

Abstract
Cytochrome P450 eicosanoids play important roles in brain function and disease through their complementary actions on cell-cell communications within the neurovascular unit (NVU) and mechanisms of brain injury. Epoxy- and hydroxyeicosanoids, respectively formed by cytochrome P450 epoxygenases and ω-hydroxylases, play opposing roles in cerebrovascular function and in pathological processes underlying neural injury, including ischemia, neuroinflammation and oxidative injury. P450 eicosanoids also contribute to cerebrovascular disease risk factors, including hypertension and diabetes. We summarize studies investigating the roles P450 eicosanoids in cerebrovascular physiology and disease to highlight the existing balance between these important lipid signaling molecules, as well as their roles in maintaining neurovascular homeostasis and in acute and chronic neurovascular and neurodegenerative disorders.
Keywords: Cytochrome P450, EETs, HETEs, she, Cerebrovascular, Neurovascular unit
Introduction
Cytochrome P450 enzymes (P450) catalyze the metabolism of a wide range of substrates including endogenous compounds to form important signaling molecules. Arachidonic acid (AA), which is highly enriched in the phospholipid of brain cell membranes, is metabolized to epoxyeicosatrienoic acids (EETs) and hydroxyeicosatetraenoic acids (HETEs) by specialized P450 enzymes collectively called epoxygenases and ω-hydroxylases, respectively (Nelson et al., 1996; Nebert and Russell, 2002). These two classes of P450 eicosanoids play important roles in the regulation of the cerebral microcirculation, neurovascular communication, neuroinflammation and neural injury and survival. Epoxy- and hydroxyeicosanoids often play opposing roles in health and disease. Maintaining a balance between the two is essential for optimal neurovascular health, whereas an imbalance predisposes to cerebrovascular disorders and exacerbates cerebrovascular injury.
P450 synthesis and metabolism
It was first reported in the liver in 1981 that AA can be metabolized by cytochrome P450 enzymes (Capdevila et al., 1981). However, the first evidence of a physiological role for EETs was provided a couple of years later when EETs were shown to be endogenous constituents of the rat brain involved in the release of peptide hormones from the hypothalamus (Capdevila et al., 1983; Junier et al., 1990). Since then a wealth of studies have demonstrated that P450 enzymes convert AA to EETs and HETEs (19- & 20-HETE) in multiple organs, including brain, where they play important physiological and pathophysiological roles.
Cellular membranes of the brain are rich in polyunsaturated fatty acids (PUFAs), particularly AA which is bound in an esterified form to phospholipids. It can be liberated by a variety of physiological and pathological stimuli activating phospholipase A2 (PLA2). Free AA is subsequently metabolized by LOX, COX and P450 enzymes to a number of biologically active metabolites collectively termed eicosanoids. Of the P450 enzymes, epoxygenases catalyze the formation of EETs by the addition of an epoxide moiety to AA, and ω-hydroxylases produce HETEs by the addition of a hydroxyl group (Roman, 2002). These two classes of AA metabolites, EETs and HETEs, and their roles in cerebrovascular physiology and disease will be discussed below.
The action of P450 enzymes on AA results in the generation of 4 regio-isomers of EETs: 5,6-, 8,9-, 11,12- and 14,15-EET and 2 regioisomers of HETEs: 19- and 20-HETE, determined by which of the 4 double bonds in the chemical structure is replaced by the epoxide or hydroxyl group; these are depicted in FIG 1. Studies using purified/recombinant cytochrome P450 enzymes from different organ systems have demonstrated that multiple P450 isoforms can metabolize AA to EETs and HETEs (for a list of isoforms identified in the brain thus far, see Table 1). The specific regioisomers of EETs formed by P450 enzymes are P450 isoform-specific; for example, CYP2C9 forms 14,15-, 11,12- and 8,9-EET in a 2.3:1:0.5 ratio, whereas CYP2C8 produces 14,15- and 11,12-EET in relatively equal amounts, in a 1.25:1 ratio, with no 8,9-EET (Daikh et al., 1994; Zeldin et al., 1995). While the majority of P450 enzymes are highly expressed in the liver, the mouse CYP2J subfamily is largely expressed outside of the liver, with Cyp2j8, Cyp2j9 and Cyp2j12 predominantly in the brain (Graves et al., 2015).
FIGURE 1. Structure of EETs and HETEs regioisomers generated from arachidonic acid by cytochrome P450 enzymes.

Cytochrome P450 enzymes generate eicosanoids from arachidonic acid; epoxygenases generate epoxyeicosatrienoic acids (EETs), ω hydroxylases generate hydroxyeicosatetraenoic acids (HETEs). The regioisomer generated is determined by insertion of epoxide or hydroxyl group in relation to the carbon placement of arachidonic acid. EETs are further metabolized to dihydroxyeicosatrienoic acids (DHETs) by soluble epoxide hydrolase (sEH).
TABLE 1.
Expression of cytochrome P450 enzymes, eicosanoid regioisomer and sEH in the brain.
| Cell or tissue type | CYP isoform | EET regioisomer | HETE regioisomer | sEH | Source |
|---|---|---|---|---|---|
| Brain homogenate | |||||
| Rat | CYP4x1 | sEH | Bylund et al., 2002; Koerner et al., 2008. | ||
| Mouse | CYP4x1, CYP2J9, CYP2J12 | sEH | Zuloaga et al., 2015; Zhang et al., 2009; Al-Anizy et al., 2006; Graves et al., 2015. | ||
| Cerebral microvessels Cat Rat |
CYP4A1, CYP4A2, CYP4A3 CYP4A1, CYP4A2, CYP4A3, CYP4A8 | 20-HETE 20-HETE |
Harder et al., 1994 Gebremedhin et al., 2000. |
||
| Middle cerebral artery, rat | CYP 4A, CYP 2C11 | Alonso-Galicia et al., 1999; Carver et al., 2014. | |||
| Basilar artery, rat | CYP4A | Alonso-Galicia et al., 1999. | |||
| Cerebral vessels Mouse |
sEH | Zhang et al., 2008. | |||
| Rat | CYP4A, CYP4A1, CYP4A3, CYP4A8. | 11,12-, 14- 15-EET | 20-HETE | Dunn et al., 2008. | |
| Smooth muscle cells | |||||
| Brain sections, rat | sEH | Iliff et al., 2007. | |||
| Arteriolar, brain slices, human | sEH | Sura et al., 2008. | |||
| Arteriolar, rat | 20-HETE | Gebremedhin et al., 2008. | |||
| Cultured, cat | CYP4A2, CYP4A3 | 20-HETE | Gebremedhin et al., 1998. | ||
| Endothelial cells | |||||
| Arterial, brain slices, human | sEH | Sura et al., 2008. | |||
| Microvascular, cultured, mouse | 8,9-, 11,12-, 14,15-EET | sEH | Gupta et al., 2012; Figure 1. | ||
| Microvascular, cultured, rat | CYP2C11, CYP4x1 | Carver et al., 2014. | |||
| Astrocytes | |||||
| Brain sections, rat | CYP2C11 | sEH | Iliff et al., 2007. | ||
| Brain sections, mouse | sEH | Marowsky et al., 2009. | |||
| Brain, cultured, rat | CYP2C11, CYP4x1, CYP4A2/3 | 5,6-, 11,12-, 14,15-EET | 20-HETE | sEH | Amruthesh et al., 1993; Alkayed et al., 1996; Rawal et al., 2009; Gebremedhin et al., 2016; Carver et al., 2014. |
| Oligodendrocytes | |||||
| Brain slices, human | sEH | Sura et al., 2008. | |||
| Neurons | |||||
| Brain slices, human | sEH | Sura et al., 2008. | |||
| Brain sections rat | CYP4x1 | Bylund et al., 2002. | |||
| Sensory nerve fibers, rat | CYP2C11, CYP2J3, CYP2J4 | sEH | Iliff et al, 2007; Iliff et al., 2009. | ||
| Parasympathetic nerve fibers, rat Putamen, pig Cerebral cortex Mouse Pig Hippocampus, CA3, pig Thalamus, VPL, pig |
CYP2C11, CYP2J3, CYP2J4 CYP4A CYP4A CYP4A CYP4A CYP4A |
sEH |
Iliff et al, 2007; Iliff et al., 2009. Zhu et al., 2015. Zhang et al., 2017c. Zhu et al., 2015. Zhu et al., 2015. Zhu et al., 2015. |
||
| Neuronal culture Mouse Rat |
CYP4A, CYP4A10, CYP4A12A | 14,15-EET | 20-HETE | sEH |
Zhang et al., 2017c. Fairbanks et al., 2012. |
The biological activity of EETs is terminated via multiple pathways including beta-oxidation, binding to fatty acid binding protein (FABP) and incorporation into membrane phospholipids (Zeldin, 2001). However, EETs levels and activity are primarily regulated by hydrolysis to their corresponding less active vicinal diols, dihydroxyeicosatrienoic acids (DHETs); a process catalyzed by soluble epoxide hydrolase (sEH, initially referred to as cytosolic epoxide hydrolase). The metabolism of EETs by sEH is highly regiospecific; 14,15-EET is the preferred substrate, which is converted to 14,15-DHET. Because sEH has a preference for epoxides distal to the carboxyl terminal, the resulting order of substrate preference for hydrolysis is 14,15- > 11,12- > 8,9- > 5,6-EET, with 5,6-EET being a poor substrate for the enzyme (Newman et al., 2005; Chacos et al., 1983; Zeldin et al., 1993). Incorporation of EETs into membrane phospholipids is also regiospecific, however with a different substrate specificity than sEH. Shivachar et al. reported that EETs are incorporated into astroglial phospholipids, including phospatidylcholine, phosphatidylinositol and phosphatidylethanolamine. In this paradigm 8,9-EETs, as well as arachidonic acid, exhibited higher incorporation rates than 14,15-EET (Shivachar et al., 1995).
Distribution of P450 eicosanoids and their synthetic enzymes in the brain
Both EETs- and HETEs-generating enzymes are widely distributed in the brain, located in both vascular and non-vascular compartments. Their pattern of distribution, and that of EETs-metabolizing sEH, is reflective of their physiological roles as discussed below, with some cells predominately generating EETs or HETEs and other cells responding to and metabolizing them.
Both EETs and EETs-synthetic enzymes are present in multiple cell types throughout the brain. In rat brain astrocytes, which express the synthetic enzyme CYP2C11, the predominant EETs regioisomer produced is 14,15-EET (Alkayed et al., 1996a). Although it was known that brain endothelial cells (ECs) have endogenous epoxygenase activity, the specific regioisomers had not previously been reported. We show here by LC-MS/MS the presence of 8,9-, 11,12- and 14,15-EET in primary mouse brain microvascular endothelial cultures (FIG 2). Interestingly, in the rat hippocampus and middle cerebral artery, CYP2C11 mRNA expression was found to be rhythmically expressed (Carver et al., 2014), peaking in the morning in both tissues, which may be related to the physiological function of EETs in regulating cerebral blood flow (CBF). Transcript and protein of CYP2C11, CYP2J3 and CYP2J4 have been detected in both sensory and parasympathetic nerve fibers, while 14,15-EET is present in neuronal cultures (Iliff et al., 2009b; Iliff et al., 2007; Fairbanks et al., 2012).
FIGURE 2. Endogenous 8,9-, 11,12- and 14,15-EET are present in brain microvascular endothelial cultures.
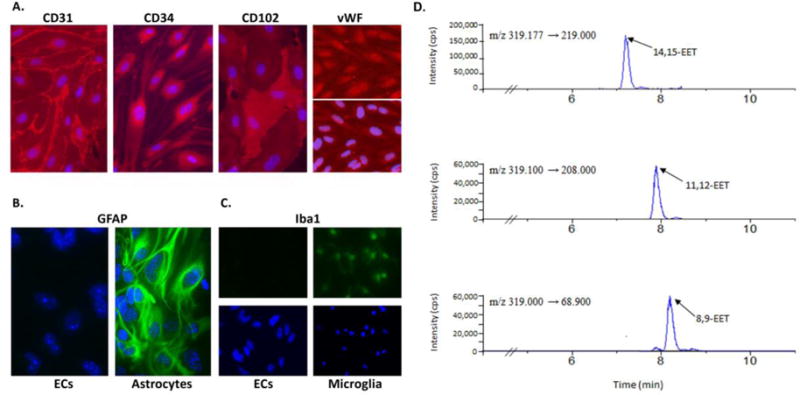
A: representative images of endothelial cultures immunolabeled in red for the endothelial markers CD31, CD34, CD102, and von Willebrand factor (vWF); cell nuclei were labeled in blue with Hoechst 33342. B and C: ECs were also immunolabeled with the astrocyte marker glial fibrillary acidic protein (GFAP; B) and microglial marker ionized Ca2+-binding adapter molecule 1 (Iba1; C) in green. Primary astrocytes and microglia were used as positive controls. D. Representative liquid chromatography-tandem MS (LC-MS/MS) chromatogram of 14,15-, 11,12- and 8,9-EET unstimulated primary mouse brain endothelial cell extracts. Culture and LC-MS/MS described in Davis et al., 2015.
Multiple members of the CYP4 family, which have hydroxylase activity, have been shown to be expressed in brain homogenates, brain sections, the basilar and middle cerebral arteries, cerebral microvessels, as well as in astrocytes, endothelial cells, vascular smooth muscle cells and neurons. All of these cells types therefore have the necessary machinery to produce 19- and 20-HETE (Alonso-Galicia et al., 1999; Bylund et al., 2002; Al-Anizy et al., 2006; Gebremedhin et al., 1998; 2000; 2016; Carver et al., 2014, Dunn et al., 2008; Zhu et al., 2015; Zhang et al., 2017c). 20-HETE, but not 19-HETE have been detected in vascular smooth muscle cells, and more recently astrocytes and neurons (Gebremedhin et al., 1998; 2008; 2016; Zhang et al., 2017c). 20-HETE has also been reported to be produced in cerebral microvessels and arteries, although the specific cell type was not identified (Gebremedhin et al., 1998; Dunn et al., 2008).
In addition to EETs-synthetic machinery, the enzyme responsible for inactivation of EETs, sEH, is also expressed in multiple cells types throughout the brain. Within the cerebral vasculature, sEH is found in both endothelial and vascular smooth muscle cells (Sura et al., 2008; Enayetallah et al., 2004), and is also highly expressed in astrocytes (Marowsky et al., 2009), a component of the neurovascular unit. We have also shown sEH expression in neuronal cell bodies and processes (Koerner et al., 2007; Zhang et al., 2007) as well as perivascular vasodilator nerve fibers innervating pial arteries in cortical surface vessels (Iliff et al., 2007). Brain sEH expression is sexually dimorphic with higher expression in male than female brain, corresponding with higher plasma 14,15-DHET in males (Zhang et al., 2009).
The biological actions of EETs and HETEs in multiple organs have been widely investigated (Imig, 2012; Spector et al., 2004; Imig, 2013). Here, the roles of these eicosanoids will be discussed only in the cerebral circulation, including discussion of cell culture studies, which will only describe work carried out on brain-derived cells.
Cellular actions of eicosanoids in the brain; a summary of in vitro studies
Astrocytes
Astrocytes are the most abundant glial cell type in the CNS, making up over 50% of the cell mass of the brain; they provide both metabolic and structural support to neurons, and are therefore critical for normal neuronal activity. Astrocytes possess the machinery necessary to produce EETs, and multiple EET regioisomers have been identified in this cell type, including 5,6- 11,12- and 14,15-EET (Amruthesh et al., 1993; Alkayed et al., 1996a). The release of AA from membrane-bound pools occurs upon astrocyte stimulation with glutamate, an excitatory neurotransmitter, thus providing a substrate for P450 enzymes to act upon. In addition to increased release of AA, glutamate also upregulates CYP2C11 levels, increasing endogenous EETs formation, an event blocked by pharmacological P450 epoxygenase inhibitor miconazole (Alkayed et al., 1996b; Stella et al., 1994; Alkayed et al., 1997). In addition to EETs, 5,6-, 8,9- and 14,15-DHET have also been identified in astrocyte cultures, formation of the latter being inhibited upon treatment with 4-phenylchalcone oxide (4-PCO), an sEH inhibitor (Amruthesh et al., 1993). As already discussed, the metabolism of EETs is regiospecific, with 14,15-EET being the preferred substrate of sEH and thus rapidly metabolized (Zeldin et al., 1993). This results in lower astroglial membrane incorporation of 14,15-EET compared to 8,9-EET, with increased 14,15-EET incorporation occurring upon sEH inhibition, in a protein kinase C (PKC)-dependent manner (Shivachar et al., 1995).
