

Abstract
Objective(s):
Development of the nervous system in human and most animals is continued after the birth. Critical role of this period in generation and specialization of the neuronal circuits is confirmed in numerous studies. Any pharmacological intervention in this period may result in structural, functional or behavioral abnormalities. In this study, sodium thiopental a GABA mimetic drug was administrated to newborn rats and their GAD65 and GAD67 expression in hippocampus was evaluated before and after puberty.
Materials and Methods:
Newborn male Wistar rats were received sodium thiopental (35 mg/kg) daily for 11 days (from 4 to 14 days after birth). Expression of GAD65 and GAD67 in their hippocampus was compared with control groups in 15 and 45 days after birth with RT-qPCR method.
Results:
Significant down regulation of GAD65 and GAD67 gene expression was observed in treated rats compared with control group in 45 days after birth animals. But no significant difference was shown between experimental and control groups 15 days after birth animals.
Conclusion:
The effect of sodium thiopental on GAD65 and GAD67 expression only at adult rats showed a latent period of influence which can be attributed to dosage or intension of sodium thiopental neurotoxicity. Significant down regulation of GAD65 and GAD67 showed unwanted effect of sodium thiopental as GABA mimetic drug in critical period of development.
Keywords: Gamma aminobutyric – acid, Glutamate decarboxylase 1, Glutamate decarboxylase 2, Real-time polymerase – chain reaction, Thiopental
Introduction
Optimal function of neural circuits is depending on balance of neurotransmitters and functions of receptors. Normal conformation of synapses and development during pre and post-gestation, takes up to 4 years in human and 2 weeks in rodent. Disruptive factors may affect neural system in any above mentioned aspects. Gama-aminobutyric acid (GABA) is one of the most abundant inhibitory neurotrans-mitter in the mammalian nervous system, where its neurons widely distributed in most regions of the brain such as the neocortex, hippocampus, thalamus, basal ganglia, brainstem, cerebellum, and hypothala-mus. This neurotransmitter represents about one-third of the brain synapses. The majority of these neurons in the neocortex are inhibitory interneurons (1-5).
During neurotransmission, GABA through allos-teric interaction with GABA receptors leads to open-ing of the chloride (Cl¯) and potassium (K+) channels which result in conductance increase of Cl¯ and K+, pre and post-synaptically (6-8). Therefore, GABA receptor activation reduce the cell excitability through the hyperpolarization of the postsynanptic neurons which proceed the inhibitory effects of GABA. Conversely, activation of such receptors is accom-panied by an increase in the amplitude of the fiber volley evoked by axon stimulation in immature rats. Furthermore, application of the GABAA receptor agonist (muscimol) enhances glutamate release from isolated boutons onto acutely dissociated pyramidal neurons (8). GABAA receptors have several binding sites for different ligands, such as muscimol (GABA agonist), bicuculline (GABA antagonist), benzodiaze-pines (BZDs), barbiturates, ethanol and neurosteroids (9, 10). These are allosteric agents which act on GABA receptors and lead to increased GABA affinity or increase in frequency of Cl¯ channel opening (11-13).
An enzymatic reaction mediated by glutamic acid decarboxylase (GAD) and pyridoxal phosphate (as
cofactor), are responsible for GABA synthesis from glutamate (14, 15). GAD is localized only in GABAergic presynaptic neurons and terminals with two common forms, GAD65 and GAD67 (16). These isoforms are encoded by two independent genes and have different subcellular localizations. Electrophysiological studies demonstrated that inhibiting GAD results in a reduction in the size of miniature synaptic events, which represent the amount of GABA released from a single synaptic vesicle (13). In contrast, knocking-out of the predominant membrane transporter responsible for GABA reuptake does not influence the size of these miniature events (17). These findings show higher importance of new synthesis in comparison with recycling of existing GABA and suggest that any factor influencing GABA synthesis is likely to play an important role in maintaining and possibly regulating inhibitory synaptic transmission.
Previous studies have shown that GAD65 knocked-out mice are more likely to develop seizures than wild type (18). A different GAD65 knocked-out mouse had an epileptic phenotype characterized by spontaneous seizures that may led to death (19). These animals also showed increase in anxiety like behaviors and diminished response to anxiolytics (20), pre-pulse inhibition deficits (21), up-regulation of the vesicular GABA transporter, and increased cytosolic GABA transport into synaptic vesicles (22). GAD67 localizes in the cell soma of inhibitory neurons. The GAD67 knocked-out mouse shows a reduction in GABA levels throughout the brain, a reduction in GAD activity, and severe cleft palate which leads to death within 24 hr after birth (23).
