

Abstract
The initial treatment of haemodynamically stable patients with pulmonary embolism (PE) has dramatically changed since the introduction of low molecular weight heparins (LMWHs). With the recent discovery of the direct oral anticoagulant drugs (DOACs), initial treatment of PE will be simplified even further. In several large clinical trials it has been demonstrated that DOACs are not inferior to standard therapy for the initial treatment of PE, and because of their practicability they are becoming the agents of first choice. However, many relative contraindications to DOACs were exclusion criteria in the clinical trials. Therefore, LMWHs will continue to play an important role in initial PE treatment and in some cases there still is a role for unfractionated heparin (UFH). In this review we will give an overview of the biophysical, pharmacokinetic and pharmacodynamic properties of anticoagulants currently available for the initial management of PE. In addition, we will provide a comprehensive overview of the indications for the use of UFH, LMWHs and DOACs in the initial management of PE from a pharmacokinetic/‐dynamic point of view.
Keywords: anticoagulation, DOAC, LMWH, pulmonary embolism, unfractionated heparin
Introduction
Untreated acute pulmonary embolism (PE) is associated with a mortality rate of up to 25% 1, and anticoagulation has been the backbone of PE treatment for decades. The goal of treatment is to reduce mortality by prevention of thrombus extension, embolization and/or formation of new thrombi. In the only randomized controlled clinical trial performed, anticoagulation decreased mortality in patients with pulmonary embolism 1, and subsequent uncontrolled trials have confirmed this finding 2, 3, 4. Although both unfractionated heparin (UFH) and vitamin K antagonists (VKAs) have been used as anticoagulants for many decades, it was not until 1990 that it was demonstrated that early initial administration of heparin is essential for survival 5, and that a 5‐day course of UFH is as effective as the formerly applied 10 days 6. Because of pharmacokinetic and biological limitations of UFH, low‐molecular‐weight heparins (LMWHs) and the indirect factor Xa (FXa) inhibitor fondaparinux have been developed and have greatly simplified the initial management of PE 7. With persisting limitations in terms of usability of LMWHs, fondaparinux and VKAs, direct oral anticoagulants (DOACs) have been developed recently 8. This new class of oral anticoagulants simplifies initial treatment, prophylaxis and long‐term management of PE even further as they are administered in fixed doses without any need for laboratory coagulation monitoring 8. However, although the beneficial effects of LMWH and fondaparinux over UFH are clear, and there is increasing evidence that DOACs have, in most patients, a similar effect on the prevention of recurrent PE as LMWHs/fondaparinux, there are still indications for the primary use of UFH in the initial management of PE based on its pharmacokinetic and pharmacodynamic properties. In this review we will translate insights in the pharmacokinetic, pharmacodynamic and relevant off‐target properties into a better understanding of the use of the old and new anticoagulants currently available for the initial management of PE.
Biophysical and pharmacokinetic properties
A schematic overview of the biophysical and pharmacokinetic properties of UFH, LMWHs, fondaparinux and DOACs is presented in Table 1.
Table 1.
Comparative pharmacokinetics
| UFH | LMWH | Fondaparinux | FII inhibitor | FXa inhibitors | |||
|---|---|---|---|---|---|---|---|
| Dabigatran | Rivaroxaban | Apixaban | Edoxaban | ||||
| Route of administration | iv | sc | sc | Oral | Oral | Oral | Oral |
| Molecular weight | 3–30 kDa | 5000 Da | 1726 Da | 627 Da | 435 Da | 459 Da | 548 Da |
| Predictable pharmacokinetics | No | Yes | Yes | Yes | Yes | Yes | Yes |
| t max (h) | Minutes | 4–6 | 2–4 | 1–3 | 2–4 | 3–4 | 1–2 |
| t 1/2 (h) | 0.5–1.5 | 3–6 | 17–21 | 12–17 | 5–13 | 9–14 | 10–14 |
| Bioavailability (%) | 100 | >90 | 100 | 3–10 | >80 | 50 | 62 |
| Volume of distribution (l kg −1 ) | 0.07 | 0.04–0.06 | 0.1–0.2 | 0.8–1 | 0.71 | 0.3 | 0.77 |
| Plasma protein binding (%) | >90 | >90 | >97 | 35 | 92–95 | 87 | 55 |
| Renal elimination (%) | Only in high dose | >80 | >80 | 80 | 33 | 27 | 50 |
| CYP metabolism (%) | None | None | None | None | 66 | 25 | <4 |
| P‐gp | None | None | None | Yes | Yes | Yes | Yes |
| Risk of HIT | Yes | Low | None | None | None | None | None |
| Pregnancy | Not‐contraindicated | Not‐contraindicated | Unknown/contraindicated | Unknown/contraindicated | Unknown/contraindicated | Unknown/contraindicated | Unknown/contraindicated |
| Reversal agent | Protamin | Protamin (partly) | Not available | Idarucizumab | Not available | Not available | Not available |
FXa, factor Xa; HIT, heparin‐induced thrombocytopenia; iv, intravenously; LMWH, low molecular weight heparin; NA, not applicable; p‐gp, p‐glycoprotein (for relevant drug interactions, see the interaction table in the online supplement); sc, subcutaneously; t 1/2, half‐life; t max, time to maximum concentration; UFH, unfractionated heparin
UFH
In contrast to LMWHs, fondaparinux and DOACs, UFH does not have predictable pharmacokinetics. UFH is a glycosaminoglycan that consists of a heterogeneous mixture of polysaccharide chains with alternating residues of D‐glucosamin and uronic acid, or glucoronic acid, or iduronic acid. The molecular weight ranges from about 3000 to 30 000 Da. UFH does not distribute into muscle or fat tissue, giving it a small volume of distribution (Vd) of 0.07 l kg−1 9 with a relatively short half‐life (about 0.5–1 h) 10. The half life of UFH is not only very variable due to its earlier described heterogeneity 11, but also due to its two‐phased and dose‐dependent elimination (the half‐life increases with increasing dose) 12. The rapid, saturable elimination phase is thought to reflect UFH binding to vascular endothelial cells, macrophages and reticuloendothelial cells 13, 14, 15, 16, 17, where it is internalized, depolymerized and metabolized into smaller and less sulphated forms 17, 18, 19. The slower phase corresponds to renal clearance. At low doses (less than 1000 IU), heparin is cleared mainly by the highly efficient saturable mechanism 18. At higher doses, the cellular binding sites are saturated, and heparin is cleared predominantly by renal elimination 12, 18. Another reason for the unpredictable pharmacokinetics of UFH is its binding to a number of endogenous plasma proteins including histidine‐rich glycoprotein (HRGP), platelet factor 4 (PF4), vitronectin, fibronectin and von Willebrand factor (vWF) 20. Binding of UFH to plasma proteins reduces its anticoagulant activity because less UFH is available for interaction with antithrombin, and the unpredictable anticoagulant response reflects the wide variability in plasma concentrations of heparin‐binding proteins. Some of these heparin‐binding proteins are acute phase reactants, the concentration of which may increase in patients, whereas others like PF4 and vWF are released during the clotting process. Non‐pharmacokinetic factors add to the unpredictable therapeutic effect of UFH: there is a high‐ and low‐affinity moiety for binding to antithrombin (see also the ‘mechanism of action’ section below), and the high‐affinity moiety has a longer half‐life than its low‐affinity counterpart 11. Because of its heterogeneity (with varying high‐ and low‐affinity moieties), half‐lives will vary. Because of the unpredictable anticoagulant response, careful/close monitoring is essential when UFH is given in therapeutic doses.
