

Abstract
Redox-active tyrosines (Ys) play essential roles in enzymes involved in primary metabolism including energy transduction and deoxynucleotide production catalyzed by ribonucleotide reductases (RNRs). Thermodynamic characterization of Ys in solution and in proteins remains a challenge due to the high reduction potentials involved and the reactive nature of the radical state. The structurally characterized α3Y model protein has allowed the first determination of formal reduction potentials (E°′) for a Y residing within a protein (Berry, B. W.; Martínez-Rivera, M. C.; Tommos, C. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9739–9743). Using Schultz’s technology, a series of fluorotyrosines (FnY, n = 2 or 3) was site-specifically incorporated into α3Y. The global protein properties of the resulting α3(3,5)F2Y, α3(2,3,5)F3Y, α3(2,3)F2Y and α3(2,3,6)F3Y variants are essentially identical to those of α3Y. A protein film square-wave voltammetry approach was developed to successfully obtain reversible voltammograms and E°’s of the very high-potential α3FnY proteins. E°′(pH 5.5; α3FnY(O•/OH)) spans a range of 1040 ± 3 mV to 1200 ± 3 mV versus the normal hydrogen electrode. This is comparable to the potentials of the most oxidizing redox cofactors in Nature. The FnY analogs, and the ability to site-specifically incorporate them into any protein of interest, provide new tools for mechanistic studies on redox-active Ys in proteins and on functional and aberrant hole-transfer reactions in metallo-enzymes. The former application is illustrated here by using the determined α3FnY ΔE°’s to model the thermodynamics of radical-transfer reactions in FnY-RNRs and to experimentally test and support the key prediction made.
TOC Image
INTRODUCTION
The basic principles of electron tunneling (ET) in biological oxidation-reduction processes, within and between proteins, have been studied in great detail because of its centrality to so many important reactions.1–3 These reactions usually involve metallo-prosthetic groups (e.g. hemes, iron-sulfur clusters, non-heme iron and copper cofactors) placed ~ 14 Å apart within the protein environment. The rate constants for electron transfer (kET) depend on the electronic coupling of the electron donor and acceptor wave functions (HAD), the energy required for the reorganization of their nuclear coordinates (λ), and the driving force (ΔG) for the reaction. kET is in general very fast and much faster than substrate redox chemistry (kcat).
Also at the heart of biology are processes that involve the coupling of electron and proton movements, proton-coupled electron transfer (PCET).4–8 These processes include among others, light-driven H2O oxidation in oxygenic photosynthesis,9 O2 reduction in cellular respiration,10 and deoxynucleotide synthesis required to make the building blocks for DNA replication and repair.11 The basic principles of PCET reactions are actively being investigated and draw heavily on our understanding of ET. In PCET, however, the differences in mass between the electron and proton require much more constrained placement of the proton, typically between a H-bonded donor–acceptor pair, and minimization of charge build up in the low dielectric protein medium. The biological examples cited above all involve stable and/or transient tyrosyl radicals (Y–O•) as redox cofactors, which is the focus of this paper. Methods to study the involvement of these species have been limited since the biochemical systems typically have turnover numbers that are governed by conformational gating (i.e. kcat ≪ kPCET). Site-specific incorporation of unnatural amino acids (UAA) presents an approach to address this experimental barrier and disclose the underlying chemistry.11–13 Recently, a tRNA/aminoacyl-tRNA synthetase (RS) pair was evolved to introduce 2,3,5-trifluorotyrosine into any protein of interest.14 This RS turned out to be polyspecific and to incorporate both di- and tri-substituted fluorotyrosines (FnY, n = 2, 3; Figure 1). These analogs are of interest for mechanistic studies of aromatic amino-acid PCET or “hole” hopping reactions since the ring substitutions alter the phenol pKa and reduction potential significantly. Studies on the zwitterionic15 and blocked forms (N-acetyl-Fn-L-tyrosinamides)16 have shown that the pKa is lowered from 10 for Y to the 6.4 – 7.8 range for the FnYs. Anodic peak potentials (Epeak) obtained from irreversible voltammograms fall both below and above the corresponding Epeak of Y.16 ΔEpeak spans ~ 150 mV for the FnY(O•/OH) redox pair and ~ 165 mV for the FnY(O•/O−) couple. These variations offer the opportunity to perturb the biochemical systems such that individual PCET and chemical steps become experimentally accessible. This is illustrated by recent mechanistic studies on E. coli RNR.17–20
Figure 1.
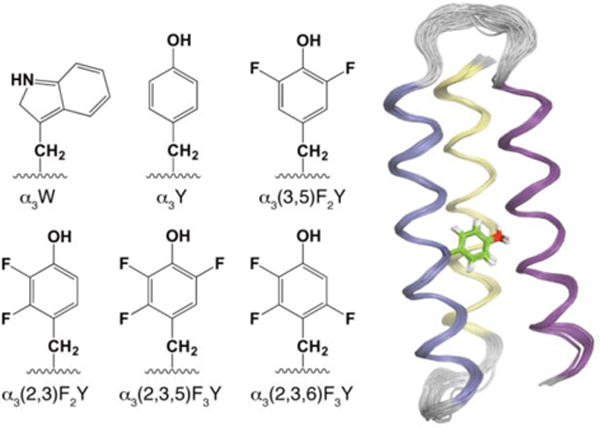
Six members of the α3X family of de novo designed proteins. The 65-residue sequence common to these proteins: GSR(1)V KALEEKVKALEEKVKA–LGGGGR–IEELKKKX(32)EELKKKIEE–LGGGGE–VKKVEEEVKKLEEEIKK–L(65), forms a three-helix bundle (α3) with an aromatic residue at a central position (site 32, labeled X).25,26 The chemical structures display the side chain of residue 32 in α3W, α3Y and four α3FnY proteins. Solution NMR structures have been obtained for α3W (PDB ID 1LQ7),27 2-mercaptophenol-α3C (2MP-α3C, 2LXY),28 and α3Y (2MI7; displayed as a ribbon diagram with the Y32 side chain in stick format).29
The broad utility of the FnYs is also apparent from recent work on other biological systems. In studies on the photosystem II (PSII) reaction center, for example, 3-FY was globally incorporated to probe changes in the PCET kinetics of the active-site YZ residue using time-resolved absorption spectroscopy.21 The active site of cytochrome c oxidase (CcO) also contains a redox-active Y species.10 Lu and coworkers used a series of unnatural Y derivatives, including 3,5-F2Y and 2,3,5-F3Y, to study their influence on the reaction mechanism of a CcO model engineered into myoglobin.22–24 Mechanistic studies revealed that pKa values of Y and the Y analogs were correlated to O2 reduction and H2O production in this model system. Thus, FnYs are emerging as an important tool to study radical-based protein PCET/hole hopping and to probe and alter active-site chemistry. An essential component currently missing with this approach is the knowledge of the formal reduction potentials (E°′) of the FnYs measured under reversible conditions and in a protein environment. The main reason the aqueous FnYs give rise to irreversible voltammograms is because the Y–O• state is unstable on the time scale of the electrochemical measurements. Epeak values reflect the thermodynamics of the redox-active species, but are also influenced by the kinetics of the Y–O• side reactions. Here we use the well-structured α3X model protein system to stabilize the oxidized state of the FnYs and render their electrochemical response reversible.
Position 32 is the dedicated radical site in the α3X proteins (Figure 1).25,26 This site is occupied by W32, Y32, (3,5)F2Y32,30 or by a 2-, 3- or 4-mercaptophenol-C32 residue.31 Nuclear magnetic resonance (NMR) and circular dichroism (CD) studies have shown that the α3X proteins are highly helical, stable and well structured across a broad pH range.25,29–32 Several of the α3X proteins have been structurally characterized and residue 32 was confirmed to be solvent sequestered in each system.27–29 The α3 scaffold does not give rise to a Faradaic current in the absence of a redox-active residue at position 32.28,31,32 Using square-wave voltammetry (SWV), Tommos and coworkers showed that Y32, 2MP-C32 and (3,5)F2Y32 can be reversibly oxidized and reduced.28,30,33 These studies demonstrate that pure thermodynamic potentials, uncompromised by Y–O• side reactions, can be obtained from the α3X proteins. A photochemical study of α3Y showed that Y32–O• is formed in a PCET process and slowly (t1/2 2–10 s) decays via intermolecular radical–radical dimerization. The protein scaffold was shown to stabilize the Y32–O• state by > 104 relative to aqueous Y–O•.29
In this report we refine the SWV approach used in earlier α3X studies to involve protein film voltammetry (PFV).34 Protein film square-wave voltammograms were collected from α3Y, α3(3,5)F2Y, α3(2,3)F2Y, α3(2,3,5)F3Y and α3(2,3,6)F3Y (Figure 1) at conditions where the global properties of the five proteins are close to identical. The obtained E°’s of the α3X proteins are compared to potentials reported for aqueous Y and FnYs and for one of Nature’s most oxidizing redox cofactor. We find that, remarkably, the 7.5 kDa α3X model protein can control a redox-active Y as oxidizing as the unique P680+/P680 redox pair, which drives the water-splitting process catalyzed by PSII. The α3X potentials are also used to construct a thermodynamic model for RNR site-specifically labeled with the FnYs shown in Figure 1. This model provides important insight into the first step in the multistep radical transfer (RT) process associated with nucleotide reduction in the class Ia RNRs.
