

Abstract
DNA methylation is an epigenetic mechanism that regulates gene transcription without changing primary nucleotide sequences. In mammals, DNA methylation involves the covalent addition of a methyl group to the 5-carbon position of cytosine by DNA methyltransferases (DNMTs). The change of DNA methylation and its pathological role in acute kidney injury (AKI) remain largely unknown. Here, we analyzed genome-wide DNA methylation during cisplatin-induced AKI by reduced representation bisulfite sequencing. This technique identified 215 differentially methylated regions between the kidneys of control and cisplatin-treated animals. While most of the differentially methylated regions were in the intergenic, intronic and coding DNA sequences, some were located in the promoter or promoter regulatory regions of 15 protein-coding genes. To determine the pathological role of DNA methylation, we initially examined the effects of DNA methylation inhibitor 5-Aza-2′-deoxycytidine and showed it increased cisplatin-induced apoptosis in a rat kidney proximal tubular cell line. We further established a kidney proximal tubule-specific DNMT1 (PT-DNMT1) knockout mouse model, which showed more severe AKI during cisplatin treatment than wild-type mice. Finally, interferon regulatory factor 8 (Irf8), a pro-apoptotic factor, was identified as a hypomethylated gene in cisplatin-induced AKI and this hypomethylation was associated with a marked induction of Irf8. In the rat kidney proximal tubular cells, knockdown of Irf8 suppressed cisplatin-induced apoptosis, supporting a pro-death role of Irf8 in renal tubular cells. Thus, DNA methylation plays a protective role in cisplatin-induced AKI by regulating specific genes, such as Irf8.
Keywords: DNA methylation, DNA methyltransferases, Cisplatin, Nephrotoxicity, Acute kidney injury
Introduction
Acute kidney injury (AKI), formerly known as acute renal failure, is generally defined as a rapid decrease in kidney function within a few hours to days. AKI is now recognized as a major public health problem with profound consequences including high mortality rates, length of hospital stay and costs.1 Clinically, major causes of AKI include renal ischemia-reperfusion, nephrotoxicity, and sepsis. Approximately 20% of AKI cases are attributed to the exposure of nephrotoxic drugs. Cisplatin (cis-diamminedichloroplatinum II - CDDP) is one of the most commonly used and highly effective chemotherapy drugs for cancer. It is used for the treatment of a wide variety of cancer types, such as testicular, ovarian, head and neck, bladder, small and non-small cell lung, cervical cancers, and many other types of cancers like sarcomas and lymphomas.2, 3 However, cisplatin is also notorious for its side-effects in normal tissues and organs, especially in the kidney, which limits its use and therapeutic efficacy.4-8 Following a single dose of cisplatin treatment, 25%-35% of the patients experience renal function deterioration. In the kidneys, cisplatin accumulates at high concentrations in renal tubular cells (about five times greater than in the blood), causing tubular cell injury and death, which is a key determinant of AKI.9 Recently, the pathogenesis of cisplatin-induced AKI has been intensively studied, and multiple signaling pathways have been implicated, such as apoptosis and necrosis of renal tubular cells, inflammation, and oxidative stress.4-8 Despite these studies, the molecular basis of cisplatin nephrotoxicity remains unclear and no effective therapies are available for renoprotection during chemotherapy.
DNA methylation is an important epigenetic mechanism, which involves the covalent addition of a methyl group to the 5-carbon position of the cytosine in CpG dinucleotide sequences by DNA methyltransferases (DNMTs).10 It is heritable during cell division and regulates gene transcription without changing its primary nucleotide sequence. There are three major DNMTs for DNA methylation—DNMT1, DNMT3a, and DNMT3b. DNMT1 is the maintenance DNA methyltransferase which is also the most abundant DNMT in various cell types, while DNMT3a and DNMT3b are de novo methyltransferases, which establish the initial DNA methylation patterns.11, 12 When DNMT1 is inhibited or absent during cell division, the newly synthesized DNA strands cannot be methylated, resulting in passive demethylation in the daughter cells and dilution of DNA methylation in the cell population. On the other hand, active demethylation can be achieved by the enzymatic replacement of methyl-cytosine to cytosine by a set of enzymes, such as thymine DNA glycosylase (TDG), activation- induced deaminase (AID) and ten-eleven translocation methylcytosine dioxygenase (TET).13, 14 The fundamental role of DNA methylation in cell biology is transcriptional regulation. Generally, hypermethylation in the promoter region of a gene leads to heterochromatin with highly packed DNA, decreased accessibility of transcription factors, and loss of gene expression.15, 16 In contrast, DNA hypomethylation may correlate with the activation of gene transcription or lead to genomic instability.17 As a result, DNA methylation plays essential roles in mammalian development, genomic integrity, X chromosome inactivation (females), and genomic imprinting. And aberrant DNA methylation has been implicated in a wide variety of disease conditions, such as cardiovascular diseases, neurological diseases, and cancer.18-23 For example, global DNA hypomethylation accompanied by hypermethylation of tumor suppressor genes is recognized as an epigenetic hallmark of cancer.24, 25
Recently, several studies suggested the involvement of DNA methylation changes in kidney diseases, including kidney fibrosis,26-28 diabetic nephropathy,29, 30 and chronic kidney disease.31, 32 However, very limited is known about the role and regulation of DNA methylation in AKI.33-35 In this study, we analyzed the global DNA methylation changes during cisplatin-induced AKI or nephrotoxicity. Functionally, inhibition of DNA methylation by 5-aza increased cisplatin-induced apoptosis in vitro and ablation of DNMT1 from kidney proximal tubules enhanced cisplatin-induced AKI in mice, suggesting a renoprotective role of DNA methylation. We further identified Irf8 as a hypomethylated gene during cisplatin treatment that was induced to contribute to tubular cell apoptosis.
Results
Genome-wide changes in DNA methylation during cisplatin-induced AKI
To determine the genome-wide DNA methylation changes in cisplatin-induced AKI, we utilized the reduced representation bisulfite sequencing (RRBS) to identify DNA methylation at singlebase resolution. Genomic DNAs were isolated from kidney cortex and outer medulla of control and cisplatin-treated mice and then subjected to RRBS analysis. Totally about 1.5 and 1.9 millions of CpG sites were analyzed in the control and cisplatin treated kidney samples, respectively. Cisplatin treatment induced obvious changes in DNA methylation as shown by the heat map (Figure 1A). Using 200 bp non-overlapping windows, we identified 215 differentially methylated regions (DMRs) between control and cisplatin-treated kidneys that showed significant (>±0.25) differences in methylation (Figure 1B). In DMR genome-wide distribution analysis, 83% of the DMRs were in the intergenic region, intron and coding DNA sequence, and only 7% of the DMRs were in the 5′ end and 5′UTR promoter or regulatory region of protein-coding genes (Figure 1C). This result is consistent with the recent study of genome-wide DNA methylation in chronic kidney disease28 that also showed the distribution of the majority of the DMRs in intronic and transcription termination regions and 3′UTRs, and not in gene promoter regions. DNA methylation in gene promoter regions is considered to be critical for transcriptional regulation. Our analysis identified 15 genes with DMRs in their 5′end and 5′UTR regulatory regions, and the functional analysis indicated these genes are involved in gene transcription, cell cycle, and apoptosis (Supplemental Figure 1). Taken together, these results indicate that cisplatin treatment induces changes in DNA methylation in kidney tissues and DNA methylation may play a role in cisplatin-induced AKI by regulating specific genes.
Figure 1. Genome-wide analysis of DNA methylation changes in cisplatin-induced AKI.