LMWH
LMWHs are fragments of UFH produced by controlled enzymatic or chemical depolymerization processes that yield chains with a mean molecular weight of about 5000 Da 21. The indirect factor Xa inhibitor fondaparinux is a synthetic analogue of the unique pentasaccharide that mediates the anticoagulant activity of both UFH and LMWHs 22. Because LMWHs do not bind to endothelial cells, macrophages or reticuloendothelial cells, the plasma half‐life is 2–4 times longer than that of UFH (3–6 vs. 0.5–1.5 h respectively) 21. Fondaparinux has an even longer half‐life of 17–21 h. In addition, because LMWHs and fondaparinux have much lower affinity for heparin‐binding plasma proteins and are mainly removed by non‐saturable renal filtration, their clearance is independent of dose and plasma concentration 22. Moreover, in contrast to UFH and LMWHs, fondaparinux rarely causes heparin‐induced thrombocytopenia (HIT), because fondaparinux does not bind PF4 (of which neo‐epitopes are recognized by HIT‐inducing antibodies) 23. However, fondaparinux is unlicensed for treatment in HIT because on rare occasions fondaparinux can cause a disorder resembling HIT 24, for which the underlying mechanism remains to be elucidated.
Of note, obese patients clear LMWHs faster than non‐obese patients due to hyperfiltration, and because LMWHs are hydrophilic, one might expect that the volume of distribution of LMWHs is not that much increased in obese patients. However, LMWHs are not dosed on lean or adjusted body weight but on total body weight. This is based on three small studies that demonstrated that the use of total body weight is as appropriate as adjusted body weight: both total body weight and adjusted body weight provided a moderate correlation with volume of distribution and clearance (a poor correlation was seen with lean body weight) 25, and mean anti‐factor Xa activity was equal in obese and non‐obese patients when dosed on total body weight 26, 27.
DOACs
DOACs are small synthetic molecules with a molecular weight ranging from 430 to 670 Da. They are either direct thrombin inhibitors or factor Xa (FXa) inhibitors. Dabigatran etexilate is the only approved oral direct thrombin inhibitor, and rivaroxaban, apixaban and edoxaban are oral FXa inhibitors. From a pharmacokinetic point of view, there are several differences in terms of bioavailability, plasma protein binding, metabolism with or without cytochrome (CYP)450 and/or P‐glycoprotein (P‐gp) handling, and mechanisms of elimination (see Table 1 for details). Dabigatran etexilate has a very low bioavailability ranging from 3–10%, in which Pg‐p handling plays an important role. Because of relatively large uptake variability, unpredictable interindividual differences in dabigatran plasma levels can occur, although it seems that this does not affect its clinical activity in the majority of patients 28, 29. In a small subset of patients this variability in plasma levels can be clinically relevant which implies that monitoring might be useful nevertheless, especially in patients ‘at risk’; for example, the elderly, patients with impaired renal function or obese patients 30, 31, 32, 33, 34, 35. Patients with (sub)total gastrectomy or gastric bypass surgery should rather avoid dabigatran or use it with caution. Because dabigatran is a P‐gp substrate, there are several drug interactions (for an overview of drug interactions, see supplementary Table S1 in the online supplement). Dabigatran is the only DOAC not metabolized by the liver, and therefore does not have CYP450 drug–drug interactions. Rivaroxaban, apixaban and edoxaban are all, at least to some extent, CYP450 and P‐gp substrates, potentially leading to drug interaction (for an overview of drug interactions, see supplementary Table S1 in the online supplement) 36. In addition, high dosed rivaroxaban (15 or 20 mg daily) must be taken with food because of higher bioavailability (from 66% to more than 80%) 37. The other DOACs do not have this requirement 38, 39. In crushed form, apixaban and rivaroxaban have similar bioavailability and therefore can be administered via a nasogastric tube 40, 41. Theoretically, DOACs could be used for treatment in HIT as they do not bind PF4, although this would be off‐label use.
Mechanism of action
A schematic representation of mechanisms of action of anticoagulation therapies is depicted in Figure 1. In summary, activated factor X (aFX) activates thrombin (factor II) which activates conversion of fibrinogen to fibrin. Both unfractionated heparin, low‐molecular‐weight heparins and fondaparinux exert their anticoagulant activity by inhibiting thrombin‐activated conversion of fibrinogen to fibrin 7, 42: binding of a unique pentasaccharide to antithrombin causes a conformational change in antithrombin that accelerates its interaction with thrombin and FXa by about 1000 times. Binding of the pentasaccharide to antithrombin results directly in inhibition of FXa, whereas inhibition of thrombin also requires binding by at least 12 saccharide units. The pentasaccharide also blocks the activation of factor IX and neutralizes aFX by activating factor X inhibitor. Fondaparinux is a synthetic analogue of this unique polysaccharide, but for UFH and LMWHs this sequence is randomly distributed along the heparin chains. Approximately one third of the chains of unfractionated heparin, and about 15–25% of the chains of LMWHs, contain this pentasaccharide sequence. Unlike UFH, which has equivalent activity against factor Xa and thrombin, LMWHs and fondaparinux have greater activity against factor Xa. This can be explained by the fact that long heparin chains can inhibit both factor IIa and Xa, while shorter chains only inhibit factor Xa 43, 44, 45. The effects of UFH, when used in pulmonary embolism, may not be fully explained by its anticoagulant actions: there is some evidence that UFH decreases bronchospasm and vasospasm associated with pulmonary embolism 46, 47. It is hypothesized that these effects result from the inhibition of serotonin release from platelets 47, 48, 49, which may be of additional value in patients with intermediate risk PE (see below). The DOACs are direct inhibitors of factor II or X (both serine proteases), thereby preventing thrombin‐activated conversion of fibrinogen to fibrin 50. Dabigatran is a potent, competitive, reversible direct thrombin inhibitor. Dabigatran also inhibits free thrombin, fibrin‐bound thrombin and thrombin‐induced platelet aggregation. Apixaban, rivaroxaban and edoxaban are highly selective, direct and reversible factor Xa inhibitors. Inhibition of factor Xa interrupts the intrinsic and extrinsic pathway of the blood coagulation cascade, inhibiting both thrombin formation and development of thrombi. The factor Xa inhibitors do not inhibit thrombin (activated factor II), and no direct effects on platelets have been demonstrated.
Figure 1.