The most schizophrenia related illness is associated with GABAergic abnormalities in hippocampal activity (24-27). Most schizophrenia studies focus on adults, but the pathogenesis may involves early stage of brain development (28). GABAA receptors activation in pre-development period leads to chloride efflux and membrane depolarization sufficient to open voltage sensitive calcium channels (VSCCs), particularly the L-type one (29-34). As development progresses, there is a gradual positive shift in the equilibrium potential for chloride (35). So, by the middle of the second postnatal week in the rat, GABAA receptor activation results in chloride influx and membrane hyperpolarization of hippocampal neurons (35).
Recent findings indicate that drugs that act by either stimulation of GABA receptors or inhibition of N-Methyl D-Aspartate(NMDA) receptors induce widespread neuronal apoptosis in immature rat brain when administered during synaptogenesis (36). in this study, we administered (daily) thiopental sodium as a GABA mimetic general anesthetics to neonatal rats in critical period of neural development repeated for 11 days (from 4 to 14 days after birth) and we focused on gene expression levels of GAD65 and GAD67 of hippocampus structure in two period of life (neonate and juvenile).
Materials and Methods
Experimental procedures
Animal preparation
16 newborn male Wistar rats were used. Animals housed in the Vivarium of Biology Department, Faculty of Science, Ferdowsi University of Mashhad. Rats were housed under a 12:12 hr light/dark cycle with free access to their mothers, food and water, in a normal temperature (22± 2 °C) and humidity (55-60%). Day of birth was assigned as postnatal day 1. All animal procedures were approved by the instructions of Animal Care and Use Committee of Ferdowsi University of Mashhad.
Experimental design
Newborn male rats were divided into experi mental and control groups (n=8). Daily administration of sodium thiopental (Sandoz, Austria) with 35 mg/kg, Intra peritoneal (37) was done for 11 days (from 4-14 days) consistently for all of the critical periods time except 3 first days after birth, because of intolerance and death under anesthetic condition. The same procedure was done for control group with placebo. Both experimental and control animals were separate from their mother’s in duration of anesthesia (about 30-45 min) and their body’s temperature was fixed in 35 °C with an special thermoregulation pad. One day after the last injection, all animals randomly were entered into two experiments: Experiment 1 and Experiment 2. Each experiment included treatment (T) and control (C) groups (n=4). In Experiment 1, animals in both groups (C1 and T1) were euthanized (day 15) and their brain was dissected and their hippocampus until RNA extraction was kept in -80 °C. Animals in Experiment 2 (C2 and T2) were euthanized at the adulthood (postnatal day 45) and similar procedure was done.
Gene expression analysis
RNA extraction, quality and quantity analysis and cDNA synthesis
Gene expression for GAD65 and GAD67 was ana-lyzed using quantitative real time PCR. Total RNA was extracted using Qiagen mini kit (Cat.Number: 74104) according to the manufacturer’s specifications. Briefly, 30 mg of target tissues with first solution of kit (RW1) in addition beta-mercaptoethanol were homogenized and centrifuged. Supernatant was moved to a new tube and ethanol 70% was added to precipitate RNA and pipetting was done. Next, these samples were moved to the column of kit and centrifuged. DNase I (fermentase) treatment for 5 min was done and RW1 was added to the column again and centrifuged. Then, RPE as the second solution of kit was added to the column twice to wash out ethanol residue. Finally, 50 µl RNase free water was added to the column to wash out the extracted RNA. Concentration and quality of RNA was determined using Nano Drop spectro-photometer (Thermo Scientific, Germany) and gel electrophoresis, respectively.
cDNA synthesis
Single-strand cDNA synthesis was performed in thin-walled PCR tubes containing 1000 ng template RNA, deoxyribonucleotide triphosphates (dNTPs), diethylpyrocarbonate (DEPC) water and oligo (dt) was mixed and followed by thermal condition 65 °C for 5 min. Next, 5X Reaction Buffer and Maxima H Minus First Strand cDNA Synthesis Kit was added (final volume 20 µl) at 37 °C for 1 hr. The samples were centrifuged slowly and recommended thermal condition was followed.
