

Abstract
Aim
To evaluate the safety, tolerability, and pharmacokinetics/pharmacodynamics of PRN1008, a novel Bruton's tyrosine kinase (BTK) inhibitor, in healthy volunteers, and thus determine the dose range for future clinical studies.
Methods
This was a two‐part randomized, placebo controlled study in healthy volunteers using a liquid formulation. Part I was a single ascending dose design with dose levels of 50–1200 mg (n = 6 active, two placebos per cohort); Part II was a multiple ascending dose design, with dose regimens ranging from 300 to 900 mg daily, either four times or twice daily for 10 days. Plasma pharmacokinetics, adverse events, vital signs, electrocardiograms and laboratory parameters were assessed. BTK occupancy in peripheral blood mononuclear cells was evaluated as a marker of target engagement.
Results
PRN1008 was rapidly absorbed following oral administration, and was safe and well tolerated in all dose regimens evaluated in both single and multiple doses. PRN1008 demonstrated a large volume of distribution, and a half‐life of approximately 3–4 h. BTK occupancy of >90% was observed within 4 h after dosing in both single and multiple dose regimens, and was closely linked to maximum plasma concentration. BTK occupancy decay was slow (–1.6% h–1), and occupancy was sustained despite drug concentrations being undetectable. No severe or serious adverse events occurred, and the most common adverse events were gastrointestinal in nature.
Conclusions
PRN1008 was safe and well‐tolerated following oral administration, and achieved high, sustained levels of BTK occupancy in peripheral blood mononuclear cells.
Keywords: autoimmune diseases, Bruton's tyrosine kinase, PRN1008
What is Already Known about this Subject
PRN1008 is a reversible, covalent BTK inhibitor with safety and disease activity in animal models.
This is the first in human clinical trial to evaluate PRN1008 as a potential drug candidate for inflammatory and autoimmune diseases.
What this Study Adds
This study characterized the early safety, tolerability and pharmacokinetic/pharmacodynamic relationship of PRN1008 following oral dosing.
The results support continued clinical development of PRN1008.
Introduction
Bruton's agammaglobulinaemia tyrosine kinase (BTK) is a nonreceptor tyrosine kinase and a member of the TEC protein tyrosine kinase family of kinases. It is an essential signalling element downstream of the B cell receptor (BCR), and is involved in activation of other haematopoietic cells including basophils, mast cells, macrophages, neutrophils and platelets. Inhibition of BTK activity in cells produces phenotypic changes consistent with blockade of the BCR and a selective BTK inhibitor has the potential to target multiple pathways and cell types involved in inflammation and autoimmunity 1, 2, 3, 4, 5.
A selective BTK inhibitor has the potential to target multiple pathways involved in inflammation and autoimmunity. These include modulation of BCR‐mediated B‐cell pathways, inhibition of FcεR‐induced cytokine release from monocytes and macrophages, FcγR‐induced mast cell degranulation, neutrophil migration and mediator release. These effects lead to the prediction that a BTK inhibitor could block the initiation and propagation of various autoimmune diseases and mitigate the resulting autoantibody mediated effects.
Pertinent to the treatment of patients with autoantibody mediated diseases, inhibitors of BTK have been shown to inhibit disease in several autoantibody dependent rodent models including arthritis 2, 6, 7, 8, 9, systemic lupus erythematosus 7, 10 and antibody mediated glomulonephritis 11. BTK inhibitors also suppress antibody mediated inflammation in rodent arthus and passive cutaneous anaphylaxis assays models via inhibition of FcγR or FcεR signalling respectively 6.
PRN1008 is a novel reversible, covalent, small molecule BTK inhibitor which has a high degree of affinity and selectivity for the BTK receptor. This selectivity offers the potential to provide efficacy benefits while minimizing adverse side effects. The in vitro biochemical 50% inhibitory concentration is 1.3 ± 0.5 nm, and the 50% inhibitory concentration in Ramos cells (human B‐cell line) is 7.4 ± 2 nm. In addition, PRN1008 has a long duration of action due to a slow off‐rate for binding to the target site, resulting in prolonged target occupancy 12 without forming permanent covalent bonds to BTK. BTK binding kinetics, as determined by fluorescence competition, determined a fast on rate of 5.1 × 104 m –1 s–1 and a slow off rate of 1.2 × 104 m –1, with BTK occupancy diminishing only modestly from 1 hour (95 ± 1%) to 6 h (91 ± 1%) to 24 h (85 ± 2%). This chemical profile is postulated to be suitable for safe, chronic administration in a broad range of diseases.
Nonclinical pharmacodynamic (PD) studies demonstrated dissociation of BTK inhibition, target occupancy and plasma pharmacokinetics (PK), with target inhibition persisting after plasma drug levels had declined demonstrating an anticlockwise hysteresis. Anticlockwise hysteresis has generally been defined as a pharmacological phenomenon where an effect can increase or be maintained for a decreasing plasma concentration leading to a sustained on‐target efficacy 13. PRN1008 manipulates this phenomenon by employing a fast association with a very slow dissociation rate. This has an advantage in reducing the prolonged systemic circulation levels of the drug that would ordinarily be required for efficacy and thus reducing the potential off target interactions of the molecule 12.
The aim of this study was to determine the safety, tolerability and PK of single and multiple oral doses of PRN1008 in healthy adult volunteers. The study was also designed to assess BTK occupancy of PRN1008 in peripheral blood mononuclear cells (PBMCs).
Methods
Study design
This study was conducted at a single centre in Australia (Linear Clinical Research, Perth, Australia). The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidance for Good Clinical Practice. The study protocol and informed consent documents received local Ethics Committee approval (Bellberry Human Research Ethics Committee), prior to subject enrolment, and all subjects provided written informed consent prior to participating in any study‐related activities (Registered on ANZCTR No. ACTRN12614000359639).
