

Abstract
Objective(s):
Apatinib recently has been used to treat patients with gastric cancer, but the function of apatinib in colon cancer remains unclear. This study was conducted to investigate the impacts of apatinib on the biological function and its potential mechanism of colon cancer cells in vitro.
Materials and Methods:
The effect of apatinib in colon cancer cells were detected by assessing cell viability, migration and invasion capabilities. Apoptosis cells and the cell cycle distribution of colon cancer cells were analyzed by flow cytometry. The potential mechanism was investigated via autophagy related proteins and pathways in vitro.
Results:
The proliferation, migration and invasion of colon cancer cells were inhibited when they were treated with different concentration of apatinib (20, 40 μM). When HCT116 and SW480 cells were treated with apatinib at the concentration of 20 μM, the apoptosis percentage were 3.7% and 5.8% respectively. As the drug concentration increased to 40μΜ, the the apoptosis percentage increased to 11.9% and 13.5%. Meanwhile, cell cycle was also altered. Furthermore, apatinib inhibited the expression of AKT-mTOR signaling pathway and increased the expression of LC3-II.
Conclusion:
Apatinib can significantly inhibit the malignant phenotype of colon cancer cells, and it was involved in regulation of autophagy.
Keywords: Apatinib, Apoptosis, Autophagy, Colon cancer, Migration, mTOR
Introduction
Colorectal cancer is not only the third largest cancer in the world, but also the fourth largest cause of cancer-related death. It is reported the highest incidence in countries of Europe, North America, and Oceania (1, 2). Currently, the clinical investigation areas are active on identifying new chemotherapy regimens and integrating new targeted therapies with cytotoxic chemotherapy (3). However, due to the heterogeneity of molecular pathogenesis of colon cancer (4), the therapeutic efficiencies of anticancer agents remain to be improved and drug resistance in cancer is still an obstacle to successful target therapy (5). Over the past few years, the therapeutic strategies against vascular endothelial growth factor (VEGF) and its receptor (VEGFR) have been extensively studied due to their important roles in carcinogenesis (6, 7). For example, Bevacizumab is designed to inhibit biologic activity of VEGF-A through blocking the subsequent binding of VEGF-A to its cognate receptors. Regorafenib is an inhibitor that targets a wide range of tyrosine kinases which have effects on the oncogenesis and angiogenesis. As for the mechanisms, the endothelial cell signaling pathways can be activated by VEGF, thereby promoting the division, migration and viability of endothelial cell precursor as well as enhancing its vascular permeability and chemotaxis (8). However, it is not known whether a different therapeutic approach, direct targeting of the receptor for example, might be of benefit in patients who develop resistance to antiangiogenic therapy. There are presently several monoclonal antibodies that are under development.
Apatinib, also known as YN968D1, is a new oral small molecule agent with antiangiogenic effect (9). It has a selectively inhibitory effect on VEGFR-2 as well as c-Kit and c-SRC tyrosine kinases (10, 11). Currently in China, the phase II/III clinical trials of apatinib for the treatment of gastric carcinoma has been finished. As a result, it is applied to treat patients with gastric cancer (12, 13). Considering the great population of colon cancer patients worldwide, it has great significance to clarify therapeutic strategies both in theory and practice. However, the anti-tumor activity of apatinib as well as its potential mechanism in colon cancer has never been studied.
In this study, we investigated the antitumor activity of apatinib in colon cancer HCT116 and SW480 cell lines. The underlying mechanism on pro-apoptotic effect and inhibitory effects of apatinib, especially the alterations of Akt/mTOR mediated pathway were evaluated. As known, the mTOR pathway is one of the signaling pathways closely involved in autophagy regulation(14). In addition, we presented the first evidence that apatinib induced autophagy in colon cancer cells, providing a novel insight into the target molecules or pathways of apatinib.
Materials and Methods
Antibodies and reagent
Antibodies we used are as follows: GAPDH, Akt, phospho-Akt (Ser473), mTOR, phospho-mTOR (Ser 2448), LC3B rabbit antibodies from Cell Signaling Technology (USA)and goat secondary antibody to rabbit (HRP-conjugated). All were purchased from Cell Signaling Technology (USA). Apatinib was dissolved in dimethylsulfoxide (DMSO). The final concentration of DMSO in cell treatment was controlled below 0.1%.