MATERIALS AND METHODS
Protein production and spectroscopic characterization
α3Y was expressed and purified as described earlier.29 2,3,5-trifluorophenol was synthesized from 2,3,5-trifluorophenylboronic acid using a published protocol, with typical yields of ~ 80%.20,35 Tyrosine phenol lyase (TPL) was purified as previously reported.36 All FnYs (n = 2, 3) were enzymatically synthesized from the corresponding phenol using TPL as the catalyst following the published protocol.37 SUMO protease was expressed from pTB145-ulp1 as a His6-tagged construct as previously reported.38 Construction of the pE-SUMO-α3TAG32 plasmid, optimization of the protein expression conditions, and the α3FnY protein purification protocol are described in the Supporting Information (SI). His6-Y731F-α2 was expressed and purified following the published protocol for wild type (wt) α2.14 2,3-F2Y122•-β2 was expressed, purified and reconstituted as previously described.18 Experimental conditions for all spectroscopic studies are described in detail in the SI.
PFV
SWV was performed using an Autolab PGSTAT12 potentiostat (Metrohm/Eco Chemie) equipped with a temperature-controlled, Faraday-cage protected three-electrode micro-cell (Princeton Applied Research). The Ag/AgCl reference electrode and the platinum wire counter electrode (Advanced Measurements Inc.) were prepared by filling the former with a 3M KCl/saturated AgCl solution and the latter with 20 mM APB, 40 mM KCl. All measurements were carried out using a 3 mm diameter pyrolytic graphite edge (PGE) working electrode (Bio-Logic, USA). CD spectroscopy was used to determine the protein concentration in the α3FnY samples used to prepare the protein films. Samples were prepared by dissolving freeze-dried protein in buffer, measuring the 222 nm ellipticity (1 mm path) and calculating the protein concentration using the [Θ]222 values listed in Table S1. The final protein concentration was 45 – 80 μM. The electrode surface was prepared for protein adsorption by manually polishing its surface for 60 s in a 1.0 μm diamond/water slurry on a diamond polishing pad (Bio-Logic, USA) followed by 60 s in a 0.05 μm alumina/water slurry on a microcloth pad (Bioanalytical systems Inc.). The electrode surface was rinsed with methanol followed by an excess of milli-Q water (18 MΩ). The pH in the protein samples and in the electrolyte buffers were matched and measured prior to and after data collection. Solution resistance was compensated for by using the Autolab positive feed-back iR compensation function. Potentials are given versus the normal hydrogen electrode (NHE). Buffers and protein samples were prepared using ultra pure chemicals (Sigma Aldrich) and data recorded under an argon atmosphere. Voltammograms were collected using the GPES software (Metrohm/Eco Chemie) and analyzed using PeakFit (Systat Software Inc.).
RESULTS
α3FnY protein expression and purification
Construction and optimization (Figure S1) of the α3FnY protein expression system are described in the SI. The α3FnY yield was typically a few mg per L culture, except for α3(2,3)F2Y where it was significantly lower. Figure S2 shows analytical C18 chromatograms and MALDI-TOF traces of purified α3(2,3)F2Y, α3(2,3,5)F3Y and α3(2,3,6)F3Y. The protein preparations are homogenous and display the correct α3FnY molecular weights. Equivalent data for purified α3(3,5)F2Y were presented in an earlier study.30
Protein characterization
A key objective for the α3X approach is to obtain a homogenous set of E°′(radical/amino acid) protein potentials, including the potentials for the FnY32 residues. Practically this translates into: (i) determining the experimental conditions where the active redox couple is well defined and the α3 scaffold structurally unperturbed, and (ii) obtaining reversible protein voltammograms at these conditions. The phenol pKa’s of aqueous N-acetyl-Fn-L-tyrosinamides (n = 2, 3) range from 6.4 to 7.8.16 α3FnY oxidation-reduction may thus involve Y32(O•/OH), Y32(O•/O−) or a pH-weighted mixture of these two redox pairs. The UV-Vis and CD studies described here were conducted to determine a common pH range where FnY32(O•/OH) is the dominant redox couple and the structural properties of the four α3FnYs (Figure 1) are minimally perturbed relative to α3Y.
The apparent pKa (pKapp) of X32 was determined to be 8.0 ± 0.1 for α3(3,5)F2Y,30 7.2 ± 0.1 for α3(2,3,5)F3Y and 7.9 ± 0.1 for α3(2,3,6)F3Y (Figure 2; Table 1). The FnY32 residues absorb poorly in the UV region16 making the absorption-monitored pH titrations very material demanding. For α3(2,3)F2Y, the amount of material precluded this measurement. The protein-induced increase in the phenol pKa was very similar for α3(3,5)F2Y, α3(2,3,5)F3Y and α3(2,3,6)F3Y, with an average ΔpKa of 0.83 ± 0.06 (Table 1). We assume that a shift of similar magnitude occurs for α3(2,3)F2Y and a pKapp of 8.6 (7.8 + 0.8) was predicted for this protein (Table 1).
Figure 2.
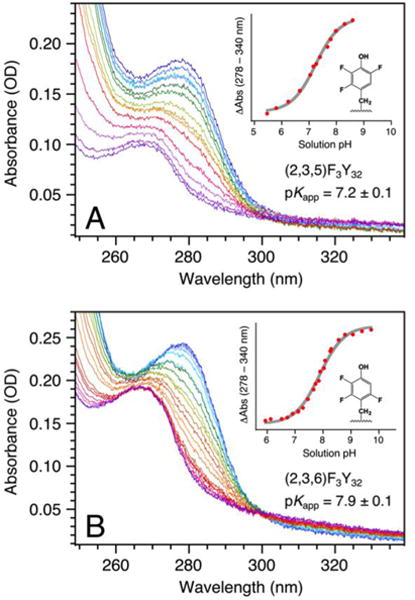
UV-Vis spectra and corresponding pH-titration curves for (A) α3(2,3,5)F3Y and (B) α3(2,3,6)F3Y. The α3(2,3,5)F3Y spectra were incrementally collected from pH 8.59 (dark blue) to pH 5.46 (dark purple) and back to pH 7.04 (red); while the α3(2,3,6)F3Y spectra were collected from pH 9.72 (dark blue)to pH 5.94 (dark purple) and back to pH 7.11 (red). The titration plots were fit to a single pKa.
Table 1.
pKapp and Enet values for α3Y and α3FnY (n = 2, 3)
| System | pKapp | Enet (mV) (pH)d | ΔEnet (mV)d | |
|---|---|---|---|---|
| α3(3,5)F2Y | 8.0a | 1040 ± 3 (5.49 ± 0.03) | − 25 | |
| α3Y | 11.b | 1065 ± 2 (5.53 ± 0.05) | 0 | |
| α3(2,3,5)F3Y | 7.2 | 1104 ± 2 (5.54 ± 0.05) | + 39 | |
| α3(2,3)F2Y | 8.6c | 1136 ± 2 (5.57 ± 0.09) | + 70 | |
| α3(2,3,6)F3Y | 7.9 | 1200 ± 3 (5.54 ± 0.05) | +135 | |
| System | pKae | DPV ΔEpeak (mV)f | CV ΔEpeak (mV)g | |
| 3,5-F2Y | 7.2 | − 51 | − 171h | |
| Y | 10 | 0 | 0h | 0i |
| 2,3,5-F3Y | 6.4 | + 5 | − 122h | −111i |
| 2,3-F2Y | 7.8 | + 44 | − 39i | |
| 2,3,6-F3Y | 7.0 | + 97 | ||
CD spectroscopy was used to determine the α-helical contents and the global stabilities of the α3FnYs. Figure 3A compares the CD spectra of α3(2,3)F2Y, α3(2,3,5)F3Y and α3(2,3,6)F3Y to the reference spectrum of α3W at pH 5.05 ± 0.01. The CD spectra are plotted in the normalized mean residue molar ellipticity form (see Fig. 3 legend) where the 222 nm amplitude ([Θ]222) scales with the helical content. The spectra show that altering the Y32 ring has little impact on the helicity of the α3 scaffold. The α-helical contents of α3(2,3)F2Y, α3(2,3,5)F3Y and α3(2,3,6)F3Y were calculated from their CD spectra using the α3W spectrum as the reference. [Θ]222 values and corresponding α-helical contents are listed in Table S1.
Figure 3.
