular genetic studies to characterize the interaction of these molecules with their target enzymes. Based on its potency, selectivity, and effectiveness in cell culture, BC54 should be a useful tool to study biological processes regulated by PDE4 and PDE7 enzymes.
Keywords: High-throughput, phosphodiesterase, PDE4, PDE7, inflammation, Schizosaccharomyces pombe
Graphical Abstract
1. Introduction
The cyclic nucleotides monophosphate (cNMPs) 3′–5′ cyclic adenosine monophosphate (cAMP) and 3′–5′ cyclic guanosine monophosphate (cGMP) are important second messengers generated by cells in response to extracellular signals in the environment. Their levels regulate a diverse array of biological processes including cell growth, apoptosis, inflammation, cognition, and steroidogenesis in a variety of cell types. cNMP levels are controlled by their synthesis by adenylyl and guanylyl cyclases and their hydrolysis by cyclic nucleotide phosphodiesterases (PDEs) [1–4]. In addition, subcellular localization of these enzymes can produce microdomains of cNMPs such that individual enzymes can be involved in the regulation of some, but not all, cNMP-regulated processes within a given cell [4].
The study of cNMP signaling in mammals is complicated by the fact that many genes encode the nucleotide cyclases and PDEs that could be involved in any particular process. For example, there are ten different genes that encode adenylyl cyclases (ACs), nine of which encode integral membrane proteins and one of which encodes a soluble adenylyl cyclase [5]. There are also 21 genes that encode PDEs, which are pharmacologically grouped into 11 families [1, 4]. Of these, 16 genes representing 8 families encode PDEs that can hydrolyze cAMP (PDE4, PDE7, and PDE8 enzymes selectively hydrolyze cAMP, while PDE1, PDE2, PDE3, PDE10, and PDE11 enzymes hydrolyze both cAMP and cGMP). Although many cells appear to produce more than one species of AC and PDE, these enzymes do not necessarily act in a redundant manner. Anchoring proteins can nucleate protein complexes that include an AC, a PDE, and a cAMP-regulated protein such as the cAMP-dependent protein kinase PKA or an EPAC (Exchange Protein Activated by cAMP; [6]), which then allows for highly localized signaling events that may be regulated independently from other cAMP-regulated processes in the same cell [4]. As such, individual ACs and PDEs can regulate specific biological processes within a cell, which supports their status as druggable targets for the treatment of diseases or conditions associated with these processes.
The PDE4 family, composed of four genes (PDE4A-4D), has long been seen as a promising drug target involved in inflammation and psychiatric disease. For example, rolipram, the drug that defines the PDE4 family, displays both anti-inflammatory and anti-depressant activity [7, 8]. However, efforts to develop rolipram, and most other PDE4 inhibitors, as a therapeutic have been hampered by common side-effects such as emesis and nausea [9], although some success has been seen with Roflumilast to treat chronic obstructive pulmonary disease and Apremilast to treat psoriasis and psoriatic arthritis [10]. It has been suggested that compounds that inhibit PDE4 enzymes together with a second PDE family could provide a therapeutic benefit at a concentration that does not cause emesis [11, 12]. Of the other PDE families, PDE7 (encoded by the PDE7A and PDE7B genes) is a leading candidate for this dual family approach as PDE7 enzymes are cAMP-specific and PDE7-selective inhibitors show some anti-inflammatory activity on their own [13–15]. However, the most commonly-used PDE7 inhibitor, BRL50481, is actually a selective PDE7A inhibitor with relatively weak activity against PDE7B [13].
Here, we describe the characterization of a potent PDE4/7 inhibitor, BC54, identified from a yeast-based high throughput screen (HTS) using fission yeast strains that express either mammalian PDE4A, PDE4B, or PDE7A as the sole source of PDE activity [16]. Both yeast-based screens and in vitro enzyme assays confirm that BC54 selectively acts on members of the PDE4 and PDE7 families, with equal potency against both PDE7A and PDE7B. BC54 also displays exceptional anti-inflammatory activity and induces apoptosis in CLL cells, consistent with its effect on PDE4 and PDE7 enzymes. To better understand the interaction between BC54 and PDE4B, we carried out a genetic screen for compound-resistant mutant alleles of the human PDE4B2 gene using a yeast-based screen. We identified two alleles that possess single amino acid changes of residues that directly interact with the adenosine group of cAMP. One mutant protein (PDE4B2Y233H) retains too little PDE activity for biochemical characterization. However, we show with both yeast-based assays and in vitro enzyme assays that the second mutant protein, PDE4B2T407A, is significantly less sensitive to inhibition by both BC54 and rolipram. These studies show that the yeast-based screening system can be used to identify and characterize novel PDE inhibitors that display potent biological activity in mammalian cell culture-based assays. In addition, strains expressing the PDE4B2T407A protein could be used in the future to screen for PDE4 inhibitors that act at a distinct site from that of rolipram and BC54.
2. Materials and methods
2.1 Yeast strains, media, and growth conditions
The genotypes of S. pombe strains used in this study are presented in Table 1. Cells were cultured at 30°C in YES-rich (yeast extract medium with supplements), EMM-defined, or 5FOA media as described previously [17–19]. Insertions of the murine PDE4A1 or PDE4B3, rat PDE4A5, human PDE4A1, PDE4B2, PDE4D3, PDE7A1 or PDE7B1 gene into the S. pombe cgs2+ (PDE) locus as well as fbp1-ura4+ and fbp1-GFP reporters have been previously described [18, 20, 21].
Table 1.