Cells and culture conditions
HCT116 and SW480 cell lines were purchased from the Cell Bank of the Chinese Academy of Science in Shanghai, China. The culture medium contained Dulbecco’s modified Eagle medium (DMEM), 10% fetal bovine serum and 1% penicillin and streptomycin. Cells culture environment is 37 °C, with 5% carbon dioxide.
Cell proliferation assay
HCT116 and SW480 cells were seeded on 96-well plates 24 hr before cells were treated with increasing concentrations of apatinib (0, 10, 20, 30, 40, 50 μM) together with 10% FBS. Then the Cell Counting Kit-8 method (Biyotime, Shanghai, China) was used to detect cell proliferation and the multifunctional microplate reader was used to measure the absorbance. Experi-ments were performed for at least three times.
Scratch wound healing assay
When 6-well plate was covered with monolayer cells, a “wounding” line was scratched in the center. After washing twice, cells were cultured in medium with no serum. Zero and 48 hr after scratch, the width of the wound was respectively measured.
In vitro migration, invasion assays
Cell migration assay was performed with transwell chambers (8 μm). 5×104 HCT116 and SW480 cells were plated in the upper chamber which is filled with 600 μl serum-free DMEM and different concentrations of apatinib. After incubation for 48 hr at 37 °C, cells in the upper membrane were removed and cells in the lower layer of membrane were fixed with 4% paraformaldehyde. Then 0.1% crystal violet dye was used to stain cells. An invert microscope was used to observe migrated cells. The invasion assay was the same as migration assay except that the upper transwell chambers (Clontech, Madison, WI) was coated with matrigel (Corning) for 1hr before plating cells.
Cell-cycle analysis and apoptosis analysis
Colon cancer cells were treated with varying concentrations of apatinib (0, 20, 40 μM) for 48 hr. As for cell-cycle analysis, treated cells were fixed with 75% ice-cold ethanol overnight at -20 °C and then incubated with propidium iodide (PI, 50 μg/ml) and RNaseA (1mg/ml) for 30 min at 37 °C. As for the apoptosis analysis, treated cells were harvested and stained Annexin V-FITC/PI. In each group, 30,000 cells were analyzed. The Scan flow cytometer (BD FACS Calibur) was used to detect the cell cycle and apoptosis respectively. Experiments were performed for three times.
Western blot
Briefly, protein extracts from HCT116 and SW480 cells were equally (30 μg per sample) electrophoresed and transferred. The membranes were blocked in 5% BSA for 1 hr. then they were incubated with diluted antibodies with gentle shaking, at 4 °C overnight. Next day, the membranes were washed and incubated with goat secondary antibody for 1 hr at room temperature. Signals were detected by chemiluminescence imaging system. GAPDH was used as a loading control.
Statistical analysis
Each experiment was performed at least three times. SPSS software version was used for statistical analysis. Student’s t-test was used to analyze statis-tical significance between different experimental groups. Results were expressed as mean values±SD. P<0.05 indicated a statistically significant difference. Graphs were created with Graphpad Prism 6.
Results
Apatinib inhibited cell proliferation in vitro
HCT116 and SW480 cells were initially treated with increasing concentrations of apatinib (0~50 μM) (Figure 1a). IC50 of apatinib treatment on HCT116 and SW480 cells were 48.61±1.68 and 43.86±1.64 μM. Therefore, the treatment with aptinib at 20 μM and 40 μM were used for all further experiments. To explore whether the capacity to inhibit cell growth of apatinib was connected with treatment time, then cells were treated in an increasing time gradient. As was shown in Figure 1b, the inhibition rate of apatinib on cell proliferation increased with the time apatinib treated.
Figure 1.