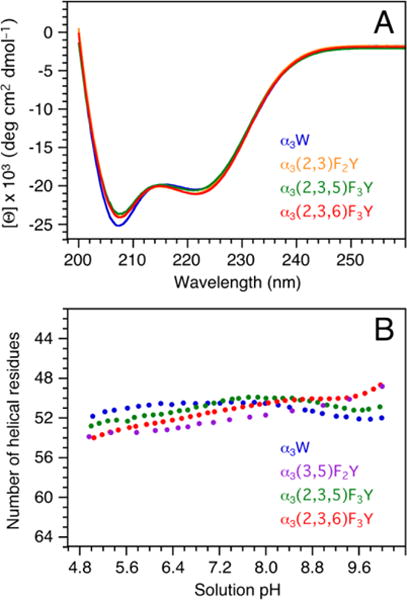
(A) Far-UV CD spectra of α3(2,3,5)F3Y (green), α3(2,3)F2Y (orange), α3(2,3,6)F3Y (red), and α3W (blue; reference spectrum). The proteins were dissolved in 40 mM sodium acetate and the final sample pH measured to 5.05 ± 0.01. The spectra are displayed in units of mean residue molar ellipticity ([Θ]) obtained by: [Θ] = θobs106/Cln where θobs is the observed ellipticity in millidegrees, C the protein concentration in μM, l the cuvette path length in mm (2), and n the number of amino-acid residues (65). C was determined using a fluorescence-based assay (SI). (B) pH-induced changes in the α-helical contents of α3(3,5)F2Y (purple; reproduced from Ref. 30), α3(2,3,5)F3Y (green), α3(2,3,6)F3Y (red) and α3W (blue; reference data25,27).
Figure 3B displays changes in the total number of α-helical residues in α3(3,5)F2Y,30 α3(2,3,5)F3Y and α3(2,3,6)F3Y as a function of pH. A pH plot of α3W is also shown (51 ± 0.6 α-helical residues, pH 4 – 10).25,27 The pH-induced structural changes are minor and the α3FnYs have the same or somewhat higher α-helical content than α3W below pH 6.8. The global stability of α3(2,3,5)F3Y and α3(2,3,6)F3Y were determined by urea denaturation at pH 5.0 and 5.5. Representative data are shown in Figure S3 and protein stability values for α3Y,29,32 α3(3,5)F2Y,30 α3(2,3,5)F3Y and α3(2,3,6)F3Y are summarized in Table S1. For the pH 5.0 – 5.5 range, there is no significant difference in the global stability of α3Y, α3(3,5)F2Y and α3(2,3,5)F3Y while α3(2,3,6)F3Y is 0.4 kcal mol−1 more stable than α3Y. The minimal variations in α-helical content and global stabilities of the α3FnYs relative to α3Y are in accordance with previous observations in other proteins.39 Fluorination of amino acids has been shown to result in minimal structural perturbations to the global protein fold.40,41 Additionally, the increased hydrophobicity resulting from fluorination usually enhance protein stability.39,42,43
We conclude that the common pH range sought for the α3FnY voltammetry studies is limited by the pKapp of α3(2,3,5)F3Y (7.2; to drive the system towards Y32(O•/OH) rather than by considerations of the structurally robust α3 scaffold.
Protein film square-wave voltammetry on the α3FnY proteins
SWV is a pulse voltammetric technique supported by well-developed theoretical models for diffusion-controlled and surface-confined electrode mechanisms.44,45 SWV was used to obtain formal E°′(radical/amino acid) protein potentials for α3Y,33 2MP-α3C,28 and α3(3,5)F2Y.30 The α3X potentials are pH dependent and measurements performed at low pH are more difficult due to the higher potentials and more pronounced background currents involved.33 SWV on α3(2,3,5)F3Y, α3(2,3)F2Y, and α3(2,3,6)F3Y was predicted to be challenging since measurements had to be done ~ pH 5.5 (≥ 1.7 pH units below the pKapp’s of the α3FnY proteins) and test studies on α3(2,3,5)F3Y placed E°′ well above +1 V. Voltammetry data from an on-going parallel project involving an α3(NH2)Y variant (Lee, Nocera, Stubbe and Tommos) suggested that a protein film approach, where the protein is introduced to the surface of the working electrode outside the electrochemical cell, improves the Faradaic response of the α3X system. A protein film protocol was developed for α3Y and applied to the α3FnY proteins. This approach allowed us to obtain high-quality reversible voltammograms in a potential range unprecedented for protein voltammetry and to determine the formal potentials of the highly oxidizing α3FnYs.
In SWV the applied potential is stepped progressively in fixed increments (Estep), and at each increment, a forward (here oxidative) potential pulse is applied followed by a reverse (reductive) pulse. The current is sampled at the end of each alternating pulse and plotted as a function of Estep. This generates a forward (Ifor), a reverse (Irev) and a net (Inet = Ifor – Irev) voltammogram. The time period (τ) over which the electrode reaction is driven in the anodic and then cathodic direction is set by the SW frequency (f = 1/τ).44,45 The apparent redox reversibility of a surface-adsorbed redox pair is described by a kinetic parameter (ω) defined as the ratio of the surface standard ET rate constant (ksur) and the SW frequency (ω = ksur/f). Theory predicts that the strongest response from an adsorbed redox pair occurs when f is close to ksur.47 In earlier reports, the Faradaic response of α3Y and 2MP-α3C were investigated over a broad frequency range of 30–960 Hz and 30–720 Hz, respectively.28,33 Figure S4 shows Inet of α3Y (blue) and 2MP-α3C (red) divided by f, and the obtained Inet/f amplitudes plotted against the frequency. The parabolic shape of the frequency-normalized data is consistent with the “quasi-reversible maximum” (QRM) feature of surface-confined redox reactions.47 The α3Y and 2MP-α3C plots are almost identical with a QRM at 440 Hz. The α3FnYs were anticipated to behave in a similar fashion and display the strongest Faradaic response around 440 Hz. Test studies on α3(2,3,5)F3Y confirmed that the most pronounced response was observed in a rather narrow frequency range of ~ 400 to 450 Hz. The response was poor above and below this range (data not shown). SWV on the α3FnY proteins were thus conducted using a SW frequency of 440 Hz.
Background-corrected protein film SW voltammograms of α3Y are displayed in Figure 4A. The forward, reverse and net currents are colored orange, purple and blue, respectively. The measurement was performed as follows: The surface of the PGE working electrode was polished (see Material and Methods for details) and introduced to a 500 μl 80 μM α3Y, 20 mM APB, 75 mM KCl, pH 5.5 sample for 30 s. The electrode was transferred to a blank electrochemical cell containing 3 mL 20 mM APB, 40 mM KCl, pH 5.5 and a voltammogram recorded. This procedure was repeated twice to collect data replicas a and b (data panels A and B) and on a separate experimental day using an independently prepared protein sample (panel C). The maximum amplitude of Inet was investigated as a function of the electrode surface/α3Y sample incubation time and found stable after about 20 s. No increase in Inet was observed at incubation times > 30 s.
Figure 4.
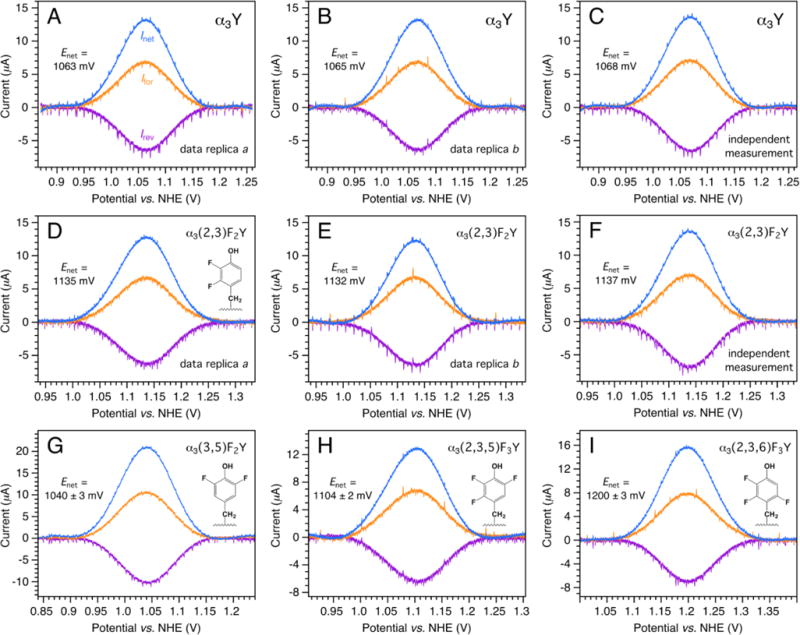
Background-corrected protein film SW voltammograms of (A–C) α3Y, (D–F) α3(2,3)F2Y, (G) α3(3,5)F2Y, (H) α3(2,3,5)F3Y, and (I) α3(2,3,6)F3Y. The proteins were adsorbed onto the surface of a PGE electrode and voltammograms recorded in 20 mM APB, 40 mM KCl, pH 5.54 ± 0.06 buffer at 25 °C. SWV settings: Equilibration time 5 s, step potential 0.15 mV, SW pulse amplitude 25 mV, SW frequency 440 Hz.