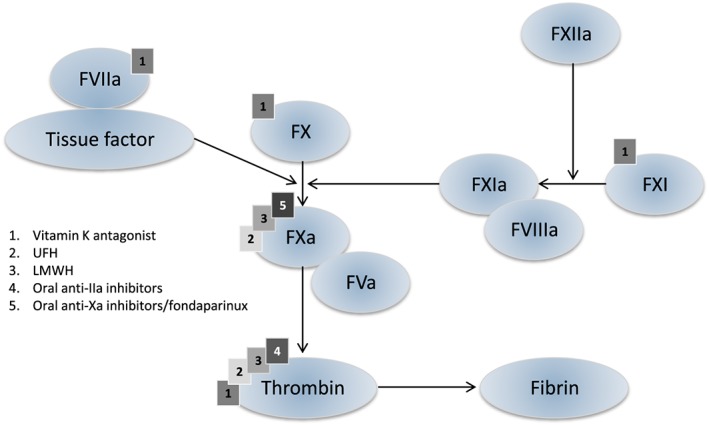
Schematic representation of the coagulation cascade and coagulation factors which are inhibited by different anticoagulation therapies UFH, unfractionated heparin; LMWH, low molecular weight heparin; DOAC, direct oral anticoagulatory drugs
Clinical implications of pharmacokinetic and ‐dynamic properties for treatment of acute PE
General
A schematic overview of the initial treatment of PE is depicted in Figure 2. In contrast to thrombolysis, there is no proof that UFH or LMWHs/fondaparinux or DOACs have a direct effect on preformed thrombi, but the discontinuation of the coagulation cascade will facilitate endogenous fibrinolysis and thereby the dissolution of the thrombus in the longer term 51. For the initial management, anticoagulants are protective by preventing further thrombus formation and subsequent thrombin‐mediated platelet aggregation. This interruption of fresh thrombus formation is crucial to short‐term PE prevention because recently formed thrombi are mechanically much more unstable and thus prone to detachment and embolization. Whether vasodilating and antibronchospastic effects of heparins in particular convey additional acute benefit has never been studied specifically.
Figure 2.
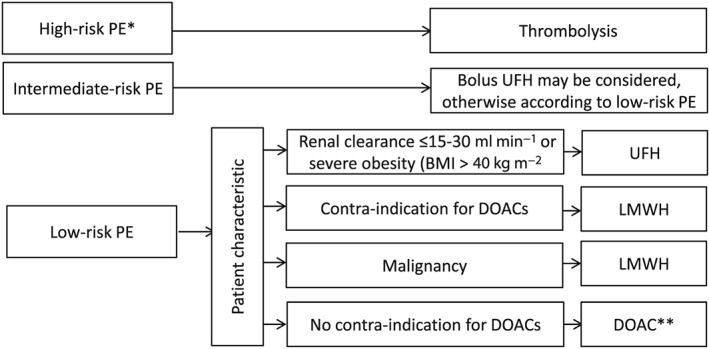
Initial pulmonary embolism treatment flowchart PE, pulmonary embolism; UFH, unfractionated heparin; LMWH, low molecular weight heparin; DOAC, direct oral anticoagulatory drugs; * haemodynamically unstable PE and/or respiratory depression; ** whether or not preceded by a short course of LMWH/fondaparinux administration. For an overview of contraindications see the online supplement
In haemodynamically unstable PE patients, thrombolysis is clearly indicated 52. Thrombolysis is usually not accompanied by concomitant anticoagulant therapy, but initiated subsequently 53. In haemodynamically stable intermediate risk PE patients, potential benefits of thrombolysis are offset by a significantly increased bleeding risk 54. In such patients, and all others at lower risk, anticoagulant therapy (with UFH, LMWH/fondaparinux or DOACs) instead of thrombolysis is recommended for the initial treatment of PE 42.
LMWHs and fondaparinux have been directly compared to UFH in a large number of trials. A recent Cochrane systematic review concluded that LMWHs/fondaparinux are preferred over (intravenous (iv) or subcutaneous (sc)) UFH for initial anticoagulation in patients with PE, as they result in fewer recurrent thromboembolic events, less major bleeding and lower mortality compared to UFH 55. In addition to these improved clinical outcomes, other advantages of LMWH or fondaparinux over UFH include more predictable pharmacokinetics with less inter‐individual variability in anticoagulant response to fixed doses, a longer plasma half‐life making once or twice daily administration possible, and a decreased likelihood of heparin‐induced thrombocytopenia 7. The preference for LMWH or subcutaneous fondaparinux should be guided by clinician familiarity, costs and availability because benefits and potential harms are similar 42. A fairly recent meta‐analysis showed that once daily LMWH is as effective and as safe as a twice daily regimen (2.2 vs. 2.9% major bleeds respectively in 1508 pooled patients), although PE patients were underrepresented 56.
Dabigatran, rivaroxaban, apixaban and edoxaban have been compared with conventional anticoagulant therapy for the treatment of acute symptomatic VTE, and compared with placebo and with VKAs for extended treatment 57, 58. In these large RCTs it was demonstrated that the DOACs are not inferior to initial LMWH therapy followed by VKA treatment in the prevention of recurrent VTE 57. Moreover, it was demonstrated that the DOACS have a lower all‐cause mortality driven primarily by a decrease in fatal intracranial bleeding risks 57, 59. Therefore, and because they are administered orally and do not need monitoring routinely, DOACs have become the agents of first choice in the treatment of acute and extended treatment of PE for most patients (fitting the clinical trial population profiles) 60. However, only rivaroxaban and apixaban are approved for the first days of treatment of PE, as dabigatran and edoxaban both require the initial treatment of LMWHs for several days. Of note, this approach is not based on the pharmacokinetic properties of the different DOACs (as they all have a similarly short t max, which is in the same range as LMWHs; see Table 1), but is guided by the different study designs in which non‐inferiority has been demonstrated. In addition, despite decreased all‐cause mortality, concerns have been raised about increased gastro‐intestinal bleeding risk for rivaroxaban, and high‐dose dabigatran and edoxaban, especially in the elderly 61, 62. The reason for this difference remains to be elucidated. For dabigatran it is suggested that the low bioavailability may cause bleeding via a luminal effect. The difference between apixaban and rivaroxaban may be explained by once vs. twice daily dosing respectively, leading to higher peak levels of rivaroxaban. However, it should be taken into account that there are no randomized clinical trials (RCTs) comparing one DOAC confirming the superiority of apixaban in this respect. Accordingly, it has been described that rivaroxaban may predispose to heavy menstrual bleeding (20–25%), which should be taken into account in premenopausal female patients 63, 64.
Another concern in clinical practice which may guide DOAC choice, is the availability of a reversal agent. For dabigatran, a recent study showed that idarucizumab (a humanized dabigatran‐specific (Fab) antibody fragment) is able to normalize coagulation tests within 10–30 min after administration without increased risk of thrombosis 65, and it has been approved for treatment of life‐threatening bleeding in dabigatran‐treated patients. Andexanet alfa (a recombinant modified human factor Xa decoy protein) was developed as a reversal agent for the FXa inhibitors. Recently it was demonstrated that andexanet alfa decreases factor Xa activity effectively in FXa inhibitor‐treated patients with life‐threatening bleeding, which resulted in good clinical haemostasis in 79% of patients 66. However, an increased risk of thrombosis was observed in patients treated with andexanet because of life‐threatening bleeding 66, and andexanet alfa is not yet registered for treatment of bleeding complications. Finally, Ciraparantag/PER977 is a small cationic molecule which has the potential to serve as an universal antidote because it binds direct Xa inhibitors, direct thrombin inhibitors, and unfractionated and LMWH through non‐covalent hydrogen bonds and charge–charge interactions.