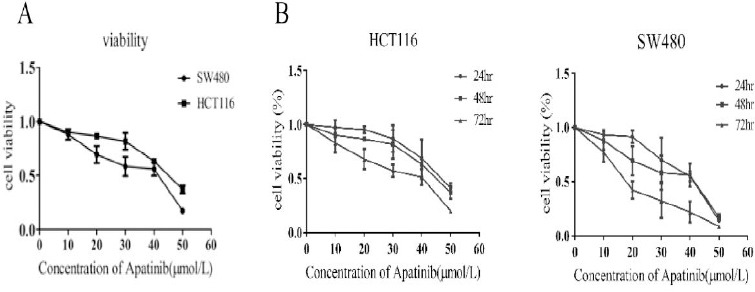
Apatinib inhibited cell proliferation in vitro
(a) Cell viability assays of cells treated with an increasing concentration of apatinib for 48 hr. (b) The cells were treated with apatinib (20 μM, 40 μM) for a varying time. The inhibitory activity was expressed as inhibition rate. n=4, P<0.05
Apatinib dose-dependently promoted cell cycle arrest and induced cell apoptosis
Cells were treated with different concentrations of apatinib. After 48 hr, a relatively normal pattern can be observed in cells treated with no apatinib. Cells (HCT116 and SW480, respectively) in the G1 phase were (41.12±1.83)%, (32.91±2.34)% and there existed a lower S phase peak (31.32±2.08)%, (41.18±1.92)%. As Figure 2a shown, after cells were treated with apatinib in the concentration of 20 and 40μM, the distributions of cell cycle of HCT116 and SW480 were as follows: the S phase was (22.22±2.73)%, (34.42± 4.07)%, G1 phase (61.38±2.08)%, (41.88±2.45)% when cells was treated with 20 μM apatinib, while the S phase was (18.36±3.45)%, (19.55±3.67)%, G1 phase(62.37±1.43)%, (71.69±2.38)% when with 40 μM apatinib. Apatinib treated HCT116 and SW480 cells exhibited that the percen-tage of G1 phase was significant increased and the S phase decreased (P<0.01). As indicated above, the increased effect of apatinib treatment on cell cycle distribution was in accordance with increased concentration. These findings clearly manifested that apatinib modulates colon cells at G0–G1 phase in progression of the mitosis. As for the effects of apatinib on cell apoptosis (Figure 2b), we found that the number of apoptotic cells was significantly increased in both HCT116 and SW480 cells which were treated with apatinib (P<0.01). In general, these results suggest that apatinib regulates both cell cycle and apoptosis in colon cells.
Figure 2.
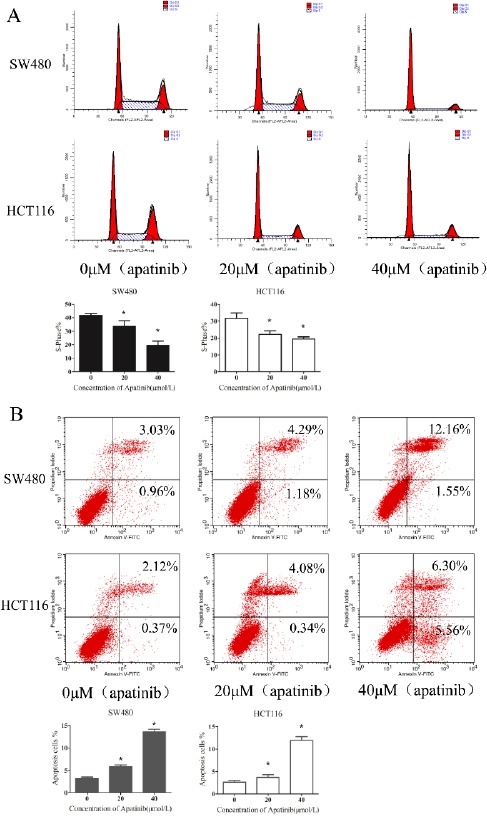
Apatinib dose-dependently promoted cell cycle arrest and induced cell apoptosis.
(a)After cells treated with apatinib (0, 20, 40 μM, respectively) for 24 hr, the cells were stained with Annexin V-FITC/PI and determined by FASC analysis. The percentage of apoptotic cell numbers is shown. (b) The distribution of the cell cycle of HCT116 and SW480 cells were determined by flow cytometry after treating with apatinib (0, 20, 40 μM, respectively) for 24 hr. The percentage of S—phase is shown. * represents P<0.05
Apatinib inhibited colon cell migration and invasion
As Figure 3a and 3b shown, the migration and invasion effects were significantly reduced in both HCT116 and SW480 cells which were treated with apatinib (P<0.01). The migration and invasion rate was dependent on the consitration of apatinib. Further-more, wound healing assay was also conducted to assess the impact of apatinib on cell movement (Figure 3c). Consistent with our previous results, apatinib treatment inhibited the mobility of colon cells and inhibition rate increased with the concentration. These results indicated that apatinib may be a promising anti-metastatic agent to reduce migration and invasion.
Figure 3.