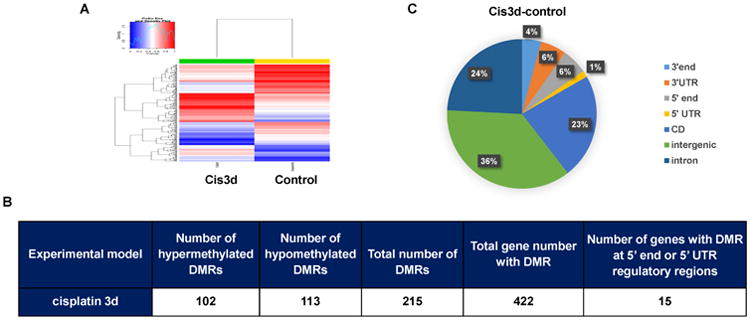
C57BL/6 mice were injected with cisplatin (cis3d) or saline (control) to collect kidney cortex and outer medulla 3 days later for the isolation of genomic DNAs, which were subjected to genome-wide DNA methylation analysis via reduced representation bisulfite sequencing (RRBS). 200 bp non-overlapping windows were used to identify differentially methylated regions (DMRs). DMRs were identified with a methylation difference over 0.25. (A) Representative heat map. Red color indicates high levels of DNA methylation and blue color indicates low levels of methylation. (B) The number of DMRs identified in cisplatin-induced AKI. (C) Genome-wide distribution of the DMRs. The results were from 2 separate experiments with 2 pairs of littermate mice.
DNA methylation inhibitor 5-Aza-2′-deoxycytidine (5-aza) increases cisplatin-induced apoptosis in RPTC cells
To determine whether DNA methylation plays a pathogenic role in cisplatin-induced AKI, we first tested the effect of 5-Aza-2′-deoxycytidine (5-aza, a pharmacological DNA methylation inhibitor) on cisplatin-induced apoptosis in RPTC, a rat renal proximal tubular cell line. As a cytidine analog, 5-aza may incorporate into DNA and irreversibly binds to DNMTs resulting in DNMT inactivation and degradation.36-38 Treatment with 1μM 5-aza could diminish DNMT1 and DNMT3a expression (Figure 2A) without noticeable toxicity in RPTC cells (Figures 2B, 2C). Importantly, 5-aza significantly increased apoptosis during cisplatin treatment (Figure 2B). Typical apoptotic cells showed cellular shrinkage and formation of apoptotic bodies viewed by phase contrast microscopy (Figure 2B, upper panel) and nuclear condensation and fragmentation viewed by Hoechst 33342 nucleus staining (Figure 2B, bottom panel). Cell counting showed that cisplatin induced 24% apoptosis, which was increased to 55% by 5-aza pretreatment (Figure 2C). The morphological results were verified by immunoblot analysis of the cleavage of caspase 3 and PARP, biochemical hallmarks of apoptosis, showing the enhancing effect of 5-aza (Figure 2A). Collectively, these results demonstrate the sensitizing effect of 5-aza to cisplatin-induced apoptosis, suggesting a cytoprotective role of DNA methylation in renal tubular cells.
Figure 2. 5-aza increases cisplatin-induced apoptosis in RPTC cells.
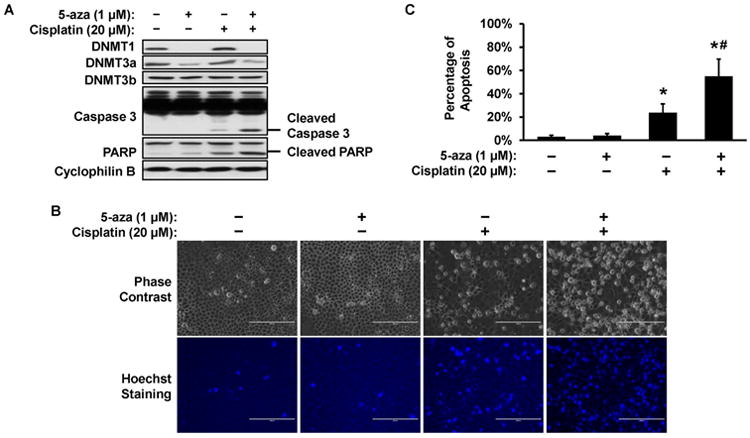
RPTC cells were treated with 20 μM cisplatin in the absence or presence of 1 μM 5-aza pretreatment. (A) Immunoblot. After 16h cisplatin treatment, whole cell lysate was collected for immunoblot analysis of indicated proteins. Cyclophilin B was used as protein loading control. (B) Cell morphology. After 16h cisplatin treatment, cells were stained with Hoechst 33342 to record cellular and nuclear morphology by phase contrast and fluorescence microscopy, respectively. Scale bar: 200 μm. (C) Percentage of cell apoptosis. The cells with typical apoptotic morphology were counted to determine the % of apoptosis. Data in (C) are presented as mean ± SD; n = 4. *P < 0.05, significantly different from control; #P < 0.05, significantly different from the cisplatin-only group.
Establishment and characterization of PT-DNMT1-KO mouse model
To further delineate the role of DNA methylation in cisplatin-induced AKI, we turned to DNMT1 knockout mouse model. DNMT1 is responsible for the maintenance of DNA methylation. Since global knockout of DNMT1 is lethal,39 we decided to generate conditional (kidney proximal tubule-specific) knockout mice via the Cre-loxp system. PEPCK-Cre mice that express the Cre-recombinase specifically in proximal tubules were crossed with DNMT1flox/flox mice to generate PT-DNMT1-KO mice (Supplemental Figure 2). In PT-DNMT1-KO mice, exon 4 and exon 5 of DNMT1 gene are deleted, including the localization domain and the entire catalytic domain.40 The deletion of DNMT1 in the proximal tubules was confirmed by PCR (Figure 3A) and immunoblot (Figure 3B). Recombinant DNMT1 alleles (∼ 300 bp) were detected only in kidney cortex of PT-DNMT1-KO and the DNMT1 protein level was suppressed in kidney cortex of PT-DNMT1-KO compared with PT-DNMT1-WT. We further verified the deletion of DNMT1 in the proximal tubules of PT-DNMT1-KO mice by immunohistochemical staining. Lotus tetragonolobus lectin (LTL) staining was used to label proximal tubules, specifically their brush border. As shown in Figure 3C, positive DNMT1 staining was detected in all nuclei of proximal tubular cells in PT-DNMT1-WT mice, but it was absent in the majority of proximal tubular cells in PT-DNMT1-KO mice. A small population of cells in the PT-DNMT1-KO kidney cortical tissue still had DNMT1, because the efficacy of PEPCK-Cre-mediated gene ablation is 70-80% in proximal tubular cells and also there are non-proximal tubular cells in renal cortex. Under normal conditions, PT-DNMT1-KO mice did not show obvious abnormalities in kidney size, renal function or histology. They had similarly low BUN level as wild-type mice at the age of 2-3 months and 6 months (Figure 3D). And there were no phenotypic alterations observed by kidney histology in these mice either (Figure 3E).
Figure 3. Establishment and characterization of PT-DNMT1-KO mouse model.