Currently, plans for Phase III trials with edoxaban have been announced. Hence, with no currently available FXa inhibitor antidote and potential severe bleeding complications in FXa inhibitor‐treated patients, antifibrinolytic agents (e.g. tranexaminic acid) should be administered, and in the case of life‐threatening bleeding, four‐factor prothrombin complex concentrates (PCCs) should be administered. Importantly, evidence from randomized trials is lacking regarding these strategies. Treatment with PCCs is based on the ability of PCCs to attenuate bleeding or correct anticoagulation tests in preclinical models 67, 68, and as it carries a substantial prothrombotic risk, it should only be used in patients with life‐threatening bleeding. In addition, unabsorbed drug should be removed from the gastrointestinal tract by administration of oral‐activated charcoal if the last dose was recent enough that unabsorbed drug is likely to be present (apixaban within 6 h, dabigatran within 2 h, edoxaban within 2 h, rivaroxaban within 8 h). Of note, active dabigatran is the only DOAC that can be removed from the circulation by haemofiltration as the FXa inhibitors are highly protein bound.
Hence, the choice between the different DOACs should be guided by patient characteristics, pharmacokinetic/pharmacodynamic properties, and possible side effects (Figure 3). The first‐day treatment (whether or not preceded by a short course of LMWH/fondaparinux administration) should be adapted accordingly. Importantly, many relative contraindications to DOACs were exclusion criteria in the clinical trials (see the online supplement for an overview of contraindications). Therefore, LMWHs will continue to play an important role in initial PE treatment and in some cases there remains a role for UFH.
Figure 3.
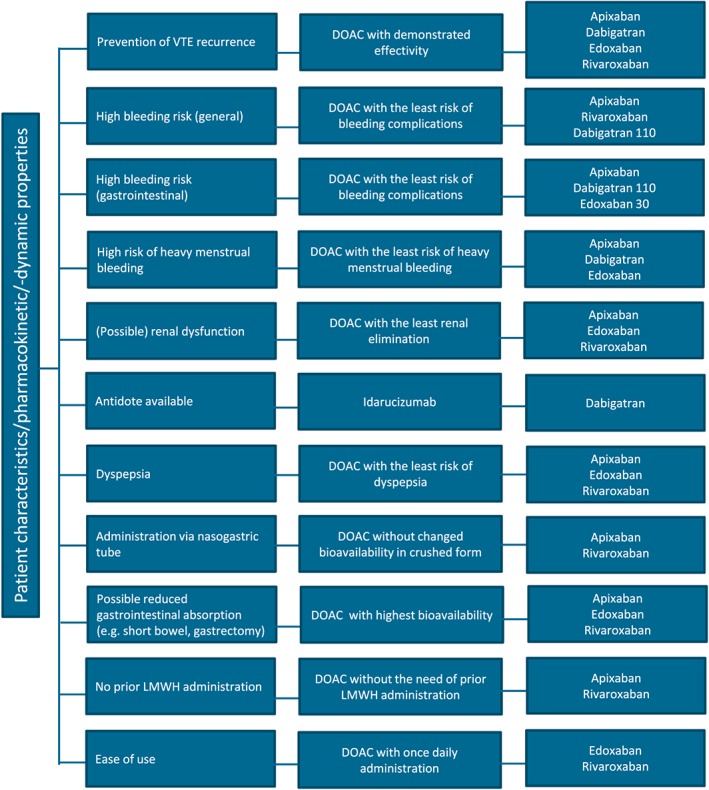
DOAC flowchart based on pharmacokinetic and pharmacodynamic properties and patient preferences
Indications for unfractionated heparin therapy
Despite the clear benefits of LMWHs/fondaparinux over UFH, and of DOACs over LMWHs/fondaparinux, UFH is not totally obsolete in the acute treatment of PE. The first two indications for iv UFH treatment are a creatinineclearance of ≤30 ml min−1 42, and severe obesity (BMI > 40 kg m−2) 69. The rationale is two‐fold. First, the efficacy of LMWHs, fondaparinux and DOACs in patients with acute PE and severe renal failure and/or severe obesity has not been well studied because most RCTs excluded such patients or failed to specify whether these patients were recruited 69, 70. Second, severe renal insufficiency and/or severe obesity alter the pharmacokinetics of the anticoagulants, requiring that activity be monitored. It is more convenient to monitor UFH than LMWH/fondaparinux/DOACs because activated partial thromboplastin time (aPTT) testing is generally more readily available and less expensive than anti‐Xa or anti‐IIa assays (dTT). Nevertheless, anti‐factor Xa assays are increasingly used to dose LMWH in patients with renal failure or severe obesity in clinical practice, although data regarding the correlation between anti‐factor Xa activity and the reduction of thromboembolic events or bleeding complications are limited. Of note, in patients with severe renal failure treated with enoxaparin, it was demonstrated that anti‐factor Xa levels poorly correlate with the occurrence of haemorrhage 71. Additional studies are clearly needed for specific dose guidance of anti‐factor Xa. Until then, in our opinion, anti‐factor Xa dose guidance should be cautiously applied. A third indication for IV UFH is the consideration of thrombolysis: because of the short half‐life of UFH, the patient is subjected to concomitant anticoagulant and thrombolytic activity for the shortest duration, during which the anticoagulant effect can be effectively interrupted with protamine.
Intermediate risk pulmonary embolism
Special concern applies to patients with intermediate risk PE 72. These patients are defined by acute right ventricular dysfunction and myocardial injury without overt haemodynamic compromise and are at a much higher risk for adverse outcome than patients with low risk PE 54, 73. In recent years several clinical trials aimed to reduce mortality in these patients by the use of thrombolysis 54, 74. However, the clear benefit of thrombolysis in terms of reduced PE‐related mortality were counterbalanced by the higher incidence of severe bleeding complications 54. Therefore, thrombolysis for these patients was ultimately not included in the clinical guidelines. Nevertheless, strategies to reduce mortality and morbidity in these patients are much needed. An initial bolus of UFH before the start of LMWHs/fondaparinux/DOACs could be considered for several reasons: firstly, intermediate risk PE patients are more likely to become haemodynamically unstable requiring secondary thrombolysis, which is contraindicated for use of anticoagulants with longer half‐lives. Secondly, with a t max ranging from 4–6 h for LMWHs, 2–3 h for fondaparinux, and 1–4 h for DOACs (Table 1), it seems rational from a pharmacokinetic point of view to start with an initial bolus of UFH in these patients in order to overcome the delay in onset of full anticoagulation. Unfortunately, the potential benefit of this approach has never been systematically studied. Finally, the earlier described preclinical evidence that UFH inhibits serotonin release from platelets associated with a decrease in vasospasm (and bronchospasm) may be of additional value in these patients (although large clinical trials investigating this approach are lacking). In all intermediate risk PE patients, despite the choice of initial therapy, careful initial monitoring is recommended for the first 24 h 72, 74.