Relative quantitative real-time RT-PCR
Based on the sequence information of GAD65, GAD67 and β-actin genes on NCBI database, specific forward and revers primers were designed using primer premier-6 (Premier Biosoft International, Palo Alto, CA, USA) and primer BLAST software (Table 1). Relative quantitative real-time RT-PCR (qPCR) was performed on cDNA prepared from isolated RNA for the reference gene β-actin and the genes of interest GAD65, GAD67. All assays were assured to detect cDNA derived from mRNA, and not genomic DNA, by performing appropriate reverse transcriptase minus controls.
Table 1.
Characteristics of primer pairs which were used in qPCR reactions
| Gene | Primer sequence | Amplication size | Annealing Temperature | Accession Number |
|---|---|---|---|---|
| GAD65 | F: 5’ CTGGAAGACAATGAAGAGAGAATG 3’ | 130 | 57 | M72422 |
| R: 5’ TGCGGAAGAAGTTGACCTTATC 3’ | ||||
| GAD67 | F: 5’ GGGACACTTGAACAGTAGAGAC 3’ | 113 | 57 | M76177 |
| R: 5’ GACGCAGGTTGGTAGTATTAGG 3’ | ||||
| β-Actin | F: 5’ CGTGCGTGACATTAAAGAGAAG 3’ | 134 | 57 | NM_031144 |
| R: 5’ CATTGCCGATAGTGATGACCTG 3’ |
First, the required standard curves were obtained and efficiency of reactions was calculated. PCRs were done in conjunction with the SYBER GREEN in an ABI 7300 Sequence Detection System (Applied Biosystems). Thermal cycle was done as 10 min pre-incubation in 95 °C, next 45 cycles of 30 sec at 95 °C and 30 sec at 57 °C and 30 sec 72 °C, and finally 15 sec denaturation step at 95°C, 1min 57 °C for annealing, again 15 sec 95 °C and 15 sec at 60 °C for melting. qPCR products were run on 1% agarose gel electrophoresis to confirm results. Obtained threshold cycle values (CT) were normalized to the reference gene (β actin) for each target gene of interest with pfaffl method.
Data analysis
Ct values from 4 samples in treatment and control groups of each experiment (with triplicate read) were analyzed by t-test using SPSS.16 software and data are presented as mean±SEM. P<0.05 were considered as significant difference. Outlier data was deleted in analysis procedure.
Results
RNA quality and expression level of reference gene
Only samples with proper quality and quantity were used for qPCR reactions. Absorbance values for all RNA samples at 260 and 280 nm (A260/280) and 260 and 230 nm (A260/230) were 2.0–2.2 and 1.8–2.2, respectively. Gel electrophoresis of RNA samples showed two clear bands related to rRNA (28s and 18s) along with 5s band which confirmed the quality of RNA. There was no evidence of contaminating genomic DNA in all runs. Moreover, negative control and RT minus samples confirmed no genomic DNA contamination in each experiment.
The expression level of reference gene (β-actin) was almost similar in all cases which is a critical criteria for reference gene (Figure 1). The melting curve analysis showed the specificity of the β-actin primers. In addition, primers were validated by amplification efficiencies (E=10-1/slope) of 100% ± 10% and the efficiency was 1.03, 1 and 0.91 for β-actin, GAD65 and GAD67 respectively.
Figure 1.
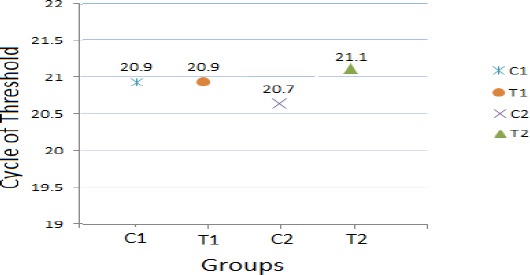
Expression pattern of reference gene (β-actin) in groups of both experiments (C = Control, T= Treatment, C1 and T1 for experiment 1, C2 and T2 for experiment 2)
Experiment 1: expression of GAD65 and GAD67 genes was not affected in treated animals with sodium thiopental in neonates
The melting curve analysis showed the specificity of the GAD65 and GAD67 primers. Subsequently, the agarose gel electrophoresis of products confirmed the size and purity of amplification products in various samples (Figure 4). There was not any significant difference in expression of GAD65 and GAD67 genes in neonatal period (postnatal day 15) between treated (T1) and control (C1) animals (Figure 2).
Figure 2.