As well as being produced by astrocytes, EETs also exert biological actions on astrocytes. Application of a synthetic EET analogue, 11-nonyloxy-undec-8(Z)-enoic acid (NUD-GA) to rat astrocytes results in elevated intracellular Ca2+ levels, with increased Ca2+-dependent outward K+ currents (Higashimori et al., 2010). These currents are significantly attenuated by inhibitors of BK channels, small conductance Ca2+-activated K+ channels, and also by inhibition of glutamate receptors, indicating involvement of all three in this phenomenon. Furthermore, blockade of the formation of endogenous EETs reduces metabotropic glutamate receptor (mGluR)-induced outward K+ currents, demonstrating the ability of EETs to act in an autocrine fashion. Therefore data obtained with both endogenous and exogenous EETs suggests that EETs are modulators of BK and small conductance Ca2+-activated K+ channels, contributing to glutamate-induced signaling; this is of relevance to neurovascular coupling, discussed below.
EETs are protective to astrocytes in the face of ischemic insults. Exogenously applied EETs reduce astrocytic cell death induced by oxygen glucose deprivation (OGD) in a dose-dependent manner, with no apparent difference among regioisomers (Liu et al., 2005; Li et al., 2012; Yuan et al., 2016). Astrocytes transfected with Cyp2J2 are protected against OGD, a result inhibited by an inhibitory analogue of EETs, 14,15-EEZE, indicating that the cells are protected by increased EETs generation (Li et al., 2012). However, although EETs are protective, the release of endogenous EETs is significantly reduced by OGD. This decrease can be blocked by sEH inhibition (Zhang et al., 2017), indicating that increased sEH activity may be responsible for decreased EETs release and increased cell death following OGD. Indeed, astrocyte viability following OGD is increased upon treatment with sEH inhibitor, 12-(3-adamantan-1-yl-ureido) dodecanoic acid (AUDA; Yuan et al, 2016).
Studies have suggested that the protective effect of 14,15-EET is in part mediated via production and release of brain derived neurotrophic factor (BDNF) from astrocytes acting in an autocrine manner via TrkB receptors, a novel, non-classical mode of BDNF regulation (Begni et al., 2017; Yuan et al., 2016). Treatment with 14,15-EET increases transcript and protein levels of BDNF after OGD, as well as its release into culture medium. Phosphorylation of both TrkB receptors and downstream Erk1/2 is increased by 14, 15-EET, blockade of which inhibits the protective effect of 14,15-EET after OGD. Other studies have indicated that the mechanism of EETs-mediated protection against OGD-induced cell death is PI3K-dependent (Li et al., 2012), with inhibition of PI3K abolishing the observed protection. Addition of exogenous EETs to astrocytes under OGD conditions increases PI3K activity and phosphorylation of Akt. OGD-induced suppression of anti-apoptotic Bcl-xl and Bcl-2 and upregulation of pro-apoptotic Bax are blocked by exogenous EETs and CYP2J2 overexpression, an effect linked to PI3K activation, suggesting that the PI3K/Akt pathway is involved in mediating the anti-apoptotic effects of EETs (Li et al., 2012). The PI3K/Akt signaling pathway is a parallel pathway to Erk1/2, with both factors potentially activated by BDNF, although these pathways have not yet been linked to 14,15-EET-mediated cytoprotection (Mitre et al., 2017).
A less severe insult, hypoxia, increases both CYP2C11 expression and EETs levels in astrocytes, in contrast to the decrease observed after OGD. Increased EETs results in increased activation of large-conductance Ca2+-activated K+ channels (KCa), which is blocked by inhibition of P450 epoxygenanse activity using miconazole or N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide (MS-PPOH), demonstrating a role for EETs in mediating the response of astrocytic KCa channel currents to hypoxia (Yamaura et al., 2006; Liu et al., 2005), which may play a significant role in changes in blood flow observed during hypoxia, as discussed later. Brief hypoxia is also protective to astrocytes by acting as a preconditioning stimulus, reducing OGD-induced cell death via increased EETs production, an effect blocked by treatment with MS-PPOH (Liu et al., 2005). Hypoxic preconditioning induces tolerance in astrocytes in part by increasing hypoxia inducible factor (HIF-1α), binds to a HIF-response element in the CYP2C11 promoter to increase its expression and EETs generation (Liu et al., 2005).
In addition to initiating protective autocrine signaling in astrocytes, 14,15-EET is also involved in paracrine signaling between astrocytes and neurons. Inhibition of sEH in astrocyte cultures, thus preventing the decrease in EETs, increases VEGF release from astrocytes into culture medium; this conditioned medium reduces cell death in neurons after both OGD and hydrogen peroxide-induced oxidative stress (Zhang et al., 2017a; Terashvili et al., 2012). Exogenously applied 14,15-EET is also protective against hydrogen peroxide-induced cell death in a dopaminergic neuronal cell line. Furthermore, treating astrocyte-neuronal co-cultures with sEH inhibitor, AUDA, increased cell viability, indicating generation of endogenous EETs that are protective against cell death, while treatment with miconazole, which inhibits EETs synthesis, increased cell death. Astrocyte-conditioned medium protects neurons, an effect enhanced by treating neurons with conditioned medium from astrocytes incubated with AUDA, indicating that endogenous EETs are released into culture medium by astrocytes, an effect that is enhanced by AUDA (Terashvili et al., 2012). Additionally, astrocyte-derived BDNF is protective to OGD-stimulated neurons, an effect that is inhibited by pharmacological inhibition of TrkB receptors (Yuan et al., 2016), indicating that EETs-stimulated BDNF is protective to both astrocytes and neurons.
In addition to EETs-synthetic enzymes, cultured rat brain astrocytes also express Cyp4a2/3 ω-hydroxylase mRNA and CYP4A protein. When incubated with AA astrocytes produce 20-HETE, which is released into culture medium, this is also enhanced upon stimulation of mGluRs. Application of exogenous 20-HETE attenuates, while pharmacological inhibition of 20-HETE synthesis using HET0016 increases, the open state probabilities of calcium-dependent potassium channel (KCa; 71 pS and 161 pS) single-channel ion currents (Gebremedhin et al., 2016). Upon glutamatergic stimulation astrocytes are therefore able to release both EETs and HETEs into the perivascular space, which have opposing roles on KCa channels and cerebrovascular function under physiological and pathophysiological conditions.
A further paracrine signaling function of astrocyte-derived P450 eicosanoids is the induction of tube formation of cerebral microvascular endothelial cells, an in vitro model of angiogenesis. Endothelial thymidine incorporation is increased upon incubation with astrocyte-conditioned medium, while tube formation is increased in co-culture with astrocytes (Munzenmaier and Harder, 2000). The latter is decreased by 17-octadecynoic acid (17-ODYA), an inhibitor of both 20-HETE and EETs synthetic enzymes, therefore implicating P450-derivatives of AA in astrocyte-induced angiogenesis. Further, exogenous 8,9-EET causes endothelial mitogenesis and is sufficient for endothelial cells to form capillary-like structures (Zhang and Harder, 2002), however in vivo studies have shown that 17-ODYA and another non-specific P450 inhibitor are able to decrease capillary formation and tumor size in glial tumors, though these inhibitors had no effect on EETs levels (Zagorac et al., 2008), indicating that 20-HETE, another eicosanoid, or other signaling mediator, may be responsible for the increased endothelial mitogenesis and tube formation upon treatment with astrocyte conditioned medium.
Endothelial cells
Endothelial cells (ECs) lining the intimal surface of the cerebrovasculature are essential for vascular function, regulating vascular tone, blood-brain barrier permeability, blood coagulation, angiogenesis and inflammatory responses. Both EETs and HETEs synthetic enzymes are expressed in cerebral endothelial cells, as well as sEH. We also show here that endogenous EETs (8,9-, 11,12- and 14,15-EET) are present in brain endothelial cells under basal conditions (FIGURE 2).
As for astrocytes, 14,15-EET is also protective against cell death following OGD in primary cultured brain ECs. Brain ECs display a sexually dimorphic response to ischemia, with ECs derived from female brain exhibiting increased survival after OGD compared to male brain ECs (Gupta et al., 2012). The sex difference has been linked to lower sEH expression and higher EETs levels in female versus male ECs. The difference in survival is abolished by either inhibiting sEH or applying 14,15-EET exogenously, which reduces male EC death to levels observed in female-derived ECs. The mechanism of EC protection by 14,15-EET is in part linked to inhibition of Rho kinase (Gupta et al., 2012). EETs therefore play a protective role to endothelial cells, the integrity of which is vital to the health of the cerebrovasculature.
Smooth muscle cells
Vascular smooth muscle cells (VSMCs) surround arterioles and large-caliber blood vessels, but not capillaries, and are the effector cells responsible for vasoconstriction and vasodilation, critical for vascular function. Brain smooth muscle cells express sEH, however it has not yet been demonstrated that they possess endogenous EETs. This is in line with their physiological role as a target cell for EETs, which they receive from surrounding cell types, such as ECs and astrocytes, the expression of sEH consistent with the need to terminate EETs following their vasodilator actions (Iliff et al., 2007). Consistent with VSMC expression of the ω-hydroxylase CYP4A, cultured feline brain VSMCs have been shown to produce 20-HETE upon incubation with AA (Gebremedhin et al., 1998).
In agreement with studies in other cerebrovascular cell types, exogenously applied EETs are also protective to VSMCs in the face of ischemic insult. 14,15-EET protects cultured rat brain VSMCs exposed to OGD in a dose-dependent manner (Qu et al., 2015), with an associated increase in phosphorylation of Akt and Kir6.1 ATP-sensitive potassium channel (KATP) subunit. This protective effect is blocked by inhibiting Akt or KATP, indicating that 14,15-EET protects against cell death via activation of Akt and opening KATP channels. EETs production has not yet been demonstrated in VSMCs, but they respond to EETs released by ECs and astrocytes. However, OGD reduces levels of EETs in both of these cell types (Zhang et al., 2017), thereby decreasing the EETs available to VSMCs. Differences in temporal regulation of EETs levels by the cell types remain to be determined and may play a role in the viability of VSMCs during ischemia.
EETs exhibit anti-inflammatory actions in multiple cell types outside of the brain and in the brain as a whole, yet there is a paucity of in vitro studies using cerebrovascular cell cultures to investigate the mechanisms involved (Node et al., 1999; Deng et al., 2010). However, EETs exogenously applied to brain microvascular SMCs do inhibit prostaglandin E2 (PGE2) production, a principal mediator of inflammation in many diseases (Fang et al., 1998). The same has been demonstrated in aortic SMCs, where EETs-induced inhibition of PGE2 potentiates platelet-derived growth factor (PDGF)-induced SMC proliferation (Fang et al., 1998).
Of particular relevance to the physiological role of VSMCs in regulating cerebral blood flow, exogenous 8,9- and 11,12-EET increase the frequency of opening, mean open time and open- state probability of K+ channels in cerebral VSMCs (Gebremedhin et al., 1992). 20-HETE, an endogenous inhibitor of KCa channels, is decreased under hypoxic conditions (Gebremedhin et al., 2008); this reduction of inhibitory 20-HETE could therefore lead to increased opening of the channel (Gebremedhin et al., 2008). Exogenously applied 20-HETE is able to block the hypoxia-induced activation of KCa channel current, suggesting that the hypoxia-induced reduction of endogenous 20-HETE may contribute to hypoxia-induced KCa channel activation, leading to hyperpolarization and relaxation cerebral arterial muscle cells (Gebremedhin et al., 2008).
Platelets
Although not a brain cell type per se, circulating platelets do come into contact with the brain vasculature and affect both physiological and pathological processes. Platelets play an important role in events following vascular injury. In injured blood vessels, they rapidly aggregate upon contact with collagen, releasing pro-aggregatory mediators to cause further aggregation, thus forming a platelet plug within the blood vessel, which then interacts with various coagulation factors.
In 1986 Fitzpatrick et al., (Fitzpatrick et al., 1986) showed that exogenous EETs inhibit COX-dependent metabolism of AA both in isolated enzyme preparations and in intact human platelets, with 14,15-EET being most potent. In washed, intact platelet preparations, 14,15-EET inhibited thromboxane B2 (TxB2) formation from AA. They also demonstrated that all of the regioisomers they tested were able to inhibit AA-induced human platelet aggregation, independent of COX activity. At lower concentrations, below 10 μM, EETs were able to inhibit AA-induced platelet aggregation, yet TxB2 levels were unaltered, indicating a dual effect of EETs on COX activity and platelet aggregation in general, independent of TxB2. This was the first report of a dissociation between TxB2 formation and platelet aggregation; the two phenomena generally correlate with each other.
Almost 10 years later (Zhu et al., 1995), it was established that EETs are actually produced in platelets; studies using isolated human platelet phospholipids treated with PLA2 demonstrated release of both EETs (14,15-, 11,12-, 8,9- and 5,6-EET in decreasing order of abundance) and 20-HETE, indicating presence of both P450 epoxygenase and hydroxylase enzymes. Activation of these platelets by thrombin or platelet-activating factor (PAF) was also able to produce low levels of EETs. In contrast to the often opposing roles of EETs and HETEs, it was demonstrated that 20-HETE is actually a potent, dose-dependent inhibitor of platelet aggregation (when stimulated by AA-, A23187 ionophore- or U46619), and also inhibitor of thromboxane B2 generation from AA; a function specific to 20-HETE, as 19-HETE does not inhibit platelet aggregation (Hill et al., 1992).
Platelet adhesion to endothelial cells in the brain, which underlies vascular inflammation and is implicated in brain pathology (Mezger et al., 2015), is inhibited by 11,12-EET under non-flow conditions. This decrease in adhesion may be due to the decrease in platelet adhesion molecule expression (P-selectin and CD41) observed upon treatment with 11,12-EET. It has further been demonstrated that in addition to exogenously applied EETs, endothelial expression of CYP2C9 inhibits adhesion of human platelets to an endothelial cell line by a mechanism that is partially dependent on membrane potential and involving platelet KCa channels (Krotz et al., 2004). EETs have therefore been proposed to serve as an endothelium-derived antiplatelet substance, in addition to nitric oxide (NO) and prostacyclin (PGI2), with different EETs regioisomers exhibiting different platelet membrane hyperpolarization potencies (11,12- being the most, and 14-15-EET the least potent) (Krotz et al., 2004). These results have been acquired in an endothelial cell line, not derived from the brain; it therefore remains to be determined whether the same mechanisms will apply to the interaction between circulating platelets and cerebrovascular endothelial cells. Exogenously applied 14,15-EET does decrease platelet aggregation in the mouse brain in vivo, an outcome that is correlated with decreased TxB2 production (Heizer et al., 1991), however adhesion or endothelial adhesion markers have not been investigated.
A further anti-inflammatory mechanism of EET involves platelet NO production, which is a signaling mediator playing an anti-platelet aggregation and anti-thrombotic role. 11,12-EET increases nitric oxide synthase (NOS) activity in platelets, resulting in increased nitrate/nitrite production as well as increased platelet uptake of L-arginine, a substrate for NOS. (Zhang et al., 2008a)
Both 5,6-EET and sEH have also been identified in red blood cells (Nakamura et al., 1997; Jiang et al., 2004) where it has been proposed that the red blood cells act as a reservoir for EETs, which are released upon ATP stimulation of the erythrocyte P2X7 receptor (Jiang et al., 2007). As well as being a releasable store for EETs, erythrocytes also possess metabolic activity, able to actively form EETs from exogenously applied AA, though the P450 enzyme has not yet been identified, and also hydrolyze EETs into DHETs by sEH (Jiang et al., 2008). Physiologically, such a function may serve a purpose in providing exogenous EETs to the endothelial cells lining cerebral blood vessels in times of pathological stress.