Cancer‐related pulmonary embolism
LMWH therapy is indicated in patients with cancer‐related PE, not only for the initial treatment but also for the long‐term treatment. It was demonstrated in large RCTs that LMWH therapy in patients with cancer‐related PE has a beneficial effect on PE‐related morbidity and mortality compared to VKA therapy 75, 76, 77, 78, possibly because of a lower time in therapeutic range due to illness and co‐medication. It is also suggested that heparin/LMWH itself has anti‐neoplastic effects which have already been extensively reviewed elsewhere 79. Heparin‐induced inhibition of angiogenesis has, for example, been an area of intense research 80, 81, 82, 83. LMWHs were shown to inhibit proliferation of endothelial cells induced by VEGF 84, 85, 86. Furthermore, the ability of LMWHs to inhibit metastasis in animal models is substantially documented 80. Antimetastatic properties of LMWHs are likely due to interference with endothelial cell adhesion. The ability of heparins to interfere with selectin binding appears to be a major pathway for their anti‐metastatic properties 87, 88, 89, 90. The possible mechanism may also involve interaction with VLA‐4/VCAM‐1 91. The importance of selectins is emphasized by findings that the anti‐metastatic effect of heparins cannot be demonstrated in animals deficient in P‐ or L‐selectin 90, 92. Based on these preclinical data, several studies with LMWHs in cancer patients were conducted. However, because conflicting clinical data have been presented, there is at present no approved use for LMWHs in cancer patients without a need for VTE prophylaxis or treatment 93, 94, 95.
Clinical trials investigating the effects of DOACs were not aimed at oncologic patients, although these patients were not excluded in the majority of studies. A trend towards reduced recurrent VTE and bleeding in favour of DOACs was observed compared to VKA therapy, although this was only in a limited number of patients 96. DOAC vs. LMWH therapy for the treatment of cancer‐related PE is currently being investigated (e.g. SELECT‐D trial, CARAVAGGIO trial, CONCO‐11 trial, CASTA‐DIVA trial, CANVAS trial). Until these results are known, LMWH therapy remains the therapy of choice in these patients.
Further considerations
In addition to the improved usability and, consequently, improved quality of life, cost‐effectiveness of the DOACs will determine whether these agents will remain the therapy of choice. However, in order to estimate the costs properly, not only the direct costs of the different anticoagulants should be taken into account, but also the costs of recurrent VTE including hospitalization monitoring anticoagulatory activity, the costs of bleeding complications and the cost of bleeding management should be weighted (including specific DOAC antidotes which became available very recently for dabigatran 65, or will be available in the near future for the oral Xa inhibitors 66). Of note, the medical costs attributable to VTE in the pre‐DOAC era in the United States were estimated to be between $7 billion and $10 billion annually 97. Other uncertainties, which will be solved in time, arise from the lack of specific tests with validated cutoff values to monitor anticoagulatory effects of DOACs, and from the absence of long‐term clinical data and long‐term side‐effects.
Conclusion
Because DOACs are equally effective as other anticoagulatory regimens, have lower bleeding risks, do not need monitoring routinely, and are administered orally, they have recently become the agents of choice in the acute and chronic treatment of PE. In patients with a contraindication to DOACs, LMWH/fondaparinux is generally preferred over UFH therapy because of a better and more predictable therapeutic effect. Nevertheless, based on pharmacokinetic, pharmacodynamic and relevant off‐target properties, there are still several indications for UFH therapy in the initial management of PE, for example in intermediate risk PE patients. There is a pathophysiological rationale for an initial bolus of UFH in LMWH‐ or DOAC‐treated patients with intermediate risk PE, but the possible benefit of this approach remains to be investigated.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 98, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 99, 100.
Competing Interests
There are no competing interests to declare.
Supporting information
Table S1 Overview of DOAC–drug interactions
Leentjens, J. , Peters, M. , Esselink, A. C. , Smulders, Y. , and Kramers, C. (2017) Initial anticoagulation in patients with pulmonary embolism: thrombolysis, unfractionated heparin, LMWH, fondaparinux, or DOACs?. Br J Clin Pharmacol, 83: 2356–2366. doi: 10.1111/bcp.13340.
References
- 1. Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism: a controlled trial. Lancet 1960; 1: 1309–1312. [DOI] [PubMed] [Google Scholar]
- 2. Kernohan RJ, Todd C. Heparin therapy in thromboembolic disease. Lancet 1966; 1: 621–623. [DOI] [PubMed] [Google Scholar]
- 3. Kanis JA. Heparin in the treatment of pulmonary thromboembolism. Thromb Diath Haemorrh 1974; 32: 519–527. [PubMed] [Google Scholar]
- 4. Alpert JS, Smith R, Carlson J, Ockene IS, Dexter L, Dalen JE. Mortality in patients treated for pulmonary embolism. JAMA 1976; 236: 1477–1480. [PubMed] [Google Scholar]
- 5. Brandjes DP, Heijboer H, Buller HR, de Rijk M, Jagt H, ten Cate JW. Acenocoumarol and heparin compared with acenocoumarol alone in the initial treatment of proximal‐vein thrombosis. N Engl J Med 1992; 327: 1485–1489. [DOI] [PubMed] [Google Scholar]
- 6. Hull RD, Raskob GE, Rosenbloom D, Panju AA, Brill‐Edwards P, Ginsberg JS, et al. Heparin for 5 days as compared with 10 days in the initial treatment of proximal venous thrombosis. N Engl J Med 1990; 322: 1260–1264. [DOI] [PubMed] [Google Scholar]
- 7. Weitz JI. Low‐molecular‐weight heparins. N Engl J Med 1997; 337: 688–698. [DOI] [PubMed] [Google Scholar]
- 8. Beyer‐Westendorf J, Ageno W. Benefit–risk profile of non‐vitamin K antagonist oral anticoagulants in the management of venous thromboembolism. Thromb Haemost 2015; 113: 231–246. [DOI] [PubMed] [Google Scholar]
- 9. Estes JW. Clinical pharmacokinetics of heparin. Clin Pharmacokinet 1980; 5: 204–220. [DOI] [PubMed] [Google Scholar]