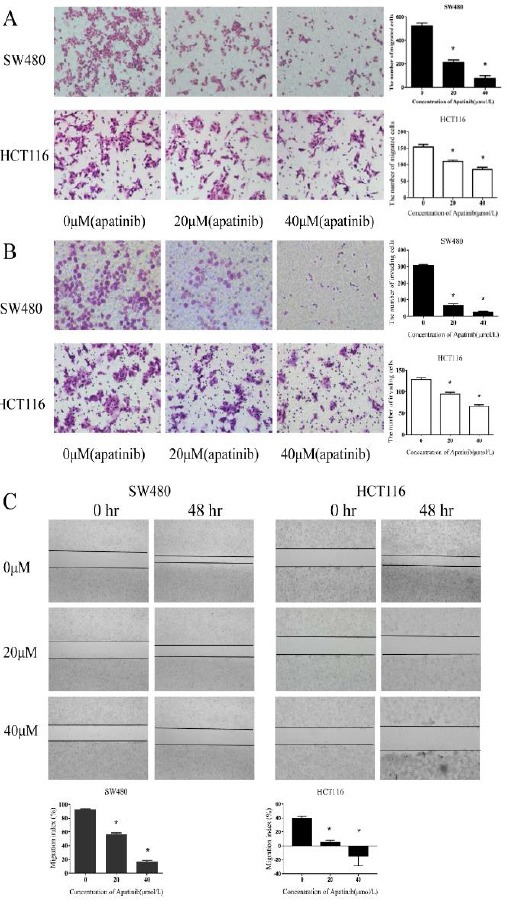
Apatinib inhibited colon cell migration and invasion
(a) HCT116 and SW480 cells after the cells were treated with apatinib (0, 20, 40 μM, respectively) for 40 hr. The migrated cells were stained. (b) HCT116 and SW480 cells after the cells were treated with apatinib (0, 20, 40μM, respectively) for 40 hr. The invaded cells were stained. (c) HCT116 and SW480 cells after the cells were treated with apatinib (0, 20, 40 μM, respectively) for 48 hr. The migration index (the ratio of migration distance to total distance) was used to measure the movement ability. * represents P<0.05
Apatinib induced apoptosis through inhibition of AKT/mTOR pathway
To determine the signaling pathway related to apatinib induced apoptosis, we examined the expression of AKT, pho-AKT, mTOR and pho-mTOR in cells after treating cells with different concentrations of apatinib (0, 20 μM and 40 μM) for 24 hr (Figure 4a). The expression of total AKT protein remained unchanged. However, treatment with apatinib at 20 μM and 40 μM resulted in a great reduction of pho-AKT in both cell lines. Concurrently, the levels of pho- mTOR protein in apatinib treated colon cells were inhibited compared to the control cells. In conclusion, these results demonstrate that regulation of the AKT/mTOR pathway is closely related to apoptosis and growth inhibition induced by apatinib in colon cancer cells.
Figure 4.
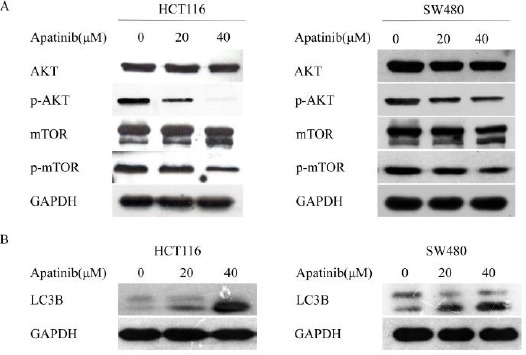
Apatinib induced apoptosis and autophagy through inhibition of AKT/mTOR pathway
(a) HCT116 and SW480 cells were treated with apatinib (0, 20, 40 μM, respectively) for 24 hr, and the alterations of phosphorylated AKT and mTOR were detected. (b) HCT116 and SW480 cells were treated with apatinib (0, 20, 40 μM, respectively) for 24 hr, and the protein level of LC3 was detected. GAPDH was included as a loading control
Apatinib induced autophagy in colon cancer cells
As above, there was a significant reduction of pho-mTOR protein in apatinib treated colon cells. To explore whether apatinib played a role in cell auto-phagy, we treated cells with apatinib for 24 hr and the expression of LC3-II was detected. It was found that the levels of LC3-II in the apatinib treated cells were obviously higher than the control group (Figure 4b). Furthermore, we detected the levels of the pro-autophagic protein Beclin-1 which is essential for autophagosome formation. Consistently, Beclin-1 was also increased in apatinib-treated cells (supplementary Figure).These data support the view that apatinib induced autophagy in colon cancer cells.