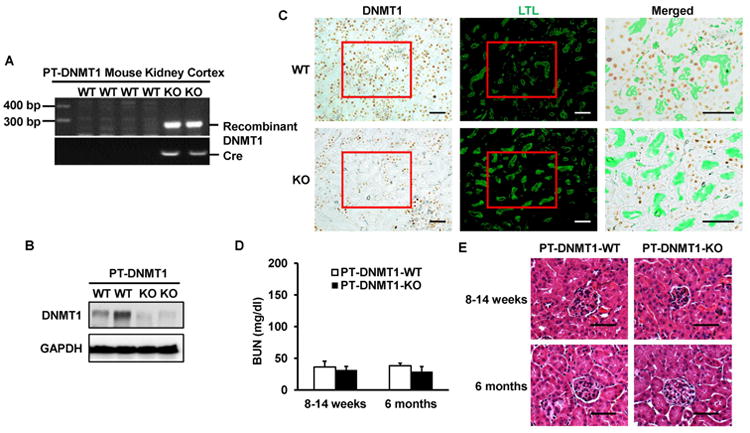
(A) PCR-based genotyping. Genomic DNA was extracted from kidney cortex for PCR amplification to detect PEPCK-Cre allele and recombinant allele of DNMT1. (B) Immunoblot analysis of renal cortical lysate confirming DNMT1 decrease in PT-DNMT1-KO tissues. GAPDH was used as protein loading control. (C) Immunohistochemical staining to verify DNMT1 ablation from proximal tubular cells in PT-DNMT1-KO mice. Kidney tissues from PT-DNMT1-WT and -KO mice were fixed and processed for immunohistochemical staining of DNMT1, followed by staining with LTL to show proximal tubules. Please note that DNMT1 staining is in cell nuclei, whereas LTL staining is on the brush border of proximal tubules. The merged images are the magnified images of the boxed areas and further show the absence of DNMT1 in many proximal tubular cells in PT-DNMT1-KO tissues. Scale bar: 50 μm. (D) Blood urea nitrogen (BUN) of PT-DNMT1-WT and -KO mice to indicate normal kidney function. (E) Representative Hematoxylin and Eosin (H&E) staining images of kidney cortex from PT-DNMT1-WT and -KO mice. Scale bar: 50 μm.
Cisplatin-induced AKI is aggravated in PT-DNMT1-KO mice
We then examined cisplatin-induced AKI in PT-DNMT1 mice. At day 4 of cisplatin injection, PT-DNMT1-WT mice showed a serum creatinine level of 1.9 ± 0.4 mg/dl, while PT-DNMT1-KO mice had 2.5 ± 0.5 mg/dl serum creatinine that was higher than that of wild-type mice with statistical significance (Figure 4A). Cisplatin induced tubular damage in both PT-DNMT1-WT and -KO mice were characterized by loss of brush border, tubular dilation, cast formation, and cell lysis in proximal tubules. Notably, consistent with serum creatinine results, significantly more tubular damage was shown in PT-DNMT1-KO mice (Figure 4B). Histo-pathological grading showed a tubular damage score of 3.7 for PT-DNMT1-KO mice, which was higher than that of PT-DNMT1-WT mice (Figure 4C). We also analyzed apoptosis in the tissues by immunohistochemical staining of active or cleaved caspase 3. As shown in Figures 4D and 4E, PT-DNMT1-KO kidney tissues had a significantly higher level of cleaved caspase 3 staining than PT-DNMT1-WT kidney tissues. Altogether, these results suggest that ablation of DNMT1 from kidney proximal tubules leads to more severe AKI following cisplatin exposure.
Figure 4. Cisplatin-induced AKI is aggravated in PT-DNMT1-KO mice.
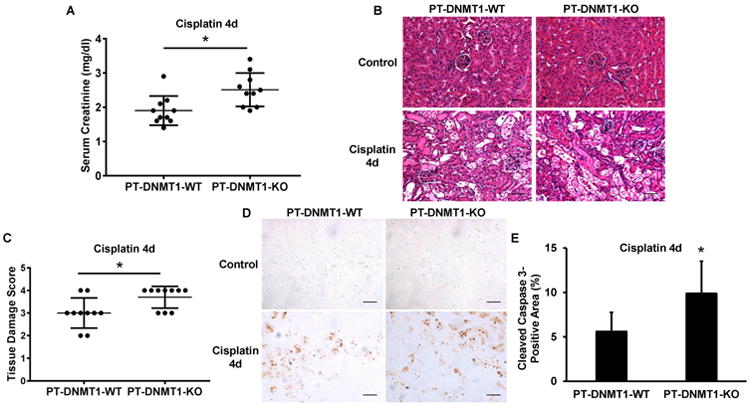
PT-DNMT1-WT and PT-DNMT1-KO littermate mice were injected with 30 mg/kg cisplatin or saline as control. Kidney tissues and blood samples were collected at day 4 after cisplatin injection for analysis. (A) Serum creatinine. (B) Representative H&E images of kidney cortex from PT-DNMT1-WT and -KO mice. Scale bar: 50 μm. (C) Kidney tissue damage score in cisplatin- treated PT -DNMT1-WT and -KO mice. (D) Representative images of cleaved caspase 3 immunohistochmical staining. Scale bar: 50 μm. (E) Quantitative analysis of cleaved caspase 3 immunohistochemical staining. Quantitative data in (A) and (C) are expressed as mean ± SD (n=10). *P < 0.05, significantly different from PT-DNMT1-WT mice.
Primary proximal tubular cells from PT-DNMT1-KO mice are more sensitive to cisplatin-induced apoptosis than wild-type cells
To further confirm the in vivo study results, we examined cisplatin-induced apoptosis using primary proximal tubular cells isolated from PT-DNM1-WT or -KO mice. As shown in Figure 5A, both DNMT1-WT and -KO tubular cells showed normal morphology with minimal apoptosis under control condition. Cisplatin treatment induced apoptosis in both groups of cells, but obviously more apoptosis was induced in DNMT1-KO tubular cells (Figure 5A). In quantification, cisplatin induced ∼18% apoptosis in DNMT1-WT tubular cells, whereas ∼40% apoptosis in DNMT1-KO tubular cells (Figure 5B). Moreover, immunoblot analysis detected much higher caspase 3 cleavage in DNMT1-KO tubular cells after cisplatin treatment (Figure 5C). Thus, cisplatin induces higher toxicity in PT-DNMT1-KO primary proximal tubular cells than wild-type cells, an observation that is consistent with above in vivo study.
Figure 5. DNMT1-KO primary proximal tubular cells are more sensitive to cisplatin-induced apoptosis.
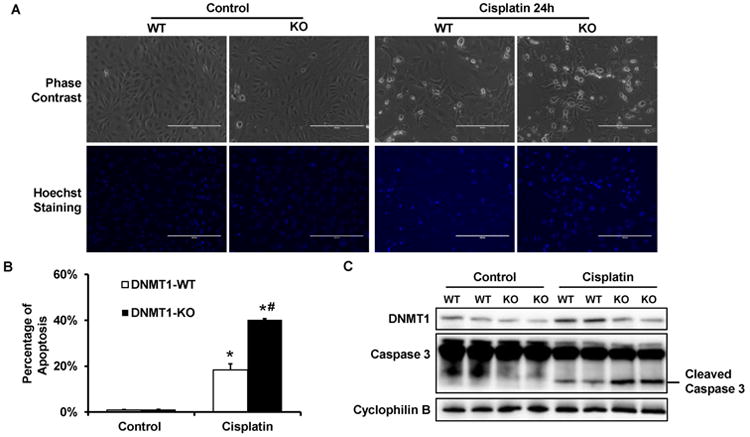
Primary proximal tubular cells were isolated from PT-DNMT1-WT or -KO mice. Then primary cells were treated with 50 μM cisplatin for 24 hours. (A) Cell morphology. After 24h cisplatin treatment, cells were stained with Hoechst 33342 to record cellular and nuclear morphology by phase contrast and fluorescence microscopy, respectively. (B) Percentage of apoptosis. The cells with typical apoptotic morphology were counted to determine the % of apoptosis. Scale bar: 200 μm. (C) Immunoblot analysis of Caspase 3 activation. After 24h cisplatin treatment, whole cell lysate was collected for immunoblot analysis of DNMT1, Caspase 3, and Cyclophilin B as protein loading control. Caspase 3 activation is indicated by the appearance of cleaved (active) fragment of caspase 3. Data in (B) are presented as mean ± SD; n= 3. *P < 0.05, significantly different from control DNMT1-WT cells; #P < 0.05, significantly different from the cisplatin treated DNMT1-WT cells.
