

Abstract
Background
The criteria for assessing upper motor neuron pathology in pure lower motor neuron disease (LMND) still remain a major issue of debate with respect to the clinical classification as an amyotrophic lateral sclerosis (ALS) variant.
Objective
The study was designed to investigate white matter damage by a hypothesis-guided tract-of-interest-based approach in patients with LMND compared with healthy controls and ´classical´ ALS patients in order to identify in vivo brain structural changes according to the neuropathologically defined ALS affectation pattern. Data were pooled from two previous studies at two different study sites (Ulm, Germany and Milano, Italy).
Methods
DTI-based white matter integrity mapping was performed by voxelwise statistical comparison and by a tractwise analysis of fractional anisotropy (FA) maps according to the ALS-staging pattern for 65 LMND patients (clinically differentiated in fast and slow progressors) vs. 92 matched controls and 101 ALS patients with a ‘classical’ phenotype to identify white matter structural alterations.
Results
The analysis of white matter structural connectivity by regional FA reductions demonstrated the characteristic alteration patterns along the CST and also in frontal and prefrontal brain areas in LMND patients compared to controls and ALS. Fast progressing LMND showed substantial involvement, like in ALS, while slow progressors showed less severe alterations. In the tract-specific analysis according to the ALS-staging pattern, fast progressing LMND showed significant alterations of ALS-related tract systems as compared to slow progressors and controls.
Conclusions
This study showed an affectation pattern for corticoefferent fibers in LMND with fast disease progression as defined for ALS, that way confirming the hypothesis that fast progressing LMND is a phenotypical variant of ALS.
Keywords: DTI, Lower motor neuron disease, ALS, PMA, MRI
Highlights
-
•
LMND is associated with brain alterations along the CST and in frontal areas.
-
•
Fast progressive LMND show cerebral tract involvement like in ALS.
-
•
DTI supports that fast progressive LMND is a phenotypical variant of ALS.
1. Introduction
Classification of motor neuron disorders (MND) is a challenge of growing importance given that the therapeutic portfolio for amyotrophic lateral sclerosis (ALS) might expand in the future, as reflected in the efforts to revise the diagnostic criteria (Ludolph et al., 2015). In this context, adult lower motor neuron disease (LMND) without clinically overt upper motor neuron (UMN) pathology accounts for about 10% of all cases of MND types and is also traditionally named progressive muscular atrophy (PMA) (Norris et al., 1993, Traynor et al., 2000).
The definition of PMA is a major issue in clinical diagnosis, as discussed in the literature since the 1990s when neuropathologists addressed the lack of evidence of UMN involvement in certain cases (Ince et al., 1998) although ALS was suspected. The challenge remained to unravel central nervous system involvement in the LMND phenotype of ALS in vivo. Neuroimaging techniques such as diffusion tensor imaging (DTI) are one of the favourite candidates (Turner et al., 2011, Filippi et al., 2015). Recently, it was possible to apply the hypothesis-guided tract-of-interest-based DTI technique which constitutes the in vivo transfer (Kassubek et al., 2014, Müller et al., 2016, Kassubek and Müller, 2016) of the neuropathological staging concept of ALS by Braak and coworkers (Braak et al., 2013, Brettschneider et al., 2015) to a sample of LMND patients. Here, a pattern of tract involvement could be demonstrated like in patients with the ‘classical’ phenotype of ALS (Rosenbohm et al., 2016). However, the CNS involvement might not be detected when MRI analyses are used which are state of the art but data-driven, as demonstrated in a recent study (Spinelli et al., 2016). Thus, a DTI study with fiber tracking techniques according to the staging hypothesis for ALS was applied to the pooled data from the two centers (Ulm, Germany and Milan, Italy) which previously have reported apparently heterogeneous results about the involvement of corticofugal tracts in LMND (Rosenbohm et al., 2016, Spinelli et al., 2016). For this study, both a data-driven whole brain based voxelwise analysis and a hypothesis-driven tract of interest-based analysis were used in order to establish a neuroimaging-based definition of disease-associated patterns of microstructural WM alterations. The following hypotheses were tested: 1) The LMND affectation pattern as previously demonstrated (Rosenbohm et al., 2016) is reproducible independent of the subject samples, the scanner, and the DTI scanning protocol; the results at the group level for LMND, ALS and controls are reproducible independent of the analysis technique (compare this study and the study by Spinelli et al., 2016); 2) fast progressive LMND patients show a pattern of disease-associated tract alterations like ALS.
2. Subjects and analysis methods
2.1. Subjects
2.1.1. Patient characteristics
All patients underwent standardized clinical, neurological, and routine laboratory examinations. All subjects gave written informed consent for the study protocol according to institutional guidelines which had been approved both by the Ethics Committee of Ulm University, Germany (No. 19/12) and the local ethical committee on human studies of Milan, Italy (No. RF-2010-2,313,220).