The protein film protocol described above was applied to α3(2,3)F2Y and the resulting voltammograms are displayed in the middle row of Figure 4. The corresponding uncorrected data for the baseline-subtracted α3Y (Figures 4A–C) and α3(2,3)F2Y (Figures 4D–F) voltammograms are shown in Figures S5 and S6, respectively. Data processing and analysis details are provided in the SI. As shown in Figures 4, S5 and S6, the voltammograms are recorded at very high positive potentials and the background currents are significant. Nonetheless, the Ifor, Irev and Inet waveforms are well defined and their position, lineshape and amplitude are highly reproducible between data replicas and independent measurements. Representative protein film voltammograms collected from α3(3,5)F2Y, α3(2,3,5)F3Y and α3(2,3,6)F3Y are shown in Figure 4 panels G, H, and I, respectively. The pH in the protein samples and in the electrolyte solutions were measured at the beginning and at the end of each experimental day. The average pH, Enet and ΔEnet values are listed in Table 1.
Summarizing the redox properties of the α3FnY proteins as Enet vs. pH diagrams
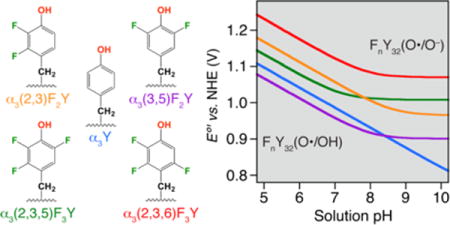
A set of Enet vs. pH diagrams was predicted for the α3FnYs based on the results described above combined with the established charge-neutral response of the α3X redox system.28,31,33 The plots shown in Figure 5 were constructed from the following data: (i) The four α3FnY Enet pH 5.5 values listed in Table 1 (single dots colored purple, green, orange and red). (ii) The α3Y Enet pH 5.53 ± 0.05 value listed in Table 1 plus three additional PFV Enet values obtained at pH 6.94 ± 0.07, 8.40 ± 0.10 and 9.94 ± 0.08 (blue dots; Figure S7). The pH dependence (55.2 mV/pH unit) of the Y32(O•/OH) potential is consistent with an overall charge-neutral redox cycle where the 1e− oxidation-reduction process driven by the electrode is coupled to the release/uptake of essentially one full H+ to/from bulk.33 (iii) α3(3,5)F2Y Enet measured between pH 4.95 ± 0.01 and 6.49 ± 0.02 (grey dots). The pH titration was done on α3(3,5)F2Y since it has a higher pKapp than α3(2,3,5)F3Y and α3(2,3,6)F3Y, and expresses better than α3(2,3)F2Y. Additionally, the potential is lower than for the other α3FnYs making the measurements less challenging. The pH titration was done with α3(3,5)F2Y in the electrochemical cell (details provided in the Figure 5 legend). The pH dependence in Enet (55.7 mV/pH unit) was used to predict the Enet vs. pH curves for all of the α3FnYs. (iv) The pKapp’s of α3(3,5)F2Y, α3(2,3,5)F3Y and α3(2,3,6)F3Y and (v) the predicted pKapp of α3(2,3)F2Y (Table 1). The Nernst equations and parameters used to model the diagrams are listed in Table S2. Table S2 also lists the α3FnY potentials at pH 7.0 as a general reference.
Figure 5.
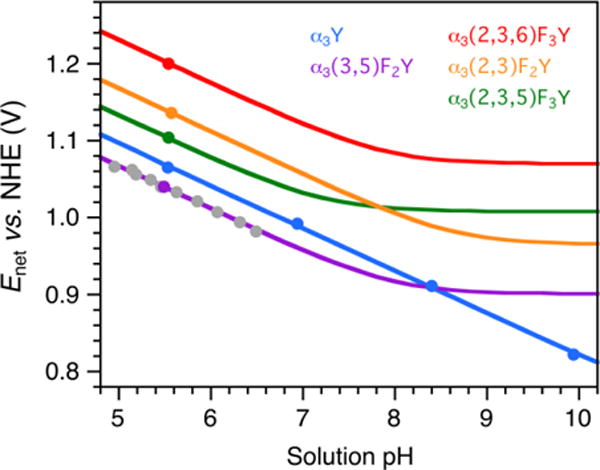
Enet vs. pH diagrams for α3Y and the α3FnY proteins. Solid dots represent Enet values obtained by PFV. Grey dots represent Enet collected from α3(3,5)F2Y using the following conditions: 70 μM protein, 20 mM APB, 75 mM KCl; pH adjusted with 1 M NaOH; step potential 0.15 mV, SW pulse amplitude 25 mV, SW frequency 190 Hz, and temperature 25 °C.
DISCUSSION
The α3X system combined with protein film SWV facilitates studies of “hot” radicals
The experimental advance made here was driven by the very “hot” nature of the FnY32 radicals and the necessity of recording reversible voltammograms at exceedingly high potentials. This issue was addressed by employing PFV.34
Voltammograms collected at high positive potentials contain capacitive currents, background currents from the oxidation of the solvent, supporting electrolytes and/or chemical groups on the surface of the working electrode (WE), in addition to the Faradaic current of the redox-active species under investigation. The effects of capacitive currents can be minimized by using pulse voltammetric techniques, such as differential pulse voltammetry (DPV) or SWV. Background currents from solvent oxidation can be reduced by using a carbon, rather than a metal-based, WE but they cannot be eliminated. Prominent background currents are unavoidable when doing high-potential voltammetry on aqueous systems and they rapidly increase above ~ 1 V. Thus, an increase in E°′ that would be insignificant when probing a 0.5 V protein oxidant may prove challenging, even prohibiting, when probing a protein oxidant in the +1.0 V range. Berry et al. used SWV and a PGE electrode to optimize the α3Y response and showed that Y32 can be reversibly oxidized and reduced.33 Here we show that the α3Y response improves even further when the protein is adsorbed onto the surface of the PGE electrode outside the electrochemical cell. The protein film samples gave rise to Faradaic currents that are easily identified above the background (Figures S5 and S6). Baseline-corrected traces with well-defined peak positions and lineshapes were reproducibly obtained despite the high potentials involved (Figure 4; Table 1). The protein film SW voltammograms collected from α3Y and the α3FnYs display an Ifor/Irev close or equal to unity and an Efor – Erev peak separation of − 2 ± 2 mV. This is consistent with a strongly adsorbed redox pair for which the surface standard ET rate constant ksur is close to the SW frequency (f = 440 s−1) and Enet equals E°′.48
The α3X potentials reflect the local protein environment
Previous DPV and SWV studies were conducted with the α3X proteins dissolved in the supporting electrolyte solution.28,30–33 The characteristics of their Faradaic response suggested that diffusion-controlled reactions dominated the electrode process.28,33 As described above, the Inet amplitude stabilizes in less than 20 s of exposing the surface of the PGE electrode to an α3Y sample. Data collected with the protein dissolved in the electrolyte solution would typically be started on this time scale after the freshly polished WE was placed in the cell. Previously published α3X voltammograms thus most likely represent a mixed contribution of surface-adsorbed and diffusional species.
Figure 5 shows a α3Y Enet vs. pH diagram constructed from PFV data. All previously reported α3Y potentials recorded over a broad range of conditions (method, SWV or DPV; WE material, pyrolytic graphite or glassy carbon; [protein] 2 – 210 μM; [KCl] 10 – 140 mM; pH 5.5 – 10; SW frequency 190 – 960 Hz)33 were plotted against values predicted from the α3Y PFV Enet vs. pH diagram in Figure 5. The plot goes through zero and displays a linear correlation of 1.001 ± 0.002 and a standard error of < ± 7 mV (not shown). Thus, there is no significant change in the observed potential when the system shifts from mixed surface/diffusional to a pure surface mechanism. This is also true for α3(3,5)F2Y (Enet(pH 5.70) 1026 ± 4 mV in Ref. 30 and 1028 ± 3 mV in Figure 5). These observation are consistent with the buried position of Y3229 where the Y32(O•/OH) potential is only influenced by local protein interactions and not by external factors such as charges on the surface of the WE.
Interestingly, there is a symmetry to the α3FnY ΔE°’s (Table 1). When the common component (2-FY = 6-FY, i.e. F attached to either of the two ring delta carbons) is isolated by pairing the four independently obtained E°’s, the same value is obtained:
We interpret this to show that the impact of the local protein environment on the FnY32(O•/OH) E°’s is equal across the four proteins. If the asymmetrically labeled FnY32 side chains have multiple rotameric states,18,49 it has little influence on their E°’s.