This was a two‐part (Part A and Part B) first‐in‐human Phase 1 study conducted in healthy adult volunteers. Both Part A and Part B were designed as double blind, placebo controlled, randomized, ascending dose studies to determine the initial safety, tolerability and PK/PD of PRN1008. Part A evaluated single ascending doses (N = 8 per cohort, 6 active/2 placebo), whereas Part B evaluated once and twice daily dosing of PRN1008 for 10 days (N = 10 per cohort, 8 active/2 placebo). All doses of PRN1008 were administered as a liquid formulation under fasting conditions with the exception of Cohort B1, Day –1, where PRN1008 was administered within 30 min of eating a moderate fat meal.
To be included in the study, subjects were required to be healthy male or surgically sterile or postmenopausal females, age 18–55 years, with a body mass index of 18–30.5 kg m–2, who had no clinically significant abnormality on clinical examination, medical history, electrocardiogram (ECG), clinical chemistry, haematology or urinalysis results (negative screen for drug abuse and alcohol use was also required). Prescription and over‐the‐counter medications and herbal supplements were prohibited, other than occasional paracetamol use.
Following confirmation of eligibility and completion of a 28‐day screening phase, participants were randomly assigned to PRN1008 or placebo to the next dose cohort. Participants were admitted to the clinical research unit on day –1, were discharged 48 h after the last dose of study drug, and completed a follow‐up safety visit 6–8 days after the last dose. As a safety precaution, two sentinel participants (one active, one placebo) were dosed in Part A 24 h prior to dosing the remainder of each cohort. Safety, tolerability, and available PK and PD data were evaluated by a Safety Monitoring Committee prior to each dose escalation step.
Safety assessments
The safety of study participants was monitored by frequent assessments of vital signs, clinical laboratory tests, physical examinations, 12‐lead ECGs and adverse event monitoring. All adverse events (AEs) were categorized using MedDRA, graded using CTCAE criteria and relationship to study drug administration.
PK
Venous blood samples for quantitation of PRN1008 in plasma were obtained before dosing (Time 0), and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12 and 24 h post‐PRN1008 administration on Day 1 in Parts A and B, and on Day 10 in Part B. In addition, single predose samples were collected in Part B on the mornings of Days 3, 5, 7, and 10. Each plasma sample was divided into two aliquots and stored at –80°C until analysed. Plasma concentrations of PRN1008 were determined by a validated liquid chromatography–tandem mass spectrometry (LC/MS/MS) method. Acetonitrile precipitation and LC/MS/MS were used to determine the concentration of PRN1008 from human K2EDTA plasma. An aliquot of the extract was injected onto a high‐performance LC/MS/MS triple quadrupole mass spectrometer (Sciex API4000). The peak area of the product ion of the compound (PRN1008) was measured against the peak area of the product ion of the internal standard. A calibration curve ranging from 0.100 to 100 ng ml–1 was used to quantify PRN1008 in the samples.
PD
Whole blood samples for PBMCs were collected in each participant in Part A at 0, 4, 12 and 24 h post dosing of study drug for determination of BTK occupancy. In Part B, samples were collected on Day 1 at 0, 4, 12 and 24 h post dose; predose on the mornings of Days 3 and 7; and on Day 10 at 0, 4, 12, 24 and 36 h postdose.
Cryopreserved PBMCs were thawed, viability determined and samples treated with a BTK selective boron dipyrromethene‐labelled probe to measure BTK occupancy. After incubation and washing, samples were lysed and clarified. Cell lysates were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and gels scanned using a fluorescence scanner (Typhoon 9410, GE Healthcare) using a fluorescein filter set to measure BTK occupancy after which proteins were transferred to a nitrocellulose membrane. The membrane was immunoblotted using a primary mouse anti‐human BTK antibody (BD Biosciences) and a secondary goat anti‐mouse IgG‐Alexa fluor 546 fluorescent antibody (Invitrogen). The membrane was scanned for total BTK. Occupancy data were normalized to total BTK and %BTK occupancy of the predose sample for each subject.
Data analysis
Pharmacokinetic parameters were estimated using standard noncompartmental methods (Phoenix WinNonlin® Version 6.2). Actual dosing and sampling times were used in the analysis. Pharmacokinetic parameters were summarized by dose group using descriptive statistics, including n, mean and coefficient of variation. For time to maximum concentration (Tmax), only n, min, median and max were reported.
Emax modelling was applied to explore the relationship between PRN1008 PK parameters and BTK occupancy (Phoenix WinNonlin® Version 6.2). BTK occupancy measures were considered as dependent variables, with PRN1008 exposure measures (maximum plasma concentration, Cmax; area under the concentration–time curve) as independent variables. Various Emax models were fit to the data, with model discrimination based on goodness‐of‐fit statistics, Akaike's information criteria, and parsimony.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 13, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 14.
Results
Study progression
Dose levels completed for Part A (Cohorts A1–A5) included 50, 150, 300, 600 and 1200 mg. Dose escalation in Part A was stopped as PRN1008 exposures did not increase between the 600 and 1200 mg dose levels.
Dose levels completed in Part B (Cohorts B1–B4) included 300 mg q. 24 h, 300 mg q. 12 h, 600 mg q. 24 h and 450 mg q. 12 h.
All study visits were conducted according to the protocol. Protocol deviations were minor, mostly relating to timing or missing assessments. All subjects completed the study, and there were no withdrawals or dose interruptions.
The maximum tolerated dose was not reached in either Part A or Part B.
Demographics
A total of 80 subjects were enrolled in the study between April and December 2014, with 62 receiving PRN1008 and 18 placebo. All subjects received their planned doses of study medication, and there were no dose interruptions, dose adjustments, dropouts or discontinuations.
Forty subjects (eight per cohort) were enrolled in Part A of the study, with 30 receiving PRN1008 and 10 placebo. The majority of subjects in Part A were white (67–100% by cohort; 85% overall), all were male (100%), with a median cohort age that ranged from 21.5 to 29.0 years. Median cohort body weight ranged from 74.0 to 79.4 kg, and the median body mass index ranged from 22.1 to 25.2 kg m–2.