Irf8 is hypomethylated and induced in cisplatin-induced AKI, and inhibition of DNA methylation by 5-aza increases its transcription
The results presented above indicate that DNA methylation is protective in cisplatin-induced AKI. To further elucidate the underlying mechanism, it is important to identify the specific gene(s) regulated by DNA methylation that contributes to renal tubular injury. Our genome-wide DNA methylation analysis identified 15 genes with DMRs in their promoter or promoter regulatory regions (Figure 1). Among these genes, interferon regulatory factor 8 (Irf8) stood out as a good candidate gene for further study, because Irf8 is a pro-apoptotic gene that is regulated by DNA methylation.41-44 In our genome-wide DNA methylation analysis, Irf8 was found to contain a DMR at 5′ UTR region with hypomethylation when mice were challenged with cisplatin (Figure 6A). Real-time PCR analysis indicated that, along with Irf8 hypomethylation, cisplatin induced Irf8 expression (Figure 6B). Immunoblot further verified Irf8 induction at the protein level in cisplatin treated mice kidneys (Figure 6C). To further confirm the role of DNA methylation in regulating Irf8 expression, we analyzed the effect of 5-aza on Irf8 expression in RPTC cells. As shown in Figure 6D, 5-aza induced Irf8 at both mRNA and protein levels with or without cisplatin treatment. We also examined Irf8 expression in PT-DNMT1-WT and -KO mice kidney tissues. Real-time PCR analysis detected similar levels of Irf8 in kidney tissues from control PT-DNMT1-KO and –WT mice. During cisplatin treatment, both PT-DNMT1-KO and –WT mice kidney tissues showed significant Irf8 induction and notably, Irf8 induction by cisplatin was indifferent in these mice (Figure 6E). Together, these results suggest that Irf8 expression during cisplatin-AKI is regulated by DNA methylation, which may not depend on DNMT1 alone, but involve other DNMTs and mechanisms.
Figure 6. Irf8 is hypomethylated and induced in cisplatin-induced AKI, and inhibition of DNA methylation by 5-aza increases its transcription.
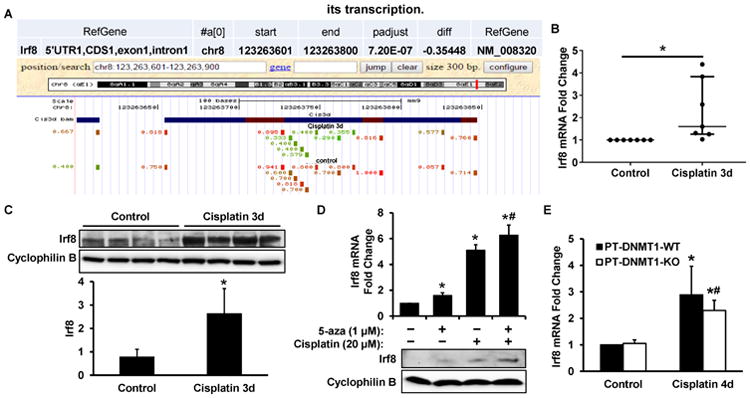
In (A), (B) and (C), male C57BL/6 mice were injected with 30 mg/kg cisplatin or saline as control. (A) The RRBS result of Irf8 gene methylation changes shown by UCSC genome browser screenshot. The hypomethylated CpG sites following cisplatin-treatment are highlighted in green. (B) Real-time PCR analysis to verify Irf8 induction in kidney cortical tissues following cisplatin treatment. (C) Immunoblot analysis of Irf8 expression in renal cortical lysate. Cyclophilin B was used as a protein loading control. Irf8 signals were then analyzed by densitometry (bottom). After normalization with cylcophilin B, the protein signal of lane 1 was arbitrarily set as 1, and the signals of other lanes were normalized with lane 1 to calculate fold changes. (D) RPTC cells were treated with 20 μM cisplatin for 16 hours in the absence or presence of 5-aza pretreatment. Irf8 expression was shown by real-time PCR (top) and immunoblot (bottom) analysis. (E) PT-DNMT1-WT or -KO littermate mice were injected with 30 mg/kg cisplatin or saline as control. Irf8 mRNA level in the cortical and outer medulla total RNA was detected by real-time PCR. Data in (B) are expressed as median ± interquartile range; n=7. Data in (C) and (D) are expressed as mean ± SD; n=4. *P < 0.05, significantly different from control; #P < 0.05, significantly different from the cisplatin-only group. Data in (E) are presented as mean ± SD; n=5. *P<0.05, significantly different from control PT-DNMT1-WT mice; #P<0.05, significantly different from control PT-DNMT1-KO mice.
Silencing Irf8 suppresses cisplatin-induced apoptosis in RPTC cells
Finally, to verify the pro-apoptotic role of Irf8 in renal tubular cells, we determined the effect of Irf8 knockdown on cisplatin-induced apoptosis in RPTC cells. Compared with scramble sequence shRNA, Irf8 shRNA significantly suppressed Irf8 expression (Figure 7A). Under control condition, both scramble cells and Irf8 knockdown cells exhibited minimal degree of apoptotic cells (Figure 7B). After 20h cisplatin treatment, ∼43% apoptosis was observed in scramble shRNA cells, but only 17% in Irf8 knockdown cells (Figure 7C). Consistently, silencing Irf8 significantly reduced caspase 3 cleavage (Figure 7D). Thus, loss of Irf8 expression protected RPTC cells from cisplatin-induced apoptosis, supporting an injurious role of Irf8 in renal tubular cells.
Figure 7. Silencing Irf8 suppresses cisplatin-induced apoptosis in RPTC cells.
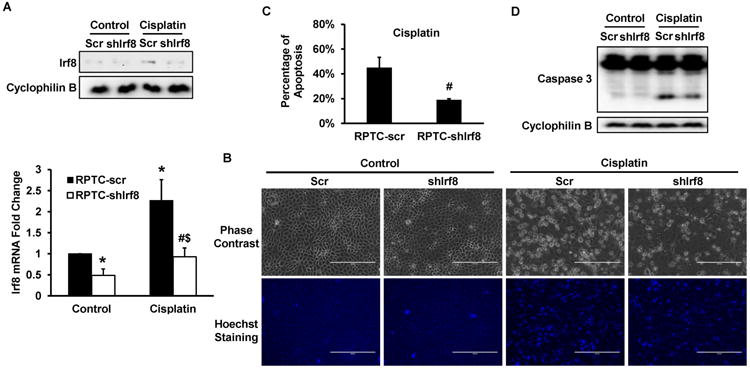
Irf8 shRNA and scrambled sequence plasmids were separately transfected into RPTC cells to select stably transfected cells. The cells were then treated with 20 μM cisplatin for 20 hours. (A) Immunoblot (top) and real-time PCR (bottom) analysis of Irf8 to confirm the silencing of Irf8. In immunoblot, cyclophilin B was used as protein loading control. (B) Cell morphology. Cells were stained with Hoechst 33342 to record cellular and nuclear morphology by phase contrast and fluorescence microscopy, respectively. Scale bar: 200 μm. (C) Percentage of cell apoptosis. The cells with typical apoptotic morphology were counted to determine the % of apoptosis. (D) Immunoblot analysis of Caspases 3 and Cyclophilin B as protein loading control. Data in (A) and (C) are presented as mean ± SD; n = 3. *P > 0.05, significantly different from control scrambled sequence-transfected cells. #P > 0.05, significantly different from cisplatin-treated scrambled sequence-transfected cells. $P>0.05, significantly different from control Irf8 shRNA sequence-transfected cells.