E0′(Y(O•/OH)) increases by about 45–65 mV when placed in a hydrophobic protein environment
Zwitterionic and blocked Y have been studied by pulse radiolysis,50–52 cyclic voltammetry (CV),23,46,53,54 DPV25,54,55 and SWV.46 The reported pH 7.0 potentials span a considerable range, 830 – 970 mV, which reflects a number of issues. In pulse radiolysis, equilibrium is established between a reference redox couple and the redox couple of interest. For accurate readings, equilibrium must be established on a time scale that is fast relative to Y–O• decay. Additionally, some of the reference potentials used in the older literature were not as well-determined as they are today.56 Aqueous Y gives rise to irreversible voltammograms since Y–O• dimerization is fast (kdim 2–7 × 108 M−1 s−1)29,57 relative to the experimental time scale. These voltammograms, which display only an anodic wave, may reflect a reversible, quasi-reversible or irreversible electrode reaction. If the electrode reaction is shown to be reversible,58 then the anodic peak potential can be corrected for kdim and an apparent formal potential derived.58,59 Harriman53 and DeFelippis et al.54 applied this approach but unfortunately used an incorrect equation to compensate for kdim.46 Recently reported (or recommended) pH 7.0 potentials for aqueous Y are 0.93 ± 0.02 V,52 0.91 ± 0.02 V,56 and 0.96 V.46 We therefore take a consensus value of 920–940 ± 20 mV for the aqueous Y(O•/OH) redox couple at pH 7.0. E°′ of Y32(O•/OH) is 986 ± 3 mV at pH 7.0 (Figure 5; Table S2). Thus, the potential of the Y(O•/OH) couple increases by ~45–65 ± 20 mV when Y is buried inside α3Y.
Comparing FnY potentials obtained at reversible and irreversible conditions
Tommos et al.25 and the Stubbe and Nocera groups11,16,55 have reported raw DPV potentials for Y, W and Y analogs. The focus of these studies was to obtain comparable peak potentials for Y vs. W,25 Y vs. NO2Y,55 and Y vs. FnYs.16 Tommos and collaborators are using the unique α3X system to refine and solidify these ΔEs by comparing the true thermodynamic E°’s of Y32,33 W32 (Tommos, Hammarström, in preparation), NO2Y32 (Tommos, on-going) and FnY32 (this study). Table 1 displays absolute and relative E°’s for α3Y and the α3FnYs proteins and compares the latter to ΔEpeak values obtained by DPV. The absolute potentials differ by ~ 168 mV (α3Y – Y)25,55 and ~ 193 mV (α3FnY – FnY).16 These differences reflect the water vs. protein medium (~ 45–65 ± 20 mV, vide supra), a DPV pulse amplitude factor (25 mV), and peak shifts due to the irreversibilty of the aqueous systems (~ 90 ± 10 mV for Y and ~ 115 ± 10 mV for the FnYs). Nonetheless, the relative potentials reported in Ref. 16 fit rather well with the α3X data. The oxidizing power of the Ys increases in the same order (3,5-F2Y < Y < 2,3,5-F3Y < 2,3-F2Y < 2,3,6-F3Y) and the overall span (E(2,3,6-F3Y) – E(3,5-F2Y)) is 160 mV and ~ 150 mV for the α3FnYs and FnYs, respectively. One significant difference between the α3X ΔE° scale and the DPV ΔEpeak scale in Table 1 is that the four Epeak(FnY)s are downshifted by ~ 30 mV relative to Epeak(Y). This does not represent a true difference in potential between the protein and aqueous systems but rather reflects the limitations of relying on irreversible voltammograms. Recently, Lu and coworkers22,23 and Mahmoudi et al.46 used CV to measure the difference in the anodic peak potential of irreversible voltammograms obtained from aqueous 3,5-F2Y, 2,3,5-F3Y and 2,3-F2Y relative to Y. Overall, there is little consensus in the values reported for the aqueous systems with ΔEpeak(pH 7.0; 3,5-F2Y – Y) ranging from −158 to −60 mV11,23 ΔEpeak(pH 7.0; 2,3,5-F2Y – Y) from −81 to +30 mV,11,23,46 and ΔEpeak(pH 7.0; 2,3-F2Y – Y) from −30 to +30 mV.11,46 Table 1 lists CV ΔEpeak values for the Y(O•/OH) redox couple (i.e. at pH < 2 units below the phenol pKa’s) as calculated from the reported pH 7.0 values.23,46 This is to provide a more direct comparison to the α3X(O•/OH) ΔE°’s. The large discrepancies observed for the ΔEpeak values highlight the importance of measuring the potentials of reactive radicals under reversible conditions such as those acheived here.
Using the α3X system to model the initial RT step in FnY122•-β2: PCET or ET?
Below we explore the general use of the α3X potentials by applying them as an analytical tool to investigate RT in E. coli RNR. This enzyme consists of two homodimeric subunits, α2 and β2, which form the active α2β2 complex.11 β2 contains a stable diferric-Y122• cofactor that reversibly generates a transient thiyl radical in α2 (C439•; Figure 6). Once formed, C439• initiates the catalytic nucleotide reduction process. Each turnover involves multistep RT via Y122–O• ⇆ [W48?] ⇆ Y356 in β2 and Y731 ⇆ Y730 ⇆ C439 in α2 (Figure 6). Slower conformational gating masks the forward and reverse RT steps and the transient Y356•, Y730• and Y731• species are not observed in wt RNR. Site-specific incorporation of NO2Y and FnY residues proved to be a successful approach to perturb the system so that the pathway Y radicals can be observed.11,61,62 The following ΔE°’s were obtained from the radical distribution patterns at equilibrium: ΔE°′(pH 7.6; 2,3,5-F3Y122• – Y356•) = 20 ± 10 mV, ΔE°′(pH 8.2; Y122• – 3,5-F2Y356•) = −70 ± 5 mV, and ΔE°′(pH 7.6; Y356• – Y731•) = ~ −100 mV at 25 °C.62,63 Taken together these ΔE°’s sketch out the thermodynamic landscape associated with the intersubunit RT pathway in E. coli RNR (Figure S8).63
Figure 6.
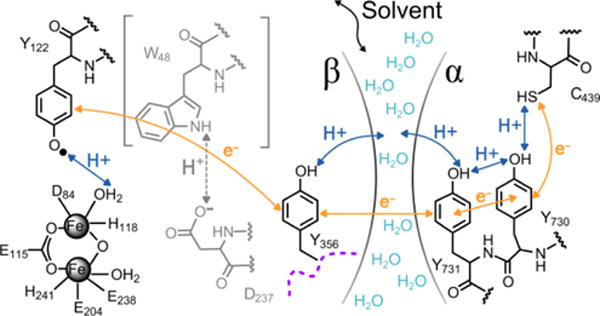
Radical transfer pathway in E. coli RNR. The arrows represent electron (orange) and proton (blue) movements within the α2 and β2 subunits. There is currently no evidence for the involvement W48 (grey) or its proposed proton acceptor D237 (grey) in RT. The water interface between α2 and β2 is suggested in Ref. 60.
In the analysis presented below, we combine the α3X potentials with two of the RNR ΔE°’s listed above to construct a thermodynamic model for sites Y122 and Y356 in wt and FnY-RNRs. The modeling addresses the question if the initial RT step in 2,3,5-F3Y122•-β2 is PCET (established for wt RNR; Figure 7A)64 or ET (established for NO2Y122•-β2: Figure 7B)61 and if this process is identical between the different FnY122•-β2 systems.
Figure 7.
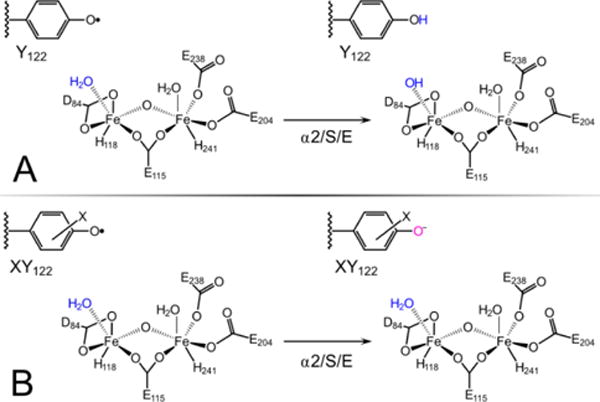
Initiation of RT when (A) wt-β2 or (B) XY-β2 (where X is NO2 or Fn) reacts with α2, substrate CDP (S) and allosteric effector ATP (E). (A) Reduction of wt Y122–O• by Y356 is coupled with proton transfer (PT) from Fe1-H2O.64 (B) Reduction of NO2Y122–O• (or FnY122–O•) by Y356 is not coupled to PT from the diiron cluster.
Modeling of RNR sites 356 and 122
Figure 8 displays a thermodynamic model for sites 356-β2 (Panel A) and 122-β2 (Panel B) in wt and FnY-RNRs. The Y356 site was modeled by using the absolute α3X potentials (Figure 5) and phenol pKa‘s that were upshifted by 0.4 relative to the aqueous FnYs (Figure 8A). The modeled potentials are pH dependent since the proton released upon Y356 oxidation is in rapid exchange with the bulk solvent.63 The pKa values are based on the 0.4 increase in the pKa of NO2Y356.65 The modeling of site 122 was more involved and is described in a stepwise manner below and in Figure 8B.