S. pombe strain list.
| Strain name
|
Genotype
|
|---|---|
| CHP1098 h+ | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::MmPDE4A1 gpa2Δ::his3+ |
| CHP1113 h− | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 his3D-1 pap1Δ::ura4− gpa2Δ::his3+ cgs2::MmPDE4B3 |
| CHP1155 h− | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::RnPDE4A5 gpa2Δ::his3+ |
| CHP1186 h+ | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::HuPDE4D3 git11Δ::kan |
| CHP1189 h+ | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::HuPDE7A1 gpa2Δ::his3+ |
| CHP1207 h− | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 his7-366 pap1Δ::ura4− cgs2-2 git2Δ::his7+ |
| CHP1209 h− | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::HuPDE7B1 git3Δ::kan |
| CHP1262 h− | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::HuPDE4A1 gpa2Δ::his3+ |
| CHP1268 h− | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::HuPDE4B2 git3Δ::kan |
| CHP1346 h+ | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2-2 |
| CHP1401 h− | fbp1::ura4+ ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::HuPDE4B2 git2Δ::his7+ |
| CHP1641 h− | fbp1::GFP ura4::fbp1-lacZ leu1-32 pap1Δ::ura4− cgs2::HuPDE4B2 git2Δ::his7+ |
| CHP1712 h− | fbp1::GFP ura4::fbp1-lacZ leu1-32 his7-366 pap1Δ::ura4− cgs2::HuPDE4B2T407A git2Δ::his7+ |
| CHP1713 h− | fbp1:: ura4+ ura4::fbp1-lacZ leu1-32 his7-366 pap1Δ::ura4− cgs2::HuPDE4B2T407A git2Δ::his7+ |
2.2 Small molecules
BC54 (ethyl (2Z)-2-(acetylimino)-1-cyclohexyl-5-oxo-1,5-dihydro-2H-dipyrido[1,2- a:2′,3′-d] pyrimidine-3-carboxylate) was purchased from ChemDiv (C098-0484). A second synthesis was carried out by Hai Le and James Morken, producing a compound with the same biochemical properties and biological activity as the original reagent.
2.3 5FOA growth assays
5FOA growth assays to screen for and to characterize PDE inhibitors were carried out using strains that express the PKA-repressed fbp1-ura4+ reporter as previously described [13, 20, 22–25]. Dose response profiling of BC54 and rolipram was carried out at concentrations of 0.05 μM to 50 μM.
2.4 High-throughput screens (HTSs)
HTSs were carried out at the Broad Institute’s Chemical Biology Program screening facility using strains expressing murine PDE4A1 (CHP1098), rat PDE4A5 (CHP1155), murine PDE4B3 (CHP1113), or human PDE7A1 (CHP1189). 100 nL of compounds (from 5 mg/mL stock solutions) were pinned into wells of 384-well microtiter dishes with a final volume of 50 μl cells. The optical density (OD600) of each well was measured after 48 h growth at 30°C using a plate reader. Composite Z scores were calculated for each compound by scaling the vector [Zscore_A, Zscore_B] by the cosine correlation with the diagonal (i.e. identical Z-scores in both replicates). HTSs were evaluated by a Z′-factor test as previously described [25, 26], using DMSO (0.2%) as a negative control, and 5 mM cAMP in the growth medium as a positive control. All Z′-factors were above 0.5.
2.5 Cell isolation and apoptosis assays
Blood samples were obtained in heparinized tubes with Institutional Review Board-approved consent from CLL patients. Whole blood was layered on Histopaque 1077 (Sigma-Aldrich) and PBMC isolated after centrifugation. PBMC were washed and resuspended in RPMI 1640 (Gibco) supplemented with 10% FBS (Sigma-Aldrich), 20 mM L-glutamine, 100 IU/ml penicillin and 100 g/ml streptomycin (Fisher), and plated at a concentration of 1 million cells per well. Cultures were treated with drug (20μM) or vehicle as indicated. Cells were incubated for 48 h at 37°C with 5% CO2. Cells were harvested, and Hoechst 33342 (Molecular Probes) was added to a final concentration of 0.25 μg/mL. After incubation at 37°C for 10 min, cells were analyzed on a LSR-II flow cytometer. Data were analyzed using FlowJo software (Tree Star).
2.6 In vitro PDE assays
In vitro PDE assays were performed as previously described [22], using the Ba(OH)2 precipitation assay [27], with recombinant human PDE1C, PDE3B, PDE5A1, PDE8A, PDE9A2, PDE10A1, PDE11A4 (BPS Bioscience Inc.), rat PDE2A (BPS Bioscience Inc.), and human PDE7A (BIOMOL International). Human PDE4A10 (Genbank accession number AF073745.2, amino acids 290-622), PDE4B2 (Genbank accession number NM_001037339.2, amino acids 152-487), PDE4D2 (Genbank accession number AF012074.1, amino acids 86-411) and PDE7B enzymes were obtained from Dr. Hengming Ke. Substrate concentrations were 120 nM cGMP for PDE1C, 1 μM cGMP for PDE2A, 30 nM cGMP for PDE3B, 40 nM cAMP for PDE4A, PDE4B, and PDE4D, 520 nM cGMP for PDE5A, 10 nM cAMP for PDE7A and PDE7B, 10 nM cAMP for PDE8A, 70 nM cGMP for PDE9A, 40 nM cAMP for PDE10A, and 120 nM cGMP for PDE11A. Additional assays were carried out using the human PDE4B2 catalytic domain (residues 152-487), either wild type PDE4B2+ or mutant PDE4B2T407A, expressed and purified from E. coli as previously described [28] (assayed using 500nM cAMP). Reactions were stopped with 200 μL 0.2 M ZnSO4 during the linear phase of the assay, and 5′-AMP was precipitated with 200 μL 0.25 N Ba(OH)2 by centrifugation for 15 min at 14000 rpm. The radioactivity remaining in the supernatant was measured in 4 mL of scintillation fluid. The amount of hydrolysis was limited to ~50% conversion.