Forty subjects (10 per cohort) were enrolled in Part B of the study, with 32 receiving PRN1008 and 8 placebo. The majority of patients in Part B were white (63–88%; 78% overall), all were male (100%), with a median age that ranged from 23 to 27.5 years. Median body weight by cohort ranged from 72.8 to 91.4 kg, and the median body mass index by cohort ranged from 21.8 to 26.7 kg m–2.
In both Part A and Part B, the demographic distribution of subjects across cohorts was reasonable. Details of the demographics characteristics of participants are provided in Table 1.
Table 1.
Demographic characteristics of study participants
| PRN1008 dose regimen: PART A | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Statistic | 50 mg | 150 mg | 300 mg | 600 mg | 1200 mg | Placebo | Total |
| Age (years) | ||||||||
| N | 6 | 6 | 6 | 6 | 6 | 10 | 40 | |
| Mean | 24.8 | 28.0 | 20.5 | 26.3 | 32.2 | 22.9 | 25.5 | |
| SD | 4.5 | 11.7 | 2.0 | 5.4 | 11.7 | 3.4 | 7.6 | |
| Median | 23.5 | 23.5 | 21.5 | 25.5 | 29.0 | 23.0 | 23.0 | |
| Race | ||||||||
| Caucasian | n (%) | 4 (67%) | 4 (67%) | 6 (100%) | 6 (100%) | 5 (83%) | 9 (90%) | 34 (85%) |
| Asian | n (%) | 1 (17%) | 1 (17%) | 1 (10%) | 3 (8%) | |||
| Black | n (%) | 1 (17%) | 1 (3%) | |||||
| Other: Arabic | n (%) | 1 (17%) | 1 (3%) | |||||
| Other: Eurasian | n (%) | 1 (17%) | 1 (3%) | |||||
| Sex | ||||||||
| Male | n (%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 10 (100%) | 40 (100%) |
| Weight (kg) | ||||||||
| Mean | 74.1 | 73.1 | 80.8 | 74.0 | 86.0 | 74.1 | 76.7 | |
| SD | 9.5 | 9.4 | 8.5 | 10.7 | 12.9 | 9.6 | 10.5 | |
| Median | 76.2 | 75.4 | 78.4 | 74.0 | 79.4 | 76.3 | 77.1 | |
| BMI | ||||||||
| Mean | 22.7 | 23.1 | 24.2 | 23.5 | 26.2 | 22.5 | 23.6 | |
| SD | 2.0 | 3.3 | 1.6 | 2.6 | 3.3 | 1.7 | 2.6 | |
| Median | 23.2 | 23.5 | 23.9 | 22.8 | 25.2 | 22.1 | 23.5 | |
| PRN 1008 dose regimen: PART B | |||||||
|---|---|---|---|---|---|---|---|
| Parameter | Statistic | 300 mg QD | 300 mg BID | 600 mg QD | 450 mg BID | Placebo | Total |
| Age (years) | |||||||
| N | 8 | 8 | 8 | 8 | 8 | 40 | |
| Mean | 30.0 | 27.0 | 23.6 | 28.9 | 24.4 | 26.8 | |
| SD | 8.5 | 7.0 | 4.2 | 7.3 | 5.0 | 6.7 | |
| Median | 27.0 | 25.5 | 23.5 | 27.5 | 23.0 | 24.0 | |
| Race | |||||||
| Caucasian | n (%) | 5 (63%) | 7 (88%) | 7 (88%) | 6 (75%) | 6 (75%) | 31 (78%) |
| Asian | n (%) | 2 (25%) | 1 (13%) | 1 (13%) | 1 (13%) | 5 (13%) | |
| Black | n (%) | 1 (13%) | 1 (13%) | 1 (13%) | 3 (8%) | ||
| Other: Eurasian | n (%) | 1 (13%) | 1 (3%) | ||||
| Sex | |||||||
| Male | n (%) | 8 (100%) | 8 (100%) | 8 (100%) | 8 (100%) | 8 (100%) | 40 (100%) |
| Weight (kg) | |||||||
| Mean | 76.0 | 75.3 | 87.0 | 86.0 | 75.5 | 79.9 | |
| SD | 10.2 | 12.3 | 15.2 | 12.7 | 10.3 | 12.8 | |
| Median | 75.6 | 72.8 | 88.1 | 91.4 | 75.7 | 76.9 | |
| BMI | |||||||
| Mean | 25.1 | 23.8 | 25.0 | 25.6 | 23.4 | 24.6 | |
| SD | 2.4 | 4.0 | 3.4 | 3.1 | 2.3 | 3.1 | |
| Median | 25.3 | 21.8 | 24.3 | 26.7 | 23.2 | 24.1 | |
SD, standard deviation; BMI, body mass index; QD = every 24 h; BID = every 12 h
PK results
Part A
PRN1008 was rapidly absorbed following oral administration under fasted conditions, with Tmax ranging from a median of 0.5 h at 50 mg to 2.5 h at 600 and 1200 mg. The mean apparent terminal elimination half‐life ranged from approximately 1.4 to 3.9 h for all cohorts, and did not appear to change with increasing doses. Plasma concentrations generally increased with dose over the range of 50–600 mg, and concentrations declined in a bi‐exponential manner after reaching Cmax. Increasing the dose from 600 to 1200 mg resulted in similar plasma exposures. The mean apparent volume of distribution ranged from 4760 to 6720 l for all cohorts. A summary of pharmacokinetic parameters is provided in Table 2, and mean concentration‐time profiles are shown in Figure 1.
Table 2.
