

Abstract
Introduction
The glymphatic system is a brain-wide perivascular network that facilitates clearance of proteins, including amyloid β, from the brain interstitium through the perivascular exchange of cerebrospinal fluid and interstitial fluid. The astrocytic water channel aquaporin-4 (AQP4) is required for glymphatic system function, and impairment of glymphatic function in the aging brain is associated with altered AQP4 expression and localization. In human cortical tissue, alterations in AQP4 expression and localization are associated with Alzheimer's disease (AD) status and pathology. Although this suggests a potential role for AQP4 in the development or progression of AD, the relationship between of naturally occurring variants in the human AQP4 gene and cognitive function has not yet been evaluated.
Methods
Using data from several longitudinal aging cohorts, we investigated the association between five AQP4 single-nucleotide polymorphisms (SNPs) and the rate of cognitive decline in participants with a diagnosis of AD.
Results
None of the five SNPs were associated with different rates of AD diagnosis, age of dementia onset in trial subjects. No association between AQP4 SNPs with histological measures of AD pathology, including Braak stage or neuritic plaque density was observed. However, AQP4 SNPs were associated with altered rates of cognitive decline after AD diagnosis, with two SNPS (rs9951307 and rs3875089) associated with slower cognitive decline and two (rs3763040 and rs3763043) associated with more rapid cognitive decline after AD diagnosis.
Discussion
These results provide the first evidence that variations in the AQP4 gene, whose gene product AQP4 is vital for glymphatic pathway function, may modulate the progression of cognitive decline in AD.
Keywords: Alzheimer's disease, Genetics, Glymphatic system, Aquaporin-4, Amyloid β, Cognitive decline, Cohort study
1. Introduction
Impaired clearance is suggested as a major cause of pathologic accumulation of amyloid β (Aβ) in the brain and subsequent development of sporadic Alzheimer's disease (AD), in contrast to familial AD in which aberrant production of Aβ is the key driver [1], [2], [3]. Emerging findings from longitudinal studies utilizing cerebrospinal fluid (CSF)–based or positron emission tomography–based Aβ biomarker measurements suggest that Aβ begins to deposit within the brains of subjects in their 40s and 50s, presumably decades before the onset of clinical symptoms [4]. Recent studies have shown that the rate of Aβ clearance is significantly slowed both with advancing age and with the presence of AD [5], [6]. However, the mechanism underlying this age-related slowing of Aβ clearance is not yet known.
Recently, a brain-wide perivascular clearance pathway has been described that facilitates the clearance of interstitial solutes and proteins, including Aβ and tau, from the brain [3], [7], [8]. Termed the “glymphatic” system, this perivascular pathway is dependent upon the astrocytic water channel aquaporin-4 (AQP4) that is localized to perivascular astrocytic endfeet that ensheathe the cerebral vasculature [8]. Genetic deletion of the mouse Aqp4 gene slows Aβ clearance [8] and accelerates the deposition of Aβ plaques in a transgenic mouse model of AD [9]. In the aging rodent brain, glymphatic function is impaired and interstitial Aβ clearance is slowed, changes that are associated with the loss of perivascular AQP4 localization [10]. In a recently published study carried out in human-autopsy tissue, loss of perivascular AQP4 localization was a strong predictor of AD status and was associated with greater Aβ plaque density and neurofibrillary pathology [11].
Although glymphatic function has only been visualized in human subjects in a single case report [12], these findings suggest that changes in AQP4 expression, localization, or function may alter glymphatic pathway function and contribute to the development of AD or other neurodegenerative conditions. If true, then naturally occurring variants in the human AQP4 gene may be associated with changes in the development of age-related cognitive decline or AD. Indeed, single-nucleotide polymorphisms (SNPs) in the human AQP4 gene have been associated with altered clinical outcomes in numerous neurological diseases including sudden infant death syndrome [13], stroke [14], leukoaraiosis [15], and traumatic brain injury (TBI) [16]. However, no studies to date have investigated the role of AQP4 SNPs in the setting of AD.
We utilized data from seven well-characterized longitudinal cohorts on aging to determine if AQP4 SNPs were associated with more rapid cognitive or functional decline in older individuals with and without AD. We employed a linear mixed modeling approach to compare the rate of cognitive and functional decline to SNP genotypes using a battery of neuropsychiatric and functional evaluations. Our findings show that AQP4 SNPs are significantly associated with altered rates of cognitive decline in AD, supporting a role for AQP4 in the development of AD and as a potential therapeutic target in the prevention or treatment of AD.
2. Methods
2.1. Database sources and study population
The protocols for the studies used for analysis were approved by the Institutional Review Board at Oregon Health & Science University, Portland, Oregon, with participants providing written informed consent across all cohorts. We conducted a retrospective cohort study using a number of existing longitudinal natural history studies of cognitive aging available through the Oregon Alzheimer's Disease Center: the Oregon Brain Aging Study (OBAS I and OBAS II; n = 130) [17], the Intelligent Systems for Assessment of Aging Changes (n = 88) [18], the African-American Dementia and Aging Project (n = 49), the Klamath Exceptional Aging Project (n = 151) [19], the Oregon Community Brain Donor Program (n = 86), Oregon Living Laboratory (n = 46), and the Layton Alzheimer's Disease Center's patient registry (n = 74) [20], leading to a final starting cohort of 634 subjects.
Different initial entry criteria were present across the seven studies leading to a potential for heterogeneity in the final data set. Although not required for study entry, the data set used for analysis required all subjects to begin their baseline visit as cognitively intact; however, a significant proportion of participants eventually developed cognitive decline and dementia. There were no further restrictions on prior comorbid illnesses beyond individual study criteria.
To appropriately contribute to the longitudinal analysis, all participants were required to have at least one follow-up visit during their study involvement and DNA available for analysis. Although the final visit content varied between studies, a set of predefined cognitive assessments were selected based on their prevalence across the annual and semi-annual assessments of the studies, with patient caregivers participating when applicable. Using the aforementioned cohorts, we derived a comprehensive longitudinal data set including demographic, outcome, and SNP genotype variables.