The diagnosis of adult LMND was based on the presence of pure lower motor neuron (LMN) findings in two or more regions (bulbar, cervical, thoracic, lumbosacral) at the first evaluation, including evidence of LMN involvement on neurological examination (weakness and muscular atrophy, absent tendon reflexes), electrophysiological evidence of LMN involvement on standardized needle EMG, and no motor nerve conduction block. Patients were required to meet the following criteria: (a) a clinical diagnosis of sporadic LMN-predominant disease (Chiò et al., 2011, van den Berg-Vos et al., 2003). (b) age of onset of 40 years or older, (c) no concomitant clinical diagnosis of frontotemporal dementia (Rascovsky et al., 2011), (d) no systemic or other neurologic diseases or substance abuse, and (e) no other causes of focal or diffuse brain and spinal cord damage at routine MR imaging, including any cerebrovascular disorders, i.e. multifocal and/or confluent white matter hyperintensity. Specific exclusion criteria for the LMN-predominant disease group were a history of syndromes that mimic LMN-predominant disease or clinical signs of definite UMN involvement such as pseudobulbar symptoms, clonus or masseter reflex, and extensor plantar response. None of the patients had a known gene mutation.
In summary, 65 patients (37 from Ulm and 28 from Milan) fulfilled these criteria and presented with the diagnosis of adult LMND. Patients with ALS (Brooks et al., 2000) with clinical signs of both UMN and LMN involvement were screened to match patients with LMN-predominant disease for age, sex, and disease severity as measured with the revised ALS Functional Rating Score. That way, 101 patients (50 from Ulm and 51 from Milan) with the diagnosis of ALS and 92 healthy controls (53 from Ulm and 39 from Milan) were selected from data bases. A summary of the subjects' characteristics is given in Table 1. The group comparison concerning gender by chi squared test revealed 0.053, thus indicating no significant differences for the different subject groups concerning gender. In addition, a test for gender differences in the control group showed no significant results in the analysis (see 2.2.3, 2.2.4) between male and female controls.
Table 1.
Subjects' characteristics. Adisease progression rate = (48-ALSFRS-R score at clinical examination)/disease duration (years).
| LMND | ALS | Controls | p | |
|---|---|---|---|---|
| Male/female | 48/17 | 58/43 | 52/40 | Chi squared: 0.053 |
| 1.5 T | 30/7 | 30/20 | 36/17 | |
| 3.0 T | 18/10 | 28/23 | 16/23 | |
| Age/years (mean ± std. dev.) |
61 ± 10 | 61 ± 11 | 60 ± 11 | Kruskal-Wallis: 0.84 |
| 1.5 T | 63 ± 10 | 58 ± 10 | 58 ± 12 | |
| 3.0 T | 60 ± 10 | 63 ± 11 | 63 ± 8 | |
| Disease duration/years (mean ± std. dev.) | 4.5 ± 6.3 | 1.9 ± 1.8 | n.a. | |
| 1.5 T | 3.9 ± 5.9 | 2.1 ± 2.0 | n.a. | |
| 3.0 T | 5.3 ± 6.8 | 1.7 ± 1.5 | n.a. | |
| Disease progression rateA (mean ± std. dev.) | 4.9 ± 6.5 | 8.7 ± 8.7 | n.a. | |
| 1.5 T | 4.5 ± 5.8 | 7.8 ± 8.4 | n.a. | |
| 3.0 T | 5.4 ± 7.5 | 8.2 ± 8.3 | n.a. |
Gross brain pathology including vascular brain alterations was excluded by conventional MRI. All healthy control subjects had no family history of neuromuscular disease and had no history of neurologic, psychiatric, or other major medical illnesses and were recruited from among spouses of patients and by word of mouth. Controls underwent a Mini Mental State Examination and were required to have a score of greater than or equal to 29.
2.1.2. MRI acquisition
DTI scanning at Ulm was performed on a 1.5 Tesla Magnetom Symphony (Siemens Medical, Erlangen, Germany); DTI scanning at Milan was performed on a 3.0 Tesla Intera (Philips Medical Systems, Best, The Netherlands).
At 1.5 T, two DTI study protocols were used. DTI study protocol A consisted of 13 volumes (45 slices, 128 × 128 pixels, slice thickness 2.2 mm, pixel size 1.5 mm × 1.5 mm) representing 12 gradient directions (b = 800 s/mm2) and one scan with gradient 0 (b = 0). The echo time (TE) and repetition time (TR) were 93 ms and 8000 ms, respectively. Five scans were averaged online by the scanner software in image space. DTI study protocol B consisted of 52 volumes (64 slices, 128 × 128 pixels, slice thickness 2.8 mm, pixel size 2.0 mm × 2.0 mm) representing 48 gradient directions (b = 1000 s/mm2) and four scans with b = 0. TE and TR were 95 ms and 8000 ms, respectively.