Physiological actions of eicosanoids in the cerebrovasculature
In order to maintain an adequate supply of oxygen and nutrients to the brain, cerebral blood flow (CBF) is regulated by an intricate interplay between autoregulation, to provide the brain with a basal supply of oxygenated blood, and neurovascular coupling (NVC), or functional hyperemia, which increases blood flow to areas of the brain with increased neuronal activity. P450 eicosanoids contribute significantly to both of these processes. In general EETs are key players in neurovascular coupling, whereas 20-HETE contributes to the autoregulatory process.
EETs are able to dilate cerebral vessels; however potencies vary between regioisomers depending on the species, the in vivo or ex vivo nature of studies and vessel type and its location (parenchymal vs pial; Ellis et al., 1990; Gebremedhin et al., 1992; Leffler and Fedinec, 1997; Blanco et al., 2008), as summarized in Table 2, with some regioisomers able to bring about large magnitudes of change in vessel diameter, while 14,15-EET appears to be minimally vasoactive in the cerebral circulation (Ellis et al., 1990). Multiple studies have demonstrated that cerebral arterial dilation in response to EETs is mediated by activation of KCa channels in VSMCs (Harder et al., 1998; Gebremedhin et al., 1992). The increase in K+ channel activity results in hyperpolarization of the cell membrane and inactivation of voltage-gated Ca2+ channels, resulting in dilation of cerebral microvessels.
TABLE 2.
Vasoactive effects of EETs and 20-HETE in cerebral blood vessels.
| Regioisomers tested | Species | Vessel | Vessel diameter: constrict / dilate | Experimental paradigm | Regioisomer potency | Source |
|---|---|---|---|---|---|---|
| 5,6-, 8,9-, 11,12-, 14,15-EET | Rabbit | Pial arteriole | Dilate | In vivo, Cranial window | 5,6- > 8,9- > 11,12- > 14,15-EET | Ellis et al., 1990. |
| 5,6-, 11,12- EET | Cat | Pial arteriole | Dilate | In vivo, Cranial window | 5,6- > 11,12-EET | Ellis et al., 1990. |
| 5,6-, 8,9-, 11,12-EET | Cat | MCA | Dilate | Ex vivo, isolated artery segments, video-microscopy | 8,9- > 5,6- > 11,12-EET | Gebremedh in et al., 1992. |
| 5,6-, 8,9-,11,12-,14,15-EET | Piglet | Pial arteriole | Dilate | In vivo, Cranial window | 5,6- > 11,12- = 14,15- > 8,9-EET | Leffler & Fedinec, 1997. |
| 11,12-EET | Rat | Cortical intraparenchy mal arteriole | Dilate (biphasic) | Ex vivo, brain slices, video- microscopy, U-46619-preconstrict ed | Blanco et al., 2008. | |
| 20-HETE | Cat | Cerebral microvessel | Constrict | Ex vivo, isolated artery segments, video-microscopy | Gebremedh in et al., 1998. |
Consistent with 20-HETE having opposing effects to EETs, 20-HETE is a potent cerebral vasoconstrictor. 20-HETE acts by inhibiting large-conductance Ca2+-activated K+ channels, increasing intracellular Ca2+, thus causing depolarization of vascular smooth muscle membranes and vasoconstriction (Gebremedhin et al., 1992); a mechanism also involving PKC activation. Independent of these inhibitory effects on KCa, 20-HETE also activate L-type Ca2+ channels to produce a concentration-dependent constriction, which is inhibited by pharmacological blockade of L-type Ca2+ channels (Gebremedhin et al., 1998; Lange et al., 1997).
As is common for many physiological processes, there is cross-talk with another vasoactive signaling mediator, NO. It has been shown that, in the rat MCA, the actions of NO as a vasodilator are in part mediated by a cGMP-related mechanism and in part by NO-induced inhibition of 20-HETE formation, via inhibition of CYP4A, thereby suppressing the 20-HETE vasoconstrictor signal. This phenomenon is however dependent on vessel type and not true of all cerebral vessels; the majority of NO-induced dilation is mediated by the cGMP mechanism in the basilar artery rather than by inhibition of 20-HETE (Alonso-Galicia et al., 1999). It has also been demonstrated that increased sEH expression specifically in the endothelium results in a blunted CBF response to acetylcholine (ACh) (Zhang et al., 2013). Since release of NO in response to ACh is measure of endothelial function, this may indicate that sEH overexpression induces endothelial dysfunction.
Regulation of CBF follows a circadian rhythm in rats. Expression of Cyp2c11 and Cyp4x1 mRNA and protein also follow a circadian rhythm in rat astrocyte cultures (Carver et al., 2014), with formation of 8,9-EET following a 12-hour cycle, while 14,15-EET was rhythmic with a 24-hour cycle. These expression patterns may potentially underlie the circadian changes in CBF.
Autoregulation
Cerebral autoregulation is the ability of blood vessels to maintain a constant cerebral blood flow over a wide range of systemic arterial pressures, ensuring adequate oxygen delivery to the brain. This is achieved by a combination of myogenic, neurogenic, and metabolic mechanisms. When perfusion pressure is not within the autoregulatory range, control of CBF is lost and is governed by mean arterial pressure. In the kidneys EETs are able to modify autoregulation, with inhibition of epoxygenase enhancing the vasoconstrictor response to increasing perfusion pressure (Imig et al., 1999). In the cerebral circulation the contribution of EETs to autoregulation has not been demonstrated, however the role of 20-HETE has been extensively studied.
20-HETE is an endogenous mediator of the generation of pressure-induced cerebrovascular vasoconstriction, thus playing an important role in cerebral autoregulation. 20-HETE levels are greatly increased in cerebral arteries in response to increasing transmural pressure, pharmacological blockade of which eliminates pressure-induced constriction of cerebral arteries in vitro and impairs autoregulation in vivo (Gebremedhin et al., 2000; Toth et al., 2011). Regulation of 20-HETE may be important in pathological or stress states. As already described, 20-HETE levels in smooth muscle cells are decreased under hypoxic conditions (Gebremedhin et al., 2008). In the context of autoregulation, this may be protective under in vivo hypoxic conditions, where the cerebral vasculature senses hypoxia, reducing 20-HETE levels, thus leading to activation of KCa channels and causing dilation in order to elevate CBF to increase the delivery of oxygen and nutrients to the brain. Since EETs and 20-HETE play opposing roles on VSMC KCa channels, reduction of 20-HETE removes inhibition of these channels, but also allows for EETs-mediated channel activation. In fact, a recent study demonstrated a role for EETs in this increase in CBF in response to reduced arterial oxygen saturation, with the response to hypoxia greatly diminished by both inhibition of EETs synthesis and by EETs antagonism, while inhibition of 20-HETE had no effect (Liu et al., 2015), most probably due to 20-HETE already being decreased by hypoxia. Inhibition of mGluRs also attenuated the increase in CBF in response to hypoxia, suggesting that activation of mGluRs may be responsible for the increased EETs observed during hypoxia, contributing to increased hypoxic responsivity (Yamaura et al., 2006; Liu et al., 2015).
Functional hyperemia (Neurovascular Coupling)
Cerebral blood flow is intricately coupled to neuronal metabolic activity by a tightly regulated and dynamic phenomenon referred to as functional hyperemia or neurovascular coupling. This process ensures sufficient local oxygen supply to active neurons.
Based on the proximity of neurons, astrocytes and blood vessels anatomically, with astrocytes bridging vessels and neurons, it was postulated in the late 1990s by Harder et al. that astrocytes may play a role as integrators of neuronal signals that can be transduced to cerebral arterioles, leading to changes in local tone and local cerebral perfusion in response to neuronal activation (Harder et al., 1998). The idea was based on the group’s demonstration that glutamate, a major excitatory neurotransmitter, was able to increase EETs formation in astrocytes, as well as increase CBF. These observations, in addition to the already known ability of EETs to dilate cerebral vessels, and the demonstration that application of 14,15-EET had direct actions on the outward K+ current in VSMCs isolated from rat cerebral microvessels, led the group to propose a model for functional hyperemia, whereby perisynaptic astrocytes sense glutamate released by active neurons into the synaptic cleft, causing the release of EETs from astrocytic endfeet surrounding adjacent arterioles; EETs then dilate these arterioles by inducing membrane hyperpolarization and VSMC relaxation (Alkayed et al., 1996a, Harder et al., 1998), as summarized in FIG 3. This paper marked a significant move forward in thinking in the field and has since been supported by several independent studies.
FIGURE 3. EETs play a prominent role in neurovascular coupling; matching cerebral blood flow to the metabolic demands of neurons.
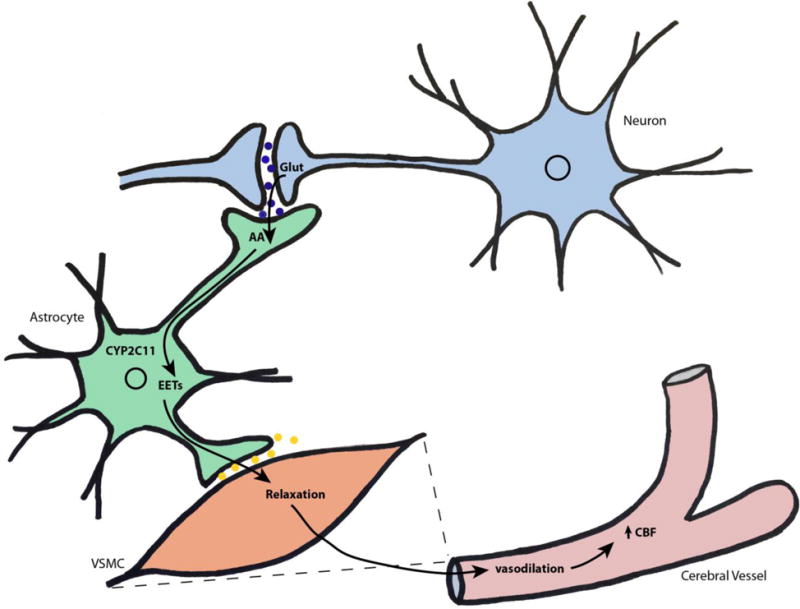
Active neurons release glutamate (Glut) into the synaptic cleft, which stimulates generation, and release, of EETs by astrocytes. Released EETs cause hyperpolarization and relaxation of surrounding vascular smooth cells (VSMC), resulting in increased vasodilation and cerebral blood flow, increasing the supply of oxygen and nutrients to neurons.
Pharmacological inhibition of cytochrome P450 epoxygenases prevents the increase in CBF observed in response to application of exogenous glutamate. Furthermore, inhibition of Cyp2c11 by use of antisense oligonucleotides also prevents glutamate-induced increases in cerebral blood flow (Alkayed et al., 1996b; Harder et al., 1998; Alkayed et al., 1997), together demonstrating regulation of glutamate-induced changes in CBF by endogenously generated EETs. Additionally, in vivo superfusion of an mGluR agonist over the cortical surface of the brain causes an increase in CBF, a response attenuated by both inhibition of EETs synthesis and by an EETs analogue (MS-PPOH and 14,15-EEZE, respectively; Liu et al., 2011). Prolonging the bioavailability of endogenous EETs, by sEH inhibition, also prolongs the response to mGluR agonism, an effect also achieved by inhibition of 20-HETE synthesis, indicating that increased CBF in vivo in response to mGluR stimulation, is primarily dependent on the release of EETs, while the duration of response is limited by both EETs breakdown by sEH and by 20-HETE accumulation.
These mechanistic studies are further supported by a physiologically relevant model of functional hyperemia, whisker stimulation. In vivo studies have demonstrated that inhibition of P450 epoxygenase, using multiple inhibitors, results in a substantial decrease in the CBF response to whisker stimulation, showing that EETs generation plays a significant role in the coupling of blood flow to physiological neuronal stimulation (Peng et al., 2002). Conversely 20-HETE plays an opposing role; inhibition of 20-HETE synthesis results in a slight increase in laser-Doppler flux in response to whisker stimulation, compared to vehicle control. Of course, due to biological redundancy, more signaling pathways are involved in functional hyperemia rather than solely relying on the balance between EETs and 20-HETE signaling. While functional hyperemia is primarily dependent on EETs, COX metabolites and NO are also implicated in the CBF response to neuronal activation; COX inhibition attenuates whisker-stimulated increase in CBF independently of EET, while NO by virtue of 20-HETE suppression is also able to elicit vasodilation, also allowing for an increased response to EET (Liu et al., 2008; Liu et al., 2012). More recent models combine pharmacological inactivation of multiple pathways including EETs, COX, and NO to achieve neurovascular uncoupling (Leithner et al., 2010; Tarantini et al., 2015), however these early studies inhibiting P450 epoxygenase were able to reveal that inhibition of EETs generation alone is sufficient to achieve neurovascular uncoupling (Peng et al., 2002).
Larger conduit arteries in the brain, such as the MCA receive inputs from extrinsic perivascular nerves, including parasympathetic and sensory vasodilator and parasympathetic vasoconstrictor nerves. This type of extrinsic control of arteries is thought to protect the brain against extreme fluctuations in CBF, such that occur under pathological situations. Iliff et al., showed that sensory and parasympathetic vasodilator perivascular fibers express both EETs synthetic machinery (CYP2C11, CYP2J3 and CYP2J4) as well as sEH, and identified a role for EETs as nerve-derived vasodilators, involved in the regulation of CBF by these extrinsic perivascular nerves(Iliff et al., 2009b; Iliff et al., 2007).
Cerebrovascular pathology and cytochrome P450 eicosanoids
Autoregulation and neurovascular coupling can both be altered under pathological conditions, with the resulting homeostatic disturbance contributing to brain dysfunction (Girouard and Iadecola, 2006; Imig et al., 2011).
Stroke
Stroke is the leading cause of serious long-term disability. In the USA, someone suffers a stroke every 40 seconds, with a person dying of a stroke every 4 minutes. Approximately 90% of these are ischemic strokes, with the rest being intracerebral or subarachnoid hemorrhagic strokes (Benjamin et al., 2017).
The benefical properties of EETs outlined above, the vasodilatory, anti-inflammatory, anti-coagulatory and pro-survival properties, make them obvious candidates as potential therapeutic targets in stroke and other cerebrovascular pathologies.
Levels of P450 eicosanoids are altered by cerebral ischemia, with both arachidonic acid and free fatty acids increasing in ischemic brain tissue. Transient ischemic attack (TIA), although a risk factor for stroke, acts as a preconditioning stimulus, and EETs have been implicated in mediating preconditioning-linked ischemic tolerance. Cerebral infarct is reduced in TIA-versus sham-preconditioned rats. Cyp2c11 mRNA and protein levels are increased in the ipsilateral hemisphere after experimental TIA (Alkayed et al., 2002), indicating that TIA may induce ischemic tolerance by upregulating the EETs synthetic Cyp2c11.
Administration of exogenous 14,15-EET reduces infarct size in mice following middle cerebral artery occlusion (MCAO; Zhang et al., 2008c). EETs are therefore protective in the face of cerebral ischemia; much of the focus of studies investigating EETs-mediated protection have centered however on the role of sEH since its identification as a potential therapeutic target (Iliff and Alkayed, 2009; Imig and Hammock, 2009). Pharmacological sEH inhibition, using AUDA, AUDA-BE, or t-AUCB. is protective following MCAO, reducing apoptosis and infarct volume as well as improving neurological function (Zhang et al., 2007; Qu et al., 2015; Zuloaga et al., 2014). This protection is likely due to increased endogenous EETs levels since protection is absent upon inhibition of P450 epoxygenase. Global sEH null mice sustain smaller brain infarcts after MCAO compared to WT control (Zhang et al., 2008c), confirming the studies carried out with the pharmacological inhibitors. These mice have been shown to have higher circulating 14,15-EET levels, supporting the notion that protection is attributed to increased EET levels (Zhang et al., 2008c; Luria et al., 2007).
While both pharmacological inhibition of sEH and its gene deletion (designated ephx2) decreases infarct volume, the mechanism of this protection is not so clear. SEH null mice display higher brain tissue perfusion compared to wild-type (WT) mice during occlusion, indicating enhanced vasodilator capacity and collateral blood flow in the presence of focal occlusion (Zhang et al., 2008c). However the study using AUDA-BE found that blood flow during occlusion was unaltered between the inhibitor- and vehicle-treated groups (Zhang et al., 2007). This may indicate that the protection by sEH inhibition may be due to a non-vascular mechanism. However, blood flow during the reperfusion period was not assessed, so a vascular mechanism cannot be ruled out. This discrepancy may also be due to the acute sEH inhibition resulting from a single injection of AUDA-BE compared to the chronic lack of sEH in the null mice. Finally, the discrepancy may be due to the fact that sEH is a bifunctional enzyme, with both hydrolase and lipid phosphatase activities; gene deletion eliminates both functions, whereas pharmacological inhibition blocks only one.