Of particular interest was whether the AQP4 SNPs had a larger effect on the rate of decline following a clinical diagnosis of AD. We hypothesized that since individuals with AD have impaired Aβ clearance [6], and AQP4 is important in Aβ clearance [8], changes in AQP4 structure and function due to SNPs may be more evident in individuals with AD than those without AD due to pathologic changes in the brain. Subjects were only considered for categorization as “AD” if their final study visit included a positive diagnosis of dementia. This was deemed a necessary criterion for pathological diagnosis when available so that all AD subjects would end their study involvement as AD. For visits before the last one, subjects were classified as “post-AD” only after two concurrent follow-up visits with a clinical diagnosis of AD based on established criteria [21]. For modeling purposes, this diagnosis was considered persistent for all subsequent time points, even if occasional follow-up visits suggested possible changes in clinical diagnosis. Of the 634 subjects that all entered cognitively intact, 163 had a diagnosis of Alzheimer's dementia and 471 remained cognitively intact at the time of their last evaluation. Frequency of AQP4 SNPs among non-AD and AD subjects is shown in Supplementary Table 1.
2.2. Neuropathologic analysis
Brain tissue for longitudinal aging study subjects that come to autopsy is maintained within the Oregon Brain Bank. At the time of autopsy, brain tissues from several regions were collected for histopathological analysis to evaluate AD, vascular, and other pathological features as described previously [22]. Briefly, brains were fixed in the neutral-buffered formaldehyde solution for at least 2 weeks and examined grossly as well as microscopically. For microscopic evaluation, tissue samples were taken from all cortical lobes bilaterally or unilaterally, frontal lobe white matter, anterior cingulate gyrus, hippocampus, amygdala, bilateral striatum and thalamus, midbrain, pons, medulla, and cerebellum. Six-micrometer sections were routinely stained with hematoxylin-eosin, Luxol fast blue, Congo red-gallocyanin, and by the modified Bielschowsky silver impregnation method. Selected sections of hippocampus and neocortical regions were immunostained with antibody to tau (tau2; Sigma, St. Louis, MO). Pathologic diagnoses were established using consensus criteria [23]. Information related to neuritic plaques and neurofibrillary tangle burdens, presence of ischemic, hemorrhagic, or vascular pathology, amyloid angiopathy, large vessel strokes, lacunes, presence of Lewy bodies, hippocampal sclerosis, and degree of arteriosclerosis was summarized using the National Alzheimer's Coordinating Center Neuropathology Data Form [24].
2.3. Outcome measures
Longitudinal changes in cognitive function were taken from the participants' annual clinical assessments, neuropsychological evaluations, and questionnaires collected during study visits. Primary outcome measures were global cognitive function, episodic memory, and executive function. Global assessments of cognitive ability included the Mini–Mental State Examination (MMSE) [25] and Clinical Dementia Rating Sum of Boxes (CDR SoB) score [26]. The Logical Memory II subtest of the Wechsler Memory Scale was used to assess episodic memory [27]. The Digit Symbol Substitution Test (Digit Symbol) [28] and part B of the Trails Making Test (Trails B) [29] were used to assess executive function.
Associations between SNP carrier status and neuropathology were evaluated using several different measures, as established previously [30]. The following binary (no vs. yes) measures were included in final analysis: presence of (1) large arterial infarcts, (2) one or more lacunes, (3) multiple infarcts, and (4) microinfarcts. In addition, the following categorical measures were included: atherosclerosis of the circle of Willis (0–2 vs. 3), Braak stage for neurofibrillary degeneration (0–IV vs. V–VI), density of neocortical neuritic plaques (0–2 vs. 3), cerebral amyloid angiopathy (0–2 vs. 3), and arteriolosclerosis (0–2 vs. 3). Finally, Lewy body pathology was evaluated as not present or unspecified versus limbic or neocortical. SNP carriers were compared with noncarriers using logistic regression.
2.4. AQP4 SNP design
Target AQP4 SNPs of interest were identified first based on SNPs that had previously been associated with altered outcomes in neurological disorders, including TBI and stroke [13], [14], [15], [16], and second based upon SNP coverage available within the gene array platform used with the present subjects' data set. Genome-wide SNP genotyping was performed through Illumina genome array platforms, including HumanExome-12, v1.0, HumanOmniExpressExome-8, HumanOmniExpressExome-8, Illumina 660, Illumina HumanCNV370, and Illumina OmniExpress. The final panel set consisted of five AQP4 SNPs: rs335929, rs3763043, rs3763040, rs9951307, and rs3875089. Those with one or two copies of the minor allele were identified as “carriers”, whereas homozygotes for the common major allele were classified as “noncarriers”. Participants carrying one or two copies of the minor allele were pooled for all SNPs because of the inadequate sample sizes of homozygous minor allele carriers in most genotyped SNPs.
2.5. Statistical analysis
All statistical analysis was carried out using R, 3.2 and R, 3.3 [31] with additional utility from the lme4 package [32]. Mixed-effect models were used to assess the relationship between genotype at the AQP4 SNPs of interest and the rate of change of the described outcome variables both before and after a clinical diagnosis of AD. Principal assessment evaluated the rate of change in outcomes for each participant over time. Known demographic confounders were selected a priori (age, sex, education level, Cumulative Illness Rating Score (CIRS) at baseline, and APOE genotype status) and were controlled for in all models, as were baseline values of outcomes when assessing rates of change. Effects of racial background on our analysis were also assessed. Although there were baseline differences in some cognitive outcomes at study entry, there was no association between race and SNP allele status, dementia status, or rate of change of the various cognitive and functional outcomes. CIRS is a validated and reliable method to measure comorbidity, and it is commonly used in clinical research [33], [34]. CIRS rates 13 different physiologic systems on a five-point severity scale [35]. The key independent variables of interest, SNP carrier status, diagnosis of AD, and their interaction, were dichotomized to give four groups for each SNP (Noncarriers pre-AD, SNP carriers pre-AD, noncarriers post-AD, and SNP carriers post-AD). An interaction between AQP4 SNP status and a clinical diagnosis of AD was the main variable used to evaluate any longitudinal relationship between genetic factors and AD and directly contrast the effects of carrier versus noncarrier status both before and after a diagnosis of AD.