Summary of PRN1008 pharmacokinetic parameters by dose following single oral doses in healthy volunteers (Part A)
| Dose | Tmax (h) | Cmax (ng ml–1) | Cmax/D | AUC0‐∞(ng.h ml–1) | AUC0‐∞/D | Half‐life (h) | Vz/F (l) | |
|---|---|---|---|---|---|---|---|---|
| 50 mg (N = 6) | Mean | 15.1 | 0.301 | 15.3 | 0.306 | 1.36 | 6720 | |
| Median | 0.50 | 14.7 | 0.293 | 16.0 | 0.319 | 1.32 | 6060 | |
| CV% | 31.0 | 37.0 | 37.0 | 19.6 | 19.6 | 21.4 | 35.7 | |
| 95% CI | 9.2–20.9 | 0.184–0.418 | 12.1–18.4 | 0.243–0.369 | 1.05–1.66 | 4200–9240 | ||
| 150 mg (N = 6) | Mean | 45.9 | 0.306 | 98.9 | 0.66 | 1.82 | 6670 | |
| Median | 1.0 | 41.8 | 0.279 | 79.7 | 0.531 | 1.66 | 5890 | |
| CV% | 31.6 | 54.6 | 54.6 | 86.0 | 86.0 | 25.1 | 79.0 | |
| 95% CI | 19.6–72.2 | 0.131–0.482 | 9.64–188 | 0.064–1.26 | 1.34–2.3 | 1140–12 200 | ||
| 300 mg (N = 6) | Mean | 103 | 0.343 | 240 | 0.799 | 2.91 | 5620 | |
| Median | 1.5 | 91.2 | 0.304 | 250 | 0.833 | 2.91 | 4910 | |
| CV% | 53.6 | 64.6 | 64.6 | 45.6 | 45.6 | 49.4 | 42.6 | |
| 95% CI | 33.2–173 | 0.111–0.576 | 125–354 | 0.417–1.18 | 1.4–4.41 | 3110–8140 | ||
| 600 mg (N = 6) | Mean | 302 | 0.503 | 1060 | 1.77 | 3.90 | 4760 | |
| Median | 2.5 | 301 | 0.501 | 1020 | 1.7 | 3.91 | 3340 | |
| CV% | 27.2 | 51.5 | 51.5 | 57.5 | 57.5 | 18.8 | 75.9 | |
| 95% CI | 139–465 | 0.231–0.775 | 139–465 | 0.70–2.83 | 139–465 | 139–465 | ||
| 1200 mg (N = 6) | Mean | 263 | 0.219 | 1070 | 0.892 | 2.53 | 4890 | |
| Median | 2.5 | 209 | 0.174 | 1090 | 0.907 | 2.27 | 3730 | |
| CV% | 57.7 | 53.0 | 53.0 | 54.3 | 54.3 | 49.2 | 60.1 | |
| 95% CI | 117–409 | 0.097–0.341 | 460–1680 | 0.383–1.40 | 1.22–3.83 | 1800–7970 |
Tmax, time to maximum plasma concentration; Cmax, maximum plasma concentration; AUC, area under the concentration–time curve; CV, coefficient of variation; CI, confidence interval
Figure 1.
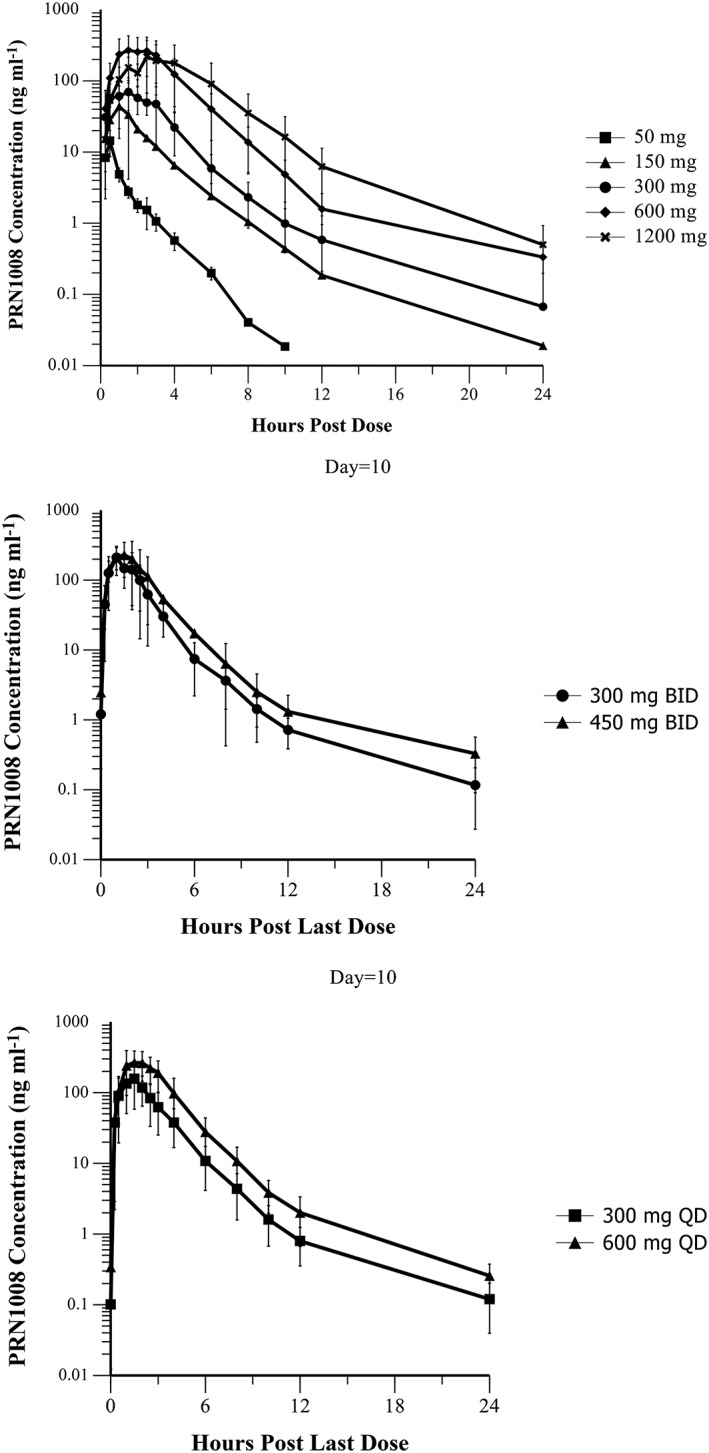
Mean (± standard deviation) concentration time profiles of PRN1008 following single (Part A) and multiple (Part B, Day 10) oral doses in healthy volunteers
Part B
The pharmacokinetics of multiple dose PRN1008 were consistent with the single dose results (Table 3). The median Tmax of PRN1008 ranged from 1 to 1.5 h and the mean Day 10 terminal half‐life was approximately 4 h. Consistent with the observed half‐life, little to no accumulation was observed with once daily administration, whereas the accumulation ratio for twice daily dosing was approximately 1.5. No evidence of autoinduction or inhibition was observed, and steady‐state appeared to be reached by Day 3 based on lack of changes in predose concentrations.