Discussion
In early studies, it has been found that the growth of adjacent tumor cells can be directly stimulated by the new vascular system which can secrete growth factors (8). However, the potentially new targets for therapy were not recognized until the important discovery of VEGF family and their homologous receptors which are the main molecular drivers of tumor angiogenesis
Apatinib is now recognized as the first generation of oral anti-angiogenesis drugs in China, which is also a potential new third-line option for refractory gastric cancer (15). Currently, a definitive conclusion of apatinib could not be given due to the insufficient number of patients recruited in clinical trials. However, in massive pretreated patients, the survival rates including overall survival and progression-free survival have been improved (16). As for the mechanism, it is proved that apatinib can inhibit migration and proliferation of endothelial cell which is stimulated by VEGF. Thus, it is considered as a promising VEGFR-2 inhibitor to inhibit tumor-induced angiogenesis, (17, 18).
In this study, we found that apatinib played a magnificent inhibition role in the proliferation, migration and invasion of colon cancer cells, and it showed a concentration-dependent manner. Furthe-rmore, we demonstrate that apatinib inhibited AKT-mTOR signaling pathway and increased the expression of LC3-II which is a marker for autophago-some number (19). In our work, we prove that apatinib is also cytotoxic to colon cancer cells. It is shown that two major mechanisms by which apatinib kill colon cancer cells are induction of apoptotic cell death and loss of cell viability. Taken together, these phenomena are in a way consistent with the clinical implications of apatinib, so as to facilitate the selection for further treatment in colon cancer patients.
The existing researches are mostly emphasized on the antiangiogenic role of apatinib (20, 21). Intriguingly, our work revealed that apatinib treat-ment could inhibit AKT/mTOR signaling pathway and induce autophagy. As we all known, autophagy is gradually noticed and considered to be an important catabolic process since 1960s (22), which is aimed to meet the change of energy requirement or to decrease the accumulation of toxic products by recycling and reallocate cellular components (14, 23, 24). It is well-known that Akt/mTOR pathway can regulate apoptosis and autophagy (25). In the process of autophagy, the Ulk1 autophagic complex can be positively regulated by the inhibition of mTORC1 and consequently promotes the autophagy and apoptosis (26). In the early 1970s, Folkman emphasized the role of neovascularization on tumor cells proliferation and metastasis and suggested that blocking the angio-genesis could finally suppress tumor cells growth. At present time, one popular notion is that autophagy promotes tumor cell death and inhibits tumor growth at the stage of tumorigenesis. In a word, autophagy has a vital effect in inhibiting tumorigenesis (27). However, there are rare studies which focus on the association between autophagy and anti-angiogenesis therapy.
We observed that LC3-II expression and apoptosis were both promoted in colon cancer cells treated with apatinib. These results indicate that apatinib may be a promising therapeutic agent to treat colon cancer and combining with autophagy is an exciting and effective new therapeutic strategy for patients with pancreatic cancer.
Conclusion
In summary, our present study not only have revealed that apatinib can significantly inhibit the biological function of colon cancer cells in vitro, but also put new insight into the regulation on autophagy and apoptosis of apatinib in colon cancer cells. These observations suggest a promising therapeutic strategy for treating colon cancer through combining apatinib with autophagy inducers. However, further animal studies or clinical trials need to be confirmed and experiments focusing on combined chemo-therapy drugs need to be explored as well.
Acknowledgment
The authors sincerely thank Mr Xia Hong for his administrative support and excellent technical assistance in this work. This study was supported by two National Natural Science Foundation of China (No. 81641110 and No.81703030) and Natural Science Foundation of Guangdong province (No. 2015A030313725).