Due to the use of multiple distinct cohorts in this study, cohort bias on the time-dependent associations of the outcomes was assessed. Although there were cohort-based population differences in the outcomes at entry into the studies, these baseline differences had no bearing on the changes in outcomes over time, which is a principal consideration of the current analysis. With no observable cohort effects on the time-dependent changes in outcomes, the multiple cohorts were pooled into a single disposition.
The mixed model allowed for an analysis framework to most appropriately leverage the repeated but varying visits among the various studies. An unstructured error covariance model was used, and parameters were estimated using restricted maximum likelihood procedures. Missing data points in the analytical sample were considered missing at random, and the above approach was sufficient under this assumption. Model fit and integrity were examined using a combination of formal fit criteria, including Cook's distance and the standardized difference of the β's, and visual inspection of the residual plots. Individual subject visits were considered for removal when they were found to be outcome outliers based on excessive model residuals as well as having undue and extensive influence on longitudinal trajectories due to large leverages. When both criteria were met, these subject visits were considered to be overly influential model outliers and were excluded globally from all analyses. Results were considered significant at P < .05 only after a two-level adjustment to P values to correct for statistical tests on multiple response variables. False discovery correction was first used across the entire cohort of assessed models for scalability to allow for only 5% of erroneous discoveries in the significant results. The significance of this selection set was then further adjusted using the more stringent Holm-Sidak family-wise error rate correction to correct for multiple comparisons. These adjusted P values were then used to determine final model significance.
Comparison of model log-likelihoods was used to evaluate both the overall goodness-of-fit of the models and the informative benefit specific to the inclusion of the principal longitudinal interactions of AD diagnosis and SNP carrier status. McFadden's R2 was calculated for the overall fit of a model to a given outcome, with the minimum log-likelihoods for the fully developed models compared with the log-likelihoods from an intercept-only design. The informativeness of the key interactions against the outcomes was assessed using a χ2 test on the change in log-likelihood of the fully developed models from the nested models without the interactions. This combined approach allowed for a determination not only of the overall fit of the model but also of the utility specific to the central hypotheses related to AD diagnosis and carrier status of the AQP4 SNPs.
3. Results
3.1. Study demographics
A total of 634 participants underwent multiple cognitive evaluations and were thus included in analysis. All participants were cognitively intact at study entry. Participant demographics are summarized in Table 1. Of the 634 participants, 471 did not receive AD diagnosis any time during the study, whereas 163 received AD diagnosis at some point. Of the 471 participants that did not receive AD diagnosis, 242 received a mild cognitive impairment (MCI) diagnosis at some point during the study. However, analysis of MCI participants did not yield any associations between SNP carrier status and cognitive decline when compared with AD or non-AD participants. Therefore, participants that received MCI diagnosis were included in the non-AD group for analysis.
Table 1.
Subject Demographics
| Status at exit | N | Time in study (years) |
Age at entry (years) |
CIRS at exit |
Years of school |
Female |
APOE ε4 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Median | Mean | SD | Median | Mean | SD | Median | Mean | SD | Median | Number | % | Number | % | ||
| Pre-AD | 471 | 8.5 | 4.8 | 7 | 80.8 | 8.4 | 84.7 | 22.0 | 3.5 | 22.0 | 14.6 | 2.8 | 14.5 | 324 | 68.8 | 83 | 17.6 |
| Post-AD | 163 | 9.0 | 4.3 | 8.2 | 81.6 | 8.8 | 85.2 | 24.0 | 3.9 | 24.0 | 14.4 | 3.4 | 14.0 | 101 | 62.0 | 57 | 35.0 |
| All | 634 | 8.6 | 4.7 | 7.4 | 81.0 | 8.5 | 84.9 | 22.5 | 3.7 | 22.0 | 14.5 | 3.0 | 14.0 | 425 | 67.0 | 140 | 22.1 |
Abbreviations: CIRS, Clinical Illness Rating Scale; SD, standard deviation.
NOTE. Pre-AD = measurements from participants that did not have AD diagnosis; Post-AD = measurements from participants with AD diagnosis.
Overall, 67% of the participants were female. 584 participants were Caucasian, whereas 50 were African-American. Similar to observed population demographics, 35.0% of participants with an AD diagnosis were carriers of the APOE ε4 allele, whereas only 17.6% of the non-AD participants were carriers (odds ration [OR] = 2.51; P < .001). No other significant differences in subject demographics were observed based on AD diagnosis at study exit.
3.2. AQP4 SNPS and cognitive and functional decline
To determine whether a minor allele in one of the five AQP4 SNPs was associated with altered cognitive and functional decline over time, we compared change in score of five different cognitive and functional tests between AQP4 SNP minor allele carriers and noncarriers. Cognitive function was quantified using the following tests: MMSE, CDR SoB, Logical Memory II, Digit Symbol, and Trails B. We separated our analysis based on AD diagnosis status (pre-AD and post-AD). The full results of our analysis are included in Supplementary Table 2. There were no significant differences for SNP genotype in rate of cognitive and functional decline for any of the five tests in the pre-AD analysis group.
However, in the post-AD analysis group, a minor allele in any of the five APQ4 SNPs was associated with altered cognitive decline compared to noncarriers. The t-statistics comparing minor allele carriers to noncarriers for the post-AD analysis group are shown in Table 2 and in detail in Supplementary Table 2. The significant effects of the SNPs on cognitive and executive function were largely consistent across different cognitive and functional tests, such that either homozygous or heterozygous possession of the minor allele was associated with either slower cognitive and functional decline (rs9951307 and rs3875089) or more rapid cognitive and functional decline (rs3763040 and rs3763043). In the case of rs335929, possession of the minor allele was associated with more rapid decline in CDR, but slower decline in Logical Memory and Digit Symbol tests performance (Table 2).