Figure 8.
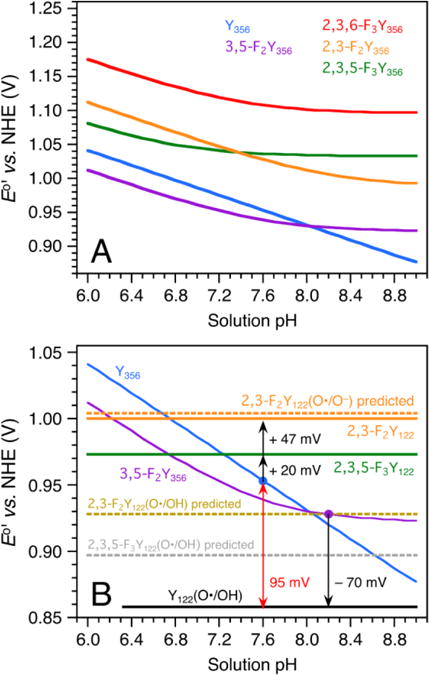
Thermodynamic model for sites Y122 and Y356 in wt and FnY-RNR. (A) Predicted E°′ vs. pH diagrams for residue Y356 in the wt RNR and in mutants containing FnY356 (n = 2, 3) residues. Y356 is modeled with a pKa of 10.4, which makes Y356(O•/OH) the dominant redox couple across the displayed pH range. The FnY356 residues are modeled with lower pKa’s (3,5-F2 7.6; 2,3,5-F3 6.8; 2,3-F2 8.2; 2,3,6-F3 7.4) and their potentials are described by a pH-weighted mixture of the FnY356(O•/OH) and FnY356(O•/O−) redox pairs. (B) Modeled E°’s for residue Y122 in the wt and FnY122-RNRs. The Y356 (blue) and 3,5-F2Y356 (purple) E°′ vs. pH diagrams are superimposed from Panel A. The black arrows represent experimentally obtained ΔE°’s (Figures S8 and S9).63 A +95 mV step is predicted for RT from Y122(O•/OH) to Y356 (O•/OH) in wt RNR (red double arrow). The E°’s are modeled for 25 °C.
(1) In wt RNR, Y122(O•/OH) is the active redox couple and the proton toggles between the phenol oxygen and the Fe1(H2O/OH) species (Figure 7A).64 The site is effectively not in communication with bulk and is therefore modeled with pH-independent potentials. The Y356 (blue) and 3,5-F2Y356 (purple) E°′ vs. pH diagrams displayed in Figure 8A are reproduced in Figure 8B. By combining these Y356 plots with the RNR ΔE°’s obtained in Ref. 63 (illustrated by black arrows), the potentials of sites 356 and 122 were linked as follows: The potential of 2,3,5-F3Y122 was obtained by taking the predicted E°′(Y356(O•/OH)) pH 7.6 value (953 mV, blue dot) and making E°′(2,3,5-F3Y122) 20 ± 10 mV more oxidizing (973 mV; green line). Likewise, the potential of Y122(O•/OH) was obtained by taking the predicted E°′(3,5-F2Y356) pH 8.2 value (928 mV, purple dot) and making E°′(Y122(O•/OH)) 70 ± 5 mV less oxidizing (858 mV; black line). Importantly, this model predicts a +95 mV uphill step between wt Y122 and Y356 (at pH 7.6 and 25 °C; red double arrow).
(2) The α3X ΔE°′(pH 5.5) scale in Table 1 mainly reflects changes in the Y32 fluorination pattern since the other key properties of the redox system (overall charge neutral redox cycle; buried hydrophobic site) are the same between α3Y and the α3FnYs. This, in turn, suggests that the α3X ΔE°’s can be used as a diagnostic tool. A RNR ΔE°′ that deviates significantly from the corresponding α3X ΔE°′ is an indicator that some property, in addition to the Y fluorination pattern, has changed. Table 1 predicts a 39 mV difference (ΔE°′(α3(2,3,5)F3Y – α3Y)) between Y122(O•/OH) (black) and 2,3,5-F3Y122(O•/OH) (grey dotted line). This is not consistent with the 115 mV difference between Y122(O•/OH) and 2,3,5-F2Y122 obtained in step (1) above (black and green lines). Below we examine the possibility that the initial RT step has changed from PCET in wt RNR to ET in 2,3,5-F3Y122-β2.
(3) Y(O•/O−) is the experimentally observed redox pair in the single turnover (NO2)Y122-β2 system.61 It appears likely that Y(O•/O−) is the active redox couple also in 2,3,5-F3Y122-β2 since the pKa of 2,3,5-F3Y (6.4)16 is lower than the pKa of NO2Y (7.1).55 In this scenario, oxidation-reduction of native Y122 involves ET between Y356 and Y122, PT between Fe1(H2O/OH) and Y122(O•/OH), and a net charge change at the diiron cluster (Figure 7A). In contrast, oxidation-reduction of 2,3,5-F3Y122-β2 involves ET between Y356 and Y122 and a net charge change at site 122 (Figure 7B). The 115 mV difference in potential between Y122(O•/OH) and 2,3,5F3Y122(O•/O−) is partly due to the ring fluorination and partly due to redox-driven electrostatic changes that are not identical between the wt system and 2,3,5-F2Y122-β2.
(4) 2,3-F2Y has a higher pKa (7.8) than the other FnYs (6.4 –7.2) and 2,3-F2Y122 may thus behave as Y122(O•/OH) or as 2,3,5-F3Y122(O•/O−). If it is the former, we predict a ΔE°′ of 70 mV (ΔE°′(α3(2,3)F2Y – α3Y); Table 1) between Y122(O•/OH) and 2,3-F2Y122(O/OH) (928 mV; tan dotted line). If it is the latter, we predict a ΔE°′ of 31 mV (ΔE°′(α3(2,3)F2Y – α3(2,3,5)F3Y); Table 1) between 2,3,5-F3Y122(O•/O−) and 2,3-F2Y122(O•/O−) (1004 mV; orange dotted line). These predictions were tested by performing EPR-monitored radical equilibration studies on 2,3-F2Y122•-β2 (Figure S9). The observed populations of 2,3-F2Y122• and Y356• correspond to a ΔE°′(2,3-F2Y122• – Y356•) of 47 ± 11 mV (pH 7.6, 25 °C). This results in a 27 ± 11 mV difference between 2,3,5-F3Y122 (green line) and 2,3-F2Y122 (1000 mV; orange solid line). Thus, the α3X ΔE°′ (31 ± 2 mV) scale is preserved between 2,3,5-F3Y122 and 2,3-F2Y122 (27 ± 11 mV). This suggests that the reactions associated with the 2,3,5-F3Y122 and 2,3-F2Y122 redox cycles are similar and that both occur via Y(O•/O−). Furthermore, since pKa(2,3,5-F3Y) < pKa(2,3,6-F3Y) < pKa(3,5-F2Y) < pKa(2,3-F2Y), we propose that Y122(O•/O−) is the operational redox couple in all four FnY-substituted RNRs.
(5) It appears counterintuitive that ΔE°′(2,3-F2Y122(O•/O−) – 2,3,5-F3Y122(O•/O−)) is well predicted by the α3X ΔE°′(pH 5.5) value and not by the ΔE°′ of the corresponding α3X(O•/O−) redox pairs (which equals −43 mV; Table S2). The simplest explanation is that FnY122(O•/O−) is not a distinct redox couple but an interacting FnY122(O•/OH)/Fe1(H2O/OH) redox couple connected by a polarized H-bond or H-bonding network. Mössbauer data from the active (α2β2/substrate/effector) wt system suggest that Y122 and Fe1 are H-bonded and that the phenol proton toggles back and forth depending on the redox state of Y122.64 In the FnY122-RNRs the phenol pKa’s are lower and we hypothesize that the proton is polarized towards Fe1 at all times. This makes the system behave spectrally as a Y–O− (as observed for reduced NO2Y122) but thermodynamically more as a Y(O•/OH) couple due to the H-bonding interactions between the phenol ring and Fe1.