Having said that, non-vascular mechanisms are likely to contribute to the protection observed since the sEH null mice exhibit a reduction in ischemia-induced cytokine expression, including TNFα, IL-6 and IFNϒ (Koerner et al., 2008), in line with EETs’ anti-inflammatory role. In rats treated with AUDA, cellular apoptosis is reduced, (Qu et al., 2015) while phosphorylation of Akt and the ATP-sensitive potassium channel subunit Kir6.1 are increased in the ischemic penumbra, both of which have been implicated in mediating the anti-apoptotic effects of EETs in cerebral VSMCs and astrocytes (Qu et al., 2015; Li et al., 2012). BDNF is also increased in astrocytes of sEH null mice, with protection being lost upon inhibition of TrkB receptors, implicating the BDNF/TrkB pathway in mediating the protection observed, mirroring in vitro studies demonstrating involvement of this pathway in astrocyte and neuronal protection from OGD (Yuan et al., 2016).
Li et al. used mice expressing human CYP2J2 specifically in endothelial cells (Tie2-CYP2J2-Tr), subjected them to 10 minutes of bilateral common carotid artery occlusion (BCCAO) as a model of transient global cerebral ischemia, which sustained a smaller infarct size than WT controls. These mice exhibited increased 14,15-DHET, both in brain tissue and plasma, and increases in DHET were inhibited in the Tie2-CYP2J2-Tr mice by pharmacological inhibition of CYP2J2, as was protection from ischemia. This protection was shown to be Akt-dependent, with increased levels in the transgenic mice compared to WT after ischemic insult and a corresponding increase in cell-survival markers and decrease in apoptotic markers in brain tissue, after ischemia. Since CYP2J2 preferentially metabolizes AA to 11,12- and 14,15-EET, these effects are thought to be dependent on 11,12- and 14,15-EET (Li et al., 2012). Tie2 is also expressed in microglia and distinct monocyte/macrophage lineages, these cell types may also therefore contribute to outcomes in such studies using Tie2 as a promoter to drive protein expression (De Palma et al., 2005; Perdiguero et al., 2015).
20-HETE levels are also regulated by cerebral ischemia in brain tissue, reaching significantly higher levels within 5.5 hours of MCAO, with plasma levels displaying a delayed reaction, increasing between 19.5–25.5 hours after insult in the rat (Tanaka et al., 2007). Not surprisingly, 20-HETE plays an opposing role to EETs following cerebral ischemia; in fact injection of 20-HETE into rat carotid arteries is itself sufficient to induce ischemic infarct and the associated neurological deficits (Miyata et al., 2005). Conversely, inhibition of 20-HETE synthesis reduces infarct following MCAO as well as improving neurological outcome (Miyata et al., 2005; Poloyac et al., 2006). Since 20-HETE is a potent vasoconstrictor, the infarct observed is likely due to reduced blood supply to the area at risk, exacerbating cellular ischemic damage. Inhibition of 20-HETE also attenuates the delayed fall in regional CBF observed after reperfusion, suggesting that it plays a role in both acute and delayed events (Poloyac et al., 2006; Renic et al., 2009). Interestingly, and of significant translational relevance, inhibition of 20-HETE synthesis by N-(3-chloro-4-morpholin-4-yl) phenyl-N′-hydroxyimido formamide (TS-011) reduces infarct size regardless of whether it is administered before, during or after vessel occlusion. Even when administered up to 4 hours post-MCAO, TS-011 is able to reduce infarct volume in the rat by approximately 25% (Omura et al., 2006). These results imply a time-window for tissue salvage after MCAO, suggesting a mechanism that is not only dependent on blocking 20-HETE’s vasoconstrictor effects, but also by increasing cell survival. Indeed an ex vivo study using rat brain slices demonstrated that inhibition of 20-HETE reduced cell death following OGD, while a 20-HETE mimetic increased cell death, by mechanisms involving reactive oxygen species (ROS) and caspase-3 (Renic et al., 2012). Further studies in young piglets support this notion, since blockade of 20-HETE resulted in increased neuronal survival, linked to suppressed Erk1/2 signaling and oxidative stress following ischemia (Yang et al., 2012a), yet no difference in CBF either during or immediately after ischemia was observed.
Recently, a study of neurologic deterioration (defined as an increase of two or more points in the National Institutes of Health Stroke Scale) in acute ischemic stroke revealed that patients with neurologic deterioration displayed higher levels of plasma 20-HETE and lower levels of EETs, compared to those without. Also, polymorphisms in cytochrome P450 genes and ephx2 were associated with neurologic deterioration and different P450 metabolite levels (Yi et al., 2016). Another study into P450 metabolite levels also revealed increased 20-HETE, EETs and DHETs plasma levels following acute ischemic stroke, with a positive correlation between 20-HETE levels and lesion size and greater neurological impairment in stroke patients (Ward et al., 2011). The duration of the ischemic insult also has bearing on levels of EETs and HETEs, and subsequent outcome; a study investigating cardiac arrest in infant rats found an increase in EETs in cerebral tissue with short insult (9 minutes), but an increase in 20-HETE after a longer ischemic insult (12 minutes), with inhibition of the latter resulting in increased CBF and reduced neurological deficits and neurodegeneration (Shaik et al., 2015).
Stroke is a sexually dimorphic disease, with premenopausal women at a lower risk compared to men, however incidence of stroke rapidly increases following menopause. Younger women are thought to be protected due to the effects of estrogen; indeed removing the influence of gonadal estrogen by ovariectomy in mice (OVX) results in larger infarct sizes following MCAO compared to intact females. Cerebrovascular expression and activity of sEH is lower in female than male mice, resulting in decreased DHETs and increased circulating EETs compared to males (Zhang et al., 2009). Estradiol has been shown to reduce both basal and post-stroke sEH expression in the brain (Koerner et al., 2008). This sexual dimorphism in sEH regulation likely contributes to underlying sex differences in stroke.
Studies using transgenic mice demonstrate that sEH is an important factor underlying sex-differences observed in cerebral ischemia, with the mechanism involving both differences in blood flow as well as neuronal cell survival. Female mice sustain smaller infarcts than males following MCAO, a sex difference which is abolished in sEH null mice. The sex difference in infarction correlates with corresponding differences in blood flow during MCAO, which is higher in females than males, a difference that is also abolished in sEH null mice. No protection is observed in sEH null female mice compared to wild-type females, presumably due to already low sEH levels in the wild-type (Zhang et al., 2009). The opposite is observed in mice with increased sEH expression specifically in the endothelium (Tie2-hsEH-Tr) where ischemic damage in female mice is exacerbated following MCAO, rendering females more susceptible to cerebral ischemia than males. Surprisingly, no increase in infarct volume is observed in male Tie2-hsEH-Tr mice, presumably due to high preexisting sEH levels (Zhang et al., 2013).
The above studies show that sEH knockdown or inhibition in rodent models is protective against cerebral ischemia. Fittingly, ephx2 polymorphisms in the human population have been linked to altered, either increased or decreased, risk of stroke depending on the nature of the variation in the gene (Fornage et al., 2005; Gschwendtner et al., 2008; Zhang et al., 2008b).
Subarachnoid hemorrhage
Both EETs and HETEs have been implicated in subarachnoid hemorrhage and its subsequent complication, vasospasm. SAH results from rupture of cerebral aneurysms or other head trauma, and is characterized by bleeding in the subarachnoid space and blood clot formation. The majority of deaths resulting from SAH occur within the first two days and are linked to the volume of the initial hemorrhage (Broderick et al., 1994). The changes in CBF that occur are biphasic (Weir et al., 1978), consisting of an acute and delayed responses. Symptomatic, or delayed, cerebral vasospasm is a major complication occurring in the cerebral vasculature 4-14 days following SAH and increases the risk of delayed cerebral ischemia (DCI), although these complications can also occur independently of each other (Crago et al., 2011).
Early studies in a rat model of SAH demonstrated a 17-fold increase in 20-HETE levels in the cerebrospinal fluid (CSF) following SAH, an increase associated with the acute decrease in CBF, consistent with 20-HETE’s role as a vasoconstrictor. This increase in 20-HETE is triggered by multiple factors after SAH, including the vasoconstrictor 5-hydroxytryptamine (5-HT), as well as other vasoactive compounds released by clotting blood (Cambj-Sapunar et al., 2003; Takeuchi et al., 2006). Other AA metabolites are also increased in SAH CSF, indicating liberation of AA from plasma membranes. The increase in 20-HETE is presumably due to increased synthesis from AA by CYP4 enzymes (Roman et al., 2006; Rodriguez et al., 1988). Indeed, inhibition of cytochrome P450 hydroxylase, by 17-ODYA, HET0016 or TS-011, preventing the generation of 20-HETE, blocks the increase in CSF 20-HETE following SAH, attenuating the acute fall in CBF observed following SAH (Kehl et al., 2002; Miyata et al., 2005; Cambj-Sapunar et al., 2003; Roman et al., 2006). Experiments have also demonstrated the converse; injection of 20-HETE agonists into the cisterna magna reduce baseline CBF as well as potentiate the fall in CBF observed after SAH, confirming the role of increased CSF 20-HETE in mediating the changes in CBF occurring acutely after SAH (Yu et al., 2004). Inhibition of 20-HETE also prevents the delayed fall in CBF in a rat dual-hemorrhage model, therefore implicating 20-HETE in both the acute and delayed cerebral vasospasm (Takeuchi et al., 2005).
Increases in CSF 20-HETE levels have been reported in SAH patients who went on to suffer complications of cerebral vasospasm, with early peaks at ~70h, followed again 250–325h after onset of SAH symptoms; these increases in 20-HETE were not observed in patients not developing vasospasm, however this association with vasospasm has not been observed in all human studies (Poloyac et al., 2005; Crago et al., 2011). While it is not known definitively what tissue or cell type produces these increased eicosanoids, due to the time courses observed clinically for increased HETEs and EETs, it is believed that the origin of both was liberation of AA from brain tissue, following brain insult, as suggested in the rat studies (Siler et al., 2015; Poloyac et al., 2005). A dog model of dual hemorrhage SAH supports this association of 20- HETE with vasospasm where levels of 20-HETE increased in CSF, corresponding to delayed reduction in basilar artery diameter, indicative of delayed vasospasm. Pharmacological inhibition of 20-HETE increased vessel diameter to levels comparable with control dogs, indicating that the development of delayed vasospasm is associated with increased CSF 20-HETE (Hacein-Bey et al., 2006).
Siler et al. sampled CSF in a cohort of SAH patients at high risk for delayed cerebral ischemia. The group confirmed previous reports of elevated 20-HETE being associated with DCI (Crago et al., 2011); however, they also reported elevated 14,15-EET in CSF of SAH patients compared to that from non-hemorrhage controls, with patients exhibiting the highest levels of both eicosanoids more likely to develop DCI. The time-course of the increase in HETEs versus EETs were different, with levels of 20-HETE rising in the first couple of days after SAH and then declining, whereas 14,15-EET only rising approximately two weeks after insult (Siler et al., 2015). A study in a model of SAH in sEH null mice was used to investigate the effect of increased EETs on DCI following SAH. These sEH null mice, which have increased 14,15-EET and decreased 14,15 DHET, with no difference in 20-HETE levels, were protected from delayed reduction of microvascular perfusion, compared to WT mice, even though initial response to SAH was no different between the two genotypes. Although the increase in EETs levels in these mice was permanent, rather than induced by SAH, this study does indicate that the delayed increase in 14,15-EET observed in humans may be protective, and also that inhibition of sEH may be a therapeutic target, to increase endogenous EETs closer to the time of SAH insult. The acute increase in 20-HETE also supports the clinical utility of inhibiting 20-HETE formation as a potential therapeutic intervention in patients who have suffered SAH. Since these changes correlate with likelihood of developing DCI, CSF eicosanoid levels may be a valuable biomarker, guiding clinicians in decision making upon admission of a patient following SAH (Siler et al., 2015).
Genetic variation in the ephx2 gene, which encodes for sEH, as well as P450 enzymes, may also serve as a predictor of outcome following SAH. A study has shown that in patients admitted with aneurysmal SAH (aSAH), those with the Lys55Arg (K55R) polymorphism, predictive of increased sEH activity, had a significantly increased likelihood of experiencing a new stroke compared to patients with the wild-type gene. No difference was observed between patients with Arg287Gln (R287Q), associated with decreased EET metabolism and increased EETs, or wild-type genotypes (Martini et al., 2014); the same pattern was also observed for mortality. However, despite the increased likelihood of stroke in the K55R group, neurological deterioration attributable to DCI is not changed by genotype, though patients with the R287Q polymorphism had a significantly reduced length of hospital stay compared to either K55R or wild-type (Martini et al., 2014). Sampling CSF after aSAH showed that CYP2C8*4 allele carriers had decreased EETs and DHETs CSF levels, and were over twice as likely to develop DCI and clinical neurologic deterioration, the latter defined in this study as a decrease of two points on the Glasgow Outcome Scale. However, surprisingly CYP2J2*7, which is expected to decrease EETs levels, also showed improved outcome. Generally these studies correlate with the animal studies, indicating that decreased sEH and increased EET may be beneficial after SAH. There is however a great degree of genetic variation in the EETs pathway; genes involved in either EETs generation or breakdown may contribute in different degrees to the clinical outcomes observed (Donnelly et al., 2015)
Subarachnoid hemorrhage has further complications for some patients; it is followed by cerebral edema and acute communicating hydrocephalus in approximately 20% of cases, both of which are life-threatening. The mechanisms underlying these complications are poorly understood. Recently using a mouse model of SAH, in both WT and sEH null mice, we showed communicating hydrocephalus forming rapidly after SAH and persisting for the duration of the study, evidenced by enlarged ventricular volume which developed in both genotypes. Cerebral edema also formed in both genotypes, specifically in the periventricular white matter. sEH null mice displayed significantly less edema at 24 and 72hr post-SAH, compared to WT. Furthermore, we assessed vascular inflammation by VCAM-1 expression levels, using VCAM-1-conjugated microparticles of iron oxide (MPIOs) and T2*-weighted MRI; we demonstrated that sEH null mice had reduced VCAM-1 MPIO uptake in the whole brain, compared to WT. These findings suggest that increased EETs levels, or decreased sEH, protects against cerebral edema following SAH. The mechanism of protection involves decreased endothelial expression of VCAM-1, a cell adhesion molecule implicated in vascular inflammation, upregulation of which often precedes blood brain barrier disruption (Siler et al., 2015). These findings are of clinical interest as plasma and CSF collected from SAH patients show increased levels of VCAM-1, therefore potentially identifying a therapeutic target against the development of edema after SAH (Kaynar et al., 2004; Frijns et al., 2006a). Endothelial cell inflammatory markers, resulting from platelet activation, have also been observed in patients who have suffered DCI (Frijns et al., 2006b), therefore identifying inflammatory processes in multiple complications of SAH. As discussed above, EETs have established anti-inflammatory activity; it would be of interest to also determine whether the protection from DCI observed in the sEH null mice was associated with decreased inflammation, in addition to the decreased edema observed. The hydrocephalus which developed in these mice was not altered by genotype, however hydrocephalus is likely mechanical in nature and therefore independent of inflammatory processes and therefore sEH or EETs levels (Siler et al., 2015).
Dementia
In the elderly, vascular cognitive impairment (VCID; also called vascular dementia) and Alzheimer’s disease (AD) are the two most common forms of cognitive impairment. Historically, these pathologies have been considered distinct from each other, even mutually exclusive (Iadecola, 2010; Roseberg et al., 2016; Erkinjutti and Gauthier, 2009). However there is a growing awareness of the similarities between the two illnesses, with a growing number of AD patients also exhibiting VCI pathology such as cerebrovascular lesions as well as reduced vascular reactivity and CBF (Calabrese et al., 2016; Binnewijzend et al., 2016; Cantin et al., 2011; Iadecola, 2016; Ruitenberg et al., 2005). While a causative relationship between the two pathologies is of debate, the Nun Study did highlight that of the participants who met the criteria of AD, those with ischemic lesions had poorer cognitive function than those with no brain lesions (Marchant et al., 2013; Wang et al., 2016; Snowdon et al., 1997).