Table 3.
Summary of PRN1008 pharmacokinetic parameters on day 10 by dose multiple oral doses in healthy volunteers (Part B)
| Dose regimen | Tmax (h) | Cmax (ng ml–1) | AUC0‐tau (ng.h ml–1 ) | Half‐life (h) | Accumulation Ratio | |
|---|---|---|---|---|---|---|
| 300 mg QD (N = 8) | Mean | 172 | 434 | 4.0 | 0.90 | |
| Median | 1.5 | 172 | 391 | 4.1 | 0.84 | |
| CV% | 25 | 52 | 52 | 49 | 36 | |
| 95% CI | 97.5–247 | 246–621 | 2.38–5.64 | 0.632–1.17 | ||
| 600 mg QD (N = 8) | Mean | 313 | 936 | 3.8 | 1.5 | |
| Median | 1.5 | 281 | 755 | 3.8 | 1.4 | |
| CV% | 46 | 35 | 39 | 13 | 57 | |
| 95% CI | 222–403 | 630–1240 | 3.41–4.23 | 0.801–2.25 | ||
| 300 mg BID (N = 8) | Mean | 223 | 477 | 4.0 | 1.8 | |
| Median | 1.0 | 209 | 442 | 4.5 | 1.6 | |
| CV% | 43 | 42 | 47 | 35 | 40 | |
| 95% CI | 146–300 | 290–664 | 2.84–5.19 | 1.22–2.43 | ||
| 450 mg BID (N = 8) | Mean | 251 | 680 | 4.5 | 1.5 | |
| Median | 1.3 | 213 | 544 | 5.1 | 1.4 | |
| CV% | 32 | 54 | 65 | 36 | 32 | |
| 95% CI | 138–364 | 309–1050 | 3.13–5.86 | 1.12–1.94 |
Accumulation Ratio: AUC0‐tau Day 10/AUC0‐tau Day 1
Tmax, time to maximum plasma concentration; Cmax, maximum plasma concentration; AUC, area under the concentration–time curve; CV, coefficient of variation; CI, confidence interval; QD = every 24 h; BID = every 12 h
PD results
Occupancy of BTK by PRN1008 in Part A occurred rapidly at doses of 150 mg and above, in a dose‐dependent manner, with the maximum occupancy observed within the first 4 h (first measured time point). As shown in Table 4, near complete occupancy of BTK was observed after 4 h with the 600 and 1200 mg doses, with a mean (standard deviation) 92% (3%), with very low interpatient variability. A dose of 50 mg resulted in little to no occupancy (< 20%), approximating the lower limit of quantitation of the assay. The observed occupancy for both 1200 and 600 mg was very similar, which is consistent with the similar plasma exposures for these two dose levels. Because the first BTK occupancy timepoint was measured at 4 h, and the Cmax of PRN1008 was typically <2 h, it may be that maximal BTK occupancy was underestimated due to the sampling timepoints.
Table 4.
Occupancy of Bruton's tyrosine kinase (BTK) following single and multiple oral doses of PRN1008
| Part A Dose | PRN1008% BTK Occupancy (Mean ± SD)** | |||||||
|---|---|---|---|---|---|---|---|---|
| Day 1 (4 h) | Day 1 (12 h) | Day 1 (24 h) | ||||||
| 50 mg | 18 ± 7.9 | 18 ± 12 | 12 ± 5.5 | |||||
| 150 mg | 55 ± 15 | 33 ± 7.1 | 21 ± 17 | |||||
| 300 mg | 75 ± 7.3 | 61 ± 9.6 | 45 ± 14 | |||||
| 600 mg | 92 ± 3.2 | 79 ± 9.7 | 57 ± 17 | |||||
| 1200 mg | 92 ± 3.0 | 83 ± 6.9 | 63 ± 14 | |||||
| Part B Dose | PRN1008% BTK Occupancy (Mean ± SD)** | |||||||
|---|---|---|---|---|---|---|---|---|
| Day 1 (4 h) | Day 1 (12 h) | Day 1 (24 h) | Day 3 (24 h) | Day 10 (Predose) | Day 10 (4 h^) | Day 10 (12 h) | Day 10 (24 h) | |
| 300 mg QD | 79 ± 7.7 | 57 ± 8.8 | 31 ± 18 | 28 ± 15 | 29 ± 24.6 | 84 ± 4.4 | 62 ± 12.7 | 27 ± 19.4 |
| 300 mg BID | 78 ± 8.6 | 59 ± 18 | 62 ± 18 | 73 ± 12 | 74 ± 4.2 | 91 ± 2.3 | 77 ± 6.7 | 60 ± 8.3 |
| 600 mg QD | 90 ± 5.8 | 78 ± 7.2 | 60 ± 9.9 | 58 ± 13 | 58 ± 11.4 | 93 ± 2.0 | 80 ± 5.6 | 59 ± 11.1 |
| 450 mg BID | 81 ± 7.4 | 73 ± 8.7 | 75 ± 8.9 | 77 ± 7.8 | 76 ± 7.4 | 91 ± 4.6 | 75 ± 8.4 | 54 ± 15.1 |
SD, standard deviation; QD = every 24 h; BID = every 12 h
The rate of occupancy decline after single doses of PRN1008 was very consistent across cohorts, with all doses exhibiting a ~30–35% reduction in occupancy between hours 4 and 24. The ‘decay rate’ in occupancy over time, as determined by linear regression of all subjects by cohort, for dose levels of 150 mg and above, was also very consistent, with a mean value of approximately –1.6% h–1 (range – 1.5 to –1.7%; Figure 2).