2.7 Isolation of compound-resistant PDE4B2 alleles
A pool of alleles of the human PDE4B2 gene (Genbank accession number L20971) were generated by PCR amplification of PDE4B2 inserted into the S. pombe cgs2 locus in plasmid pKG3-PDE4B2 [20]. PCR was carried out using the FailSafe™ PCR kit with 2X buffers B, C, J and L (Epicentre Biotechnologies), as these conditions produce both transition and transversion mutations (unpublished data). 20 cycles of amplification were performed using oligonucleotides cgsmut5′ (5′TCATAGCATACTTCTTCACCAAGC3′) and cgsmut3′ (3′AAAGTGTCCGATGAGAAAAGCGTG). The PCR products containing PDE4B2 were co-transformed together with StuI-linearized plasmid pKG3-dropout vector (Hoffman lab, unpublished) by gap-repair transformation [29] into strain CHP1346 (fbp1-ura4+ cgs2-2; the cgs2-2 frameshift mutation eliminates endogenous PDE activity [19] leading to a defect in stationary phase entry [30]). Cells were plated to EMM-leucine to select for transformants. After colonies formed, cells were enriched and screened for expression of a BC54-resistant PDE4B enzyme as described in the text. Plasmids were rescued from Ura+ colonies to E. coli for DNA sequencing, leading to the identification of the PDE4B2Y233H and PDE4B2T407A alleles.
2.8 Expression of wild type and mutant PDE4B2 catalytic domains
A clone expressing wild type human PDE4B2 catalytic domain (residues 152 to 487; GenBank accession number NM_001037339.2; [27]) from the pET15b vector was obtained from Dr. Hengming Ke. Two mutant derivatives were constructed by site-directed mutagenesis using the QuickChange II kit (Agilent Technologies), using primers T407A-F (5′AATTGTACGGCAATGGGCAGACCGCATCATGGAGGA3′), T407A-R (5′TCCTCCATGATGCGGTCTGCCCATTGCCGATACAATT3′), Y233H-F (5′CCATTCTGACGTGGCACATCACAACAGCCTGAC3′), and Y233H-R (5′GTGCAGGCTGTTGTGATGTGCCACGTCAGAATGG3′). The mutations were confirmed by DNA sequencing.
PDE4B2 catalytic domains were expressed in E. coli BL21 (DE3) cells (New England BioLabs Inc.) grown at 16°C using an overnight IPTG induction. Purification of the catalytic domains was carried out using Talon beads (Clontech) followed by a Q-Sepharose ion exchange column (GE Healthcare Sciences) as previously described [27]. Coomassie staining of SDS PAGE gels of the purified protein extracts showed a single band at the expected size of ~43kDa (data not shown).
3. Results
3.1 Identification of PDE4/7 inhibitor BC54 via a fission yeast-based growth assay
PKA activity in the fission yeast Schizosaccharomyces pombe can be monitored using a PKA-repressed fbp1-ura4+ reporter that confers 5FOA-sensitivity due to low PKA activity [18]. Using strains deleted for the gpa2+ gene that encodes the Gα that activates the Git2/Cyr1 adenylyl cyclase [31–33], we constructed a series of strains that are 5FOAS due to the expression of mammalian cAMP-specific PDE4 and PDE7 enzymes expressed in place of the S. pombe Cgs2 PDE [13, 25]. We screened strains expressing either murine PDE4A1 (CHP1098), rat PDE4A5 (CHP1155), murine PDE4B3 (CHP1113), or human PDE7A1 (CHP1189) for small molecules that confer 5FOAR growth, presumably by inhibiting the PDE to elevate cAMP levels and activate PKA. One compound, BC54 (ethyl (2Z)-2-(acetylimino)-1-cyclohexyl-5-oxo-1,5-dihydro-2H-dipyrido[1,2- a:2′,3′-d] pyrimidine-3-carboxylate), showed remarkable potency in all four screens. It was one of the most effective PDE7A inhibitors and generally outperformed PDE4 inhibitors rolipram and zardaverine present in the Prestwick Bioactive compound library (Fig. 1A, B). In other previously-published screens, BC54 was not a hit against PDE8A (composite Z score −2.35, ranking 225,000 of 242,028 compounds; [34]) and a very weak hit compound against PDE11A (composite Z score 7.74, ranking 9840 of 193,561 compounds; [22]).
Fig. 1.

Structure and HTS data for PDE4/7 inhibitor BC54 and PDE4 inhibitors rolipram and zardaverine. A) Chemical structures of BC54, rolipram, and zardaverine. B) Comparison of PDE inhibitory activity of BC54, rolipram, and zardaverine as judged by HTSs using strains expressing murine PDE4A1, rat PDE4A5, murine PDE4B3 or human PDE7A1. Composite Z scores were determined from duplicate plates as described in Materials and Methods. Using an arbitrary Z score cut-off of 40, there were 227 hit compounds in the PDE4A1 screen, 335 hit compounds in the PDE4A5 screen, 1610 hit compounds in the PDE4B3 screen, and 163 hit compounds in the PDE7A1 screen. BC54 was one of only three compounds to produce Z scores of >40 in all four screens. C) Inhibition of PDE4B, PDE7A and PDE7B by BC54 as measured by in vitro enzyme assays. Assays were carried out as described in Materials and Methods with substrate concentrations of 40nM cAMP for PDE4B and 10nM cAMP for PDE7A and PDE7B. One representative assay is shown for each enzyme.