Table 2.
Post-AD t-statistics summary for five AQP4 SNPs
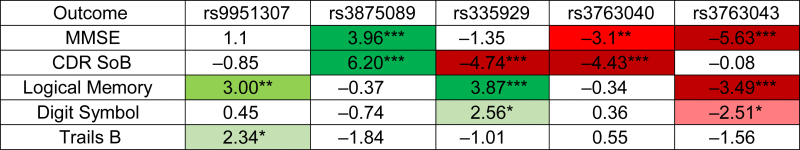 |
Abbreviations: CDR SoB, Clinical Dementia Rating Sum of Boxes; MMSE, Mini–Mental State Examination; SNP, single-nucleotide polymorphism.
NOTE. t-statistics for the difference in rate of change over time for the five functional and cognitive tests between AQP4 SNP carriers and noncarriers, post-AD diagnosis. Cells shaded in green represent a significant association with slowed cognitive or functional decline with possession of the SNP. Cells shaded in red represent a significant association with more rapid cognitive or functional decline with possession of the SNP. * = P < .05, ** = P < .01, *** = P < .001.
3.3. AQP4 SNPS associated with slower cognitive and functional decline after AD diagnosis
Homozygous or heterozygous possession of the minor allele at rs3875089 was associated with significantly slower decrease in MMSE score (t = 3.96, P < .001—all reported P values are adjusted as described in the Methods) and significantly slower increase in CDR SoB score (t = 6.20, P < .001) compared with noncarriers. Individual scatter plots used for calculating slopes for all five functional and cognitive tests to compare rs3875089 carriers with noncarriers post-AD are shown in Fig. 1. In addition, homozygous or heterozygous possession of the minor allele at rs9951307 was associated with significantly slower decline in Logical Memory II score (t = 3.00, P < .01) and significantly slower increase in Trails B time (t = 2.34, P < .05) post-AD (Table 2, Supplementary Table 2).
Fig. 1.

Plots of individual data points and annualized rate of change for rs3875089 carriers and noncarriers. Graphs (A-E) show the score recorded for all five functional and cognitive test for every individual visit (after AD diagnosis) in all of the studies. From these data points, slopes are calculated for rs3875089 carriers (both homozygous and heterozygous carriers of the minor allele) and noncarriers (shown in the table). Abbreviations: CDR SoB, Clinical Dementia Rating Sum of Boxes; MMSE, Mini–Mental State Examination.
3.4. AQP4 SNPS associated with faster cognitive and functional decline after AD diagnosis
Homozygous or heterozygous possession of the minor allele at rs3763043 was associated with significantly faster decline in MMSE (t = −5.63, P < .001), Logical Memory II (t = −3.49, P < .001), and Digit Symbol (t = −2.51, P < .05) scores compared with noncarriers. Individual scatter plots used for calculating slopes for all five functional and cognitive tests to compare rs3763043 carriers with noncarriers post-AD are shown in Fig. 2. In addition, homozygous or heterozygous possession of the minor allele at rs3763040 was associated with significantly faster decline in MMSE score (t = −3.1, P < .01) and significantly faster increase in CDR SoB score (t = −4.43, P < .001) compared with noncarriers (Table 2, Supplementary Table 2).
Fig. 2.

Plots of individual data points and annualized rate of change for rs3763043 carriers and noncarriers. Graphs (A-E) show the score recorded for all five functional and cognitive test for every individual visit (after AD diagnosis) in all of the studies. From these data points, slopes are calculated for rs763043 carriers (both homozygous and heterozygous carriers of the minor allele) and noncarriers (shown in the table). Abbreviations: CDR SoB, Clinical Dementia Rating Sum of Boxes; MMSE, Mini–Mental State Examination.
Homozygous or heterozygous possession of the minor allele at rs335929 was associated with significantly faster increase in CDR SoB score (t = −4.74, P < .001) compared with noncarriers. However, homozygous or heterozygous possession of the minor allele at rs335929 was associated with significantly slower decline in Logical Memory II (t = 3.87, P < .01) and Digit Symbol (t = 2.56, P < .05) scores compared to noncarriers (Table 2, Supplementary Table 2).
In our analysis, we included subjects diagnosed with MCI in the “non-AD” group because subjects with MCI have a distinct clinical progression from those diagnosed with AD. To confirm that the analysis was not influenced by the spurious combination of cognitively intact and MCI subjects, we repeated the above analysis after removing MCI subjects (Supplementary Table 3). With removal of the MCI subjects and the limited coverage in the cohort of specific neurocognitive tests (Digit Symbol and Trails B), several of these associations were underpowered. For the MMSE and CDR SoB, no difference was observed in the associations with individual AQP4 SNPs with the exclusion of MCI subjects. For the individual neurocognitive tests that were sufficiently powered after removal of MCI subjects (Logical Memory across all AQP4 SNPs, Digit Symbol, and Trails B for rs9951307 and rs335929), results did not change with removal of MCI subjects. Thus, to maximize statistical power, we included MCI subjects in the non-AD cohort.
3.5. Enrichment of functional decline effects in double carriers of AQP4 SNPS after AD diagnosis
Because the minor allele frequency was quite high for many of our SNPs (Supplementary Table 2), we surmised that there would be a large number of participants carrying multiple SNPs. Furthermore, since SNPs were associated with either slower cognitive decline (rs3875089 and rs9951307—“protective” SNPs) or faster cognitive decline (rs3763043 and rs3763040—“deleterious” SNPs), we hypothesized that there might be an additive effect of these protective or deleterious SNPs. We compared rates of cognitive decline between subjects carrying a single protective or deleterious SNP with subjects carrying a second protective or deleterious SNP, respectively. In addition, we compared rates of cognitive decline between noncarrier subjects and subjects carrying two protective or two deleterious SNPs. All analysis was performed in subjects with AD diagnosis.