VCID
The most common cause of VCID is cerebral small vessel ischemic disease, an age-related vascular pathology (Yang et al., 2016). Post mortem samples have demonstrated increased sEH, localized to brain microvascular endothelial cells, and a corresponding increase in DHETs, specifically 14,15-DHET in cortical brain tissue from VCI subjects (Nelson et al., 2014). SEH is more prominent in small than large vessels, particularly in microvessels near regions of microinfarction, indicating that sEH, and therefore decreased bioavailable EETs, may play a role in the small vessel ischemic damage underlying VCID.
Alterations in the cerebral microvasculature have a critical role in the age-related decline in cognitive function. There is increasing evidence that NVC, or functional hyperemia, is impaired in aging (Zlokovic, 2011; Stefenova et al., 2013; Toth et al., 2017). Tarantini et al. tested whether NVC uncoupling itself leads to cognitive deficit; pharmacological neurovascular uncoupling was modeled in mice by simultaneous inhibition of P450 epoxygenase, NO synthase and COX. CBF in the whisker barrel cortex in response to whisker stimulation was decreased in vivo, signifying neurovascular uncoupling. This uncoupling was sufficient to cause impaired cognitive function, with deficits in learning and memory and spatial memory, presumably caused by impaired delivery of nutrients via the blood to active areas of the brain (Tarantini et al., 2015). As discussed above, EETs play a prominent role in NVC; it would therefore be pertinent to assess the role of EETs or sEH in this phenomenon, perhaps leading to the identification of a therapeutic target, such as inhibiting sEH or increasing EETs, to alleviate age-related decline in cognitive function due to NVC uncoupling.
AD
The pathogenesis of AD is unclear; it is likely a complex interaction between genetic and environmental factors. The hallmark of AD is an excessive accumulation of cytotoxic amyloid beta (Aβ), however vascular abnormalities are also prominent in this disease. Recently it has been demonstrated that Cyp2j2 polymorphisms, rs890293 and rs1155002, are associated with susceptibility to late-onset AD (LOAD) in humans (Yan et al., 2015; Geng et al., 2016), associations independent of ApoE-∊4. While Cyp2j2 rs890293 is associated with decreased EETs production, this is not known for Cyp2j2 rs1155002. AD is often accompanied by chronic inflammation, since EETs have known anti-inflammatory roles, inflammation due to decreased EETs may therefore be a contributing factor to this susceptibility to LOAD. It has been shown that microglial-mediated inflammation is associated with AD progression, and AD brains show activated microglia and pro-inflammatory cytokines (Mandrekar-Colucci and Landreth, 2010; Rao et al., 2011). Although, PLA2 is upregulated in human postmortem AD brains, both P450 epoxygenase transcript and protein are reduced, while inflammatory markers are increased (Rao et al., 2011) suggesting that decreased EETs may play a role in the pathogenesis of AD.
In addition to any potential effects on chronic inflammation, EETs protect against Aβ-induced mitochondrial dysfunction in astrocytes in vitro, a phenomenon suggested to be a contributing factor to neurodegeneration in both aging and AD (Shetty et al., 2011; Hirai et al., 2001; Sarkar et al., 2014). Inhibition of P450 epoxygenase activity has detrimental effects, decreasing mitochondrial membrane potential and increasing mitochondrial fragmentation, whereas pre-incubation with 11,12- and 14,15-EET prevents both Aβ-induced membrane depolarization and fragmentation, and reduces reactive oxygen species generation. Although EETs are able to protect against this amyloid β-induced mitochondrial dysfunction (Sarkar et al., 2011), Aβ reduces P450 epoxygenase activity in cultured astrocytes and neurons, resulting in decreased EET synthesis. This reduction in EETs further exacerbates Aβ-induced injury, both to astrocytes and presumably neurons as astrocyte-derived EETs are reduced, which are protective to neurons. Decreased EETs levels in AD brains may therefore contribute to neurologic deterioration both by making cells more vulnerable to Aβ-induced cellular toxicity, and also by disrupting NVC.
Vascular Risk factors
The diseases discussed above have common mechanisms, namely reduced blood flow, leading to a level of ischemia, inflammation, and cell loss. It is not surprising therefore that they also share multiple vascular risk factors that are associated with pathogenesis of disease and outcome, and in which P450 eicosanoids play important roles.
Hypertension
Hypertension exerts damaging actions on the cerebrovasculature, altering the structure of the cerebral vessels by vascular hypertrophy and remodeling and by the deposition of atherosclerotic plaques (Imig et al., 2011; Faraci et al., 1990; Alexander, 1995), thus impairing the function of cerebral blood vessels. Hypertension consequently can result in impaired autoregulation and NVC, which are essential to normal brain function, and as already discussed, uncoupling of the latter is sufficient to cause neurological deficits (Girouard and Iadecola, 2006; Tarantini et al., 2015). Hypertension is therefore a risk factor for stroke, AD, VCI, SAH and other vascular pathologies (Li et al. 2016; Levine and Langa, 2011; Bromberg et al., 1996). Rodent genetic models of hypertension indicate roles for CYP2C and CYP4A in the pathophysiology of hypertension; CYP4a14 null mice develop hypertension as well as increased 20-HETE, and CYP4a10 and CYP2c44 null mice develop salt-sensitive hypertension (Capdevila et al., 2015), and sEH also plays a role in blood pressure regulation (Sinal et al., 2000).
The spontaneously hypertensive stroke-prone rat (SHRSP) is a genetic model of spontaneous hypertension, which exhibits increased sensitivity to cerebral ischemia compared to normotensive strains. Production of 20-HETE by cerebral vessels, as well as Cyp4a1 and Cyp4a8 mRNA levels are elevated in these rats compared to WKY rats. This increase is not a result of the hypertension itself, therefore also implicating 20-HETE in the development of hypertension. Infarct in SHRSP is increased compared to WKY, an effect completely abolished upon 20-HETE inhibition. These rats also demonstrate a loss of CBF autoregulation in the affected region during reperfusion; hypoperfusion which is prevented by 20-HETE blockade (Dunn et al., 2008). The differences in stroke outcome in SHRSP, compared to normotensive rats is therefore attributable to increased 20-HETE signaling. Conversely increasing EETs levels by sEH inhibition is protective against cerebral ischemia in SHRSP (Simpkins et al., 2009).
Hypertension in the elderly increases the risk of stroke, partly due to impaired autoregulation in response to high blood pressure. While initially these autoregulatory responses adapt, they eventually become dysfunctional (Castellani et al., 2006). A study comparing pressure-induced increases in intracellular smooth muscle calcium levels and myogenic tone in MCAs from young and old hypertensive mice demonstrated that the adaptive responses in young vessels, pressure-induced myogenic constriction, which protect the thin walled arterioles and capillaries in the distal portion of the cerebral circulation from pressure overload, were inhibited by pharmacological inhibition of 20-HETE, whereas administration of 20-HETE increased calcium levels and caused constriction of the MCA. Vessels from the aged mice, did not show these protective adaptive increases in pressure-induced calcium and myogenic tone, and their response to alteration of 20-HETE levels is blunted, demonstrating that this functional maladaptation observed in the cerebral arteries of the aged mice to hypertension is, in part, due to a dysregulation of pressure-induced 20-HETE-mediated SMC calcium signaling (Toth et al., 2013).
Diabetes
Type 1 and type 2 diabetes are characterized by hyperglycemia. Type 2 is commonly associated with obesity and hypertension. Diabetes is a risk factor for many cerebrovascular diseases including stroke, AD and VCI (Zhang et al., 2017b; Rincon and Wright, 2013), and hyperglycemia exacerbates ischemic injury in the brain (Bruno et al., 2010).
Mice treated with streptozotocin (STZ) to induce hyperglycemia, a model of type 1 diabetes, have increased EPHX2 levels specifically in the cerebral vessels, though not in the brain as a whole (Jouihan et al., 2013). These mice have decreased EETs levels in the brain, which are restored by pharmacological sEH inhibition. Not only is diabetes a risk factor, outcome is worse following cerebral ischemia; infarct size in STZ-treated mice is doubled, a result that is completely reversed upon sEH inhibition. sEH therefore modulates the hyperglycemia-induced increased ischemic injury. A high-fat diet in mice, a model of pre-diabetes, is also sufficient to increase sEH expression in the brain. Type 2 diabetic mice, induced by a high fat diet in addition to STZ, display decreased CBF during reperfusion and increased infarct after MCAO; inhibition of sEH is able to both increase CBF and decrease infarct to non-diabetic control levels (Zuloaga et al., 2014). These studies therefore demonstrate the involvement of sEH in mediating the detrimental effects of diabetes following cerebral ischemic insult.
In addition to exacerbating ischemic damage after stroke, a high fat diet in mice causes impaired cognitive performance in naïve mice, as well as augmenting cognitive impairment in an experimental model of VCI compared to mice fed a low fat diet (Zuloaga et al., 2016). Although these mice have elevated sEH levels, a mechanistic link is yet to be shown. The high-fat fed mice have increased markers of glial activation, indicating that increased sEH may cause a chronic inflammatory state, which is known to be present in both VCI and AD (Zuloaga et al., 2016).
Atherosclerosis
sEH has been implicated in the pathogenesis of atherosclerosis and studies have shown that the Arg287Gln sEH polymorphism, predictive of decreased EET levels, is associated with increased pathogenesis in humans (Fornage et al., 2004). It is characterized, similar to VCI and AD, by chronic inflammation (Cochain and Zernecke, 2017), thereby making sEH inhibition an attractive therapeutic target. Atherosclerosis is also characterized by aberrant proliferation of VSMCs; sEH has been implicated in this proliferation, with blockade of sEH inhibiting the proliferation of a human aortic cell line (Davis et al., 2002).
Conclusions
There are many additional risk factors contributing to cerebrovascular pathology. Many of these pathologies have in common chronic inflammation, as do the risk factors. Studies have demonstrated that mice with endothelial-specific expression of human CYP4F2 exhibit 2-fold increases in 20-HETE levels in both tissue and endothelial cells. Among other effects outside of the cerebrovascular system, a 3-fold increase in basal serum IL-6 levels is observed, indicating a systemic pro-inflammatory phenotype (Cheng et al., 2014). This study therefore supports previous reports of pro-inflammatory actions of CYP4A-derived 20-HETE in human umbilical cord ECs (HUVEC) (Ishizuka et al., 2008). As discussed, EETs possess anti-inflammatory properties, while 20-HETE is pro-inflammatory; once again demonstrating the opposing roles of EETs and 20-HETE. Since inflammation appears to underlie many of the risk factors of these cerebrovascular pathologies, improving the balance of EETs and HETEs may serve as an attractive interventional target, both prophylactically, as well as therapeutically in the face of acute inflammation observed in acute ischemic attacks.
A further common mechanism of many cerebrovascular pathologies are alterations in CBF homeostasis, either in NVC or autoregulation, resulting in insufficient nutrient delivery to target tissues. Again, maintaining an optimal balance between EETs and HETEs to preserve vascular function will improve CBF and neurological disease progression. Increasing EETs bioavailability has the potential to protect the brain against both acute insults and in reducing the impact of the contribution of vascular risk factors to the pathogenesis of disease. However, multiple reports have agreed that delivering EETs is impractical. As fatty acids, EETs promptly bind to plasma proteins and are also rapidly converted to DHETs by the action of sEH; it has been demonstrated in human aorta and coronary artery segments that 93-96% of 14,15-EET was converted into 14,15-DHET by sEH after 6 hours of incubation (Fang et al., 2004). It is for this reason that sEH has been identified as a therapeutic target of cerebrovascular diseases and associated risk factors (Iliff and Alkayed, 2009; Imig and Hammock, 2009).
The identification and characterization of sEH inhibitors is an active field. Two sEH inhibitors have made it to clinical trial, AR9281 and GSK2256294. AR9281 was tested first in healthy human subjects and was well tolerated in both sexes, despite a largely male cohort (Chen et al., 2012). The drug dose-dependently inhibited blood sEH activity which was sustained by multiple doses, demonstrating both safety and target inhibition, required before moving onto further trials. According to www.clinicaltrials.gov AR9281 did subsequently enter a further clinical trial in patients with mild to moderate hypertension and impaired glucose tolerance, risk factors outlined above; however although the trial was completed in 2009, no results have been reported, indicating lack of efficacy or the occurrence of serious adverse reactions. More recently, GSK2256294 was given to healthy males, obese smokers as well as healthy elderly males and females; this drug also showed dose-dependent inhibition of blood sEH activity, was well tolerated, with no significant effect of age or gender on pharmacokinetics. The drug increased forearm blood flow in response to bradykinin in overweight smokers, which is blunted compared to control group, demonstrating improved endothelial function, therefore demonstrating promising viability for further testing (Lazaar et al., 2015; Yang et al., 2017). Further sEH inhibitor drug discovery is ongoing, with the identification of further drugs, both peripherally restricted, as well as peripherally and centrally active, already identified, as well as virtual screening to identify new drugs, potentially leading to future clinical trials (McReynolds et al., 2016; Waltenberger et al., 2016). The multiple therapeutic applications of inhibitors of sEH have been reviewed (Shen and Hammock, 2012). Since sEH is a target for multiple diseases in multiple organs (Harris et al., 2008; Lopez-Vicario et al., 2015), it is not known whether this will be problematic therapeutically. The implied increased organ-specificity may be a benefit of the newer-identified peripherally restricted drug, and these many sites of action may or may not have contributed to the lack of output in the 2009 AR9281 clinical trial. Whether sEH inhibitors will be effective in females of reproductive age is uncertain due to the sEH-inhibiting effects of estrogen already discussed (Koerner et al., 2008; Zhang et al., 2009). Non-pharmacological routes may also be effective at increasing endogenous EETs; we have reported recently that ultrasound stimulation of cerebrovascular endothelial cells increases EETs production. It remains to be determined whether this may provide a non-pharmacological means of increasing endogenous EETs levels in an organ-targeted manner (Davis et al., 2015).