Discussion
DNA methylation is a major form of epigenetic modification that regulates gene expression in mammalian development and disease pathogenesis. In kidneys, DNA methylation changes have been implicated in kidney fibrosis, diabetic nephropathy, and chronic kidney disease. 26-32 However, the information of DNA methylation in AKI is very limited. In this regard, Rabb and colleagues33 demonstrated the first evidence of hypermethylation of calcitonin gene promoter in the urine samples from kidney transplant patients, suggesting its potential as a biomarker of ischemic AKI during transplantation. Pratt et al.34 reported that a specific cytosine in C3 promoter was de-methylated in rat kidney during renal ischemia-reperfusion. More recently, Huang et al.35 showed a decrease in global 5-hydroxymethylcytosine (5hmC) during ischemic AKI in mice, whereas global 5-methylcytosine (5mC) did not change. It is noteworthy that these studies are mainly descriptive and the role of DNA methylation in the pathogenesis of AKI remains elusive and the regulation of specific pathogenic genes by DNA methylation in AKI is largely unknown. In the current study, we have provided the first analysis of genome-wide DNA methylation changes in cisplatin-induced AKI. We further determined the pathological role of DNA methylation in this nephrotoxic AKI model. 5-aza increased cisplatin-induced apoptosis in RPTC cells and genetic ablation of DNMT1 from proximal tubules worsened cisplatin-induced AKI in vivo, suggesting a renoprotective role for DNA methylation. Moreover, we identified Irf8 that was hypomethylated and induced during cisplatin treatment to contribute to tubular cell injury and death.
Genome-wide DNA methylation analysis provides a profile of changes in DNA methylation that may pinpoint the hyper- or hypo-methylation of specific genes in diseases uncovering novel therapeutic targets. In the present study, we profiled DNA methylation changes in cisplatin-induced AKI using RRBS, an efficient and high-throughput technology with single nucleotide resolution. As shown in the heat map (Figure 1A), cisplatin treatment induced widespread DNA methylation alterations throughout the genome in kidney tissues. The analysis identified 102 hypermethylated DMRs and 113 hypomethylated DMRs following cisplatin treatment (Figure 1B). Thus, although cisplatin induced profound changes in DNA methylation in kidneys, it did not induce an overall shift to DNA hypermethylation or hypomethylation. This observation suggests that the DMRs in specific genes or sequences, other than overall shift of DNA methylation status, may contribute to cisplatin-induced AKI. Indeed, we identified 15 genes with DMRs in their promoters or promoter regulatory regions that are likely regulated by DNA methylation during cisplatin treatment. Of note, these are only a small portion (7%) of the DMRs shown in cisplatin-induced AKI, and the DMRs in non-promoter regions are also valuable, because DNA methylation in the enhancer and gene body may also contribute to transcription modulation.28, 45-48
Our study investigated the role of DNA methylation in cisplatin-induced AKI using both in vitro cell culture and in vivo mouse models. In RPTC cells, we examined the effect of 5-aza on cisplatin-induced apoptosis, an important mechanism of cisplatin-induced AKI or nephrotoxicity. 5-aza is one of the most commonly used DNA methylation inhibitors in experimental study. 5-aza decreased DNMT1 and DNMT3a expression in RPTC cells (Figure 2A), indicative of its inhibitory effect. Importantly, 5-aza markedly increased RPTC apoptosis during cisplatin treatment (Figures 2B, 2C). In vivo, we established conditional DNMT1 knockout mouse model, in which DNMT1 was ablated specifically in kidney proximal tubules. After cisplatin treatment, PT-DNMT1-KO mice showed more severe AKI than wild-type littermates (Figure 4). Altogether, the effect of 5-aza in RPTC cells and the effect of PT-DNMT1-KO in mice support a renoprotective role of overall DNA methylation in cisplatin-induced AKI.
Although PT-DNMT1-KO mice developed significantly more injury in renal tubules following cisplatin treatment, the effect is not dramatic. This may be caused by several factors. First, DNMT1 was not ablated from all proximal tubular cells in our conditional KO model (Figure 3C), which would lead to a partial effect on phenotype. Second, it is possible that DNMT3a and DNMT3b may function as maintenance DNA methyltransferases to compensate for the loss of DNMT1. This possibility is supported by the recent evidence implicating DNMT3a and DNMT3b in the maintenance of DNA methylation.49-51 Finally, it is possible that DNMT1 ablation may not significantly block DNA methylation in some of the proximal tubular cells in our model. This is because DNMT1 is the maintenance DNMT that mainly catalyzes DNA methylation in dividing or proliferating cells. In our model, DNMT1 ablation was driven by PEPCK-Cre at ∼3 weeks after birth in mice,52 a time-point when kidney development has mostly completed. Under this condition, the role of DNMT1 in DNA methylation may be limited due to the low rate of cell proliferation. However, upon AKI and especially during kidney repair after AKI, there is a remarkable increase of proliferation in tubule cells.53 Thus, DNMT1 ablation may specifically affect these proliferative cells. In line with this possibility, primary tubular cells from PT-DNMT1-KO mice (that were proliferative) had more than two times increase in apoptosis during cisplatin treatment than wild-type cells (Figure 5).
From the genome-wide DNA methylation analysis, we identified Irf8 as a good candidate that is regulated by DNA methylation and contributes to cisplatin-induced apoptosis (Figure 6 and 7). Irf8 is also known as interferon consensus sequence-binding protein (ICSBP), belonging to the IRF transcription factor family. In 2007, Irf8 was reported to be regulated by DNA methylation.41 Hypermethylation in Irf8 promoter region repressed Irf8, which was attributed to the apoptotic resistance and metastatic phenotype in tumor cells.41 After that, several studies verified the regulation of Irf8 expression by DNA methylation.42, 44, 54 In the present study, we showed hypomethylation in Irf8 at its 5′UTR during cisplatin treatment (Figure 6A), which was associated with Irf8 induction (Figure 6B and 6C). Previously, Irf8 was thought to be exclusively expressed in myeloid and lymphoid cells,55, 56 but recently it has been found that Irf8 is expressed in a variety of cell and tissue types, such as kidney, liver, lung, and colon.44 We also detected Irf8 expression in mouse kidney and rat proximal tubular cells. Under control condition, Irf8 was expressed at a low level, but after cisplatin treatment, Irf8 was induced markedly (Figure 6C and 6D). Also treatment of RPTC cells with 5-aza resulted in dramatic increase of Irf8 expression (Figure 6D), further confirming the regulation of Irf8 by DNA methylation in renal tubular cells. Together, these data support a role of Irf8 hypomethylation and induction in cisplatin-induced kidney injury.