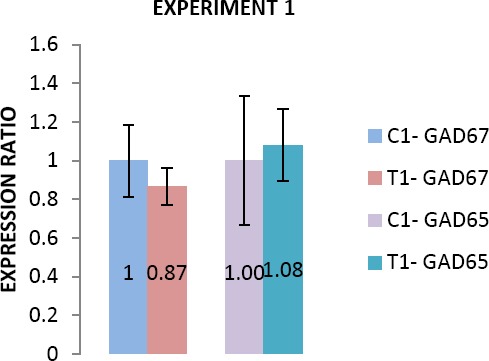
Comparison of GAD65 and GAD67 expression between control and treatment (C=Control, T=Treatment) subgroups in neonate animals (experiment 1). Data are presented as mean±SEM
Figure 3.
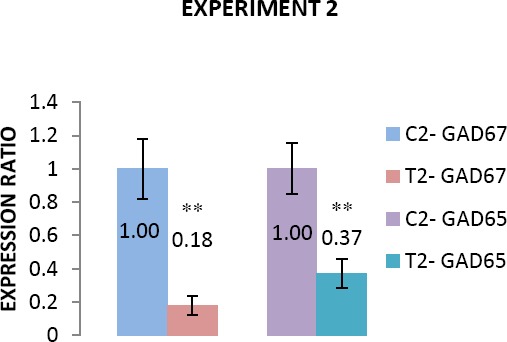
Comparison of GAD65 and GAD67 expression between control and treatment (C=Control, T=Treatment) subgroups in puberty (experiment 2). Data are presented as mean±SEM
Figure 4.

Representative ethidium bromide-stained gel electro-phoresis confirmed the size of all generated PCR products was at 134 bp for β-actin in 1 and 2 columns, 130 bp for GAD65 in 3 and 4 columns and 113 bp for GAD67 in 5 and 6 columns. Lane C- is negative control and lane M100 is 100-bp DNA marker
Experiment 2: GAD65 and GAD67 genes were down regulated in pubertal age in treated animal with sodium thiopental
Expression of GAD65 and GAD67 genes in pubertal age (post-natal day 45) in treated group compared with control, showed a significant down regulation (P<0.05). Expression of GAD65 and GAD67 were declined 63 and 82 percent in treated animals compared with control ones (Figure 3).
Discussion
This study showed that the application of sodium thiopental a GABA mimetic drug in critical period of development results in significant down regulation of mRNA expression of GAD65 and GAD67 genes in adulthood rats without any effect in neonatal period.
GABA mimetic drugs have a wide functional effect from sedation to general anesthesia. Biophysical effect of GABA depends on two cation-chloride cotransporters -NKCC1 and KCC2- that mediate the polarization of postsynaptic membrane by regulation of intracellular Cl− concentration. The direction and degree of GABAA receptor-mediated Cl− current, among other things, depend on the chloride gradient across the neuronal membrane. This phenomenon, in turns, depends on the relative expression and activity of NKCC1 and KCC2 which cause a higher intracellular concentration of chloride versus a higher extracellular concentration of chloride, respectively. Accordingly, activation of GABAA receptors during higher expression of NKCC1 (early stage of life) results in depolarization, and conversely can lead to hyperpolarization when KCC2 expression is up (post developmental period) (24, 38-41). In fact, NKCC1/KCC2 expression ratio change parallels the change of GABA from an excitatory to an inhibitory neurotransmitter (24, 40, 41). Therefore, drugs can act on the GABAergic system in two opposite manners: excitatory (in embryonic period) and inhibitory (in adulthood). Excitatory action of GABA can cause inward current of calcium that is crucial for morphological changes, synaptogenesis and plasticity of neurons. Thus, administration of these drugs in pregnancy will affect pregnant mother as sedative or anesthetic whereas lead to excitatory effect drug on fetal nervous system. Administration of these drugs in neonates can induce adverse effects on nervous system and its development.
Development of neural system include not only neuroanatomical structures, quantity of nerve cells, but also formation of neural circuits, synaptic patterns, and qualitative and quantitative balance between receptors and ligands, that pharmacological intervention can change this developmental process. Down regulation in expression of GAD65 gene as the principle enzyme in vesicular GABA synthesis and reduction in inhibitory neurotransmitter source can effect neural transmission in GABAergic circuits of the brain. Balance between excitation and inhibition is tightly regulated in the normal condition. Disturbed excitatory/inhibitory balance is associated with numerous neuropsychological disorders, such as autism, epilepsy and schizophrenia (42). Expression of GAD67 gene the principle enzyme responsible for tonic production of GABA is required for its trophic effect on formation of neural system (41). Therefore, its reduction can enhance the possibility of abnormality in any aspect of morphometric, neuroanatomical, neurochemical or other characteristics of treated animals.