List of abbreviations
- AA
Arachidonic acid
- ACh
Acetylcholine
- AD
Alzheimer’s disease
- ADP
Adenosine diphosphate
- AUDA
12-(3-adamantan-1-yl-ureido) dodecanoic acid
- aSAH
Aneurysmal subarachnoid hemorrhage
- BDNF
Brain derived neurotrophic factor
- CBF
Cerebral blood flow
- CSF
Cerebrospinal fluid
- CYP
Cytochrome P450
- COX
Cyclooxygenase
- DHET
Dihydroxyeicosatrienoic acid
- EC
Endothelial cell
- EDHF
Endothelial derived hyperpolarizing factor
- EET
Epoxyeicosatrienoic acid
- HETE
Hydroxyeicosatetraenoic acid
- KCa
Large-conductance Ca2+-activated K+ channels
- LOX
Lipoxygenase
- MCA
Middle cerebral artery
- MCAO
Middle cerebral artery occlusion
- mGluR
Metabotropic glutamate receptor
- MS-PPOH
N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide
- NO
Nitric oxide
- NOS
Nitric oxide synthase
- NVC
Neurovascular coupling
- OGD
Oxygen glucose deprivation
- OVX
Ovariectomy
- P450
Cytochrome P450
- PGE2
Prostaglandin E2
- PI3K
Phosphoinositide 3-kinase
- PKC
Protein kinase C
- PLA2
Phospholipase A2
- PUFA
Polyunsaturated fatty acid
- ROS
Reactive oxygen species
- SAH
Subarachnoid hemorrhage
- SMC
Smooth muscle cell
- STZ
Streptozotocin
- TS-011
N-(3-chloro-4-morpholin-4-yl) phenyl-N′-hydroxyimido formamide
- TxB2
Thromboxane B2
- VCID
Vascular cognitive impairment and dementia
- VSM
Vascular smooth muscle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
- Al-Anizy M, Horley NJ, Kuo CW, Gillett LC, Laughton CA, Kendall D, et al. Cytochrome P450 Cyp4x1 is a major P450 protein in mouse brain. FEBS J. 2006;273(5):936–47. doi: 10.1111/j.1742-4658.2006.05119.x. [DOI] [PubMed] [Google Scholar]
- Alexander RW. Hypertension and the Pathogenesis of Atherosclerosis. Oxidative Stress and the Mediation of Arterial Inflammatory Response: A New Perspective. Hypertension. 1995;25:155–161. doi: 10.1161/01.hyp.25.2.155. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996a;27:971–9. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Birks EK, Hudetz AG, Roman RJ, Henderson L, Harder DR. Inhibition of brain P-450 arachidonic acid epoxygenase decreases baseline cerebral blood flow. Am J Physiol. 1996b;271(4 Pt 2):H1541–6. doi: 10.1152/ajpheart.1996.271.4.H1541. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Birks EK, Narayanan J, Petrie KA, Kohler-Cabot AE, Harder DR. Role of P-450 arachidonic acid epoxygenase in the response of cerebral blood flow to glutamate in rats. Stroke. 1997;28(5):1066–72. doi: 10.1161/01.str.28.5.1066. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Goyagi T, Joh HD, Klaus J, Harder DR, Traystman RJ, et al. Neuroprotection and P450 2C11 upregulation after experimental transient ischemic attack. Stroke. 2002;33(6):1677–84. doi: 10.1161/01.str.0000016332.37292.59. [DOI] [PubMed] [Google Scholar]
- Alonso-Galicia M, Hudetz AG, Shen H, Harder DR, Roman RJ. Contribution of 20-HETE to vasodilator actions of nitric oxide in the cerebral microcirculation. Stroke. 1999;30(12):2727–34. doi: 10.1161/01.str.30.12.2727. [DOI] [PubMed] [Google Scholar]
- Amruthesh SC, Boerschel MF, McKinney JS, Willoughby KA, Ellis EF. Metabolism of arachidonic acid to epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and prostaglandins in cultured rat hippocampal astrocytes. J Neurochem. 1993;61(1):150–9. doi: 10.1111/j.1471-4159.1993.tb03550.x. [DOI] [PubMed] [Google Scholar]
- Begni V, Riva MA, Cattaneo A. Cellular and molecular mechanisms of the brain-derived neurotrophic factor in physiological and pathological conditions. Clin Sci (Lond) 2017;131(2):123–138. doi: 10.1042/CS20160009. [DOI] [PubMed] [Google Scholar]
- Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017 doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewijzend MAA, Benedictus MR, Kuijer JPA, van der Flier WM, Teunissen CE, Prins ND, et al. Cerebral perfusion in the predementia stages of Alzheimer’s disease. European Radiology. 2016;26(2):506–14. doi: 10.1007/s00330-015-3834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnie M, Morrison R, Camara R, Strauss KI. Temporal changes of cytochrome P450 (Cyp) and eicosanoid-related gene expression in the rat brain after traumatic brain injury. BMC Genomics. 2013;14:303. doi: 10.1186/1471-2164-14-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco VM, Stern JE, Filosa JA. Tone-dependent vascular responses to astrocyte-derived signals. Am J Physiol Heart Circ Physiol. 2008;294(6):H2855–63. doi: 10.1152/ajpheart.91451.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke. 1994;25(7):1342–7. doi: 10.1161/01.str.25.7.1342. [DOI] [PubMed] [Google Scholar]
- Bromberg JE, Rinkel GJ, Algra A, van den Berg UA, Tjin-A-Ton ML, van Gijn J. Hypertension, stroke, and coronary heart disease in relatives of patients with subarachnoid hemorrhage. Stroke. 1996;27(1):7–9. doi: 10.1161/01.str.27.1.7. [DOI] [PubMed] [Google Scholar]
- Bruno A, Liebeskind D, Hao Q, Raychev R. Diabetes mellitus, acute hyperglycemia, and ischemic stroke. Curr Treat Options Neurol. 2010;12(6):492–503. doi: 10.1007/s11940-010-0093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylund J, Zhang C, Harder DR. Identification of a novel cytochrome P450, CYP4X1, with unique localization specific to the brain. Biochem Biophys Res Commun. 2002;296(3):677–84. doi: 10.1016/s0006-291x(02)00918-x. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Giordano J, Signorile A, Laura Ontario M, Castorina S, De Pasquale C, et al. Major pathogenic mechanisms in vascular dementia: Roles of cellular stress response and hormesis in neuroprotection. J Neurosci Res. 2016;94(12):1588–1603. doi: 10.1002/jnr.23925. [DOI] [PubMed] [Google Scholar]
- Cambj-Sapunar L, Yu M, Harder DR, Roman RJ. Contribution of 5-hydroxytryptamine1B receptors and 20-hydroxyeiscosatetraenoic acid to fall in cerebral blood flow after subarachnoid hemorrhage. Stroke. 2003;34(5):1269–75. doi: 10.1161/01.STR.0000065829.45234.69. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78(3):415–23. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol Sci. 2000;21:125–7. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- Cantin S, Villien M, Moreaud O, Tropres I, Keignart S, Chipon E, et al. Impaired cerebral vasoreactivity to CO2 in Alzheimer’s disease using BOLD fMRI. NeuroImage. 2011;58(2):579–587. doi: 10.1016/j.neuroimage.2011.06.070. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW. Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc Natl Acad Sci U S A. 1981;78(9):5362–6. doi: 10.1073/pnas.78.9.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J, Chacos N, Falck JR, Manna S, Negro-Vilar A, Ojeda SR. Novel hypothalamic arachidonate products stimulate somatostatin release from the median eminence. Endocrinology. 1983;113(1):421–3. doi: 10.1210/endo-113-1-421. [DOI] [PubMed] [Google Scholar]
- Capdevila JH, Wang W, Falck JR. Arachidonic acid monooxygenase: Genetic and biochemical approaches to physiological/pathophysiological relevance. Prostaglandins Other Lipid Mediat. 2015;120:40–9. doi: 10.1016/j.prostaglandins.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver KA, Lourim D, Tryba AK, Harder DR. Rhythmic expression of cytochrome P450 epoxygenases CYP4x1 and CYP2c11 in the rat brain and vasculature. Am J Physiol Cell Physiol. 2014;307(11):C989–98. doi: 10.1152/ajpcell.00401.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani S, Bacci M, Ungar A, Prati P, Di Serio C, Geppetti P, et al. Abnormal pressure passive dilatation of cerebral arterioles in the elderly with isolated systolic hypertension. Hypertension. 2006;48(6):1143–50. doi: 10.1161/01.HYP.0000248533.58693.c4. [DOI] [PubMed] [Google Scholar]
- Chacos N, Capdevila J, Falck JR, Manna S, Martin-Wixtrom C, Gill SS, et al. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch Biochem Biophys. 1983;223(2):639–48. doi: 10.1016/0003-9861(83)90628-8. [DOI] [PubMed] [Google Scholar]
- Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, et al. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol. 2012;52(3):319–28. doi: 10.1177/0091270010397049. [DOI] [PubMed] [Google Scholar]
- Cheng J, Edin ML, Hoopes SL, Li H, Bradbury JA, Graves JP, et al. Vascular characterization of mice with endothelial expression of cytochrome P450 4F2. FASEB J. 2014;28(7):2915–31. doi: 10.1096/fj.13-241927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochain C, Zernecke A. Macrophages in vascular inflammation and atherosclerosis. Pflugers Arch. 2017 doi: 10.1007/s00424-017-1941-y. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Crago EA, Thampatty BP, Sherwood PR, Kuo CW, Bender C, Balzer J, et al. Cerebrospinal fluid 20-HETE is associated with delayed cerebral ischemia and poor outcomes after aneurysmal subarachnoid hemorrhage. Stroke. 2011;42(7):1872–7. doi: 10.1161/STROKEAHA.110.605816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikh BE, Lasker JM, Raucy JL, Koop DR. Regio- and stereoselective epoxidation of arachidonic acid by human cytochromes P450 2C8 and 2C9. J Pharmacol Exp Ther. 1994;271(3):1427–33. [PubMed] [Google Scholar]
- Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2002;99(4):2222–7. doi: 10.1073/pnas.261710799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CM, Ammi AY, Alkayed NJ, Kaul S. Ultrasound stimulates formation and release of vasoactive compounds in brain endothelial cells. Am J Physiol Heart Circ Physiol. 2015;309(4):H583–91. doi: 10.1152/ajpheart.00690.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Theken KN, Lee CR. Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J Mol Cell Cardiol. 2010;48(2):331–41. doi: 10.1016/j.yjmcc.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8(3):211–26. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Donnelly MK, Conley YP, Crago EA, Ren D, Sherwood PR, Balzer JR, et al. Genetic markers in the EET metabolic pathway are associated with outcomes in patients with aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2015;35(2):267–76. doi: 10.1038/jcbfm.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KM, Renic M, Flasch AK, Harder DR, Falck J, Roman RJ. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295(6):H2455–65. doi: 10.1152/ajpheart.00512.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis EF, Police RJ, Yancey L, McKinney JS, Amruthesh SC. Dilation of cerebral arterioles by cytochrome P-450 metabolites of arachidonic acid. Am J Physiol. 1990;259(4 Pt 2):H1171–7. doi: 10.1152/ajpheart.1990.259.4.H1171. [DOI] [PubMed] [Google Scholar]
- Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. 2004;52(4):447–54. doi: 10.1177/002215540405200403. [DOI] [PubMed] [Google Scholar]
- Erkinjuntti T, Gauthier S. The concept of vascular cognitive impairment. Front Neurol Neurosci. 2009;24:79–85. doi: 10.1159/000197886. [DOI] [PubMed] [Google Scholar]
- Fairbanks SL, Young JM, Nelson JW, Davis CM, Koerner IP, Alkayed NJ. Mechanism of the sex difference in neuronal ischemic cell death. Neuroscience. 2012;219:183–91. doi: 10.1016/j.neuroscience.2012.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Moore SA, Stoll LL, Rich G, Kaduce TL, Weintraub NL, et al. 14,15-Epoxyeicosatrienoic acid inhibits prostaglandin E2 production in vascular smooth muscle cells. Am J Physiol. 1998;275(6 Pt 2):H2113–21. doi: 10.1152/ajpheart.1998.275.6.H2113. [DOI] [PubMed] [Google Scholar]
- Fang X, Weintraub NL, McCaw RB, Hu S, Harmon SD, Rice JB, et al. Effect of soluble epoxide hydrolase inhibition on epoxyeicosatrienoic acid metabolism in human blood vessels. Am J Physiol Heart Circ Physiol. 2004;287(6):H2412–20. doi: 10.1152/ajpheart.00527.2004. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Baumbach GL, Heistad DD. Cerebral circulation: humoral regulation and effects of chronic hypertension. J Am Soc Nephrol. 1990;1:53–57. doi: 10.1681/ASN.V1153. [DOI] [PubMed] [Google Scholar]
- Farias SE, Heidenreich KA, Wohlauer MV, Murphy RC, Moore EE. Lipid mediators in cerebral spinal fluid of traumatic brain injured patients. J Trauma. 2011;71(5):1211–8. doi: 10.1097/TA.0b013e3182092c62. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick FA, Ennis MD, Baze ME, Wynalda MA, McGee JE, Liggett WF. Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids. Influence of stereochemistry. J Biol Chem. 1986;261(32):15334–8. [PubMed] [Google Scholar]
- Fornage M, Boerwinkle E, Doris PA, Jacobs D, Liu K, Wong ND. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation. 2004;109(3):335–9. doi: 10.1161/01.CIR.0000109487.46725.02. [DOI] [PubMed] [Google Scholar]
- Fornage M, Lee CR, Doris PA, Bray MS, Heiss G, Zeldin DC, et al. The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet. 2005;14(19):2829–37. doi: 10.1093/hmg/ddi315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns CJ, Fijnheer R, Algra A, van Mourik JA, van Gijn J, Rinkel GJ. Early circulating levels of endothelial cell activation markers in aneurysmal subarachnoid haemorrhage: associations with cerebral ischaemic events and outcome. J Neurol Neurosurg Psychiatry. 2006a;77(1):77–83. doi: 10.1136/jnnp.2005.064956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns CJ, Kasius KM, Algra A, Fijnheer R, Rinkel GJ. Endothelial cell activation markers and delayed cerebral ischaemia in patients with subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2006b;77(7):863–7. doi: 10.1136/jnnp.2005.081539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol. 1992;263(2 Pt 2):H519–25. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Narayanan J, Aebly MR, Jacobs ER, Harder DR. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J Physiol. 1998;507(Pt 3):771–81. doi: 10.1111/j.1469-7793.1998.771bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, et al. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ Res. 2000;87(1):60–5. doi: 10.1161/01.res.87.1.60. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Yamaura K, Harder DR. Role of 20-HETE in the hypoxia-induced activation of Ca2+-activated K+ channel currents in rat cerebral arterial muscle cells. Am J Physiol Heart Circ Physiol. 2008;294(1):H107–20. doi: 10.1152/ajpheart.01416.2006. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Zhang DX, Carver KA, Rau N, Rarick KR, Roman RJ, et al. Expression of CYP 4A ω-hydroxylase and formation of 20-hydroxyeicosatetreanoic acid (20-HETE) in cultured rat brain astrocytes. Prostaglandins Other Lipid Mediat. 2016;124:16–26. doi: 10.1016/j.prostaglandins.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng S, Wang Y, Sun Y, Li J, Yin H, Zeng Z, et al. Gene- gene interaction between CYP2J2 and PPAR -γ gene on late-onset Alzheimer’s disease in the eastern Chinese Han population. Behav Brain Res. 2016;(16):S0166–4328. 30438–7. doi: 10.1016/j.bbr.2016.07.010. [DOI] [PubMed] [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100(1):328–35. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Graves JP, Gruzdev A, Bradbury JA, DeGraff LM, Li H, House JS, et al. Quantitative Polymerase Chain Reaction Analysis of the Mouse Cyp2j Subfamily: Tissue Distribution and Regulation. Drug Metab Dispos. 2015;43(8):1169–80. doi: 10.1124/dmd.115.064139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwendtner A, Ripke S, Freilinger T, Lichtner P, Müller-Myhsok B, Wichmann HE, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with an increased risk of ischemic stroke in white Europeans. Stroke. 2008;39(5):1593–6. doi: 10.1161/STROKEAHA.107.502179. [DOI] [PubMed] [Google Scholar]
- Gupta NC, Davis CM, Nelson JW, Young JM, Alkayed NJ. Soluble epoxide hydrolase: sex differences and role in endothelial cell survival. Arterioscler Thromb Vasc Biol. 2012;32(8):1936–42. doi: 10.1161/ATVBAHA.112.251520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey L, Harder DR, Meier HT, Varelas PN, Miyata N, Lauer KK, et al. Reversal of delayed vasospasm by TS-011 in the dual hemorrhage dog model of subarachnoid hemorrhage. AJNR Am J Neuroradiol. 2006;27(6):1350–4. [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508(7494):55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder DR, Gebremedhin D, Narayanan J, Jefcoat C, Falck JR, Campbell WB, et al. Formation and action of a P-450 4A metabolite of arachidonic acid in cat cerebral microvessels. Am J Physiol. 1994;266(5 Pt 2):H2098–107. doi: 10.1152/ajpheart.1994.266.5.H2098. [DOI] [PubMed] [Google Scholar]