Limitations of adequate coverage for carriers were immediately apparent. As all SNP effects were seen after AD diagnosis, sufficient numbers of post-AD single and double carriers were necessary to create models capable of developing generalizable interpretations. In particular, the specific cognitive assessments such as Logical Memory and Trails B testing were too underpowered to be able to draw meaningful conclusions. Therefore, only the functional assessments of MMSE and CDR were considered under the double-carrier designs.
For carriers of the protective SNPS, both carriers of the rs3875089 minor allele and double carriers of rs3875089 and rs9951307 both showed significantly reduced functional decline after an AD diagnosis compared with noncarriers for both MMSE score (rs3875089 single carrier: t = 4.60, P < .001; double carrier: t = 2.64, P < .01) and CDR SoB (rs3875089 single carrier: t = 2.85, P < .01; double carrier: t = −6.25; P < .001). However, when comparing noncarriers with rs9951307 post-AD, there were no observed differences in rates of decline for either MMSE (t = 1.79, not significant [NS]) or CDR SoB (t = 0.02, NS). Finally, contrasting rs3875089 single carriers to the double carriers observed no significant difference in their SNP effects for either MMSE or CDR SoB.
Evaluation of the carriers of the deleterious SNPS was further complicated for allele frequencies as it was found that all but one of the subjects with an AD diagnosis who were minor carriers of the rs3763040 minor allele were also carriers of the rs3763043 allele. As there were no AD subjects who were solely rs3763040 minor allele carriers, we compared noncarriers with rs3763043 single carriers and rs3763043 single carriers to subjects who were carriers of the minor allele at rs3763040 and at rs3763043. When evaluating MMSE score, increased rates of functional decline were observed for both rs3763043 single carriers (t = −4.03, P < .01) and the double carriers (t = −6.46, P < .001) when compared with noncarriers post-AD. Further, the increased rate of decline seen in double carriers was significantly greater than the rate observed in rs3763043 single carriers (t = −2.49, P < .05). A similar pattern was observed with CDR SoB, with double carriers showing significantly faster rates of decline compared with both noncarriers (t = −4.71, P < .001) and single carriers of the rs3763043 minor allele (t = −5.25, P < .001). However, with this enriched cohort definition of rs3763043 carriers, single carriers were not observed to be significantly different from noncarriers (t = 1.15, NS).
3.6. Associations between AQP4 SNP status and neuropathology measures
Among subjects that had come to autopsy, none of the five AQP4 SNPs were associated with altered measurements of AD pathology, including Braak stage, density of neuritic plaques, or the presence of amyloid angiopathy (Table 3). Homozygous or heterozygous possession of the minor allele at rs9951307 was associated with fewer large arterial infarcts compared with noncarriers in AD subjects (z = −2.34, P < .05). Homozygous or heterozygous possession of the minor allele at rs3763040 or rs3763043 was each associated with the presence of lacunar infarcts compared to noncarriers in AD subjects (z = 2.26, P < .05; z = 2.26, P < .05; respectively, Table 3).
Table 3.
Relationship between AQP4 SNPs and brain pathology post-AD
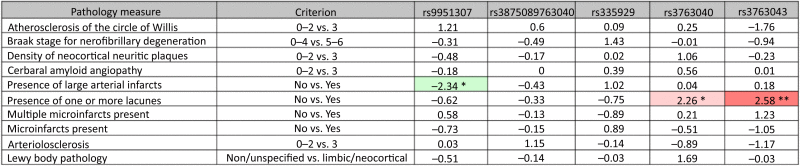 |
Abbreviation: SNP, single-nucleotide polymorphism.
NOTE. Z-statistics for the difference in neurohistopathological measures between AQP4 SNP carriers and noncarriers, post-AD diagnosis. Cells shaded in green represent a significant association with slowed cognitive or functional decline with possession of the SNP. Cells shaded in red represent a significant association with more rapid cognitive or functional decline with possession of the SNP. * = P < .05, ** = P < .01.
4. Discussion
We utilized patient data from several longitudinal cohorts to determine if AQP4 SNPs were associated with altered progression of age-related cognitive or functional impairment. We identified five SNPs in the AQP4 gene where homozygous or heterozygous possession of the minor allele was associated with altered rate of cognitive decline after AD diagnosis compared with noncarriers. rs9951307 and rs3875089 were associated with slower decline or dementia progression, whereas rs3763040 and rs3763043 were associated with faster decline or dementia progression. In addition, we found that subjects carrying the minor allele at both rs3763040 and rs3763043 had faster cognitive decline compared with noncarriers and subjects carrying the minor allele only at rs3763043. Taken together, these data suggest that in persons with AD, SNPs in the AQP4 loci may lead to altered disease progression.
Our results are consistent with previous studies showing that AQP4 SNPs alter clinical outcomes in various central nervous system (CNS) disorders [13], [14], [15], [16]. AQP4 variants have been associated with altered clinical outcomes in TBI [16], sudden infant death syndrome [13], leukoaraiosis [15], and stroke [14]. In all of these disorders, impaired brain fluid homeostasis was thought to lead to neurological damage and dysfunction. In a study of 363 patients with TBI, Dardiotis et al. [16] found that AQP4 SNP rs3763043 was associated with poorer outcomes, as quantified by Glasgow Coma Scale and Glasgow Outcome Scale. In this study, the authors proposed that changes in the formation or resolution of cerebral edema might underlie these reported associations. In agreement with these results, we found that rs3763043 was associated with faster cognitive decline after AD diagnosis. This suggests rs3763043 may be associated with altered AQP4 protein expression, localization and/or function, which subsequently could alter CSF-interstitial fluid dynamics and ultimately brain function.