The exceptional potency of BC54 was confirmed in dose response assays using yeast strains that express human PDE4A1, PDE4B2, PDE4D3, PDE7A1 or PDE7B1 enzymes (Supplemental Fig. 1). With the exception of the strain expressing PDE4D3, BC54 is more effective than rolipram at inducing 5FOAR growth. In addition, BC54 is a potent inhibitor of both PDE7A and PDE7B, unlike BRL50481 [15], which is a highly selective inhibitor of PDE7A with significantly less activity against PDE7B as determined by both 5FOA growth assays and in vitro enzyme assays [13]. While many PDE4, PDE7, and PDE4/7 inhibitors were identified in these screens, BC54 was the only compound to show significant potency at sub-micromolar concentrations in the 5FOA growth assay, thus warranting further investigation of its activity. In vitro enzyme assays confirm that BC54 is a potent inhibitor of PDE4 and PDE7 (Fig. 1C). IC50 values range from 50nM to 110nM for PDE4A, PDE4B, and PDE4D (tested at a substrate concentration of 40nM cAMP), and were 140nM for both PDE7A and PDE7B (tested at a substrate concentration of 10nM cAMP). BC54 showed only modest activity (IC50 ~3μM) against PDE2A (tested at 1μM cGMP) and PDE11A (tested at 120nM cGMP), and little to no activity (IC50 >10μM) against PDE1C (at 120nM cGMP), PDE3B (at 30nM cGMP), PDE5A (at 520nM cGMP), PDE8A (at 10nM cAMP), PDE9A (at 70nM cGMP), and PDE10A (at 40nM cAMP) (data not shown). The activity against PDE11A is consistent with the observation that it came up as a very weak hit in our screen for PDE11A inhibitors [22] as mentioned above.
3.2 BC54 displays biologically-relevant activity in cell culture
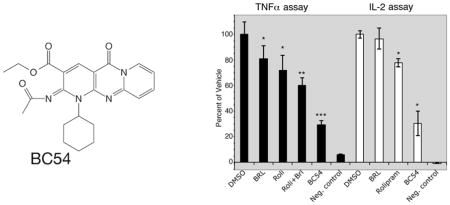
One argument for using a yeast cell-based assay as a small molecule discovery platform is that compounds identified in such screens are cell permeable and fairly specific for binding their target protein (compounds that interact with many proteins will likely prevent yeast growth by inhibiting an essential protein). We therefore tested BC54 in mammalian cell culture for activities associated with the inhibition of PDE4 and/or PDE7 proteins. Consistent with its PDE4/7 inhibitory activity, BC54 significantly reduces TNFα production by LPS-stimulated macrophage and IL-2 production by PMA-PHA stimulated Jurkat T cells, outperforming a combined treatment with rolipram (PDE4 inhibitor) and BRL50481 (PDE7A inhibitor) in the TNFα assay (Fig. 2). Similarly, BC54 is superior to a combination of rolipram and BRL50481 for the induction of apoptosis in CLL cells (Table 2; Supplemental Fig. 2) from three different patients. These results are consistent with previous studies that identify PDE4 and PDE7 proteins as the prominent PDEs in CLL cells [35–37].
Fig. 2.

Anti-inflammatory activity of BC54 in TNFα and IL-2 assays. The effect of the indicated compounds (at 20μM) was measured in TNFα assays using U937 cells and in IL-2 assays using Jurkat T cells. Values represent averages and SDs of three TNFα assays or two IL-2 assays. *- P value < 0.05 compared to the DMSO vehicle control. **-P value < 0.01 compared to the DMSO vehicle control. ***- P value < 0.01 compared to DMSO vehicle control and to all other compound-treated cells. Negative control values are that of unstimulated cells.
Table 2.
CLL apoptosis in response to compound treatment
| Untreated | DMSO | Roli | BRL | Roli/BRL | BC54 | P value (BC54 vs Roli/BRL) | |
|---|---|---|---|---|---|---|---|
| Patient 1 | 46.6±1.37 | 47.5±1.12 | 76.7±0.76 | 52.5±0.78 | 78.7±0.86 | 87.3±0.76 | 0.0001 |
| Patient 2 | 23.7±0.67 | 22.6±0.89 | 35.6±0.7 | 26.3±1.02 | 34.7±0.46 | 58.8±3.12 | 0.0001 |
| Patient 3 | 4.9±0.47 | 4.9±0.31 | 20.3±2.18 | 5.1±0.72 | 27.2±0.83 | 39.8±1.79 | 0.0002 |
All compounds were tested at 20μM final concentrations. Each value represents the median and standard deviation of three independent assays (see Materials and Methods for details).
3.3 A yeast-based genetic screen for BC54-resistant alleles of PDE4B2
To study the interaction between BC54 and its target PDEs, we developed a genetic screen for mutations in PDE4B2 that produce an enzyme with reduced sensitivity to BC54. As shown in Fig. 3A, BC54 inhibits growth of S. pombe cells that express PDE4B2 and the PKA-repressed fbp1-ura4+ reporter in SC-ura (lacking uracil) medium. This is due to PDE4B2 inhibition leading to PKA activation, to repress fbp1-ura4+ expression. In addition, as cells with elevated PKA activity such as cgs1− (encoding the PKA regulatory subunit) and cgs2− (encoding the only S. pombe PDE) mutants display a defect in stationary phase entry [30], we used a cgs2-2 mutant host strain, CHP1346. This allowed us to enrich for transformants expressing a functional PDE4B2 enzyme by growing cells to stationary phase in the presence of BC54. As seen in Fig. 3B, BC54 reduces stationary phase viability in a strain that expresses the S. pombe Git2 adenylyl cyclase and the human PDE4B2 by approximately ten-fold, but has no effect on strains that lack either adenylyl cyclase or PDE activity. To identify BC54-resistant PDE4B2 alleles, we PCR amplified PDE4B2 under conditions previously-determined to introduce a modest level of mutations (see Materials and Methods). PCR using the FailSafe™ PCR kit with buffers B, C, J or L and 20 cycles of amplification leads to a low percentage of mutant alleles, most of which carry a single amino acid change due to either a transition or transversion mutation ([38] and unpublished data). These PCR products were cloned by gap repair transformation into an expression vector plasmid, thus simultaneously inserting the PCR products into the vector and expressing the candidate proteins in a strain lacking PDE activity. Transformants were originally plated on medium lacking leucine to select for receipt of the LEU2-marked expression vector and subjected to three consecutive rounds of enrichment by growing transformants to stationary phase for three days in YES medium containing 10μM BC54. Cells lacking PDE activity, either due to the absence of a functional PDE4B2 gene on the plasmid or due to BC54 inhibition of the plasmid-expressed PDE4B2 protein, rapidly die in stationary phase, which enriches for transformants that express a PDE4B2 protein that remains active in the presence of BC54. These cells were then plated to SC-ura medium (also lacking leucine to select for transformants) onto which BC54 had been spotted to inhibit growth of cells that express a BC54-sensitive PDE as such inhibition would stimulate PKA activity to repress transcription of the fbp1-ura4+ reporter. From colonies that formed under these conditions, we identified two plasmids that, when rescued to E. coli and reintroduced to strain CHP1346, conferred Ura+ growth on SC-ura plates in the presence of BC54. Sequence analysis of these alleles showed that one carries a single missense mutation converting tyrosine 233 to histidine (PDE4B2Y233H) while the other carries a mutation converting threonine 407 to alanine (PDE4B2T407A). These residues have been shown to be in direct contact with cAMP in a crystal of PDE4D (PDB ID: 2PW3; [39]) and with rolipram in a crystal of PDE4B (PDB ID: 1RO6; [40]) (Fig. 4).