- 10. Middeldorp S. Heparin: from animal organ extract to designer drug. Thromb Res 2008; 122: 753–762. [DOI] [PubMed] [Google Scholar]
- 11. Caranobe C, Petitou M, Dupouy D, Gabaig AM, Sie P, Buchanan MR, et al. Heparin fractions with high and low affinities to antithrombin III are cleared at different rates. Thromb Res 1986; 43: 635–641. [DOI] [PubMed] [Google Scholar]
- 12. Boneu B, Caranobe C, Sie P. Pharmacokinetics of heparin and low molecular weight heparin. Bailliere's Clin Haematol 1990; 3: 531–544. [DOI] [PubMed] [Google Scholar]
- 13. Hiebert LM, Jaques LB. The observation of heparin on endothelium after injection. Thromb Res 1976; 8: 195–204. [DOI] [PubMed] [Google Scholar]
- 14. Barzu T, Molho P, Tobelem G, Petitou M, Caen JP. Binding of heparin and low molecular weight heparin fragments to human vascular endothelial cells in culture. Nouv Rev Franc Hemat 1984; 26: 243–247. [PubMed] [Google Scholar]
- 15. Jaques LB, Mahadoo J, Riley JF. The mast cell/heparin paradox. Lancet 1977; 1: 411–413. [DOI] [PubMed] [Google Scholar]
- 16. Palm M, Mattsson C. Pharmacokinetics of heparin and low molecular weight heparin fragment (Fragmin) in rabbits with impaired renal or metabolic clearance. Thromb Haemost 1987; 58: 932–935. [PubMed] [Google Scholar]
- 17. Barzu T, van Rijn JL, Petitou M, Tobelem G, Caen JP. Heparin degradation in the endothelial cells. Thromb Res 1987; 47: 601–609. [DOI] [PubMed] [Google Scholar]
- 18. Dawes J, Papper DS. Catabolism of low‐dose heparin in man. Thromb Res 1979; 14: 845–860. [DOI] [PubMed] [Google Scholar]
- 19. McAllister BM, Demis DJ. Heparin metabolism: isolation and characterization of uroheparin. Nature 1966; 212: 293–294. [DOI] [PubMed] [Google Scholar]
- 20. Manson L, Weitz JI, Podor TJ, Hirsh J, Young E. The variable anticoagulant response to unfractionated heparin in vivo reflects binding to plasma proteins rather than clearance. J Lab Clin Med 1997; 130: 649–655. [DOI] [PubMed] [Google Scholar]
- 21. Hirsh J, Levine MN. Low molecular weight heparin. Blood 1992; 79: 1–17. [PubMed] [Google Scholar]
- 22. Donat F, Duret JP, Santoni A, Cariou R, Necciari J, Magnani H, et al. The pharmacokinetics of fondaparinux sodium in healthy volunteers. Clin Pharmacokinet 2002; 41 (Suppl 2): 1–9. [DOI] [PubMed] [Google Scholar]
- 23. Greinacher A. Heparin‐induced thrombocytopenia. N Engl J Med 2015; 373: 1883–1884. [DOI] [PubMed] [Google Scholar]
- 24. Warkentin TE, Maurer BT, Aster RH. Heparin‐induced thrombocytopenia associated with fondaparinux. N Engl J Med 2007; 356: 2653–2655 discussion 53–5. [DOI] [PubMed] [Google Scholar]
- 25. Yee JY, Duffull SB. The effect of body weight on dalteparin pharmacokinetics: a preliminary study. Eur J Clin Pharmacol 2000; 56: 293–297. [DOI] [PubMed] [Google Scholar]
- 26. Bazinet A, Almanric K, Brunet C, Turcotte I, Martineau J, Caron S, et al. Dosage of enoxaparin among obese and renal impairment patients. Thromb Res 2005; 116: 41–50. [DOI] [PubMed] [Google Scholar]
- 27. Sanderink GJ, Le Liboux A, Jariwala N, Harding N, Ozoux ML, Shukla U, et al. The pharmacokinetics and pharmacodynamics of enoxaparin in obese volunteers. Clin Pharmacol Ther 2002; 72: 308–318. [DOI] [PubMed] [Google Scholar]
- 28. Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, et al., RE‐LY Steering Committee and Investigators . Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361: 1139–1151. [DOI] [PubMed] [Google Scholar]
- 29. Schulman S, Kearon C, Kakkar AK, Mismetti P, Schellong S, Eriksson H, et al., RE‐COVER Study Group . Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361: 2342–2352. [DOI] [PubMed] [Google Scholar]
- 30. Breuer L, Ringwald J, Schwab S, Kohrmann M. Ischemic stroke in an obese patient receiving dabigatran. N Engl J Med 2013; 368: 2440–2442. [DOI] [PubMed] [Google Scholar]
- 31. Cohen D. Dabigatran: how the drug company withheld important analyses. BMJ 2014; 349: g4670. [DOI] [PubMed] [Google Scholar]
- 32. Douros A, Schlemm L, Bolbrinker J, Ebinger M, Kreutz R. Insufficient anticoagulation with dabigatran in a patient with short bowel syndrome. Thromb Haemost 2014; 112: 419–420. [DOI] [PubMed] [Google Scholar]
- 33. Sargento‐Freitas J, Silva F, Pego J, Duque C, Cordeiro G, Cunha L. Cardioembolic stroke in a patient taking dabigatran etexilate: the first case report of clinical and pharmacologic resistance. J Neurol Sci 2014; 346: 348–349. [DOI] [PubMed] [Google Scholar]
- 34. Safouris A, Demulder A, Triantafyllou N, Tsivgoulis G. Rivaroxaban presents a better pharmacokinetic profile than dabigatran in an obese non‐diabetic stroke patient. J Neurol Sci 2014; 346: 366–367. [DOI] [PubMed] [Google Scholar]
- 35. Lee MJ, Jang HM, Jeong WK, Bang OY. The need for a coagulation assay after initiation of new oral anticoagulants in patients with renal dysfunction: a case report. J Clin Neurol 2015; 11: 395–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirsh J, O'Donnell M, Eikelboom JW. Beyond unfractionated heparin and warfarin: current and future advances. Circulation 2007; 116: 552–560. [DOI] [PubMed] [Google Scholar]
- 37. Mueck W, Stampfuss J, Kubitza D, Becka M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin Pharmacokinet 2014; 53: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blech S, Ebner T, Ludwig‐Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008; 36: 386–399. [DOI] [PubMed] [Google Scholar]
- 39. Stangier J, Rathgen K, Stahle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 2007; 64: 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Song Y, Wang X, Perlstein I, Wang J, Badawy S, Frost C, et al. Relative bioavailability of apixaban solution or crushed tablet formulations administered by mouth or nasogastric tube in healthy subjects. Clin Ther 2015; 37: 1703–1712. [DOI] [PubMed] [Google Scholar]
- 41. Moore KT, Krook MA, Vaidyanathan S, Sarich TC, Damaraju CV, Fields LE. Rivaroxaban crushed tablet suspension characteristics and relative bioavailability in healthy adults when administered orally or via nasogastric tube. Clin Pharmacol Drug Dev 2014; 3: 321–327. [DOI] [PubMed] [Google Scholar]