Concluding remarks
Site-specific incorporation of UAAs provides a powerful tool to study and engineer proteins and enzymes.13 As the methodologies for UAA incorporation continue to improve, this approach is expanding in multiple directions in both basic science and applied research. e.g.66 This report focuses on the use of FnYs to perturb and characterize ET/PCET reactions in radical- and metallo-enzymes that use Y–O• in catalytic or long-range radical/hole transfer processes. Figure 1 shows FnYs that have been used to study these types of reactions in enzymes.11,17–23,63 Here we contribute data necessary for FnY-based approaches to study protein redox chemistry. Until now, true thermodynamic potentials of the FnYs have not been available for any medium, water or protein. Such data could be obtained by incorporating the FnYs at site 32 in the well-structured α3Y model protein (Figure 1). The FnY32–O• species are powerful oxidants. This issue was addressed by developing a protein film SW voltammetry approach that enabled collection of fully reversible voltammograms at uniquely high potentials. This approach generated α3FnY voltammograms of excellent quality and reproducibility. E°’s as high as +1200 mV vs. the NHE could be determined with a precision of 2–3 mV. To provide a relevant comparison, the potential of the transient P680+/P680 redox pair in the PSII reaction center is estimated at ~ 1170–1210 mV.67 Thus, E°’s representing the very oxidizing edge of the biological redox scale could be reproduced in the 7.5 kDa α3X model protein.
It has recently been suggested that aberrant reactive intermediates formed at buried metallo-cofactors may be removed via hole transfer along intramolecular Y/W chains linking the active site to the protein surface.68 The FnY32(O•/OH) and FnY32(O•/O−) redox couples span a considerable ΔE°′ range and, in combination with protein engineering,13,14 could be used to test this hypothesis by altering the potentials of Ys along the proposed hole-transfer chains. These Y analogs could potentially also be used to map the thermodynamic profiles of radical migration processes observed in enzymes such as CcO, cyclooxygenase I and II and various peroxidases. e.g.69 If engineered at active sites, the FnYs could serve to estimate the potentials of metallo-oxidants too hot or too short-lived to be assessed by any other method. Additionally, and as demonstrated here, the α3X-derived potentials can be used as a mechanistic tool to investigate the PCET vs. ET nature of Y-based redox reactions.
Supplementary Material
Acknowledgments
Funding was provided by National Institutes of Health grants GM47274 (D.G.N), GM29595 (J.S.) and GM079190 (C.T.)
Footnotes
Supporting Information
Construction of the α3FnY expression system; protein expression and purification protocols; experimental conditions and settings for spectroscopic studies; analytical HPLC and MALDI-TOF evaluation of purified α3FnYs; representative urea-induced denaturation plots; CD and structural properties of the α3FnY proteins; SWV quasi-reversible maxima of α3Y and 2MP-α3C; representative protein film SW voltammograms and data processing details; Nernst equations and parameters used to model the α3X Enet vs. pH diagrams; current thermodynamic landscape of the RT pathway in E. coli class Ia RNR; radical equilibrium studies on FnY-labeled RNRs. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 2.(a) Beratan DN, Onuchic JN, Winkler JR, Gray HB. Science. 1992;258:1740–1741. doi: 10.1126/science.1334572. [DOI] [PubMed] [Google Scholar]; (b) Gray HB, Winkler JR. Quart Rev Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]; (c) Winkler JR, Gray HB. Chem Rev. 2014;114:3369–3380. doi: 10.1021/cr4004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]; (b) Page CC, Moser CC, Chen X, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]; (c) Moser CC, Anderson JLR, Dutton PL. Biochim Biophys Acta. 2010;1797:1573–1586. doi: 10.1016/j.bbabio.2010.04.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Cukier RI, Nocera DG. Annu Rev Phys Chem. 1998;49:337–369. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]; (b) Reece SY, Nocera DG. Annu Rev Biochem. 2009;78:673–699. doi: 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Huynh MH, Meyer TJ. Chem Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Chem Rev. 2012;112:4016–4093. doi: 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- 6.Hammes-Schiffer S, Stuchebrukhov AA. Chem Rev. 2010;110:6939–6960. doi: 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Mayer JM. Annu Rev Phys Chem. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]; (b) Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961–697001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Migliore A, Polizzi NF, Therien MJ, Beratan DN. Chem Rev. 2014;114:3381–3465. doi: 10.1021/cr4006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Vinyard DJ, Ananyev GM, Dismukes GC. Annu Rev Biochem. 2013;82:577–606. doi: 10.1146/annurev-biochem-070511-100425. [DOI] [PubMed] [Google Scholar]; (b) Shen JR. Annu Rev Plant Biol. 2015;66:23–48. doi: 10.1146/annurev-arplant-050312-120129. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kaila VRI, Verkhovsky MI, Wikström M. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]; (b) Blomberg MRA. Biochemistry. 2016;55:489–500. doi: 10.1021/acs.biochem.5b01205. [DOI] [PubMed] [Google Scholar]
- 11.Minnihan EC, Nocera DG, Stubbe J. Acc Chem Res. 2013;46:2524–2535. doi: 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 13.Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 14.Minnihan EC, Young DD, Schultz PG, Stubbe J. J Am Chem Soc. 2011;133:15942–15945. doi: 10.1021/ja207719f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Cole PA. J Am Chem Soc. 1998;120:6851–6858. [Google Scholar]
- 16.Seyedsayamdost MR, Reece SY, Nocera DG, Stubbe J. J Am Chem Soc. 2006;128:1569–1579. doi: 10.1021/ja055926r. [DOI] [PubMed] [Google Scholar]
- 17.Ravichandran KR, Minnihan EC, Wei Y, Nocera DG, Stubbe J. J Am Chem Soc. 2015;137:14387–14395. doi: 10.1021/jacs.5b09189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oyala PH, Ravichandran KR, Funk MA, Stucky PA, Stich TA, Drennan CL, Britt RD, Stubbe J. J Am Chem Soc. 2016;138:7951–7964. doi: 10.1021/jacs.6b03605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Holder PG, Pizano AA, Anderson BL, Stubbe J, Nocera DG. J Am Chem Soc. 2012;134:1172–1180. doi: 10.1021/ja209016j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Olshansky L, Pizano AP, Wei Y, Stubbe J, Nocera DG. J Am Chem Soc. 2014;136:16210–16216. doi: 10.1021/ja507313w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Song DY, Pizano AA, Holder PG, Stubbe J, Nocera DG. Chem Sci. 2015;6:4519–4524. doi: 10.1039/c5sc01125f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olshansky L, Stubbe J, Nocera DG. J Am Chem Soc. 2016;138:1196–1205. doi: 10.1021/jacs.5b09259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rappaport F, Boussac A, Force DA, Peloquin J, Brynda M, Sugiura M, Un S, Britt RD, Diner BA. J Am Chem Soc. 2009;131:4425–4433. doi: 10.1021/ja808604h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu Y, Lv X, Li J, Zhou Q, Cui C, Hosseinzadeh P, Mukherjee A, Nilges MJ, Wang J, Lu Y. J Am Chem Soc. 2015;137:4594–4597. doi: 10.1021/ja5109936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y, Zhou Q, Wang L, Liu X, Zhang W, Hu M, Dong J, Li J, Lv X, Ouyang H, Li H, Gao F, Gong W, Lu Y, Wang J. Chem Sci. 2015;6:3881–3885. doi: 10.1039/c5sc01126d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosseinzadeh P, Lu Y. Biochim Biophys Acta. 2016;1857:557–581. doi: 10.1016/j.bbabio.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tommos C, Skalicky JJ, Pilloud DL, Wand AJ, Dutton PL. Biochemistry. 1999;38:9495–9507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 26.Westerlund K, Berry BW, Privett HK, Tommos C. Biochim Biophys Acta. 2005;1707:103–116. doi: 10.1016/j.bbabio.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Dai QH, Tommos C, Fuentes EJ, Blomberg MRA, Dutton PL, Wand AJ. J Am Chem Soc. 2002;124:10952–10953. doi: 10.1021/ja0264201. [DOI] [PubMed] [Google Scholar]