PT-DNMT1-KO and -WT mice kidney tissues showed similar levels of Irf8 induction during cisplatin treatment (Figure 6E), indicating that DNMT1 deficiency does not significantly affect Irf8 expression under this condition. This observation also suggests that Irf8 may not be a key to the cisplatin injury sensitivity of PT-DNMT1-KO mice. A previous study also showed that knockdown of DNMT1 alone in HCT116 cells did not lead to Irf8 induction, which required the knockdown of both DNMT1 and DNMT3b.42 Thus, in the absence of DNMT1, other DNMTs or mechanisms may regulate Irf8 methylation to control its expression. Moreover, the injury sensitivity of PT-DNMT1-KO mice may involve the methylation regulation of other genes than Irf8.
Irf8 is a central mediator of IFN-γ signaling pathway.42, 57 As a transcription factor, Irf8 can act as either transcription activator or repressor. The target genes of Irf8 include Fas, Bax, FLIP, STAT1 and iNOS, suggesting an essential role of Irf8 in apoptosis.58-62 Consistently, knockdown of Irf8 attenuated cisplatin-induced apoptosis in RPTC cells (Figure 7). Thus, hypomethylation of Irf8 during cisplatin-induced AKI may lead to the up-regulation of this pro-apoptotic transcription factor for tubular cell injury and death. However, it should be noted that other genes regulated by DNA methylation may contribute significantly to the pathogenesis of AKI as well. As presented in Supplemental Figure 1, these genes may function in the regulation of gene transcription, cell cycle, and apoptosis. For example, Hdac5, a class IIa HDAC, was hypomethylated in cisplatin-induced AKI. As a transcriptional repressor, Hdac5 has been implicated in disease conditions including AKI. Induction of Hdac5 has been reported in cisplatin-induced AKI63 and septic AKI64, which is associated with BMP7 suppression, leading to the inhibition of tubular cell proliferation and repair. Future investigation should extend to these genes to have a comprehensive understanding of DNA methylation in AKI.
Materials and Methods
Antibodies and special reagents
Antibodies used in this study were from the following sources: rabbit monoclonal anti-DNMT1 (5032), rabbit monoclonal anti-Caspase-3 (9665), rabbit polyclonal anti-Cleaved caspase 3 (9661), rabbit monoclonal anti-PARP (9532), and rabbit monoclonal anti-GAPDH (5174s) from Cell Signaling Technology (Danvers, MA); rabbit polyclonal anti-Cyclophilin B from Abcam Inc. (Cambridge, MA); and rabbit polyclonal anti-DNMT3a (sc-20703), mouse monoclonal anti-DNMT3b (sc-81252), goat polyclonal anti-ICSBP (Irf8, sc-6058) from Santa Cruz Biotechnology Inc. (Dallas, TX). Secondary antibodies for immunoblotting were from Thermo Scientific (Rockford, IL) and for immunohistochemical staining from Dako (Carpinteria, CA), respectively. Fluorescein-labeled lotus tetragonolobus lectin (LTL) was bought from Vector Labs (FL-1321). cis-Diammineplatinum(II) dichloride crystalline (Cisplatin, P4394-1G) and 5-aza-2′-deoxycytidine (A3656-50MG) were purchased from Sigma-Aldrich (St. Louis, MO). Enhanced chemiluminescence kit was purchased from Thermo Scientific (Rockford, IL). All the primers were purchased from Integrated DNA Technologies (Coralville, IA). Unless specifically indicated, all other reagents were from Sigma.
Animals and animal model of cisplatin-induced AKI
DNMT1flox/flox mice were generated in Dr. Guoping Fan's laboratory at University of California at Los Angeles as described previously.40, 65 PEPCK-Cre mice were originally obtained from Dr. Volker Haase (Vanderbilt University).66 To generate kidney proximal tubule-specific DNMT1 knockout mice, DNMT1flox/flox mice were crossed with PEPCK-Cre mice according to the breeding protocol shown in Supplemental Figure 2. In these experiments, littermate wild-type and knockout mice were compared. For the animal work not involving gene knockout models, C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Male mice of 8 to 12 weeks were used for cisplatin treatment. Briefly, the mice were injected with a single dose of 30 mg/kg cisplatin intraperitoneally, while control animals received saline injection. After indicated time, kidneys and blood samples were collected for analysis. All animals were maintained in the animal facility of Charlie Norwood Veterans Affairs Medical Center at Augusta. All animal experiments were conducted following a protocol approved by the Institutional Animal Care and Use Committee.
Reduced representation bisulfite sequencing (RRBS)
RRBS was performed by Integrated Genomics Core and data was analyzed by Bioinformatics Core at Augusta University as previously described.67 In brief, genomic DNA was extracted from kidney cortex and outer medulla using DNeasy Blood & Tissue Kit from Qiagen (Hilden, Germany) and digested with a methylation-insensitive enzyme MspI (New England Biolabs, Ipswich, MA). The fragment ends were filled in and adapters were ligated. Next, the fragments were size selected with size of 150 bp-450 bp. Size-selected DNA fragments were then subjected to bisulfite conversion using EpiTect Fast DNA Bisulfite Kit from Qiagen. The libraries were generated using the PfuTurbo Cx Hotstart polymerase (Agilent Technologies) and sequenced by an Illumina Genome Analyzer IIx (Ilumina).
Renal function analysis
Renal function was analyzed by measuring blood urea nitrogen (BUN) and serum creatinine as described previously.68 In brief, at indicated time, blood samples were collected and centrifuged to collect serum. Then the BUN and serum creatinine levels were determined by using the analytical kit from Stanbio Laboratory (Boerne, TX).
Histological examination
Kidneys were harvested, fixed with 4% paraformaldehyde, embedded in paraffin and sectioned at 4 μm for histological examination. Renal histology was examined by hematoxylin and eosin (H&E) staining to analyze kidney tissue damage following a standard protocol. Loss of brush border, tubular dilation and disruption, protein/cell cast formation and cell lysis (nuclear loss) were considered as renal tubular damage. Kidney tissue damage was examined by two experimental pathologists in a blind manner and scored according to the percentage of renal tubular damage as follows: 0, no damage; 1, <25%; 2, 26–50%; 3, 51–75%; 4, >75%.
Immunohistochemical staining
Paraffin embedded section preparation was the same as the sections used in H&E staining. After rehydration, antigen retrieval was performed in 10 mM EDTA (pH8.0) for DNMT1 or 10 mM sodium citrate (pH6.0) for cleaved caspase 3 at 100°C for 1 hour, followed by 1 hour blocking. Then tissue sections were exposed to primary antibody anti-DNMT1 or anti-cleaved caspase 3 overnight at 4°C. After washing, tissue sections were incubated with secondary antibody with HRP labelled polymer from Dako (Carpinteria, CA) for 1 hour and then the slides were developed with ImmPACT DAB Peroxidase Substrate (Vector Laboratories, Burlingame, CA). For DNMT1 staining, the tissue sections were further stained with fluorescein isothiocyanate (FITC)-labeled lotus tetragonolobus lectin (LTL) (Vector Laboratories, Burlingame, CA), which is proximal tubule specific marker; and nuclei were counterstained with Hoechst33342 (Molecular Probes, Eugene, OR). Finally, the slides were mounted with Prolong Antifade Kit from Molecular Probes.