- Harder DR, Alkayed NJ, Lange AR, Gebremedhin D, Roman RJ. Functional hyperemia in the brain: hypothesis for astrocyte-derived vasodilator metabolites. Stroke. 1998;29:229–34. doi: 10.1161/01.str.29.1.229. [DOI] [PubMed] [Google Scholar]
- Harris TR, Li N, Chiamvimonvat N, Hammock BD. The potential of soluble epoxide hydrolase inhibition in the treatment of cardiac hypertrophy. Congest Heart Fail. 2008;14(4):219–24. doi: 10.1111/j.1751-7133.2008.08430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heizer ML, McKinney JS, Ellis EF. 14,15-Epoxyeicosatrienoic acid inhibits platelet aggregation in mouse cerebral arterioles. Stroke. 1991;22(11):1389–93. doi: 10.1161/01.str.22.11.1389. [DOI] [PubMed] [Google Scholar]
- Higashimori H, Blanco VM, Tuniki VR, Falck JR, Filosa JA. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. Am J Physiol Cell Physiol. 2010;299:C1068–78. doi: 10.1152/ajpcell.00225.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E, Fitzpatrick F, Murphy RC. Biological activity and metabolism of 20-hydroxyeicosatetraenoic acid in the human platelet. Br J Pharmacol. 1992;106(2):267–74. doi: 10.1111/j.1476-5381.1992.tb14327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun P, Parkins NE, Soblosky J, Carey ME, Rodriguez de Turco EB, Bazan NG. Cortical impact injury in rats promotes a rapid and sustained increase in polyunsaturated free fatty acids and diacylglycerols. Neurochem Res. 2000;25(2):269–76. doi: 10.1023/a:1007583806138. [DOI] [PubMed] [Google Scholar]
- Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120(3):287–96. doi: 10.1007/s00401-010-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Vascular and Metabolic Factors in Alzheimer’s Disease and Related Dementias: Introduction. Cell Mol Neurobiol. 2016;36(2):151–4. doi: 10.1007/s10571-015-0319-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Close LN, Selden NR, Alkayed NJ. A novel role for P450 eicosanoids in the neurogenic control of cerebral blood flow in the rat. Exp Physiol. 2007;92(4):653–8. doi: 10.1113/expphysiol.2006/036889. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Alkayed NJ. Soluble Epoxide Hydrolase Inhibition: Targeting Multiple Mechanisms of Ischemic Brain Injury with a Single Agent. Future Neurol. 2009a;4(2):179–199. doi: 10.2217/14796708.4.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang R, Zeldin DC, Alkayed NJ. Epoxyeicosanoids as mediators of neurogenic vasodilation in cerebral vessels. Am J Physiol Heart Circ Physiol. 2009b;296:H1352–63. doi: 10.1152/ajpheart.00950.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Falck JR, Inscho EW. Contribution of cytochrome P450 epoxygenase and hydroxylase pathways to afferent arteriolar autoregulatory responsiveness. Br J Pharmacol. 1999;127(6):1399–405. doi: 10.1038/sj.bjp.0702662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov, 1251. 2009;8(10):794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Simpkins AN, Renic M, Harder DR. Cytochrome P450 eicosanoids and cerebral vascular function. Expert Rev Mol Med. 2011;13:e7. doi: 10.1017/S1462399411001773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92(1):101–30. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD. Epoxyeicosatrienoic acids, 20-hydroxyeicosatetraenoic acid, and renal microvascular function. Prostaglandins Other Lipid Mediat. 2013;104–105:2–7. doi: 10.1016/j.prostaglandins.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, et al. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther. 2008;324(1):103–10. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- Jeffs B, Clark JS, Anderson NH, Gratton J, Brosnan MJ, Gauguier D, Reid JL, Macrae IM, et al. Sensitivity to cerebral ischaemic insult in a rat model of stroke is determined by a single genetic locus. Nat Genet. 1997;16(4):364–7. doi: 10.1038/ng0897-364. [DOI] [PubMed] [Google Scholar]
- Jiang H, McGiff JC, Quilley J, Sacerdoti D, Reddy LM, Falck JR, et al. Identification of 5,6-trans-epoxyeicosatrienoic acid in the phospholipids of red blood cells. J Biol Chem. 2004;279(35):36412–8. doi: 10.1074/jbc.M403962200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zhu AG, Mamczur M, Falck JR, Lerea KM, McGiff JC. Stimulation of rat erythrocyte P2X7 receptor induces the release of epoxyeicosatrienoic acids. Br J Pharmacol. 2007;151(7):1033–40. doi: 10.1038/sj.bjp.0707311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zhu AG, Mamczur M, Morisseau C, Hammock BD, Falck JR, et al. Hydrolysis of cis- and trans-epoxyeicosatrienoic acids by rat red blood cells. J Pharmacol Exp Ther. 2008;326(1):330–7. doi: 10.1124/jpet.107.134858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouihan SA, Zuloaga KL, Zhang W, Shangraw RE, Krasnow SM, Marks DL, et al. Role of soluble epoxide hydrolase in exacerbation of stroke by streptozotocin-induced type 1 diabetes mellitus. J Cereb Blood Flow Metab. 2013;33(10):1650–6. doi: 10.1038/jcbfm.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junier MP, Dray F, Blair I, Capdevila J, Dishman E, Falck JR, et al. Epoxygenase products of arachidonic acid are endogenous constituents of the hypothalamus involved in D2 receptor-mediated, dopamine-induced release of somatostatin. Endocrinology. 1990;126(3):1534–40. doi: 10.1210/endo-126-3-1534. [DOI] [PubMed] [Google Scholar]
- Kaynar MY, Tanriverdi T, Kafadar AM, Kacira T, Uzun H, Aydin S, et al. Detection of soluble intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in both cerebrospinal fluid and serum of patients after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2004;101(6):1030–6. doi: 10.3171/jns.2004.101.6.1030. [DOI] [PubMed] [Google Scholar]
- Kehl F, Cambj-Sapunar L, Maier KG, Miyata N, Kametani S, Okamoto H, et al. 20-HETE contributes to the acute fall in cerebral blood flow after subarachnoid hemorrhage in the rat. Am J Physiol Heart Circ Physiol. 2002;282(4):H1556–65. doi: 10.1152/ajpheart.00924.2001. [DOI] [PubMed] [Google Scholar]
- Kenney K, Amyot F, Haber M, Pronger A, Bogoslovsky T, Moore C, et al. Cerebral Vascular Injury in Traumatic Brain Injury. Experimental Neurology. 2016;275(3):353–366. doi: 10.1016/j.expneurol.2015.05.019. [DOI] [PubMed] [Google Scholar]
- Koerner IP, Jacks R, DeBarber AE, Koop D, Mao P, Grant DF, et al. Polymorphisms in the human soluble epoxide hydrolase gene EPHX2 linked to neuronal survival after ischemic injury. J Neurosci. 2007;27(17):4642–9. doi: 10.1523/JNEUROSCI.0056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerner IP, Zhang W, Cheng J, Parker S, Hurn PD, Alkayed NJ. Soluble epoxide hydrolase: regulation by estrogen and role in the inflammatory response to cerebral ischemia. Front Biosci. 2008;13:2833–41. doi: 10.2741/2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krötz F, Riexinger T, Buerkle MA, Nithipatikom K, Gloe T, Sohn HY, et al. Membrane-potential-dependent inhibition of platelet adhesion to endothelial cells by epoxyeicosatrienoic acids. Arterioscler Thromb Vasc Biol. 2004;24(3):595–600. doi: 10.1161/01.ATV.0000116219.09040.8c. [DOI] [PubMed] [Google Scholar]
- Lange A, Gebremedhin D, Narayanan J, Harder D. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J Biol Chem. 1997;272(43):27345–52. doi: 10.1074/jbc.272.43.27345. [DOI] [PubMed] [Google Scholar]
- Lazaar AL, Yang L, Boardley RL, Goyal NS, Robertson J, Baldwin SJ, et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016;81(5):971–9. doi: 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler CW, Fedinec AL. Newborn piglet cerebral microvascular responses to epoxyeicosatrienoic acids. Am J Physiol. 1997;273(1 Pt 2):H333–8. doi: 10.1152/ajpheart.1997.273.1.H333. [DOI] [PubMed] [Google Scholar]
- Leithner C, Royl G, Offenhauser N, Füchtemeier M, Kohl-Bareis M, Villringer A, et al. Pharmacological uncoupling of activation induced increases in CBF and CMRO2. J Cereb Blood Flow Metab. 2010;30(2):311–22. doi: 10.1038/jcbfm.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine DA, Langa KM. Vascular cognitive impairment: disease mechanisms and therapeutic implications. Neurotherapeutics. 2011;8(3):361–73. doi: 10.1007/s13311-011-0047-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Xu X, Chen C, Yu X, Edin ML, Degraff LM, et al. Cytochrome P450 2J2 is protective against global cerebral ischemia in transgenic mice. Prostaglandins Other Lipid Mediat. 2012;99(3–4):68–78. doi: 10.1016/j.prostaglandins.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JQ, Tan L, Wang HF, Tan MS, Tan L, Xu W, et al. Risk factors for predicting progression from mild cognitive impairment to Alzheimer’s disease: a systematic review and meta-analysis of cohort studies. J Neurol Neurosurg Psychiatry. 2016;87(5):476–84. doi: 10.1136/jnnp-2014-310095. [DOI] [PubMed] [Google Scholar]
- Liu M, Alkayed NJ. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) 1alpha-linked induction of P450 2C11 epoxygenase in astrocytes. J Cereb Blood Flow Metab. 2005;25(8):939–48. doi: 10.1038/sj.jcbfm.9600085. [DOI] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Roman RJ, Harder DR, Koehler RC. Interaction of nitric oxide, 20-HETE, and EETs during functional hyperemia in whisker barrel cortex. Am J Physiol Heart Circ Physiol. 2008;295(2):H619–31. doi: 10.1152/ajpheart.01211.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li C, Gebremedhin D, Hwang SH, Hammock BD, Falck JR, et al. Epoxyeicosatrienoic acid-dependent cerebral vasodilation evoked by metabotropic glutamate receptor activation in vivo. Am J Physiol Heart Circ Physiol. 2011;301(2):H373–81. doi: 10.1152/ajpheart.00745.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Harder DR, Koehler RC. Relative contribution of cyclooxygenases, epoxyeicosatrienoic acids, and pH to the cerebral blood flow response to vibrissal stimulation. Am J Physiol Heart Circ Physiol. 2012;302(5):H1075–85. doi: 10.1152/ajpheart.00794.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gebremedhin D, Harder DR, Koehler RC. Contribution of epoxyeicosatrienoic acids to the cerebral blood flow response to hypoxemia. J Appl Physiol. 2015;119(10):1202–9. doi: 10.1152/japplphysiol.01043.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Vicario C, Alcaraz-Quiles J, García-Alonso V, Rius B, Hwang SH, Titos E, et al. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides. Proc Natl Acad Sci U S A. 2015;112(2):536–41. doi: 10.1073/pnas.1422590112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang H, et al. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem. 2007;282(5):2891–8. doi: 10.1074/jbc.M608057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, et al. The aging brain and cognition: contribution of vascular injury and aβ to mild cognitive dysfunction. JAMA Neurol. 2013;70(4):488–95. doi: 10.1001/2013.jamaneurol.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):156–67. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A, Burgener J, Falck JR, Fritschy JM, Arand M. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience. 2009;163(2):646–61. doi: 10.1016/j.neuroscience.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Martini RP, Ward J, Siler DA, Eastman JM, Nelson JW, Borkar RN, et al. Genetic variation in soluble epoxide hydrolase: association with outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2014;121(6):1359–66. doi: 10.3171/2014.7.JNS131990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McReynolds C, Schmidt WK, Wagner K, Hammock BD. Advancing soluble epoxide hydrolase inhibitors through the valley of death into phase 1 clinical trials for treating painful diabetic neuropathy by utilizing university partnerships, collaborations and NIH support. FASEB J. 2016;30(1):6. Supplement 1272. [Google Scholar]
- Meyer CH, Lowe D, Meyer M, Richardson PL, Neil-Dwyer G. Progressive change in cerebral blood flow during the first three weeks after subarachnoid hemorrhage. Neurosurgery. 1983;12(1):58–76. doi: 10.1227/00006123-198301000-00010. [DOI] [PubMed] [Google Scholar]
- Mezger M, Göbel K, Kraft P, Meuth SG, Kleinschnitz C, Langer HF. Platelets and vascular inflammation of the brain. Hamostaseologie. 2015;35(3):244–51. doi: 10.5482/HAMO-14-11-0071. [DOI] [PubMed] [Google Scholar]
- Miyata N, Seki T, Tanaka Y, Omura T, Taniguchi K, Doi M, et al. Beneficial effects of a new 20-hydroxyeicosatetraenoic acid synthesis inhibitor, TS-011 [N-(3-chloro-4-morpholin-4-yl) phenyl-N′-hydroxyimido formamide], on hemorrhagic and ischemic stroke. J Pharmacol Exp Ther. 2005;314(1):77–85. doi: 10.1124/jpet.105.083964. [DOI] [PubMed] [Google Scholar]
- Munzenmaier DH, Harder DR. Cerebral microvascular endothelial cell tube formation: role of astrocytic epoxyeicosatrienoic acid release. Am J Physiol Heart Circ Physiol. 2000;278(4):H1163–7. doi: 10.1152/ajpheart.2000.278.4.H1163. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Bratton DL, Murphy RC. Analysis of epoxyeicosatrienoic and monohydroxyeicosatetraenoic acids esterified to phospholipids in human red blood cells by electrospray tandem mass spectrometry. J Mass Spectrom. 1997;32(8):888–96. doi: 10.1002/(SICI)1096-9888(199708)32:8<888::AID-JMS548>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360(9340):1155–62. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
- Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6(1):1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Young JM, Borkar RN, Woltjer RL, Quinn JF, Silbert LC, et al. Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat. 2014;113–115:30–7. doi: 10.1016/j.prostaglandins.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44(1):1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285(5431):1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Tanaka Y, Miyata N, Koizumi C, Sakurai T, Fukasawa M, et al. Effect of a new inhibitor of the synthesis of 20-HETE on cerebral ischemia reperfusion injury. Stroke. 2006;37(5):1307–13. doi: 10.1161/01.STR.0000217398.37075.07. [DOI] [PubMed] [Google Scholar]
- Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, et al. Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. American Journal of Physiology - Heart & Circulatory Physiology. 2002;283:H2029–37. doi: 10.1152/ajpheart.01130.2000. [DOI] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilitsis JG, Coplin WM, O’Regan MH, Wellwood JM, Diaz FG, Fairfax MR, et al. Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci Lett. 2003;349(2):136–8. doi: 10.1016/s0304-3940(03)00803-6. [DOI] [PubMed] [Google Scholar]
- Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol. 2013;304(12):H1598–614. doi: 10.1152/ajpheart.00490.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloyac SM, Reynolds RB, Yonas H, Kerr ME. Identification and quantification of the hydroxyeicosatetraenoic acids, 20-HETE and 12-HETE, in the cerebrospinal fluid after subarachnoid hemorrhage. J Neurosci Methods. 2005;144(2):257–63. doi: 10.1016/j.jneumeth.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Poloyac SM, Zhang Y, Bies RR, Kochanek PM, Graham SH. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J Cereb Blood Flow Metab. 2006;26(12):1551–61. doi: 10.1038/sj.jcbfm.9600309. [DOI] [PubMed] [Google Scholar]
- Qu YY, Yuan MY, Liu Y, Xiao XJ, Zhu YL. The protective effect of epoxyeicosatrienoic acids on cerebral ischemia/reperfusion injury is associated with PI3K/Akt pathway and ATP-sensitive potassium channels. Neurochem Res. 2015;40(1):1–14. doi: 10.1007/s11064-014-1456-2. [DOI] [PubMed] [Google Scholar]
- Rao JS, Rapoport SI, Kim HW. Altered neuroinflammatory, arachidonic acid cascade and synaptic markers in postmortem Alzheimer’s disease brain. Transl Psychiatry. 2011;1:e31. doi: 10.1038/tp.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rawal S, Morisseau C, Hammock BD, Shivachar AC. Differential subcellular distribution and colocalization of the microsomal and soluble epoxide hydrolases in cultured neonatal rat brain cortical astrocytes. J Neurosci Res. 2009;87(1):218–27. doi: 10.1002/jnr.21827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renic M, Klaus JA, Omura T, Kawashima N, Onishi M, Miyata N, et al. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009;29(3):629–39. doi: 10.1038/jcbfm.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renic M, Kumar SN, Gebremedhin D, Florence MA, Gerges NZ, Falck JR, et al. Protective effect of 20-HETE inhibition in a model of oxygen-glucose deprivation in hippocampal slice cultures. Am J Physiol Heart Circ Physiol. 2012;302(6):H1285–93. doi: 10.1152/ajpheart.00340.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon F, Wright CB. Vascular cognitive impairment. Curr Opin Neurol. 2013;26(1):29–36. doi: 10.1097/WCO.0b013e32835c4f04. [DOI] [PubMed] [Google Scholar]