Figure 2.
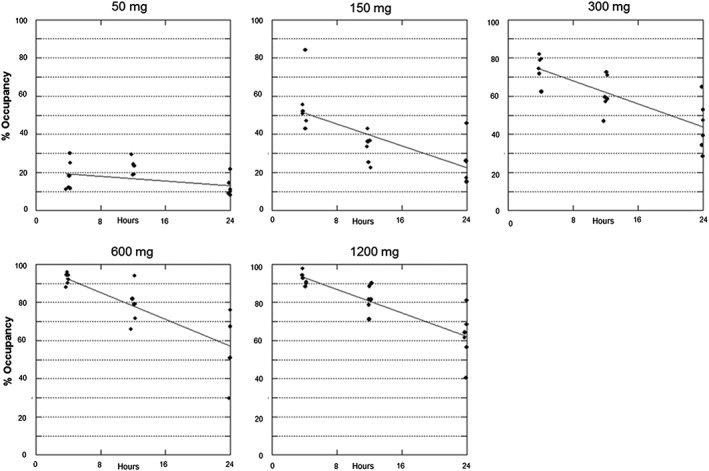
Individual BTK occupancy by PRN1008 dose level (Part A). Solid line represents fit of a linear regression model to estimate loss of occupancy over time
BTK occupancy for Part B is also shown in Table 4. Day 1 occupancy by dose in Part B was very similar to the observed results for single doses administered in Part A.
With multiple dosing, steady‐state occupancy levels were observed by Day 3, exemplified by stable predose occupancy levels thereafter.
With multiple doses of PRN1008, twice daily dosing regimens resulted in higher predose occupancy compared to the same total daily dose administered once daily, as expected. Both the 300 mg twice daily and the 600 mg once daily regimens achieved >90% occupancy 4 h after dosing, whereas the predose occupancy was 77% for the twice daily regimen and 59% for the once daily regimen, respectively. At dose levels which achieved <90% occupancy following the first dose (450 and 300 mg), some accumulation in measured occupancy was observed with multiple dosing, which was probably a result of subsequent doses being administered when a portion of the BTK enzyme remains occupied by PRN1008 from prior doses.
It is interesting to note that, despite the moderate variability in PK observed with PRN1008, variability in BTK occupancy was low. This may be due, at least in part, to many subjects achieving near maximal occupancy with multiple dosing, and a low variability in BTK turnover between subjects.
PK/PD results
Despite rapid clearance of PRN1008, occupancy of BTK was sustained, which is consistent with a slow target‐off rate. Figure 3 illustrates the relationship between PRN1008 plasma concentrations, and BTK occupancy.
Figure 3.
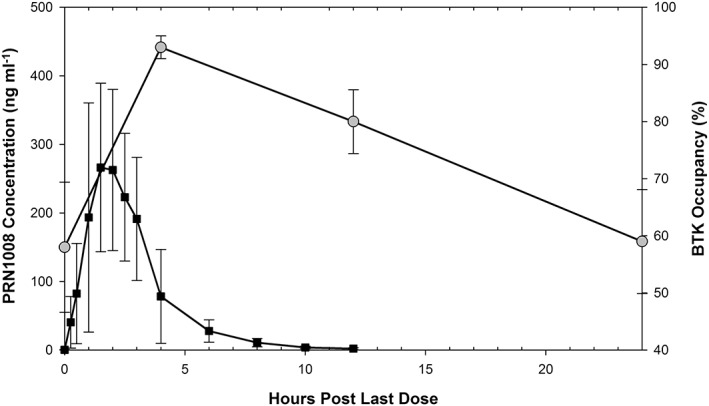
Duration of BTK occupancy (squares) in relation to the plasma concentration profile of PRN1008 (circles), following final dose on day 10 of a 600 mg once daily dosing regimen in the multiple ascending dose study
Cmax was highly correlated with the magnitude of BTK occupancy 4 h post‐PRN1008 dosing (Figure 4). A sigmoidal Emax model, with a fitted γ and E0 provided the best fit of PRN1008 Cmax vs. occupancy data. The parameter estimates (% coefficient of variation) were: Emax 76.7 (13) %; EC50 46.5 (15) ng ml–1; E0 17.8 (42) %, and γ 2.1 (31). These results suggest a fairly steep exposure–response relationship, with low variability, and with 80% of maximal occupancy achieved at Cmax concentrations of approximately 100 ng ml–1 and above. The E0 value of 17.8% is consistent with the lower quantifiable range of the assay.
Figure 4.
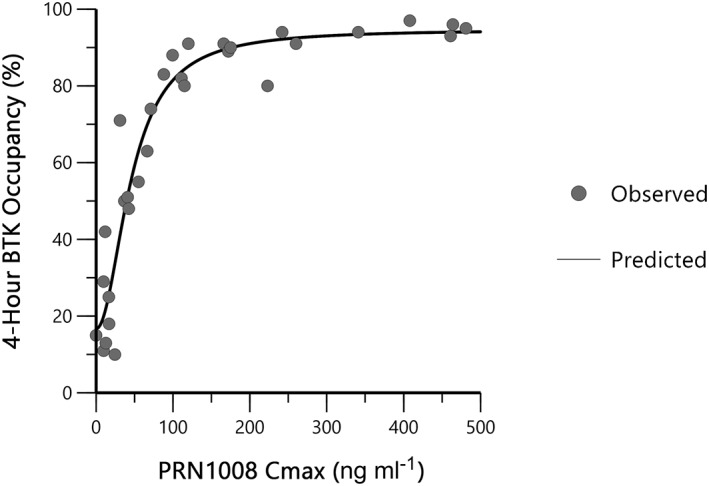
Exposure–response relationship between 4‐hour BTK occupancy and PRN1008 maximum observed concentration (Part A)
The robust relationship between PRN1008 Cmax and 4‐h occupancy, along with the very consistent occupancy decay rate of ~1.6% h–1 should allow for design of dosing regimens (dose and dose intervals) which precisely target various levels of occupancy over the course of a dose interval. It is noted that the nature of the PK and PK/PD relationships of PRN1008 may differ between healthy volunteers and patient populations, and should be further evaluated in future studies in patients.