Although all of the SNPs we investigated are located in the noncoding regions of the AQP4 gene, two SNPs, rs3875089 and rs3763040, are located in the putative promoter region of one of the two isoforms of AQP4, AQP4-M23 (Fig. 3). AQP4 is a 3-kb gene located on chromosome 18 that can be translated either into the AQP4-M1 or the AQP4-M23 isoform based upon which of two alternative transcription-initiation sites are employed. The larger AQP4-M1 isoform moves freely within the astrocyte plasma membrane, whereas the smaller AQP4-M23 isoform complexes in crystalline arrays within the membrane [36]. In our recent study, we observed that the ratio of AQP4-M23:AQP4-M1 increased in subjects with AD and reported that an increase in this proportion was associated with the loss of perivascular AQP4 localization [11]. The fact that rs3875089 and rs3763040 are associated with altered cognitive decline in AD suggests that they may result in differential AQP4-M23 expression, which in turn could lead to altered AQP4 localization and altered Aβ clearance. Future experimental studies are needed to investigate the association between AQP4 genotype and AQP4 isoform expression and localization.
Fig. 3.

Schematic of AQP4 gene and AQP4 protein perivascular localization in the brain. (A) AQP4 consists of five exons (exons 0–4, shown as solid colored rectangles) and can be translated into two different isoforms. The AQP4-M1 isoform consists of exons 0–4 and is 323 amino acids long. The AQP4-M23 isoform consists of exons 1–4 and is 301 amino acids long. When the APQ4 gene is translated into the AQP4-M23 isoform, the subsequent protein aggregates into crystalline arrays that localize to astrocyte endfeet that ensheathe the cerebral vasculature. AQP4 SNPs rs3875089 and rs3763040 are located in the putative promoter of exon 1, which is the first exon of the AQP4-M23 isoform. The other SNPs are located in either the 3′ untranslated region (UTR) (rs335929 and rs3763043) or in the 3′ intronic region. (B) Immunohistochemistry for AQP4 in human cerebral cortex tissue section. AQP4 (labeled in red) is expressed on astrocyte endfeet that ensheathe cerebral blood vessels, including capillaries (B1) and large vessels (B2). Abbreviations: AQP4, aquaporin-4; SNP, single-nucleotide polymorphism.
Neuropathologic analysis of brain-autopsy samples showed that AQP4 SNPs were associated with changes in brain pathology in AD subjects. rs9951307 was associated with lower probability of large arterial infarcts, whereas rs3763040 and rs3763043 were associated with greater probability of multiple microinfarcts compared with noncarriers. Although large arterial infarcts and microinfarcts are typically not classic histopathological features of AD, these results are in alignment with our neurocognitive data. rs9951307 was associated with slower cognitive decline, whereas rs3763040 and rs3763043 were associated with faster cognitive decline. Although experimental studies suggest that AQP4 plays a key role in perivascular Aβ clearance along the glymphatic pathway [8], [9], [10], no associations were observed between AQP4 SNPs and measures of AD pathology such as Braak stage or neuritic plaque density. One possible explanation for this finding is that differences in the pathological progression between SNP carriers and noncarriers are obscured with the approach of AD subjects to the end stage of disease. This might explain why these associations were only evident when evaluating longitudinal neurocognitive data collected through the course of clinical disease. Future studies measuring biomarkers of AD pathology, including CSF Aβ and amyloid positron emission tomography, throughout the course of preclinical, early, and late disease stages will be necessary to conclusively define whether these naturally occurring variants in the human AQP4 gene alter the course of AD pathology.
The limitations in this study are those inherent to its secondary nature and retrospective design. However, the longitudinal nature of measures used in our analysis is a major strength. We report data from 634 subjects that underwent multiple cognitive and functional evaluations over the course of several years, in some cases for nearly 3 decades. Furthermore, this is one of the first studies to investigate the effect of genetic mutations on the progression of AD and rates of cognitive decline. Erten-Lyons et al. [37] conducted an analysis on association of 97 SNPs on brain volume in AD. They found that only the FAS gene, a member of the tumor necrosis factor receptor superfamily, was associated with smaller brain volumes and larger ventricular volumes in AD patients. However, they did not include SNPs in the AQP4 gene in their analysis. In addition, our results are complicated by the fact that all but one of the AD subjects carrying the minor allele at rs3763040 were also carriers of the minor allele at rs3763043. However, we did not observe linkage disequilibrium with these alleles. Furthermore, our results suggest these genes may have a synergistic effect with one another, in that increased rate subjects carrying both the minor allele at rs3763043 and rs7363040 had faster cognitive decline compared with noncarriers and rs3763043 single carriers. Future mechanistic studies are needed to determine the structural and functional effects of these SNPs during AD.
Our results are noteworthy in that associations held true over multiple distinct cognitive and functional domains. In four of the five SNPs we investigated, SNP associations were either “protective” (associated with slower cognitive or functional decline across multiple tests) or “deleterious” (associated with more rapid cognitive or functional decline across multiple tests). However, rs335929 was associated with slower cognitive decline in two different tests, yet was associated with faster functional decline, as indicated by an association with faster increase in CDR SoB score. The reason for this discrepant finding is not clear. Future studies are needed to validate and replicate the associations and the direction of the associations observed in this study. In addition, these results can be further verified and expanded upon by utilizing larger databases (Alzheimer's Disease Neuroimaging Initiative [ADNI], etc.) to investigate the associations between variants in the AQP4 gene and additional structural and functional parameters (magnetic resonance imaging, histopathology, etc.). Furthermore, we selected only five APQ4 SNPs based upon their prior identification as being associated with CNS pathology [13], [14], [15], [16]. Nevertheless, our findings suggest that variation in the AQP4 gene alters the clinical course of AD, and thus, a more comprehensive analysis that better samples the genetic landscape surrounding the AQP4 gene is needed.
We incorporated a rigorous statistical analysis, utilizing a linear mixed modeling paradigm and incorporated stringent multiple comparison corrections to account for the multitude of tests that were run. As such, these results provide a foundation for future functional studies in both rodents and humans. These investigations should assess the effect of identified SNPs on perivascular AQP4 localization, expression levels, AQP4-M1:AQP4-M23 expression ratio, and glymphatic function.