References
- 1.Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1437. doi: 10.1016/S0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- 2.bParkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001;2:533–543. doi: 10.1016/S1470-2045(01)00486-7. [DOI] [PubMed] [Google Scholar]
- 3.Gill S, Thomas RR, Goldberg RM. Colorectal cancer chemotherapy. Aliment Pharmacol Ther. 2003;18:683–692. doi: 10.1046/j.1365-2036.2003.01735.x. [DOI] [PubMed] [Google Scholar]
- 4.Hu T, Li Z, Gao CY, Cho CH. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J Gastroenterol. 2016;22:6876–6889. doi: 10.3748/wjg.v22.i30.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas H, Coley HM. Overcoming multidrug resistance in cancer:an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S, Heukamp LC, Siobal M, Schottle J, Wieczorek C, Peifer M, et al. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J Clin Invest. 2013;123:1732–1740. doi: 10.1172/JCI65385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fontanella C, Ongaro E, Bolzonello S, Guardascione M, Fasola G, Aprile G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann Translat Med. 2014;2:123. doi: 10.3978/j.issn.2305-5839.2014.08.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology:current status and future directions. Lancet. 2016;388:518–529. doi: 10.1016/S0140-6736(15)01088-0. [DOI] [PubMed] [Google Scholar]
- 9.Roviello G, Ravelli A, Polom K, Petrioli R, Marano L, Marrelli D, et al. Apatinib:A novel receptor tyrosine kinase inhibitor for the treatment of gastric cancer. Cancer Lett. 2016;372:187–191. doi: 10.1016/j.canlet.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Ding J, Chen X, Gao Z, Dai X, Li L, Xie C, et al. Metabolism and pharmacokinetics of novel selective vascular endothelial growth factor receptor-2 inhibitor apatinib in humans. Drug Metab. 2013;41:1195–1210. doi: 10.1124/dmd.112.050310. [DOI] [PubMed] [Google Scholar]
- 11.Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. 2009;6:569–579. doi: 10.1038/nrclinonc.2009.130. [DOI] [PubMed] [Google Scholar]
- 12.Ding J, Chen X, Dai X, Zhong D. Simultaneous determination of apatinib and its four major metabolites in human plasma using liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study. J Chromatogr B. 2012;895-896:108–115. doi: 10.1016/j.jchromb.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 13.Scott AJ, Messersmith WA, Jimeno A. Apatinib:a promising oral antiangiogenic agent in the treatment of multiple solid tumors. Drugs Today. 2015;51:223–229. doi: 10.1358/dot.2015.51.4.2320599. [DOI] [PubMed] [Google Scholar]
- 14.Yang J, Carra S, Zhu WG, Kampinga HH. The regulation of the autophagic network and its implications for human disease. Int J Biol Sci. 2013;9:1121–1133. doi: 10.7150/ijbs.6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aoyama T, Yoshikawa T. Targeted therapy:Apatinib - new third-line option for refractory gastric or GEJ cancer. Nat Rev Clin Oncol. 2016;13:268–2670. doi: 10.1038/nrclinonc.2016.53. [DOI] [PubMed] [Google Scholar]
- 16.Roviello G, Ravelli A, Fiaschi AI, Cappelletti MR, Gobbi A, Senti C, et al. Apatinib for the treatment of gastric cancer. Exp Rev Gastroenterol Hepatol. 2016;10:887–892. doi: 10.1080/17474124.2016.1209407. [DOI] [PubMed] [Google Scholar]
- 17.Tian S, Quan H, Xie C, Guo H, Lu F, Xu Y, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potentactivity in vitro and in vivo. Cancer Sci. 2011;102:1374–1380. doi: 10.1111/j.1349-7006.2011.01939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Zhao X, Chen L, Guo H, Lv F, Jia K, et al. Safety and pharmacokinetics of novel selective vascular endothelial growth factor receptor-2 inhibitor YN968D1 in patients with advanced malignancies. BMC Cancer. 2010;10:529. doi: 10.1186/1471-2407-10-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaaf MB, Keulers TG, Vooijs MA, Rouschop KM. LC3/GABARAP family proteins:autophagy-(un)related functions. FASEB J. 2016;30:3961–3978. doi: 10.1096/fj.201600698R. [DOI] [PubMed] [Google Scholar]
- 20.Chan MM, Sjoquist KM, Zalcberg JR. Clinical utility of ramucirumab in advanced gastric cancer. Biologics. 2015;9:93–105. doi: 10.2147/BTT.S62777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fornaro L, Vasile E, Falcone A. Apatinib in Advanced Gastric Cancer:A Doubtful Step Forward. J Clin Oncol. 2016:JCO686931. doi: 10.1200/JCO.2016.68.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahlberg J, Glaumann H. Uptake--microautophagy--and degradation of exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Exp Mol Pathol. 1985;42:78–88. doi: 10.1016/0014-4800(85)90020-6. [DOI] [PubMed] [Google Scholar]
- 23.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JJ, Chu E. Sequencing of antiangiogenic agents in the treatment of metastatic colorectal cancer. Clin Colorectal Cancer. 2014;13:135–144. doi: 10.1016/j.clcc.2014.02.001. [DOI] [PubMed] [Google Scholar]