Safety results
Data from all 80 enrolled subjects who received study drug (either PRN1008 or placebo) were included in the safety population. All participants were assessed for AEs for the duration of the study. No serious AEs or deaths were reported during the study, and no participants discontinued treatment due to an AE in either Part A or Part B.
At PRN1008 single doses of 50 to 600 mg, safety and tolerability was similar to placebo. In these four cohorts, there was only one subject of the six treated in each cohort who experienced a treatment‐emergent AE (TEAE). Of these four TEAEs, only one was considered related to study drug (nausea in Cohort A4), and one was graded as moderate (toothache in Cohort A2, unrelated to study drug). This compares with two TEAEs reported in two of the 10 subjects who received placebo (both graded as mild, not drug related).
In Cohort A5 (1200 mg), the primary AEs observed were gastrointestinal (GI) in nature, and were reported by each of the six subjects receiving PRN1008. The drug‐related AEs included diarrhoea (coded as loose stools; n = 6, 3 mild and 3 moderate severity), nausea (n = 3, 2 mild and 1 moderate severity), vomiting (n = 1), throat irritation (n = 3), and oropharyngeal discomfort (n = 1).
There was no apparent relationship between GI AEs and PRN1008 pharmacokinetics. As described above, both Cmax and area under the concentration–time curve for PRN1008 were similar at the 600 mg and 1200 mg doses. Despite similar plasma PK, the increase in GI AEs for the 600 mg vs. 1200 mg dose levels would suggest a localized effect related to total administered dose, and not to plasma exposure.
Following 10 or 11 days of dosing, PRN1008 was generally safe and well tolerated. TEAEs were reported in 7/8, 4/8, 8/8, 7/8 and 4/8 subjects in the 300 mg QD, 300 mg twice daily (BID), 600 mg QD, 450 mg BID and placebo groups, respectively. TEAEs classified as treatment‐related appeared to be more frequent in PRN1008 receiving subjects, reported in 6/8, 3/8, 8/8, 6/8 and 1/8 subjects in the 300 mg QD, 300 mg BID, 600 mg QD, 450 mg BID and placebo groups, respectively. All TEAEs classified as related to study drug were mild in intensity, with the exception of one subject in the 450 mg BID cohort who reported moderate diarrhoea.
There were no clinically significant or dose‐dependent changes observed in haematology, biochemistry or coagulation laboratory parameters in either Part A or Part B of the study. Similarly, no clinically significant changes were observed in vital signs, ECGs or urinalysis evaluations.
Discussion
PRN1008 was safe and well tolerated when administered orally as a liquid formulation to healthy volunteers in single doses up to 1200 mg, and over 10 days of multiple dosing. The most common AEs were GI in nature, and included nausea/vomiting and diarrhoea, which were observed most frequently in the highest dose levels. In particular, the 1200 mg single oral dose (Part A) was associated with more GI‐related AEs compared to lower doses administered in either both Part A or Part B. In addition, due to the plateau in PRN1008 exposure observed in this study, plasma exposures in the 1200 mg dose were similar to plasma exposures following a 600 mg dose. In Part B, tolerability was similar when 300 mg doses were given twice daily compared to once daily. These results suggest that the GI‐related adverse effects of liquid PRN1008 are likely to be a result of a local process driven by the total oral dose administered, and not driven by plasma exposures or by frequency of dosing. It is anticipated that administering PRN1008 via a solid dosage form will improve the GI tolerability of PRN1008 due to the nature of the liquid formulation.
PRN1008 when administered as a liquid formulation exhibited rapid absorption, moderate pharmacokinetic variability and a short plasma half‐life. Near complete occupancy of the BTK enzyme in PBMCs was achieved with the higher doses of PRN1008. Consistent with its reversible covalent chemistry, PRN1008 demonstrated a prolonged duration of occupancy despite the short plasma half‐life (anticlockwise hysteresis). Consistent with its slow target off‐rate mechanism, BTK occupancy in PBMCs was sustained despite the rapid plasma clearance of PRN1008, which is in contrast to reversible inhibitors such as dasatinib where BTK occupancy declines rapidly when drug is removed 15. It is hypothesized that this hit and run approach to engaging the target with a slow‐off rate compound with rapid plasma clearance, may lead to effective drugs which are potentially safer via limiting systemic exposure and reducing off‐target effects 12.
Typically, PD variability is greater than PK variability. This was, however, not observed for PRN1008, as the variability in PD as measured by BTK occupancy was quite low. This low variability in occupancy may represent the rate of BTK turnover, rather than the off‐rate of the PRN1008 from the target. This could potentially explain the low observed variability stemming from the biological process of BTK turnover, rather than being a drug‐related phenomenon. In vitro, Koff for PRN1008 is slow, and may be longer than the turnover rate of BTK in PBMCs. While the turnover rate of BTK in humans has not been fully characterized to our knowledge, Evans and colleagues 16 reported a BTK synthesis half‐life of approximately 48–72 h following administration of the irreversible BTK inhibitor CC‐292. In the current study, BTK occupancy recovered towards baseline at a rate of approximately 1.6% (range 1.5–1.7%) h–1 after PRN1008 dosing, which is generally consistent with the prior findings.