AQP4 function is critical to glymphatic pathway function and the clearance of Aβ [3], [6], [8]. In animal models of aging, Alzheimer's disease, and post-traumatic neurodegeneration, slowing Aβ clearance and increasing tau pathology are linked to changes in AQP4 expression and localization and impairment of glymphatic function [3], [9], [10]. Although these experimental studies suggest that impairment of glymphatic pathway function may contribute to the development and progression of AD pathology, a role for these processes in human AD has not yet been conclusively demonstrated. This is in part due to the absence of clinically relevant imaging approaches to visualizing and evaluating glymphatic function. A recent analysis of human frontal cortical tissue from a cohort of 10 young (25–40 yrs.), 20 aged cognitively intact subjects (>65 years), and 20 aged AD subjects (>65 years) showed that loss of perivascular AQP4 localization was significantly associated with both worsening Aβ plaque burden and neurofibrillary pathology, even when controlling for the impact of age. Similarly, logistic regression analysis showed that perivascular AQP4 localization was a strong predictor of AD status [38]. Though strictly correlative, this recent histopathological study and the present human genetics study provide key data from human AD subjects that support the notion that changes in AQP4 and glymphatic pathway function may contribute to AD pathogenesis. If validated with future studies, and substantiated by the direct clinical imaging approaches, then these studies suggest that glymphatic pathway dysfunction and AQP4 may represent novel therapeutic targets for the prevention and treatment of neurodegenerative diseases like AD.
In summary, we identified five SNPs in the AQP4 gene associated with the rate of cognitive and functional decline in AD. AQP4 is the key astroglial water channel in the brain and is important in the clearance of solutes and proteins from the CNS, including Aβ. This is the first study to investigate the association between mutations in AQP4 and functional outcomes in AD. Although future mechanistic studies are needed to assess functional consequences of AQP4 SNPs, these results provide a potential biomarker for cognitive decline in AD, as well as a potential therapeutic target.
Research in Context.
-
1.
Systematic review: The glymphatic system is a brain-wide perivascular network that facilitates clearance of amyloid β from the brain interstitium and depends on the astrocytic water channel aquaporin-4 (AQP4).
-
2.
Interpretation: We evaluated whether variants in the human AQP4 gene are associated with changes in the progression of AD. Using genetic and cognitive data from longitudinal cohorts, we identified five AQP4 single-nucleotide polymorphisms associated with altered rate of cognitive decline after AD diagnosis; two associated with slower decline, two associated with more rapid decline.
-
3.
Future directions: These findings suggest that AQP4 function may influence trajectory of cognitive decline in AD and support the role of AQP4 and the glymphatic system in the development of AD. Future studies should (1) evaluate whether AQP4 single-nucleotide polymorphisms are associated with AD biomarkers such as CSF amyloid β or amyloid positron emission tomography imaging and (2) determine the molecular basis for the association between noncoding AQP4 variants and AQP4 function.
Acknowledgments
This work was supported by funding from the Oregon Partnership for Alzheimer's Research (J.J.I), the Paul G. Allen Family Foundation (J.J.I), the Research & Development Office of the Department of Veterans Affairs (Merit Review Grant, J.A.K) and the NIH (AG054456, NS089709, J.J.I), including an Alzheimer's Disease Center grant from the National Institute on Aging (NIH, P30-AG08017, J.A.K), which supported the longitudinal follow-up used in this study. J.J.I reports receiving lecture fees from GlaxoSmithKline, Shire, Genentech, and Neurim Pharmaceuticals.
Footnotes
The authors have declared that no conflict of interest exists.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.trci.2017.05.001.
Supplementary data
References
- 1.Tarasoff-Conway J.M., Carare R.O., Osorio R.S., Glodzik L., Butler T., Fieremans E. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wildsmith K.R., Holley M., Savage J.C., Skerrett R., Landreth G.E. Evidence for impaired amyloid beta clearance in Alzheimer's disease. Alzheimers Res Ther. 2013;5:33. doi: 10.1186/alzrt187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iliff J.J., Chen M.J., Plog B.A., Zeppenfeld D.M., Soltero M., Yang L. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180–16193. doi: 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutphen C.L., Jasielec M.S., Shah A.R., Macy E.M., Xiong C., Vlassenko A.G. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 2015;72:1029–1042. doi: 10.1001/jamaneurol.2015.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patterson B.W., Elbert D.L., Mawuenyega K.G., Kasten T., Ovod V., Ma S. Age and amyloid effects on human central nervous system amyloid-beta kinetics. Ann Neurol. 2015;78:439–453. doi: 10.1002/ana.24454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mawuenyega K.G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J.C. Decreased clearance of CNS amyloid-β in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iliff J.J., Lee H., Yu M., Feng T., Logan J., Nedergaard M. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. 2013;123:1299–1309. doi: 10.1172/JCI67677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iliff J.J., Wang M., Liao Y., Plogg B.A., Peng W., Gundersen G.A. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra11. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Z., Xiao N., Chen Y., Huang H., Marshall C., Gao J. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol Neurodegener. 2015;10:58. doi: 10.1186/s13024-015-0056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kress B.T., Iliff J.J., Xia M., Wang M., Wei H.S., Zeppenfeld D. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeppenfeld D.M., Simon M., Haswell J.D., D'Abreo D., Murchison C., Quinn J.F. Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 2017;74:91–99. doi: 10.1001/jamaneurol.2016.4370. [DOI] [PubMed] [Google Scholar]