This study was focused on gene expression at mRNA level. Study on expression of GAD65 and GAD67 genes in neonates showed non-significant difference between control and treated animals that maybe due to a latent response. Generally, traditional study on gene expression is based on the source of transcript and not transcription. Therefore, these results cannot be a conclusive cause for lack of any effect on transcription.
Conclusion
Our results in this study suggested that there is a significant effect for use of sodium thiopental as a GABA mimetic drug in critical period of neural development on GAD65 and GAD67 expression in puberty and non-significant effect on neonatal age. Future studies can clarify more detailed of sodium thiopental side effects.
Acknowledgment
This work was financially supported by the Ferdowsi University of Mashhad, Iran No. 3/13465).
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Zachmann M TP, Nyhan WL. The occurrence of gammaaminobutyric acid in human tissues other than brain. J Biol Chem. 1966;241:1355–1358. [PubMed] [Google Scholar]
- 2.Otsuka M, Iversen LL, Hall ZW, Kravitz EA. Release of gamma-aminobutyric acid from inhibitory nerves of lobster. Proc Natl Acad Sci U S A. 1966;56:1110–1115. doi: 10.1073/pnas.56.4.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meldrum B. Pharmacology of GABA. Clin Neuro-pharmacol. 1982;5:293–316. doi: 10.1097/00002826-198205030-00004. [DOI] [PubMed] [Google Scholar]
- 4.Guidotti A, Corda MG, Wise BC, Vaccarino F, Costa E. GABAergic synapses. Supramolecular organization and biochemical regulation. Neuropharmacol. 1983;22:1471–1479. doi: 10.1016/0028-3908(83)90115-6. [DOI] [PubMed] [Google Scholar]
- 5.Ouellet L, de Villers-Sidani E. Trajectory of the main GABAergic interneuron populations from early develop-ment to old age in the rat primary auditory cortex. Front Neuroanat. 2014;8:40. doi: 10.3389/fnana.2014.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bormann J. Electrophysiology of GABAA and GABAB receptor subtypes. Trends Neurosci. 1988;11:112–116. doi: 10.1016/0166-2236(88)90156-7. [DOI] [PubMed] [Google Scholar]
- 7.Macdonald RL, Twyman RE, Ryan-Jastrow T, Angelotti TP. Regulation of GABAA receptor channels by anticonvulsant and convulsant drugs and by phosphorylation. Epilepsy Res. 1992;9:265–277. [PubMed] [Google Scholar]
- 8.Ruiz A, Campanac E, Scott RS, Rusakov DA, Kullmann DM. Presynaptic GABAA receptors enhance transmission and LTP induction at hippocampal mossy fiber synapses. Nat Neurosci. 2010;13:431–438. doi: 10.1038/nn.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicoll RA, Eccles JC, Oshima T, Rubia F. Prolongation of hippocampal inhibitory postsynaptic potentials by barbiturates. Nature. 1975;258:625–627. doi: 10.1038/258625a0. [DOI] [PubMed] [Google Scholar]
- 10.Narahashi T, Arakawa O, Brunner EA, Nakahiro M, Nishio M, Ogata N, et al. Modulation of GABA receptor-channel complex by alcohols and general anesthetics. Adv Biochem Psychopharmacol. 1992;47:325–334. [PubMed] [Google Scholar]
- 11.Mhatre M, Ticku MK. Chronic ethanol treatment upregulates the GABA receptor beta subunit expression. Brain Res Mol Brain Res. 1994;23:246–252. doi: 10.1016/0169-328x(94)90231-3. [DOI] [PubMed] [Google Scholar]
- 12.Curtis DR, Duggan AW, Felix D, Johnston GAR. Bicuculline and central GABA receptors. Nature. 1970;228:676–677. doi: 10.1038/228676a0. [DOI] [PubMed] [Google Scholar]
- 13.Engel D, Pahner I, Schulze K, Frahm C, Jarry H, Ahnert-Hilger G, et al. Plasticity of rat central inhibitory synapses through GABA metabolism. J Physiol. 2001;535:473–482. doi: 10.1111/j.1469-7793.2001.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng L, Hertz L, Huang R, Sonnewald U, Petersen SB, Westergaard N, et al. Utilization of Glutamine and of TCA Cycle Constituents as Precursors for Transmitter Glutamate and GABA. Dev Neurosci. 1993;15:367–377. doi: 10.1159/000111357. [DOI] [PubMed] [Google Scholar]
- 15.Schousboe A, Westergaard N, Sonnewald U, Petersen SB, Huang R, Peng L, et al. Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev Neurosci. 1993;15:359–366. doi: 10.1159/000111356. [DOI] [PubMed] [Google Scholar]