- 42. Garcia DA, Baglin TP, Weitz JI, Samama MM, American College of Chest Physicians . Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines. Chest 2012; 141: e24S–e43S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olson ST, Swanson R, Raub‐Segall E, Bedsted T, Sadri M, Petitou M, et al. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems: comparison with heparin and low‐molecular‐weight heparin. Thromb Haemost 2004; 92: 929–939. [DOI] [PubMed] [Google Scholar]
- 44. Buller HR, Davidson BL, Decousus H, Gallus A, Gent M, Piovella F, et al. Subcutaneous fondaparinux versus intravenous unfractionated heparin in the initial treatment of pulmonary embolism. N Engl J Med 2003; 349: 1695–1702. [DOI] [PubMed] [Google Scholar]
- 45. Buller HR, Davidson BL, Decousus H, Gallus A, Gent M, Piovella F, et al. Fondaparinux or enoxaparin for the initial treatment of symptomatic deep venous thrombosis: a randomized trial. Ann Intern Med 2004; 140: 867–873. [DOI] [PubMed] [Google Scholar]
- 46. Gurewich V, Thomas D, Stein M, Wessler S. Bronchoconstriction in the presence of pulmonary embolism. Circulation 1963; 27: 339–345. [DOI] [PubMed] [Google Scholar]
- 47. Sasahara AA, Cannilla JE, Morse RL, Sidd JJ, Tremblay GM. Clinical and physiologic studies in pulmonary thromboembolism. Am J Cardiol 1967; 20: 10–20. [DOI] [PubMed] [Google Scholar]
- 48. Lee SL, Wang WW, Joseph PM, Hales CA, Fanburg BL. Inhibitory effect of heparin on serotonin‐induced hyperplasia and hypertrophy of smooth muscle cells. Am J Respir Cell Mol Biol 1997; 17: 78–83. [DOI] [PubMed] [Google Scholar]
- 49. Kuga T, Ohara Y, Hata H, Hirakawa Y, Tomoike H, Takeshita A. Inhibitory effects of heparin, aspirin and ketanserin on coronary artery vasoconstriction after arterial balloon injury in hypercholesterolemic miniature pigs. J Am Coll Cardiol 1993; 22: 291–295. [DOI] [PubMed] [Google Scholar]
- 50. Mega JL, Simon T. Pharmacology of antithrombotic drugs: an assessment of oral antiplatelet and anticoagulant treatments. Lancet 2015; 386: 281–291. [DOI] [PubMed] [Google Scholar]
- 51. Deykin D. Current concepts: the use of heparin. N Engl J Med 1969; 280: 937–938. [DOI] [PubMed] [Google Scholar]
- 52. Jerjes‐Sanchez C, Ramirez‐Rivera A, de Lourdes Garcia M, Arriaga‐Nava R, Valencia S, Rosado‐Buzzo A, et al. Streptokinase and heparin versus heparin alone in massive pulmonary embolism: a randomized controlled trial. J Thromb Thrombolysis 1995; 2: 227–229. [DOI] [PubMed] [Google Scholar]
- 53. Hao Q, Dong BR, Yue J, Wu T, Liu GJ. Thrombolytic therapy for pulmonary embolism. Cochrane Database Syst Rev 2015; 9: CD004437. [DOI] [PubMed] [Google Scholar]
- 54. Meyer G, Vicaut E, Danays T, Agnelli G, Becattini C, Beyer‐Westendorf J, et al., PEITHO Investigators . Fibrinolysis for patients with intermediate‐risk pulmonary embolism. N Engl J Med 2014; 370: 1402–1411. [DOI] [PubMed] [Google Scholar]
- 55. Erkens PM, Prins MH. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2010; 4: CD001100. [DOI] [PubMed] [Google Scholar]
- 56. Bhutia S, Wong PF. Once versus twice daily low molecular weight heparin for the initial treatment of venous thromboembolism. Cochrane Database Syst Rev 2013; 7: CD003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van Es N, Coppens M, Schulman S, Middeldorp S, Buller HR. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: evidence from phase 3 trials. Blood 2014; 124: 1968–1975. [DOI] [PubMed] [Google Scholar]
- 58. Hokusai VTEI, Buller HR, Decousus H, Grosso MA, Mercuri M, Middeldorp S, et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med 2013; 369: 1406–1415. [DOI] [PubMed] [Google Scholar]
- 59. Chai‐Adisaksopha C, Hillis C, Isayama T, Lim W, Iorio A, Crowther M. Mortality outcomes in patients receiving direct oral anticoagulants: a systematic review and meta‐analysis of randomized controlled trials. J Thromb Haemost 2015; 13: 2012–2020. [DOI] [PubMed] [Google Scholar]
- 60. Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149: 315–352. [DOI] [PubMed] [Google Scholar]
- 61. Providencia R, Grove EL, Husted S, Barra S, Boveda S, Morais J. A meta‐analysis of phase III randomized controlled trials with novel oral anticoagulants in atrial fibrillation: comparisons between direct thrombin inhibitors vs. factor Xa inhibitors and different dosing regimens. Thromb Res 2014; 134: 1253–1264. [DOI] [PubMed] [Google Scholar]
- 62. Sharma M, Cornelius VR, Patel JP, Davies JG, Molokhia M. Efficacy and harms of direct oral anticoagulants in the elderly for stroke prevention in atrial fibrillation and secondary prevention of venous thromboembolism: systematic review and meta‐analysis. Circulation 2015; 132: 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bryk AH, Pirog M, Plens K, Undas A. Heavy menstrual bleeding in women treated with rivaroxaban and vitamin K antagonists and the risk of recurrent venous thromboembolism. Vascul Pharmacol 2016; 87: 242–247. [DOI] [PubMed] [Google Scholar]
- 64. Myers B, Webster A. Heavy menstrual bleeding on rivaroxaban – comparison with apixaban. Br J Haematol 2017; 176: 833–835. [DOI] [PubMed] [Google Scholar]
- 65. Pollack CV Jr, Reilly PA, Eikelboom J, Glund S, Verhamme P, Bernstein RA, et al. Idarucizumab for dabigatran reversal. N Engl J Med 2015; 373: 511–520. [DOI] [PubMed] [Google Scholar]
- 66. Connolly SJ, Milling TJ Jr, Eikelboom JW, Gibson CM, Curnutte JT, Gold A, et al., ANNEX‐A4 Investigators . Andexanet alfa for acute major bleeding associated with factor Xa inhibitors. N Engl J Med 2016; 375: 1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zahir H, Brown KS, Vandell AG, Desai M, Maa JF, Dishy V, et al. Edoxaban effects on bleeding following punch biopsy and reversal by a 4‐factor prothrombin complex concentrate. Circulation 2015; 131: 82–90. [DOI] [PubMed] [Google Scholar]
- 68. Perzborn E, Gruber A, Tinel H, Marzec UM, Buetehorn U, Buchmueller A, et al. Reversal of rivaroxaban anticoagulation by haemostatic agents in rats and primates. Thromb Haemost 2013; 110: 162–172. [DOI] [PubMed] [Google Scholar]
- 69. Patel JP, Roberts LN, Arya R. Anticoagulating obese patients in the modern era. Br J Haematol 2011; 155: 137–149. [DOI] [PubMed] [Google Scholar]
- 70. Lim W, Cook DJ, Crowther MA. Safety and efficacy of low molecular weight heparins for hemodialysis in patients with end‐stage renal failure: a meta‐analysis of randomized trials. J Am Soc Nephrol 2004; 15: 3192–3206. [DOI] [PubMed] [Google Scholar]