Fig. 3.

PDE4 inhibition prevents colony formation on SC-ura medium and reduces stationary phase viability. A) Approximately 5×105 cells of strain CHP1268 (PDE4B2 git3Δ) were plated to SC-ura medium after which four 5μl spots (10μM) of either the PDE4 inhibitor rolipram or the PDE4/7 inhibitor BC54 were added. Plates were incubated at 30°C for four days and photographed. B) Transformants of strains CHP1346 (git2+ cgs2-2; expressing either no PDE or PDE4B2) and CHP1207 (git2Δ cgs2-2; expressing PDE4B2) were grown to stationary phase for three days in EMM-leucine medium in the presence or absence of 10μM BC54. Cells were diluted 100-fold in fresh YES medium and further subjected to four 10-fold serial dilutions. Five microliters of cells were spotted to YES and grown at 30°C before photographing. BC54 alters viability only in cells that express adenylyl cyclase and PDE4B2. C) Halo assay of strains expressing wild type and mutant PDE4B as performed in panel A. SC-ura plates with transformants expressing either wild type PDE4B or PDE4BT407A were incubated for 4 days. SC-ura plate with transformant expressing PDE4BY233H required a longer incubation period, indicating relatively low PDE activity.
Fig. 4.

Residues altered in BC54-resistant variants of PDE4B2 are located in the active site. A) Y233 (in red) and T407 (in blue) contact the adenosine ring of cAMP in a crystal of PDE4D bound to cAMP (PDB ID: 2PW3) [39]. B) Y233 (in red) and T407 (in blue) contact rolipram in a crystal of PDE4B bound to rolipram (note that both the R- and S-enantiomers of rolipram are shown) (PDB ID: 1RO6 [40]).
3.4 Cell-based assays confirm that PDE4B2Y233H and PDE4B2T407A display reduced sensitivity to BC54
To further characterize the PDE4B2Y233H and PDE4B2T407A alleles, we first integrated them into the cgs2 locus by homologous recombination so as to increase mitotic stability and reduce copy number, both of which can produce artifacts due to overexpression from autonomously-replicating plasmids in S. pombe. We then examined strains expressing either wild type or mutant PDE4B2 enzymes together with the fbp1-ura4+ reporter for sensitivity to BC54 using an SC-ura halo assay. As seen in Fig. 3C, strains expressing either mutant allele of PDE4B2 show a significant reduction in the BC54 growth inhibition halo. The strain expressing PDE4B2Y233H displays relatively weak Ura+ growth, indicating low PDE activity, thus requiring a longer incubation period to determine the effect of BC54. Due to the weak activity of the PDE4B2Y233H enzyme, further studies were only carried out on the PDE4B2T407A enzyme. Consistent with the halo assay, a strain expressing PDE4B2T407A displays significant resistance to both BC54 and rolipram in a 5FOA growth assay when compared to a strain expressing wild type PDE4B2 (Fig. 5A). Expression of a PKA-repressed fbp1-GFP reporter for which PDE inhibition represses GFP expression [21, 24] confirmed the reduced sensitivity of PDE4B2T407A to BC54 and rolipram (Fig. 5B), although PDE4BT407A appears to be inhibited by high levels of both compounds. Consistent with both 5FOA assays and in vitro enzyme assays, the GFP data indicate that BC54 is a more potent PDE4 inhibitor than rolipram.
Fig. 5.

Cell based assays confirm reduced sensitivity to BC54 and rolipram by mutant PDE4B2T407A enzyme. A) 5FOA growth assays of strains CHP1401 (PDE4B2wt) and CHP1713 (PDE4B2T407A) expressing the fbp1-ura4+ reporter. PDE inhibition leads to 5FOAR growth. B) GFP assays of strains CHP1641 (PDE4B2wt) and CHP1712 (PDE4B2T407A) expressing the fbp1-GFP reporter. PDE inhibition leads to a reduction in the GFP signal.
3.5 The PDE4B2T407A catalytic domain displays reduced sensitivity to BC54 and rolipram in in vitro enzyme assays
To further characterize the effect of the T407A substitution in PDE4B2 on inhibition by BC54 and rolipram, wild type and T407A mutant forms of the PDE4B catalytic domain (residues 152-487 of the 564aa protein, which has been used to characterize enzyme kinetics and inhibition; [27]) were expressed in E. coli and purified for in vitro enzyme assays. The Y233H mutant form of the catalytic domain was also constructed and purified, however it displayed too little activity for use in in vitro assays (data not shown). In vitro enzyme assays produced IC50 values for the wild type PDE4B2 catalytic domain of 6.5 ± 8.6 nM for BC54 and 503 ± 6.7 nM for rolipram, while the IC50 values for the mutant PDE4B2T407A catalytic domain are 124.3 ± 8.8 nM for BC54 and 1858 ± 4.9 nM for rolipram (Fig. 6; tested at 500nM cAMP). Thus, the mutant enzyme is more resistant to both BC54 and rolipram compared to the wild type enzyme, although the magnitude of the effect is much greater for BC54. However, consistent with the GFP assay data (Fig. 5B), the mutant enzyme retains some sensitivity to both compounds.