- 16.Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100. doi: 10.1016/0896-6273(91)90077-d. [DOI] [PubMed] [Google Scholar]
- 17.Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice:differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol. 2003;90:2690–701. doi: 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- 18.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, et al. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- 19.Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, et al. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid-decarboxylase. Proc Natl Acad Sci U S A. 1997;94:14060–14065. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kash SF, Tecott LH, Hodge C, Baekkeskov S. Increased anxiety and altered responses to anxiolytics in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1999;96:1698–1703. doi: 10.1073/pnas.96.4.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heldt SA, Green A, Ressler KJ. Prepulse inhibition deficits in GAD65 knockout mice and the effect of antipsychotic treatment. Neuropsychopharmacology. 2004;29:1610–1619. doi: 10.1038/sj.npp.1300468. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Jin Y, Buddhala C, Osterhaus G, Cohen E, Jin H, et al. Role of glutamate decarboxylase (GAD) isoform, GAD65, in GABA synthesis and transport into synaptic vesicles-Evidence from GAD65-knockout mice studies. Brain Res. 2007;1154:80–83. doi: 10.1016/j.brainres.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 23.Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, et al. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charych EI, Liu F, Moss SJ, Brandon NJ. GABA(A) receptors and their associated proteins:implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology. 2009;57:481–495. doi: 10.1016/j.neuropharm.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akbarian S, Huntsman MM, Kim JJ, Tafazzoli A, Potkin SG, Bunney WE Jr, et al. GABAA receptor subunit gene expression in human prefrontal cortex:comparison of schizophrenics and controls. Cereb Cortex. 1995;5:550–560. doi: 10.1093/cercor/5.6.550. [DOI] [PubMed] [Google Scholar]
- 27.Heckers S, Konradi C. GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophr Res. 2015;167:4–11. doi: 10.1016/j.schres.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marenco S, Weinberger DR. The neurodevelopmental hypothesis of schizophrenia:following a trail of evidence from cradle to grave. Dev Psychopathol. 2000;12:501–527. doi: 10.1017/s0954579400003138. [DOI] [PubMed] [Google Scholar]
- 29.Leinekugel X, Tseeb V, Ben-Ari Y, Bregestovski P. Synaptic GABAA activation induces Ca2+rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. J Physiol. 1995;487:319–329. doi: 10.1113/jphysiol.1995.sp020882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 31.Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus:developmental reversal from Ca2+elevating to depressing. J Neurosci. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–532. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- 33.Ben-Ari Y. Excitatory actions of gaba during development:the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 34.Perrot-Sinal TS, Auger AP, McCarthy MM. Excitatory actions of GABA in developing brain are mediated by l-type Ca2+channels and dependent on age, sex, and brain region. Neuroscience. 2003;116:995–1003. doi: 10.1016/s0306-4522(02)00794-7. [DOI] [PubMed] [Google Scholar]
- 35.Nuñez JL, McCarthy MM. Evidence for an extended duration of GABA-mediated excitation in the developing male versus female hippocampus. Dev neurobiol. 2007;67:1879–1890. doi: 10.1002/dneu.20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 37.Gargiulo S, Greco A, Gramanzini M, Esposito S, Affuso A, Brunetti A, et al. Mice anesthesia, analgesia, and care, Part I:anesthetic considerations in preclinical research. ILAR J. 2012;53:E55–E69. doi: 10.1093/ilar.53.1.55. [DOI] [PubMed] [Google Scholar]
- 38.Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 39.Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Z, Sun QQ. The balance between excitation and inhibition and functional sensory processing in the somatosensory cortex. Int Rev Neurobiol. 2011;97:305–333. doi: 10.1016/B978-0-12-385198-7.00012-6. [DOI] [PubMed] [Google Scholar]