- 71. Brophy DF, Martin EJ, Gehr TW, Carr ME Jr. Enhanced anticoagulant activity of enoxaparin in patients with ESRD as measured by thrombin generation time. Am J Kidney Dis 2004; 44: 270–277. [DOI] [PubMed] [Google Scholar]
- 72. Konstantinides S. Clinical practice: acute pulmonary embolism. N Engl J Med 2008; 359: 2804–2813. [DOI] [PubMed] [Google Scholar]
- 73. Torbicki A, Perrier A, Konstantinides S, Agnelli G, Galie N, Pruszczyk P, et al., Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC) . Guidelines on the diagnosis and management of acute pulmonary embolism. Eur Heart J 2008; 29: 2276–2315. [DOI] [PubMed] [Google Scholar]
- 74. Sanchez O, Planquette B, Meyer G. Management of massive and submassive pulmonary embolism: focus on recent randomized trials. Curr Opin Pulm Med 2014; 20: 393–399. [DOI] [PubMed] [Google Scholar]
- 75. Lee AY, Levine MN, Baker RI, Bowden C, Kakkar AK, Prins M, et al., Randomized Comparison of Low‐Molecular‐Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer Investigators . Low‐molecular‐weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349: 146–153. [DOI] [PubMed] [Google Scholar]
- 76. Meyer G, Marjanovic Z, Valcke J, Lorcerie B, Gruel Y, Solal‐Celigny P, et al. Comparison of low‐molecular‐weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med 2002; 162: 1729–1735. [DOI] [PubMed] [Google Scholar]
- 77. Hull RD, Pineo GF, Brant RF, Mah AF, Burke N, Dear R, et al., LITE Trial Investigators . Long‐term low‐molecular‐weight heparin versus usual care in proximal‐vein thrombosis patients with cancer. Am J Med 2006; 119: 1062–1072. [DOI] [PubMed] [Google Scholar]
- 78. Deitcher SR, Kessler CM, Merli G, Rigas JR, Lyons RM, Fareed J, et al., ONCENOX Investigators . Secondary prevention of venous thromboembolic events in patients with active cancer: enoxaparin alone versus initial enoxaparin followed by warfarin for a 180‐day period. Clin Appl Thromb Hemost 2006; 12: 389–396. [DOI] [PubMed] [Google Scholar]
- 79. Mousa SA, Petersen LJ. Anti‐cancer properties of low‐molecular‐weight heparin: preclinical evidence. Thromb Haemost 2009; 102: 258–267. [DOI] [PubMed] [Google Scholar]
- 80. Smorenburg SM, Van Noorden CJ. The complex effects of heparins on cancer progression and metastasis in experimental studies. Pharmacol Rev 2001; 53: 93–105. [PubMed] [Google Scholar]
- 81. Noonan DM, De Lerma Barbaro A, Vannini N, Mortara L, Albini A. Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Rev 2008; 27: 31–40. [DOI] [PubMed] [Google Scholar]
- 82. Rickles FR, Falanga A. Molecular basis for the relationship between thrombosis and cancer. Thromb Res 2001; 102: V215–V224. [DOI] [PubMed] [Google Scholar]
- 83. Norrby K. Low‐molecular‐weight heparins and angiogenesis. Acta Pathol Microbiol Immunol Scand 2006; 114: 79–102. [DOI] [PubMed] [Google Scholar]
- 84. Khorana AA, Sahni A, Altland OD, Francis CW. Heparin inhibition of endothelial cell proliferation and organization is dependent on molecular weight. Arterioscler Thromb Vasc Biol 2003; 23: 2110–2115. [DOI] [PubMed] [Google Scholar]
- 85. Takahashi H, Ebihara S, Okazaki T, Asada M, Sasaki H, Yamaya M. A comparison of the effects of unfractionated heparin, dalteparin and danaparoid on vascular endothelial growth factor‐induced tumour angiogenesis and heparanase activity. Br J Pharmacol 2005; 146: 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Marchetti M, Vignoli A, Russo L, Balducci D, Pagnoncelli M, Barbui T, et al. Endothelial capillary tube formation and cell proliferation induced by tumor cells are affected by low molecular weight heparins and unfractionated heparin. Thromb Res 2008; 121: 637–645. [DOI] [PubMed] [Google Scholar]
- 87. Niers TM, Klerk CP, DiNisio M, Van Noorden CJ, Buller HR, Reitsma PH, et al. Mechanisms of heparin induced anti‐cancer activity in experimental cancer models. Crit Rev Oncol Hematol 2007; 61: 195–207. [DOI] [PubMed] [Google Scholar]
- 88. Stevenson JL, Choi SH, Varki A. Differential metastasis inhibition by clinically relevant levels of heparins – correlation with selectin inhibition, not antithrombotic activity. Clin Cancer Res 2005; 11: 7003–7011. [DOI] [PubMed] [Google Scholar]
- 89. Stevenson JL, Varki A, Borsig L. Heparin attenuates metastasis mainly due to inhibition of P‐ and L‐selectin, but non‐anticoagulant heparins can have additional effects. Thromb Res 2007; 120 (Suppl 2): S107–S111. [DOI] [PubMed] [Google Scholar]
- 90. Borsig L. Antimetastatic activities of heparins and modified heparins: experimental evidence. Thromb Res 2010; 125 (Suppl 2): S66–S71. [DOI] [PubMed] [Google Scholar]
- 91. Fritzsche J, Simonis D, Bendas G. Melanoma cell adhesion can be blocked by heparin in vitro: suggestion of VLA‐4 as a novel target for antimetastatic approaches. Thromb Haemost 2008; 100: 1166–1175. [PubMed] [Google Scholar]
- 92. Ludwig RJ, Boehme B, Podda M, Henschler R, Jager E, Tandi C, et al. Endothelial P‐selectin as a target of heparin action in experimental melanoma lung metastasis. Cancer Res 2004; 64: 2743–2750. [DOI] [PubMed] [Google Scholar]
- 93. Lyman GH, Bohlke K, Falanga A. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Oncol Pract 2015; 11: e442–e444. [DOI] [PubMed] [Google Scholar]
- 94. Zhang J, Zhang YL, Ma KX, Qu JM. Efficacy and safety of adjunctive anticoagulation in patients with lung cancer without indication for anticoagulants: a systematic review and meta‐analysis. Thorax 2013; 68: 442–450. [DOI] [PubMed] [Google Scholar]
- 95. Kuderer NM, Khorana AA, Lyman GH, Francis CW. A meta‐analysis and systematic review of the efficacy and safety of anticoagulants as cancer treatment: impact on survival and bleeding complications. Cancer 2007; 110: 1149–1161. [DOI] [PubMed] [Google Scholar]
- 96. Larsen TB, Nielsen PB, Skjoth F, Rasmussen LH, Lip GY. Non‐vitamin K antagonist oral anticoagulants and the treatment of venous thromboembolism in cancer patients: a semi systematic review and meta‐analysis of safety and efficacy outcomes. PLoS One 2014; 9: e114445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: a review of estimated attributable healthcare costs. Thromb Res 2016; 137: 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 2015; 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Overview of DOAC–drug interactions