Total RNA isolation and Real-time PCR
Total RNA was isolated from cells or kidney cortex and outer medulla using mirVana™ miRNA Isolation Kit (Life Technologies, Grand Island, NY) according to the manufacturer's instructions. 1 μg of total RNA for each sample was then reverse transcribed into cDNA using iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-Time PCR was performed using iTaq™ Universal SYBR® Green Supermix (Bio-Rad, Hercules, CA) with StepOne Real-Time System (Applied Biosystems, Carlsbad, USA) and each reaction was done in triplicates. For normalization, β-actin was used as internal control, followed by normalization to the control group. Real-time PCR was conducted using the following primers: Irf8-mouse (forward: 5′-TGTCTCCCTCTTTAAACTTCCC-3′ reverse: 5′-GAAGACCATGTTCCGTATCCC-3′), Irf8-rat (forward: 5′-TGTCTCCCTCTTTAAACTTCCC-3′ reverse: 5′-GAAGACCCTGTTCCGCATCCC-3′), β-actin-mouse (forward; 5′-GACTCATCGTACTCCTGCTTG-3′ reverse: 5′-GATTACTGCTCTGGCTCCTAG-3′), β-actin-rat (forward: 5′-GGCATAGAGGTCTTTACGGAT G-3′ reverse: 5′-TCACTATCGGCAATGAGCG-3′)
Cells and cisplatin treatment
The immortalized rat kidney proximal tubular cell (RPTC) line was originally obtained from Dr. Ulrich Hopfer (Case Western Reserve University, Cleveland, OH) and cultured as described before.69 RPTC Irf8 knockdown or scramble cells were generated by transfection of Irf8 shRNA plasmids or scramble sequence shRNA plasmids with GFP tag from Origene (Rockville, MD) with Lipofectamine 2000 and selected by 1 μg/ml puromycin (Clontech Laboratories, Mountain View, CA) for two weeks to obtain cells with stable shRNA expression. Then puromycin-resistant and GFP positive cell colonies were selected and Irf8 knockdown efficiency was determined by real-time PCR and immunoblotting. Primary proximal tubular cells isolation and maintenance were performed as described previously.70 After 6-7 days of growth, the isolated primary proximal tubular cells were plated at 0.5 ×106 cells per 35 mm dish. For cisplatin treatment, RPTC cells and primary proximal tubular cells were incubated with 20 μM or 50 μM cisplatin in culture medium, respectively. To determine the effects of DNA methylation inhibitor, the cells were pretreated with 1 μM 5-aza-2′-deoxycytidine for 3 hours, followed by cisplatin treatment for 16 hours. After incubation for indicated time, apoptosis was determined by morphologic criteria and cells were harvested to collect cell lysates for various biochemical analysis.
Morphological analysis of apoptosis in cultured cells
Cell apoptosis was examined by morphology as described previously.69 In brief, after cisplatin treatment, cell nuclei were stained with 10 μg/mL Hoechst 33342. Then cellular and nuclear morphology were observed by phase contrast and fluorescence microscopy, respectively. Cellular shrinkage, formation of apoptotic bodies and nuclear condensation and fragmentation are the typical apoptotic morphology. For each condition, 3 to 5 fields with approximately 200 cells per field were randomly selected, and the cells showing typical apoptotic morphology were counted to determine the percentage of apoptosis. Each experiment was repeated three times independently.
Immunoblot analysis
Proteins were extracted from cells or renal cortex and outer medulla using 2% SDS lysis buffer as previously described. Equal amounts (20 μg for cell lysate and 100 μg for tissue lysate) of protein for each sample were resolved by 10% sodium dodecyl sulfate-polyacrylamide electrophoresis gel, and then transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% fat-free milk for 1 hour at room temperature and subsequently incubated with primary antibody overnight at 4 °C. After washing with PBST (1XPBS with 0.1% Tween 20 (MP Biomedicals)), the membranes were incubated with horseradish peroxidase (HRP) conjugated secondary antibody for 1 hour, and antigen specific signals were visualized with Clarity™ Western ECL Blotting Substrates from Bio-Rad (Hercules, CA).
Statistical Analysis
Quantitative data was expressed as means ± standard deviation (SD). GraphPad Prism software was used to determine the statistical difference. Statistical differences between two groups were determined by 2-tailed paired t-test and statistical differences between multiple groups were determined by multi-factor ANOVA. p<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Kebin Liu at Augusta University (Augusta, GA) for providing the Irf8 antibody in initial experiments. This study was supported in part by grants from the National Natural Science Foundation of China (81528004, 81430017), the National Institutes of Health (DK058831, DK087843) and Department of Veterans Administration of USA (BX000319).
Footnotes
Disclosure: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mehta RL, Burdmann EA, Cerda J, et al. Recognition and management of acute kidney injury in the International Society of Nephrology 0by25 Global Snapshot: a multinational cross-sectional study. Lancet. 2016;387:2017–2025. doi: 10.1016/S0140-6736(16)30240-9. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 3.Cepeda V, Fuertes MA, Castilla J, et al. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7:3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 4.Launay-Vacher V, Rey JB, Isnard-Bagnis C, et al. Prevention of cisplatin nephrotoxicity: state of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer chemotherapy and pharmacology. 2008;61:903–909. doi: 10.1007/s00280-008-0711-0. [DOI] [PubMed] [Google Scholar]
- 5.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney international. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 6.Finkel M, Goldstein A, Steinberg Y, et al. Cisplatinum nephrotoxicity in oncology therapeutics: retrospective review of patients treated between 2005 and 2012. Pediatric nephrology. 2014;29:2421–2424. doi: 10.1007/s00467-014-2935-z. [DOI] [PubMed] [Google Scholar]
- 7.dos Santos NA, Carvalho Rodrigues MA, Martins NM, et al. Cisplatin-induced nephrotoxicity and targets of nephroprotection: an update. Archives of toxicology. 2012;86:1233–1250. doi: 10.1007/s00204-012-0821-7. [DOI] [PubMed] [Google Scholar]
- 8.Miller RP, Tadagavadi RK, Ramesh G, et al. Mechanisms of Cisplatin nephrotoxicity. Toxins. 2010;2:2490–2518. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linkermann A, Chen G, Dong G, et al. Regulated cell death in AKI. Journal of the American Society of Nephrology : JASN. 2014;25:2689–2701. doi: 10.1681/ASN.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 13.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piccolo FM, Fisher AG. Getting rid of DNA methylation. Trends Cell Biol. 2014;24:136–143. doi: 10.1016/j.tcb.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Song F, Smith JF, Kimura MT, et al. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3336–3341. doi: 10.1073/pnas.0408436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber M, Davies JJ, Wittig D, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature genetics. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez J, Frigola J, Vendrell E, et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Research. 2006;66:8462–8468. doi: 10.1158/0008-5472.CAN-06-0293. [DOI] [PubMed] [Google Scholar]
- 18.Robertson KD. DNA methylation and human disease. Nature reviews Genetics. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 19.Kanai Y, Hirohashi S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis. 2007;28:2434–2442. doi: 10.1093/carcin/bgm206. [DOI] [PubMed] [Google Scholar]
- 20.Stenvinkel P, Karimi M, Johansson S, et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease? J Intern Med. 2007;261:488–499. doi: 10.1111/j.1365-2796.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- 21.Murphy TM, Perry AS, Lawler M. The emergence of DNA methylation as a key modulator of aberrant cell death in prostate cancer. Endocr-Relat Cancer. 2008;15:11–25. doi: 10.1677/ERC-07-0208. [DOI] [PubMed] [Google Scholar]
- 22.Weng YL, An R, Shin J, et al. DNA modifications and neurological disorders. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2013;10:556–567. doi: 10.1007/s13311-013-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20:274–281. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- 24.Kulis M, Esteller M. DNA Methylation and Cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 25.Robert MF, Morin S, Beaulieu N, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nature genetics. 2003;33:61–65. doi: 10.1038/ng1068. [DOI] [PubMed] [Google Scholar]