- Rodriguez y Baena R, Gaetani P, Paoletti P. A study on cisternal CSF levels of arachidonic acid metabolites after aneurysmal subarachnoid hemorrhage. J Neurol Sci. 1988;84(2–3):329–35. doi: 10.1016/0022-510x(88)90136-0. [DOI] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82(1):131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Roman RJ, Renic M, Dunn KM, Takeuchi K, Hacein-Bey L. Evidence that 20-HETE contributes to the development of acute and delayed cerebral vasospasm. Neurol Res. 2006;28(7):738–49. doi: 10.1179/016164106X152016. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Wallin A, Wardlaw JM, Markus HS, Montaner J, Wolfson L, et al. Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab. 2016;36(1):6–25. doi: 10.1038/jcbfm.2015.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruitenberg A, Heijer den T, Bakker SLM, van Swieten JC, Koudstaal PJ, Hofman A, et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Annals of Neurology. 2005;57(6):789–794. doi: 10.1002/ana.20493. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Narayanan J, Harder DR. Differential effect of amyloid β on the cytochrome P450 epoxygenase activity in rat brain. Neuroscience. 2011;194:241–9. doi: 10.1016/j.neuroscience.2011.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar P, Zaja I, Bienengraeber M, Rarick KR, Terashvili M, Canfield S, et al. Epoxyeicosatrienoic acids pretreatment improves amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am J Physiol Heart Circ Physiol. 2014;306(4):H475–84. doi: 10.1152/ajpheart.00001.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik JS, Poloyac SM, Kochanek PM, Alexander H, Tudorascu DL, Clark RS, et al. 20-Hydroxyeicosatetraenoic Acid Inhibition by HET0016 Offers Neuroprotection, Decreases Edema, and Increases Cortical Cerebral Blood Flow in a Pediatric Asphyxial Cardiac Arrest Model in Rats. J Cereb Blood Flow Metab. 2015;35(11):1757–63. doi: 10.1038/jcbfm.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HC, Hammock BD. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem. 2012;55(5):1789–808. doi: 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty PK, Galeffi F, Turner DA. Age-Induced Alterations in Hippocampal Function and Metabolism. Aging Dis. 2011;2(3):196–218. [PMC free article] [PubMed] [Google Scholar]
- Shivachar AC, Willoughby KA, Ellis EF. Effect of protein kinase C modulators on 14,15-epoxyeicosatrienoic acid incorporation into astroglial phospholipids. J Neurochem. 1995;65(1):338–46. doi: 10.1046/j.1471-4159.1995.65010338.x. [DOI] [PubMed] [Google Scholar]
- Siler DA, Martini RP, Ward JP, Nelson JW, Borkar RN, Zuloaga KL, et al. Protective role of p450 epoxyeicosanoids in subarachnoid hemorrhage. Neurocrit Care. 2015;22(2):306–19. doi: 10.1007/s12028-014-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siler DA, Berlow YA, Kukino A, Davis CM, Nelson JW, Grafe MR, et al. Soluble Epoxide Hydrolase in Hydrocephalus, Cerebral Edema, and Vascular Inflammation After Subarachnoid Hemorrhage. Stroke. 2015;46(7):1916–22. doi: 10.1161/STROKEAHA.114.008560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, et al. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol. 2009;174(6):2086–95. doi: 10.2353/ajpath.2009.080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275(51):40504–10. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- Singh H, Cheng J, Deng H, Kemp R, Ishizuka T, Nasjletti A, et al. Vascular cytochrome P450 4A expression and 20-hydroxyeicosatetraenoic acid synthesis contribute to endothelial dysfunction in androgen-induced hypertension. Hypertension. 2007;50(1):123–9. doi: 10.1161/HYPERTENSIONAHA.107.089599. [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277(10):813–7. [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43(1):55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Stefanova I, Stephan T, Becker-Bense S, Dera T, Brandt T, Dieterich M. Age-related changes of blood-oxygen-level-dependent signal dynamics during optokinetic stimulation. Neurobiol Aging. 2013;34(10):2277–86. doi: 10.1016/j.neurobiolaging.2013.03.031. [DOI] [PubMed] [Google Scholar]
- Stella N, Tencé M, Glowinski J, Prémont J. Glutamate-evoked release of arachidonic acid from mouse brain astrocytes. J Neurosci. 1994;14(2):568–75. doi: 10.1523/JNEUROSCI.14-02-00568.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KI, Gruzdev A, Zeldin DC. Altered Behavioral Phenotypes In Soluble Epoxide Hydrolase Knockout Mice: Effects of Traumatic Brain Injury. Prosta glandins Other Lipid Mediat. 2013;104–105:18–24. doi: 10.1016/j.prostaglandins.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sura P, Sura R, Enayetallah AE, Grant DF. Distribution and expression of soluble epoxide hydrolase in human brain. J Histochem Cytochem. 2008;56(6):551–9. doi: 10.1369/jhc.2008.950659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Renic M, Bohman QC, Harder DR, Miyata N, Roman RJ. Reversal of delayed vasospasm by an inhibitor of the synthesis of 20-HETE. Am J Physiol Heart Circ Physiol. 2005;289(5):H2203–11. doi: 10.1152/ajpheart.00556.2005. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Miyata N, Renic M, Harder DR, Roman RJ. Hemoglobin, NO, and 20-HETE interactions in mediating cerebral vasoconstriction following SAH. Am J Physiol Regul Integr Comp Physiol. 2006;290(1):R84–9. doi: 10.1152/ajpregu.00445.2005. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Omura T, Fukasawa M, Horiuchi N, Miyata N, Minagawa T, et al. Continuous inhibition of 20-HETE synthesis by TS-011 improves neurological and functional outcomes after transient focal cerebral ischemia in rats. Neurosci Res. 2007;59(4):475–80. doi: 10.1016/j.neures.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Tarantini S, Hertelendy P, Tucsek Z, Valcarcel-Ares MN, Smith N, Menyhart A, et al. Pharmacologically-induced neurovascular uncoupling is associated with cognitive impairment in mice. J Cereb Blood Flow Metab. 2015;35(11):1871–81. doi: 10.1038/jcbfm.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashvili M, Sarkar P, Nostrand MV, Falck JR, Harder DR. The protective effect of astrocyte-derived 14,15-epoxyeicosatrienoic acid on hydrogen peroxide-induced cell injury in astrocyte-dopaminergic neuronal cell line co-culture. Neuroscience. 2012;223:68–76. doi: 10.1016/j.neuroscience.2012.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Rozsa B, Springo Z, Doczi T, Koller A. Isolated human and rat cerebral arteries constrict to increases in flow: role of 20-HETE and TP receptors. J Cereb Blood Flow Metab. 2011;31(10):2096–105. doi: 10.1038/jcbfm.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Csiszar A, Tucsek Z, Sosnowska D, Gautam T, Koller A, et al. Role of 20-HETE, TRPC channels, and BKCa in dysregulation of pressure-induced Ca2+ signaling and myogenic constriction of cerebral arteries in aged hypertensive mice. Am J Physiol Heart Circ Physiol. 2013;305(12):H1698–708. doi: 10.1152/ajpheart.00377.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. 2017;312(1):H1–H20. doi: 10.1152/ajpheart.00581.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltenberger B, Garscha U, Temml V, Liers J, Werz O, Schuster D, et al. Discovery of Potent Soluble Epoxide Hydrolase (sEH) Inhibitors by Pharmacophore-Based Virtual Screening. J Chem Inf Model. 2016;56(4):747–62. doi: 10.1021/acs.jcim.5b00592. [DOI] [PubMed] [Google Scholar]
- Wang L, Du Y, Wang K, Xu G, Luo S, He G. Chronic cerebral hypoperfusion induces memory deficits and facilitates Aβ generation in C57BL/6J mice. Exp Neurol. 2016;283(Pt A):353–64. doi: 10.1016/j.expneurol.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Ward NC, Croft KD, Blacker D, Hankey GJ, Barden A, Mori TA, et al. Cytochrome P450 metabolites of arachidonic acid are elevated in stroke patients compared with healthy controls. Clin Sci (Lond) 2011;121(11):501–7. doi: 10.1042/CS20110215. [DOI] [PubMed] [Google Scholar]
- Weir B, Grace M, Hansen J, Rothberg C. Time course of vasospasm in man. J Neurosurg. 1978;48(2):173–8. doi: 10.3171/jns.1978.48.2.0173. [DOI] [PubMed] [Google Scholar]
- Yamaura K, Gebremedhin D, Zhang C, Narayanan J, Hoefert K, Jacobs ER, et al. Contribution of epoxyeicosatrienoic acids to the hypoxia-induced activation of Ca2+ -activated K+ channel current in cultured rat hippocampal astrocytes. Neuroscience. 2006;143(3):703–16. doi: 10.1016/j.neuroscience.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Yan H, Kong Y, He B, Huang M, Li J, Zheng J, et al. CYP2J2 rs890293 polymorphism is associated with susceptibility to Alzheimer’s disease in the Chinese Han population. Neurosci Lett. 2015;593:56–60. doi: 10.1016/j.neulet.2015.03.024. [DOI] [PubMed] [Google Scholar]
- Yang S, Ma Y, Liu Y, Que H, Zhu C, Liu S. Arachidonic acid: a bridge between traumatic brain injury and fracture healing. J Neurotrauma. 2012;29(17):2696–705. doi: 10.1089/neu.2012.2442. [DOI] [PubMed] [Google Scholar]
- Yang ZJ, Carter EL, Kibler KK, Kwansa H, Crafa DA, Martin LJ, et al. Attenuation of neonatal ischemic brain damage using a 20-HETE synthesis inhibitor. J Neurochem. 2012a;121(1):168–79. doi: 10.1111/j.1471-4159.2012.07666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Sun Y, Lu Z, Leak RK, Zhang F. The impact of cerebrovascular aging on vascular cognitive impairment and dementia. Ageing Res Rev. 2016:S1568-1637(16)30118-0. doi: 10.1016/j.arr.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Cheriyan J, Gutterman DD, Mayer RJ, Ament Z, Griffin JL, et al. Mechanisms of Vascular Dysfunction in COPD and Effects of a Novel Soluble Epoxide Hydrolase Inhibitor in Smokers. Chest. 2017;151(3):555–563. doi: 10.1016/j.chest.2016.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Yi X, Wang J, Zhou CQ. CYP Genetic Variants, CYP Metabolite Levels, and Neurologic Deterioration in Acute Ischemic Stroke in Chinese Population. J Stroke Cerebrovasc Dis. 2016;(16):S1052–3057. 30452–9. doi: 10.1016/j.jstrokecerebrovasdis.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Yu M, Cambj-Sapunar L, Kehl F, Maier KG, Takeuchi K, Miyata N, et al. Effects of a 20-HETE antagonist and agonists on cerebral vascular tone. Eur J Pharmacol. 2004;486(3):297–306. doi: 10.1016/j.ejphar.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Yuan L, Liu J, Dong R, Zhu J, Tao C, Zheng R, et al. 14,15-epoxyeicosatrienoic acid promotes production of brain derived neurotrophic factor from astrocytes and exerts neuroprotective effects during ischaemic injury. Neuropathol Appl Neurobiol. 2016;42(7):607–620. doi: 10.1111/nan.12291. [DOI] [PubMed] [Google Scholar]
- Zagorac D, Jakovcevic D, Gebremedhin D, Harder DR. Anti-Angiogenic Effect of Inhibitors of Cytochrome P450 on Rats with Glioblastoma Multiforme. J Cereb Blood Flow Metab. 2008;28(8):1431–1439. doi: 10.1038/jcbfm.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldin DC, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, et al. Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. J Biol Chem. 1993;268(9):6402–7. [PubMed] [Google Scholar]
- Zeldin DC, DuBois RN, Falck JR, Capdevila JH. Molecular cloning, expression and characterization of an endogenous human cytochrome P450 arachidonic acid epoxygenase isoform. Arch Biochem Biophys. 1995;322(1):76–86. doi: 10.1006/abbi.1995.1438. [DOI] [PubMed] [Google Scholar]
- Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276(39):36059–62. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- Zhang C, Harder DR. Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic. Acid Stroke. 2002;33(12):2957–64. doi: 10.1161/01.str.0000037787.07479.9a. [DOI] [PubMed] [Google Scholar]
- Zhang W, Koerner IP, Noppens R, Grafe M, Tsai HJ, Morisseau C, et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27(12):1931–40. doi: 10.1038/sj.jcbfm.9600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Cui Y, Geng B, Zeng X, Tang C. 11,12-Epoxyeicosatrienoic acid activates the L-arginine/nitric oxide pathway in human platelets. Mol Cell Biochem. 2008a;308(1–2):51–6. doi: 10.1007/s11010-007-9611-6. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ding H, Yan J, Hui R, Wang W, Kissling GE, et al. Genetic variation in cytochrome P450 2J2 and soluble epoxide hydrolase and risk of ischemic stroke in a Chinese population. Pharmacogenet Genomics. 2008b;18(1):45–51. doi: 10.1097/FPC.0b013e3282f313e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, et al. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008c;39(7):2073–8. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Iliff JJ, Campbell CJ, Wang RK, Hurn PD, Alkayed NJ. Role of soluble epoxide hydrolase in the sex-specific vascular response to cerebral ischemia. J Cereb Blood Flow Metab. 2009;29(8):1475–81. doi: 10.1038/jcbfm.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Davis CM, Edin ML, Lee CR, Zeldin DC, Alkayed NJ. Role of endothelial soluble epoxide hydrolase in cerebrovascular function and ischemic injury. PLoS One. 2013;8(4):e61244. doi: 10.1371/journal.pone.0061244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hong G, Lee KS, Hammock BD, Gebremedhin D, Harder DR, et al. Inhibition of soluble epoxide hydrolase augments astrocyte release of vascular endothelial growth factor and neuronal recovery after oxygen-glucose deprivation. J Neurochem. 2017a;140(5):814–825. doi: 10.1111/jnc.13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen C, Hua S, Liao H, Wang M, Xiong Y, et al. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract. 2017b;124:41–47. doi: 10.1016/j.diabres.2016.10.024. [DOI] [PubMed] [Google Scholar]
- Zhang H, Falck JR, Roman RJ, Harder DR, Koehler RC, Yang ZJ. Upregulation of 20-HETE Synthetic Cytochrome P450 Isoforms by Oxygen-Glucose Deprivation in Cortical Neurons. Neurons Cell Mol Neurobiol. 2017c doi: 10.1007/s10571-017-0462-8. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Schieber EB, McGiff JC, Balazy M. Identification of arachidonate P-450 metabolites in human platelet phospholipids. Hypertension. 1995;25(4 Pt 2):854–9. doi: 10.1161/01.hyp.25.4.854. [DOI] [PubMed] [Google Scholar]
- Zhu J, Wang B, Lee JH, Armstrong JS, Kulikowicz E, Bhalala US, et al. Additive Neuroprotection of a 20-HETE Inhibitor with Delayed Therapeutic Hypothermia after Hypoxia-Ischemia in Neonatal Piglets. Dev Neurosci. 2015;37(4–5):376–89. doi: 10.1159/000369007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–38. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuloaga KL, Krasnow SM, Zhu X, Zhang W, Jouihan SA, Shangraw RE, et al. Mechanism of protection by soluble epoxide hydrolase inhibition in type 2 diabetic stroke. PLoS One. 2014;9(5):e97529. doi: 10.1371/journal.pone.0097529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuloaga KL, Zhang W, Roese NE, Alkayed NJ. Soluble epoxide hydrolase gene deletion improves blood flow and reduces infarct size after cerebral ischemia in reproductively senescent female mice. Front Pharmacol. 2015a;5:290. doi: 10.3389/fphar.2014.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuloaga KL, Zhang W, Yeiser LA, Stewart B, Kukino A, Nie X, et al. Neurobehavioral and imaging correlates of hippocampal atrophy in a mouse model of vascular cognitive impairment. Transl Stroke Res. 2015b;6(5):390–8. doi: 10.1007/s12975-015-0412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuloaga KL, Johnson LA, Roese NE, Marzulla T, Zhang W, Nie X, et al. High fat diet-induced diabetes in mice exacerbates cognitive deficit due to chronic hypoperfusion. J Cereb Blood Flow Metab. 2016;36(7):1257–70. doi: 10.1177/0271678X15616400. [DOI] [PMC free article] [PubMed] [Google Scholar]