Fig. 6.

In vitro enzyme assays confirm that PDE4BT407A is less sensitive to BC54 and rolipram as compared to wild type PDE4B. In vitro enzyme assays using the catalytic domains of wild type PDE4B and PDE4BT407A expressed and purified from E. coli. Values represent averages of inhibition ± S.E.M. from three independent assays carried out at 500nM cAMP substrate concentration.
4. Discussion
In this study, we identified BC54 as a new chemical probe to study the biological roles of PDE4 and PDE7 enzymes. It displays a greater than 20-fold selectivity for PDE4 and PDE7 relative to PDE2A and PDE11A, and is relatively inert against enzymes from the other PDE families. One expectation when developing this platform was that compounds identified by their ability to inhibit heterologously-expressed PDEs in S. pombe would likely display biologically-relevant effects on mammalian cells as these compounds must be cell permeable and relatively stable so as to inhibit the target PDE during the 48 hour growth period. In addition, the compounds must be fairly selective in their binding specificity as promiscuously-binding molecules would likely affect some of the many endogenous yeast proteins required for growth. In our previous studies, we detected both rolipram and zardaverine in PDE4 inhibitor screens (Fig. 1; [25]), which are known anti-inflammatory drugs. We also identified the first-in-class PDE11A inhibitor BC11-38 that elevates cAMP levels, PKA activity, and cortisol release in H295R adrenocortical cells [22], as well as the PDE4/8 dual-specificity inhibitor BC8-15 that elevates steroidogenesis in Leydig cells [34]. Thus, we were not surprised to see that BC54 displays remarkably potent anti-inflammatory activity in both TNFα assays in macrophage and IL-2 assays in T cells (Fig. 2), as well as in CLL apoptosis assays (Table 2, Supplemental Fig. 2). Consistent with activity against PDE4 and PDE7, BC54 outperforms the PDE4 inhibitor rolipram in anti-inflammatory assays for TNFα production by macrophage and IL-2 production by T cells (Fig. 2), and a combination of rolipram and the PDE7A inhibitor BRL50481 in TNFα assays (Fig. 2) and in CLL apoptosis assays (Table 2, Supplemental Fig. 2). Based on the tissue-specific expression of the various PDE genes [1, 4], it seems unlikely that the modest activity of BC54 against PDE2A and PDE11A contributes to its potency in these immune cell-related assays.
The relative efficacy of BC54 when compared to rolipram plus BRL50481 highlights a significant problem with PDE7 studies that use BRL50481 as a PDE7 or PDE7B inhibitor rather than a PDE7A inhibitor. BRL50481 development was guided by its activity against PDE7A and it had not been tested against PDE7B [15], although it is marketed as an inhibitor of both PDE7A and PDE7B. Our own studies using both a yeast-based assessment of PDE activity and in vitro enzyme assays show that BRL50481 is a relatively weak PDE7B inhibitor (IC50 = 12.1μM in comparison to 0.15μM for PDE7A; [13]). As such, studies that use BRL50481 as an inhibitor of both PDE7A and PDE7B or of PDE7B alone may fail to identify the importance of these enzymes in a biological process or may push researchers to use concentrations that promote inhibition of enzymes from other PDE families [41]. This is not to say that BC54 is the ideal PDE4/7 inhibitor as it retains some activity against other PDEs that could be relevant in certain assays. However, the PDE7 field lacks truly selective inhibitors such as those available to study PDE4 enzymes. While other PDE7 or PDE4/7 inhibitors have been described in the literature [42], none have been fully characterized for their activity against the 11 PDE families.
Along with identifying a potent PDE4/7 inhibitor, this study demonstrates the utility of the yeast-based system to characterize the interaction of a compound with its target via a genetic screen for compound resistance. The high PKA phenotype of the failure to enter stationary phase allowed us to enrich for transformants expressing rare PCR-induced mutations that confer resistance to BC54. Similarly, PKA repression of fbp1-ura4+ expression to inhibit growth on SC-ura medium [18, 43] allowed us to identify individual transformants expressing PDE4B2 enzymes that are partially-resistant to BC54. Of the two mutant alleles we obtained in our screen, the Y233H substitution causes a dramatic reduction in activity, while the T407A substitution leads to a significant reduction in inhibition by BC54 or by rolipram. These results are consistent with a previous study by Houslay and Adams [44], that shows the Y233 residue taking part in substrate binding and the T407 residue taking part in rolipram binding. In addition, Pillai and co-workers identified the equivalent mutation in a rat PDE4B clone (mutant A5; T405A) among a collection of rolipram-resistant alleles obtained using a heat shock survival screen in S. cerevisiae [45].
In summary, we have used a yeast-based screen to identify a potent PDE4/7 inhibitor that could be a useful tool in the study of enzymes from these two families as it is very effective in three different mammalian cell-based assays. In addition, we have developed a genetic screen for compound-resistant PDEs that identify residues important for the action of a PDE inhibitor. This work also underscores the need for even better and more fully characterized compounds for the study of the biological roles of PDE7A and PDE7B.
Supplementary Material
Highlights.