- 28.Tommos C, Valentine KG, Martínez-Rivera MC, Liang L, Moorman VR. Biochemistry. 2013;52:1409–1418. doi: 10.1021/bi301613p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glover SD, Jorge C, Liang L, Valentine KG, Hammarström L, Tommos C. J Am Chem Soc. 2014;136:14039–14051. doi: 10.1021/ja503348d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravichandran KR, Liang L, Stubbe J, Tommos C. Biochemistry. 2013;52:8907–8915. doi: 10.1021/bi401494f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hay S, Westerlund K, Tommos C. Biochemistry. 2005;44:11891–11902. doi: 10.1021/bi050901q. [DOI] [PubMed] [Google Scholar]
- 32.Martinéz-Rivera MC, Berry BW, Valentine KG, Westerlund K, Hay S, Tommos C. J Am Chem Soc. 2011;133:17786–17795. doi: 10.1021/ja206876h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berry BW, Martinéz-Rivera MC, Tommos C. Proc Natl Acad Sci USA. 2012;109:9739–9743. doi: 10.1073/pnas.1112057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Armstrong FA, Wilson GS. Electrochim Acta. 2000;45:2623–2645. [Google Scholar]; (b) Armstrong FA, Camba R, Heering HA, Hirst J, Jeuken LJC, Jones AK, Léger C, McEvoy JP. Faraday Discuss. 2000;116:191–203. doi: 10.1039/b002290j. [DOI] [PubMed] [Google Scholar]; (c) Léger C, Bertrand P. Chem Rev. 2008;108:2379–2438. doi: 10.1021/cr0680742. [DOI] [PubMed] [Google Scholar]
- 35.Simon J, Salzbrunn S, Prakash GKS, Petasis NA, Olah GA. J Org Chem. 2001;66:633–634. doi: 10.1021/jo0015873. [DOI] [PubMed] [Google Scholar]
- 36.Chen H, Gollnick P, Phillips RS. Eur J Biochem. 1995;229:540–549. [PubMed] [Google Scholar]
- 37.Seyedsayamdost MR, Yee CS, Stubbe J. Nat Protoc. 2007;2:1225–1235. doi: 10.1038/nprot.2007.159. [DOI] [PubMed] [Google Scholar]
- 38.Bendezú FO, Hale CA, Bernhardt TG, de Boer PAJ. EMBO J. 2009;28:193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salwiczek M, Nyakatura EK, Gerling UIM, Ye S, Koksch B. Chem Soc Rev. 2012;41:2135–2171. doi: 10.1039/c1cs15241f. [DOI] [PubMed] [Google Scholar]
- 40.Buer BC, Meagher JL, Stuckey JA, Marsh ENG. Proc Natl Acad Sci U S A. 2012;109:4810–4815. doi: 10.1073/pnas.1120112109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mortenson DE, Satyshur KA, Guzei IA, Forest KT, Gellman SH. J Am Chem Soc. 2012;134:2473–2476. doi: 10.1021/ja210045s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Y, Ghirlanda G, Vaidehi N, Kua J, Mainz DT, Goddard WA, III, DeGrado WF, Tirrell DA. Biochemistry. 2001;40:2790–2796. doi: 10.1021/bi0022588. [DOI] [PubMed] [Google Scholar]
- 43.Clark GA, Baleja JD, Kumar K. J Am Chem Soc. 2012;134:17912–17921. doi: 10.1021/ja212080f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osteryoung J, O’Dea JJ. In: Electroanalytical Chemistry. Bard AJ, editor. Vol. 14. Marcel Dekker; New York: 1986. pp. 209–308. 1986. [Google Scholar]
- 45.(a) Mirčeski V, Komorsky-Lovrić Š, Lovrić M. Square-wave voltammetry: Theory and applications. In: Scholz F, editor. Monographs in electrochemistry. Springer-Verlag; Berlin, Germany: 2007. [Google Scholar]; b Mirčeski V, Gulaboski R, Lovrić M, Bogeski I, Kappl R, Hoth M. Electroanalysis. 2013;25:2411–2422. [Google Scholar]
- 46.Mahmoudi L, Kissner R, Nauser T, Koppenol WH. Biochemistry. 2016;55:2849–2856. doi: 10.1021/acs.biochem.6b00019. [DOI] [PubMed] [Google Scholar]
- 47.(a) Lovrić M, Komorsky-Lovrić Š. J Electroanal Chem. 1988;248:239–253. [Google Scholar]; (b) Komorsky-Lovrić Š, Lovrić M. J Electroanal Chem. 1995;384:115–122. [Google Scholar]; (c) Komorsky-Lovrić Š, Lovrić M. Anal Chim Acta. 1995;305:248–255. [Google Scholar]
- 48.(a) Reeves JH, Song S, Bowden EF. Anal Chem. 1993;65:683–688. [Google Scholar]; (b) O’Dea JJ, Osteryoung JG. Anal Chem. 1993;65:3090–3097. [Google Scholar]
- 49.Wu Y, Fried SD, Boxer SG. Biochemistry. 2015;54:7110–7119. doi: 10.1021/acs.biochem.5b00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.(a) Jovanovic SV, Harriman A, Simic MG. J Phys Chem. 1986;90:1935–1939. [Google Scholar]; (b) Lind J, Eriksen TE, Merényi G. J Am Chem Soc. 1990;112:479–482. [Google Scholar]
- 51.DeFelippis MR, Murthy CP, Faraggi M, Klapper MH. Biochemistry. 1989;28:4847–4853. doi: 10.1021/bi00437a049. [DOI] [PubMed] [Google Scholar]
- 52.Folkes LK, Trujillo M, Bartesaghi S, Radi R, Wardman P. Arch Biochem Biophys. 2011;506:242–249. doi: 10.1016/j.abb.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 53.Harriman A. J Phys Chem. 1987;91:6102–6104. [Google Scholar]
- 54.DeFelippis MR, Murthy CP, Broitman F, Weinraub D, Faraggi M, Klapper MH. J Phys Chem. 1991;95:3416–3419. [Google Scholar]
- 55.Yee CS, Seyedsayamdost MR, Chang MCY, Nocera DG, Stubbe J. Biochemistry. 2003;42:14541–14552. doi: 10.1021/bi0352365. [DOI] [PubMed] [Google Scholar]
- 56.Armstrong DA, Huie RE, Koppenol WH, Lymar SV, Merényi G, Neta P, Ruscic B, Stanbury DM, Steen Steenken S, Wardman P. Pure Appl Chem. 2015;87:1139–1150. [Google Scholar]
- 57.(a) Prütz WA, Butler J, Land EJ. Int J Radiat Biol. 1983;44:183–196. doi: 10.1080/09553008314550981. [DOI] [PubMed] [Google Scholar]; (b) Hawkins CL, Davies MJ. Biochim Biophys Acta. 2001;1504:196–219. doi: 10.1016/s0005-2728(00)00252-8. [DOI] [PubMed] [Google Scholar]
- 58.Costentin C, Louault C, Robert M, Savéant JM. Proc Nat Acad Sci USA. 2009;106:18143–18148. doi: 10.1073/pnas.0910065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nicholson RS. Anal Chem. 1965;37:667–671. [Google Scholar]
- 60.Kasanmascheff M, Lee W, Nick TU, Stubbe J, Bennati M. Chem Sci. 2016;7:2170–2178. doi: 10.1039/c5sc03460d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yokoyama K, Uhlin U, Stubbe J. J Am Chem Soc. 2010;132:15368–15379. doi: 10.1021/ja1069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yokoyama K, Smith AA, Corzilius B, Griffin RG, Stubbe J. J Am Chem Soc. 2011;133:18420–18432. doi: 10.1021/ja207455k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ravichandran KR, Taguchi AT, Wei Y, Tommos C, Nocera DG, Stubbe J. J Am Chem Soc. 2016;138:13706–13716. doi: 10.1021/jacs.6b08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wörsdörfer B, Conner DA, Yokoyama K, Livada J, Seyedsayamdost M, Jiang W, Silakov A, Stubbe J, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2013;135:8585–8593. doi: 10.1021/ja401342s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yokoyama K, Uhlin U, Stubbe J. J Am Chem Soc. 2010;132:8385–8397. doi: 10.1021/ja101097p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.(a) Kim CH, Axup JY, Schultz PG. Curr Opin Chem Biol. 2013;17:412–419. doi: 10.1016/j.cbpa.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hallam TJ, Wold E, Wahl A, Smider VV. Mol Pharm. 2015;12:1848–1862. doi: 10.1021/acs.molpharmaceut.5b00082. [DOI] [PubMed] [Google Scholar]; (c) Quast RB, Mrusek D, Hoffmeister C, Sonnabend A, Kubick S. FEBS Lett. 2015;589:1703–1712. doi: 10.1016/j.febslet.2015.04.041. [DOI] [PubMed] [Google Scholar]
- 67.(a) Shibamoto T, Kato Y, Sugiura M, Watanabe T. Biochemistry. 2009;48:10682–10684. doi: 10.1021/bi901691j. [DOI] [PubMed] [Google Scholar]; (b) Nilsson H, Cournac L, Rappaport F, Messinger J, Lavergne J. Biochim Biophys Acta. 2016;1857:23–33. doi: 10.1016/j.bbabio.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 68.(a) Gray HB, Winkler JR. Proc Natl Acad Sci USA. 2015;112:10920–10925. doi: 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Winkler JR, Gray HB. Quart Rev Biophys. 2015;48:411–420. doi: 10.1017/S0033583515000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.(a) Yu MA, Egawa T, Shinzawa-Itoh K, Yoshikawa S, Guallar V, Yeh SR, Rousseau DL, Gerfen GJ. J Am Chem Soc. 2012;134:4753–4761. doi: 10.1021/ja210535w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tsai AL, Kulmacz RJ. Arch Biochem Biophys. 2010;493:103–124. doi: 10.1016/j.abb.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ranguelova K, Girotto S, Gerfen GJ, Yu S, Suarez J, Metlitsky L, Magliozzo RS. J Biol Chem. 2007;282:6255–6264. doi: 10.1074/jbc.M607309200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Miner KD, Pfister TD, Hosseinzadeh P, Karaduman N, Donald LJ, Loewen PC, Lu Y, Ivancich A. Biochemistry. 2014;53:3781–3789. doi: 10.1021/bi500353p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.