The maximum BTK occupancy following PRN1008 dosing was closely correlated with Cmax concentrations, and followed an Emax relationship. Maximal occupancy was achieved at Cmax concentrations of greater than 100 ng ml–1, suggesting that higher doses or higher Cmax concentrations would not result in additional benefit. In designing PRN1008 dosing regimens for study in therapeutic trials, these characteristics should allow for the selection of doses to achieve Cmax concentrations associated with complete or near complete BTK occupancy, with the dosing interval selected based on the desired occupancy level at minimum plasma concentration. Based on the current results with the liquid formulation, formulation‐equivalent doses >300 mg twice daily are unlikely to produce additional benefit in terms of occupancy. This assumes that BTK occupancy, a measure of target engagement, also translates into alteration of BTK function and clinical efficacy.
How much BTK occupancy is required for clinical efficacy in human trials for conditions such as rheumatoid arthritis and other inflammatory and autoimmune diseases is currently unknown. In CIA rodent models with PRN1008, predose BTK occupancy levels of approximately 50% were associated with significant improvements in disease scores, whereas predose levels of approximately 70% resulted in reversal and normalization of disease, similar to corticosteroid controls 17. Future clinical trials will evaluate dose regimens designed to achieve specific occupancy targets, with the goal of exploring the link between occupancy and efficacy.
In summary, PRN1008 in this phase 1 healthy volunteer study has demonstrated acceptable safety, tolerability, pharmacokinetics, and BTK occupancy to warrant continued clinical development. Proof of target engagement, and characterization of the PK/PD relationship suggests that PRN1008 administered once or twice daily may be effective in inflammatory and autoimmune diseases. By design using reversible covalent chemistry, BTK occupancy was shown to be prolonged long after plasma concentrations of PRN1008 reached the lower limit of detection. This study represents the first demonstration that the goal of achieving low plasma exposures for a short duration, while maintaining target engagement is feasible. This approach allowed for rapid characterization of PK/PD in healthy volunteers, and will facilitate clinical proof of concept studies in patients, whereby doses can be chosen based to evaluate clinical activity of specific levels of target BTK occupancy (e.g. 50, 70, 90%).
Competing Interests
This work was funded by Principia Biopharma.
This manuscript is written in memory of Dr Jens Oliver Funk, former Chief Scientific Officer of Principia Biopharma.
These results, in part, were presented previously in abstract form at the 2015 American College of Rheumatology Annual Meeting, San Francisco, CA, and the 2015 European League Against Rheumatism Annual Meeting, Rome Italy.
Smith, P. F. , Krishnarajah, J. , Nunn, P. A. , Hill, R. J. , Karr, D. , Tam, D. , Masjedizadeh, M. , Funk, J. O. , and Gourlay, S. G. (2017) A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton's tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol, 83: 2367–2376. doi: 10.1111/bcp.13351.
References
- 1. Crofford LJ, Nyhoff LE, Sheehan JH, Kendall PL. The role of Bruton's tyrosine kinase in autoimmunity and implications for therapy. Expert Rev Clin Immunol 2016; 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, et al. Specific Btk inhibition suppresses B cell‐ and myeloid cell‐mediated arthritis. Nat Chem Biol 2011; 7: 41–50. [DOI] [PubMed] [Google Scholar]
- 3. Lopez‐Herrera G, Vargas‐Hernandez A, Gonzalez‐Serrano ME, Berron‐Ruiz L, Rodriguez‐Alba JC, Espinosa‐Rosales F, et al. Bruton's tyrosine kinase–an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol 2014; 95: 243–250. [DOI] [PubMed] [Google Scholar]
- 4. McAtee CP. The many faces of Bruton's tyrosine kinase. Nat Biotechnol 2012; 30: 394. [DOI] [PubMed] [Google Scholar]
- 5. Pan Z. Bruton's tyrosine kinase as a drug discovery target. Drug News Perspect 2008; 21: 357–362. [DOI] [PubMed] [Google Scholar]
- 6. Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther 2011; 13: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 blocks B‐cell activation and is efficacious in models of autoimmune disease and B‐cell malignancy. Proc Natl Acad Sci U S A 2010; 107: 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu D, Kim Y, Postelnek J, Vu MD, Hu DQ, Liao C, et al. RN486, a selective Bruton's tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J Pharmacol Exp Ther 2012; 341: 90–103. [DOI] [PubMed] [Google Scholar]
- 9. Tan SL, Liao C, Lucas MC, Stevenson C, DeMartino JA. Targeting the SYK‐BTK axis for the treatment of immunological and hematological disorders: recent progress and therapeutic perspectives. Pharmacol Ther 2013; 138: 294–309. [DOI] [PubMed] [Google Scholar]
- 10. Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end‐organ disease in murine lupus. Arthritis Res Ther 2012; 14: R243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rankin AL, Seth N, Keegan S, Andreyeva T, Cook TA, Edmonds J, et al. Selective inhibition of BTK prevents murine lupus and antibody‐mediated glomerulonephritis. J Immunol 2013; 191: 4540–4550. [DOI] [PubMed] [Google Scholar]
- 12. Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol 2015; 11: 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hantschel O, Rix U, Schmidt U, Burckstummer T, Kneidinger M, Schutze G, et al. The Btk tyrosine kinase is a major target of the Bcr‐Abl inhibitor dasatinib. Proc Natl Acad Sci U S A 2007; 104: 13283–13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, et al. Inhibition of Btk with CC‐292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther 2013; 346: 219–228. [DOI] [PubMed] [Google Scholar]
- 17. Hill RJ, Bradshaw JM, Bisconte A, Tam D, Owens TD, Brameld KA, et al. Preclinical characterization of PRN1008, a novel reversible covalent inhibitor of BTK that shows efficacy in a RAT model of collagen‐induced arthritis. Ann Rheum Dis 2015; 74 (Suppl 2): 216. [Google Scholar]