- 12.Eide P.K., Ringstad G. MRI with intrathecal MRI gadolinium contrast medium administration: a possible method to assess glymphatic function in human brain. Acta Radiol Open. 2015;4 doi: 10.1177/2058460115609635. 2058460115609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opdal S.H., Vege A., Stray-Pedersen A., Rognum T.O. Aquaporin-4 gene variation and sudden infant death syndrome. Pediatr Res. 2010;68:48–51. doi: 10.1203/PDR.0b013e3181df4e7c. [DOI] [PubMed] [Google Scholar]
- 14.Kleffner I., Bungeroth M., Schiffbauer H., Schabitz W.R., Ringelstein E.B., Kuhlenbaumer G. The role of aquaporin-4 polymorphisms in the development of brain edema after middle cerebral artery occlusion. Stroke. 2008;39:1333–1335. doi: 10.1161/STROKEAHA.107.500785. [DOI] [PubMed] [Google Scholar]
- 15.Yadav B.K., Oh S.Y., Kim N.K., Shin B.S. Association of rs2075575 and rs9951307 polymorphisms of AQP-4 gene with leukoaraiosis. J Stroke Cerebrovasc Dis. 2014;23:1199–1206. doi: 10.1016/j.jstrokecerebrovasdis.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 16.Dardiotis E., Paterakis K., Tsivgoulis G., Tsintou M., Hadjigeorgiou G.F., Dardioti M. AQP4 tag single nucleotide polymorphisms in patients with traumatic brain injury. J Neurotrauma. 2014;31:1920–1926. doi: 10.1089/neu.2014.3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payami H., Grimslid H., Oken B., Camicioli R., Sexton G., Dame A. A prospective study of cognitive health in the elderly (Oregon Brain Aging Study): effects of family history and apolipoprotein E genotype. Am J Hum Genet. 1997;60:948–956. [PMC free article] [PubMed] [Google Scholar]
- 18.Kaye J.A., Maxwell S.A., Mattek N., Hayes T.L., Dodge H., Pavel M. Intelligent Systems for Assessing Aging Changes: Home-Based, Unobtrusive, and Continuous Assessment of Aging. J Gerontol B Psychol Sci Soc Sci. 2011;66B:i180–i190. doi: 10.1093/geronb/gbq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaye J., Michael Y., Calvert J., Leahy M., Crawford D., Kramer P. Exceptional Brain Aging in a Rural Population-Based Cohort. J Rural Health. 2009;25:320–325. doi: 10.1111/j.1748-0361.2009.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howieson D.B., Holm L.A., Kaye J.A., Oken B.S., Howieson J. Neurologic function in the optimally healthy oldest old. Neuropsychological evaluation. Neurology. 1993;43:1882–1886. doi: 10.1212/wnl.43.10.1882. [DOI] [PubMed] [Google Scholar]
- 21.McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R., Jr., Kawas C.H. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green M.S., Kaye J.A., Ball M.J. The Oregon brain aging study: neuropathology accompanying healthy aging in the oldest old. Neurology. 2000;54:105–113. doi: 10.1212/wnl.54.1.105. [DOI] [PubMed] [Google Scholar]
- 23.Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer's Disease” The Ronald and Nancy Reagan Research Institute of the Alzheimer's Association and the National Institute on Aging Working Group. Neurobiol Aging. 1998;19:109–116. [PubMed] [Google Scholar]
- 24.Beekly D.L., Ramos E.M., van Belle G., Deitrich W., Clark A.D., Jacka M.E. The National Alzheimer's Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18:270–277. [PubMed] [Google Scholar]
- 25.Folstein M.F., Folstein S.E., McHugh P.R. “Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 26.O'Bryant S.E., Waring S.C., Cullum C.M., Hall J., Lacritz L., Massman P.J., Texas Alzheimer's Research Consortium Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer's research consortium study. Arch Neurol. 2008;65:1091–1095. doi: 10.1001/archneur.65.8.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wechsler D. Pscyhological Corporation; New York: 1987. Wechsler Memory Scale–Revised. [Google Scholar]
- 28.Wechsler D. 1st ed. Williams and Wilkins Corporation; Baltimore: 1939. The Measurement and Appraisal of Adult Intelligence. [Google Scholar]
- 29.Dikmen S.S., Heaton R.K., Grant I., Temkin N.R. Test–retest reliability and practice effects of expanded Halstead–Reitan Neuropsychological Test Battery. J Int Neuropsychol Soc. 1999;5:346–356. [PubMed] [Google Scholar]
- 30.Brenowitz W.D., Keene C.D., Hawes S.E., Hubbard R.A., Longstreth W.T., Jr., Woltjer R.L. Alzheimer's disease neuropathologic change, Lewy body disease, and vascular brain injury in clinic- and community-based samples. Neurobiol Aging. 2017;53:83–92. doi: 10.1016/j.neurobiolaging.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Team R.C. R Foundation for Statistical Computing; Vienna, Austria: 2016. R: A Language and Environment for Statistical Computing.https://www.R-project.org Available at: Accessed May 30, 2017. [Google Scholar]
- 32.Bates D., Mächler M., Bolker B., Walker S. Fitting Linear Mixed-Effects Models Using lme4. J Stat Softw. 2015;1:2015. [Google Scholar]
- 33.de Groot V., Beckerman H., Lankhorst G.J., Bouter L.M. How to measure comorbidity. a critical review of available methods. J Clin Epidemiol. 2003;56:221–229. doi: 10.1016/s0895-4356(02)00585-1. [DOI] [PubMed] [Google Scholar]
- 34.Parmelee P.A., Thuras P.D., Katz I.R., Lawton M.P. Validation of the Cumulative Illness Rating Scale in a geriatric residential population. J Am Geriatr Soc. 1995;43:130–137. doi: 10.1111/j.1532-5415.1995.tb06377.x. [DOI] [PubMed] [Google Scholar]
- 35.Linn B.S., Linn M.W., Gurel L. Cumulative illness rating scale. J Am Geriatr Soc. 1968;16:622–626. doi: 10.1111/j.1532-5415.1968.tb02103.x. [DOI] [PubMed] [Google Scholar]
- 36.Nagelhus E.A., Ottersen O.P. Physiological roles of aquaporin-4 in brain. Physiol Rev. 2013;93:1543–1562. doi: 10.1152/physrev.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erten-Lyons D., Jacobson A., Kramer P., Grupe A., Kaye J. The FAS gene, brain volume, and disease progression in Alzheimer's disease. Alzheimers Dement. 2010;6:118–124. doi: 10.1016/j.jalz.2009.05.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeppenfeld D., Simon M., Haswell J.D., D'Abreo D., Murchison C., Quinn J.F. Preservation of perivascular aquaporin-4 localization in the cognitively-healthy elderly. JAMA Neurol. 2017;74:91–99. doi: 10.1001/jamaneurol.2016.4370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.