Compound BC54 was identified in parallel cell-based PDE4 and PDE7 inhibitor HTSs
BC54 shows potent activity in TNFα and IL-2 anti-inflammatory assays
BC54 outperforms rolipram (PDE4) plus BRL50481 (PDE7A) in a CLL apoptosis assay
A plasmid-based screen identified BC54-resistant allele of PDE4B2
The PDE4B2T407A protein is partially resistant to both BC54 and rolipram
Acknowledgments
This work was supported by grants from Boston College and by the National Institutes of Health [R21 GM079662] to C.S.H.. We thank Hengming Ke for the generous gift of recombinant PDE4 enzymes and the pET15b-PDE4B clone, and Nicola Tolliday, Lynn VerPlank, Jason Burbank, and Stephanie Norton for guidance on high throughput screening. The project has been funded in part with Federal funds from the National Cancer Institute’s Initiative for Chemical Genetics, National Institutes of Health, under Contract No. N01-CO-12400 and was performed with the assistance of the Chemical Biology Platform of the Broad Institute of Harvard and MIT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58(3):488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 2.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 3.Lerner A, Epstein PM. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem J. 2006;393(Pt 1):21–41. doi: 10.1042/BJ20051368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13(4):290–314. doi: 10.1038/nrd4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadana R, Dessauer CW. Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals. 2009;17(1):5–22. doi: 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31(12):680–6. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Sommer N, Loschmann PA, Northoff GH, Weller M, Steinbrecher A, Steinbach JP, Lichtenfels R, Meyermann R, Riethmuller A, Fontana A, et al. The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nat Med. 1995;1(3):244–8. doi: 10.1038/nm0395-244. [DOI] [PubMed] [Google Scholar]
- 8.Wachtel H. Potential antidepressant activity of rolipram and other selective cyclic adenosine 3′,5′-monophosphate phosphodiesterase inhibitors. Neuropharmacology. 1983;22(3):267–72. doi: 10.1016/0028-3908(83)90239-3. [DOI] [PubMed] [Google Scholar]
- 9.Heaslip RJ, Evans DY. Emetic, central nervous system, and pulmonary activities of rolipram in the dog. Eur J Pharmacol. 1995;286(3):281–90. doi: 10.1016/0014-2999(95)00457-2. [DOI] [PubMed] [Google Scholar]
- 10.Gavalda A, Roberts RS. Phosphodiesterase-4 inhibitors: a review of current developments (2010 – 2012) Expert Opin Ther Pat. 2013;23(8):997–1016. doi: 10.1517/13543776.2013.794789. [DOI] [PubMed] [Google Scholar]
- 11.Castro A, Jerez MJ, Gil C, Martinez A. Cyclic nucleotide phosphodiesterases and their role in immunomodulatory responses: advances in the development of specific phosphodiesterase inhibitors. Med Res Rev. 2005;25(2):229–44. doi: 10.1002/med.20020. [DOI] [PubMed] [Google Scholar]
- 12.Giembycz MA. Life after PDE4: overcoming adverse events with dual-specificity phosphodiesterase inhibitors. Curr Opin Pharmacol. 2005;5(3):238–44. doi: 10.1016/j.coph.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Alaamery MA, Wyman AR, Ivey FD, Allain C, Demirbas D, Wang L, Ceyhan O, Hoffman CS. New classes of PDE7 inhibitors identified by a fission yeast-based HTS. J Biomol Screen. 2010;15(4):359–67. doi: 10.1177/1087057110362100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo J, Watson A, Kempson J, Carlsen M, Barbosa J, Stebbins K, Lee D, Dodd J, Nadler SG, McKinnon M, Barrish J, Pitts WJ. Identification of potent pyrimidine inhibitors of phosphodiesterase 7 (PDE7) and their ability to inhibit T cell proliferation. Bioorg Med Chem Lett. 2009;19(7):1935–8. doi: 10.1016/j.bmcl.2009.02.060. [DOI] [PubMed] [Google Scholar]
- 15.Smith SJ, Cieslinski LB, Newton R, Donnelly LE, Fenwick PS, Nicholson AG, Barnes PJ, Barnette MS, Giembycz MA. Discovery of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a selective inhibitor of phosphodiesterase 7: in vitro studies in human monocytes, lung macrophages, and CD8+ T-lymphocytes. Mol Pharmacol. 2004;66(6):1679–89. doi: 10.1124/mol.104.002246. [DOI] [PubMed] [Google Scholar]
- 16.Demirbas D, Ceyhan O, Wyman AR, Hoffman CS. A fission yeast-based platform for phosphodiesterase inhibitor HTSs and analyses of phosphodiesterase activity. Handb Exp Pharmacol. 2011;(204):135–49. doi: 10.1007/978-3-642-17969-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutz H, Heslot H, Leupold U, Loprieno N. Schizosaccharomyces pombe. In: King RC, editor. Handbook of Genetics. Plenum Press; New York: 1974. pp. 395–446. [Google Scholar]
- 18.Hoffman CS, Winston F. Isolation and characterization of mutants constitutive for expression of the fbp1 gene of Schizosaccharomyces pombe. Genetics. 1990;124(4):807–16. doi: 10.1093/genetics/124.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Griffiths K, Zhang YH, Ivey FD, Hoffman CS. Schizosaccharomyces pombe adenylate cyclase suppressor mutations suggest a role for cAMP phosphodiesterase regulation in feedback control of glucose/cAMP signaling. Genetics. 2005;171:1523–33. doi: 10.1534/genetics.105.047233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demirbas D, Ceyhan O, Wyman AR, Ivey FD, Allain C, Wang L, Sharuk MN, Francis SH, Hoffman CS. Use of a Schizosaccharomyces pombe PKA-repressible reporter to study cGMP metabolising phosphodiesterases. Cell Signal. 2011;23(3):594–601. doi: 10.1016/j.cellsig.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Medeiros AS, Magee A, Nelson K, Friedberg L, Trocka K, Hoffman CS. Use of PKA-mediated phenotypes for genetic and small-molecule screens in Schizosaccharomyces pombe. Biochem Soc Trans. 2013;41(6):1692–5. doi: 10.1042/BST20130159. [DOI] [PubMed] [Google Scholar]