- 26.Bechtel W, McGoohan S, Zeisberg EM, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tampe B, Tampe D, Muller CA, et al. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J Am Soc Nephrol. 2014;25:905–912. doi: 10.1681/ASN.2013070723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko YA, Mohtat D, Suzuki M, et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013;14:R108. doi: 10.1186/gb-2013-14-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marumo T, Yagi S, Kawarazaki W, et al. Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. J Am Soc Nephrol. 2015;26:2388–2397. doi: 10.1681/ASN.2014070665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennan EP, Ehrich M, O'Donovan H, et al. DNA methylation profiling in cell models of diabetic nephropathy. Epigenetics. 2010;5:396–401. doi: 10.4161/epi.5.5.12077. [DOI] [PubMed] [Google Scholar]
- 31.Smyth LJ, McKay GJ, Maxwell AP, et al. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics. 2014;9:366–376. doi: 10.4161/epi.27161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wing MR, Devaney JM, Joffe MM, et al. DNA methylation profile associated with rapid decline in kidney function: findings from the CRIC study. Nephrol Dial Transplant. 2014;29:864–872. doi: 10.1093/ndt/gft537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehta TK, Hoque MO, Ugarte R, et al. Quantitative detection of promoter hypermethylation as a biomarker of acute kidney injury during transplantation. Transplant Proc. 2006;38:3420–3426. doi: 10.1016/j.transproceed.2006.10.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pratt JR, Parker MD, Affleck LJ, et al. Ischemic epigenetics and the transplanted kidney. Transplant Proc. 2006;38:3344–3346. doi: 10.1016/j.transproceed.2006.10.112. [DOI] [PubMed] [Google Scholar]
- 35.Huang N, Tan L, Xue Z, et al. Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem Biophys Res Commun. 2012;422:697–702. doi: 10.1016/j.bbrc.2012.05.061. [DOI] [PubMed] [Google Scholar]
- 36.Patel K, Dickson J, Din S, et al. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010;38:4313–4324. doi: 10.1093/nar/gkq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 38.Michalowsky LA, Jones PA. Differential nuclear protein binding to 5-azacytosine-containing DNA as a potential mechanism for 5-aza-2′-deoxycytidine resistance. Mol Cell Biol. 1987;7:3076–3083. doi: 10.1128/mcb.7.9.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 40.Jackson-Grusby L, Beard C, Possemato R, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 41.Yang D, Thangaraju M, Greeneltch K, et al. Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res. 2007;67:3301–3309. doi: 10.1158/0008-5472.CAN-06-4068. [DOI] [PubMed] [Google Scholar]
- 42.McGough JM, Yang D, Huang S, et al. DNA methylation represses IFN-gamma-induced and signal transducer and activator of transcription 1-mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol Cancer Res. 2008;6:1841–1851. doi: 10.1158/1541-7786.MCR-08-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tshuikina M, Jernberg-Wiklund H, Nilsson K, et al. Epigenetic silencing of the interferon regulatory factor ICSBP/IRF8 in human multiple myeloma. Exp Hematol. 2008;36:1673–1681. doi: 10.1016/j.exphem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Q, Zhang L, Li L, et al. Interferon regulatory factor 8 functions as a tumor suppressor in renal cell carcinoma and its promoter methylation is associated with patient poor prognosis. Cancer Lett. 2014;354:227–234. doi: 10.1016/j.canlet.2014.07.040. [DOI] [PubMed] [Google Scholar]
- 45.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 46.Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maunakea AK, Chepelev I, Cui K, et al. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23:1256–1269. doi: 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magnusson M, Lu EX, Larsson P, et al. Dynamic Enhancer Methylation--A Previously Unrecognized Switch for Tissue-Type Plasminogen Activator Expression. PLoS One. 2015;10:e0141805. doi: 10.1371/journal.pone.0141805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang G, Chan MF, Tomigahara Y, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DNMT3A and TET2 Cooperate and Compete to Maintain HSC DNA Methylation. Cancer Discov. 2016;6:944. [Google Scholar]
- 52.Wei Q, Bhatt K, He HZ, et al. Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2010;21:756–761. doi: 10.1681/ASN.2009070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basile DP, Bonventre JV, Mehta R, et al. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. Journal of the American Society of Nephrology : JASN. 2016;27:687–697. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee KY, Geng H, Ng KM, et al. Epigenetic disruption of interferon-gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene. 2008;27:5267–5276. doi: 10.1038/onc.2008.147. [DOI] [PubMed] [Google Scholar]
- 55.Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. 2002;22:145–152. doi: 10.1089/107999002753452755. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen H, Hiscott J, Pitha PM. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997;8:293–312. doi: 10.1016/s1359-6101(97)00019-1. [DOI] [PubMed] [Google Scholar]
- 57.Contursi C, Wang IM, Gabriele L, et al. IFN consensus sequence binding protein potentiates STAT1-dependent activation of IFNgamma-responsive promoters in macrophages. Proc Natl Acad Sci U S A. 2000;97:91–96. doi: 10.1073/pnas.97.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang D, Thangaraju M, Browning DD, et al. IFN regulatory factor 8 mediates apoptosis in nonhemopoietic tumor cells via regulation of Fas expression. J Immunol. 2007;179:4775–4782. doi: 10.4049/jimmunol.179.7.4775. [DOI] [PubMed] [Google Scholar]
- 59.Yang J, Hu X, Zimmerman M, et al. Cutting edge: IRF8 regulates Bax transcription in vivo in primary myeloid cells. J Immunol. 2011;187:4426–4430. doi: 10.4049/jimmunol.1101034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang D, Wang S, Brooks C, et al. IFN regulatory factor 8 sensitizes soft tissue sarcoma cells to death receptor-initiated apoptosis via repression of FLICE-like protein expression. Cancer Res. 2009;69:1080–1088. doi: 10.1158/0008-5472.CAN-08-2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kubosaki A, Lindgren G, Tagami M, et al. The combination of gene perturbation assay and ChIP-chip reveals functional direct target genes for IRF8 in THP-1 cells. Mol Immunol. 2010;47:2295–2302. doi: 10.1016/j.molimm.2010.05.289. [DOI] [PubMed] [Google Scholar]
- 62.Simon PS, Sharman SK, Lu C, et al. The NF-kappaB p65 and p50 homodimer cooperate with IRF8 to activate iNOS transcription. BMC Cancer. 2015;15:770. doi: 10.1186/s12885-015-1808-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sakao Y, Kato A, Tsuji T, et al. Cisplatin induces Sirt1 in association with histone deacetylation and increased Werner syndrome protein in the kidney. Clin Exp Nephrol. 2011;15:363–372. doi: 10.1007/s10157-011-0421-5. [DOI] [PubMed] [Google Scholar]
- 64.Hsing CH, Lin CF, So E, et al. alpha2-Adrenoceptor agonist dexmedetomidine protects septic acute kidney injury through increasing BMP-7 and inhibiting HDAC2 and HDAC5. Am J Physiol Renal Physiol. 2012;303:F1443–1453. doi: 10.1152/ajprenal.00143.2012. [DOI] [PubMed] [Google Scholar]
- 65.Fan G, Beard C, Chen RZ, et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pei L, Choi JH, Liu J, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012;7:567–578. doi: 10.4161/epi.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang M, Wei Q, Dong G, et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–1283. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo C, Wei Q, Su Y, et al. SUMOylation occurs in acute kidney injury and plays a cytoprotective role. Biochim Biophys Acta. 2015;1852:482–489. doi: 10.1016/j.bbadis.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei Q, Dong G, Franklin J, et al. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007;72:53–62. doi: 10.1038/sj.ki.5002256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.