- 22.Ceyhan O, Birsoy K, Hoffman CS. Identification of Biologically Active PDE11-Selective Inhibitors Using a Yeast-Based High-Throughput Screen. Chem Biol. 2012;19(1):155–63. doi: 10.1016/j.chembiol.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 23.de Medeiros AS, Kwak G, Vanderhooft J, Rivera S, Gottlieb R, HCS . Fission yeast-based high-throughput screens for PKA pathway inhibitors and activators. In: Hempel JE, Williams CH, Hong CC, editors. Chemical Biology: Methods and Protocols. Springer Humana Press; 2015. in press. [DOI] [PubMed] [Google Scholar]
- 24.de Medeiros AS, Kwak G, Vanderhooft J, Rivera S, Gottlieb R, Hoffman CS. Fission yeast-based high-throughput screens for PKA pathway inhibitors and activators. Methods Mol Biol. 2015;1263:77–91. doi: 10.1007/978-1-4939-2269-7_6. [DOI] [PubMed] [Google Scholar]
- 25.Ivey FD, Wang L, Demirbas D, Allain C, Hoffman CS. Development of a fission yeast-based high-throughput screen to identify chemical regulators of cAMP phosphodiesterases. J Biomol Screen. 2008;13(1):62–71. doi: 10.1177/1087057107312127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Peng MS, Chen Y, Geng J, Robinson H, Houslay MD, Cai J, Ke H. Structures of the four subfamilies of phosphodiesterase-4 provide insight into the selectivity of their inhibitors. Biochem J. 2007;408(2):193–201. doi: 10.1042/BJ20070970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Liu Y, Chen Y, Robinson H, Ke H. Multiple elements jointly determine inhibitor selectivity of cyclic nucleotide phosphodiesterases 4 and 7. J Biol Chem. 2005;280(35):30949–55. doi: 10.1074/jbc.M504398200. [DOI] [PubMed] [Google Scholar]
- 29.Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14(10):943–51. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 30.DeVoti J, Seydoux G, Beach D, McLeod M. Interaction between ran1+ protein kinase and cAMP dependent protein kinase as negative regulators of fission yeast meiosis. Embo J. 1991;10(12):3759–68. doi: 10.1002/j.1460-2075.1991.tb04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isshiki T, Mochizuki N, Maeda T, Yamamoto M. Characterization of a fission yeast gene, gpa2, that encodes a G alpha subunit involved in the monitoring of nutrition. Genes Dev. 1992;6(12B):2455–62. doi: 10.1101/gad.6.12b.2455. [DOI] [PubMed] [Google Scholar]
- 32.Ivey FD, Hoffman CS. Direct activation of fission yeast adenylate cyclase by the Gpa2 Gα of the glucose signaling pathway. Proc Natl Acad Sci U S A. 2005;102(17):6108–13. doi: 10.1073/pnas.0502270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nocero M, Isshiki T, Yamamoto M, Hoffman CS. Glucose repression of fbp1 transcription of Schizosaccharomyces pombe is partially regulated by adenylate cyclase activation by a G protein α subunit encoded by gpa2 (git8) Genetics. 1994;138(1):39–45. doi: 10.1093/genetics/138.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demirbas D, Wyman AR, Shimizu-Albergine M, Cakici O, Beavo JA, Hoffman CS. A Yeast-Based Chemical Screen Identifies a PDE Inhibitor that Elevates Steroidogenesis in Mouse Leydig Cells Via PDE8 and PDE4 Inhibition. PLoS One. 2013;8(8):e71279. doi: 10.1371/journal.pone.0071279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim DH, Lerner A. Type 4 cyclic adenosine monophosphate phosphodiesterase as a therapeutic target in chronic lymphocytic leukemia. Blood. 1998;92(7):2484–94. [PubMed] [Google Scholar]
- 36.Zhang L, Murray F, Zahno A, Kanter JR, Chou D, Suda R, Fenlon M, Rassenti L, Cottam H, Kipps TJ, Insel PA. Cyclic nucleotide phosphodiesterase profiling reveals increased expression of phosphodiesterase 7B in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2008;105(49):19532–7. doi: 10.1073/pnas.0806152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee R, Wolda S, Moon E, Esselstyn J, Hertel C, Lerner A. PDE7A is expressed in human B-lymphocytes and is up-regulated by elevation of intracellular cAMP. Cell Signal. 2002;14(3):277–84. doi: 10.1016/s0898-6568(01)00250-9. [DOI] [PubMed] [Google Scholar]
- 38.Ivey FD, Taglia FX, Yang F, Lander MM, Kelly DA, Hoffman CS. Activated alleles of the Schizosaccharomyces pombe gpa2+ Galpha gene identify residues involved in GDP-GTP exchange. Eukaryot Cell. 2010;9(4):626–33. doi: 10.1128/EC.00010-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Robinson H, Ke H. The molecular basis for different recognition of substrates by phosphodiesterase families 4 and 10. J Mol Biol. 2007;371(2):302–7. doi: 10.1016/j.jmb.2007.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Nolte RT. Crystal structures of the catalytic domain of phosphodiesterase 4B complexed with AMP, 8-Br-AMP, and rolipram. J Mol Biol. 2004;337(2):355–65. doi: 10.1016/j.jmb.2004.01.040. [DOI] [PubMed] [Google Scholar]
- 41.Strahm E, Rane A, Ekstrom L. PDE7B is involved in nandrolone decanoate hydrolysis in liver cytosol and its transcription is up-regulated by androgens in HepG2. Front Pharmacol. 2014;5:132. doi: 10.3389/fphar.2014.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vergne F, Bernardelli P, Chevalier E. PDE7 inhibitors: Chemistry and potential therapeutic utilites. Annu Rep Med Chem. 2005;40:227–241. [Google Scholar]
- 43.Hoffman CS, Winston F. Glucose repression of transcription of the Schizosaccharomyces pombe fbp1 gene occurs by a cAMP signaling pathway. Genes Dev. 1991;5(4):561–71. doi: 10.1101/gad.5.4.561. [DOI] [PubMed] [Google Scholar]
- 44.Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003;370(Pt 1):1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pillai R, Kytle K, Reyes A, Colicelli J. Use of a yeast expression system for the isolation and analysis of drug-resistant mutants of a mammalian phosphodiesterase. Proc Natl Acad Sci U S A. 1993;90(24):11970–4. doi: 10.1073/pnas.90.24.11970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.