At 3.0 T, the DTI study protocol consisted of 34 volumes (55 slices, 96 × 96 pixels, slice thickness 2.5 mm, pixel size 0.94 mm × 0.94 mm) representing 32 gradient directions (b = 1000 s/mm2) and two scans with b = 0. TE and TR were 80 ms and 8986 ms, respectively. Two scans were averaged online by the scanner software in image space.
2.2. Data analysis
The postprocessing and statistical analysis was performed by use of the analysis software Tensor Imaging and Fiber Tracking (TIFT) (Müller et al., 2007a). Fig. 1 provides a schematical workflow for the analysis of DTI data of 140 data sets from 1.5 T and 118 data sets from 3.0 T. In short, after stereotaxic normalization and calculation of subjects' FA maps a merging of centers' FA maps was performed followed by corrections for the covariates age and disease duration. These center-harmonized FA maps were then used to compared ALS patients, low and fast LMND progressors and controls in an unbiased approach (by whole brain-based statistical analysis – WBSS) as well as tractwise (by Tractwise FA statistics – TFAS).
Fig. 1.
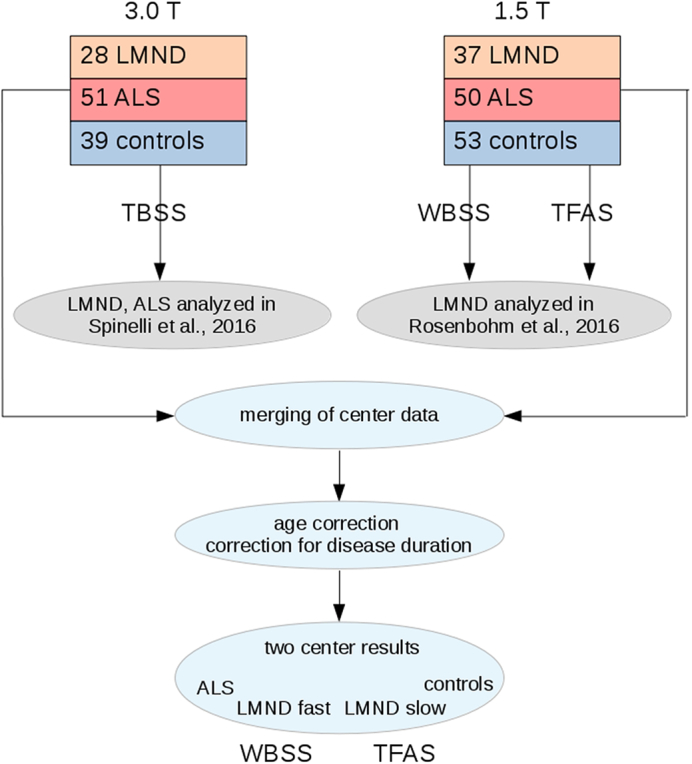
Schematical workflow for the analysis of DTI data. 3.0 T: DTI data of 28 LMND patients, 51 ALS patients, and 39 controls – data were partially analyzed by (Spinelli et al., 2016), analysis methods was hypothesis free tract-based spatial statistics (TBSS – Smith et al., 2006). 1.5 T: DTI data of 37 LMND patients and 53 controls – data were partially analyzed by (Rosenbohm et al., 2016), analysis methods were hypothesis free whole brain-based statistical analysis (WBSS) as well as tractwise fractional anisotropy statistics (TFAS). After stereotaxic normalization and calculation of subjects' FA maps a merging of centers' FA maps was performed followed by corrections for the covariates age and disease duration. These center-harmonized FA maps were then used to compared ALS patients, low and fast LMND progressors and controls hypothesis free (WBSS) as well as tractwise (TFAS).
2.2.1. Stereotaxic normalization
In order to spatially normalize the data to the Montreal Neurological Institute (MNI) stereotaxic space, study-specific templates were created and MNI normalization was performed iteratively (Müller and Kassubek, 2013). From the stereotaxically normalized DTI data sets, fractional anisotropy (FA) maps were calculated for quantitative mapping of structural connectivity (Le Bihan et al., 2001).
In a consecutive step, an 8 mm (FWHM) Gaussian filter was applied for smoothing of FA maps in order to achieve a good balance between sensitivity and specificity. For smoothing, the fact that filter size influences results of DTI data analysis (Jones et al., 2005) requires application of the matched filter theorem which states that the width of the filter used to process the data should be tailored to the size of the difference one expects to see (Rosenfeld and Kak, 1982). Variations on the filter size between 6 mm and 10 mm did not show significant differences to the selected filter size of 8 mm, as previously reported (Unrath et al., 2010).
2.2.2. Harmonisation of FA-maps according to different acquisition protocols
FA maps of controls recorded with the different protocols were used for calculation of 3D–correction matrices according to a previously reported protocol (Rosskopf et al., 2015, Müller et al., 2016). Then, FA maps of LMND and ALS patients and controls were harmonized by application of the respective 3D–correction matrix (linear first order correction). In the final step, FA maps of all subjects were corrected for the covariate age.
2.2.3. Whole brain-based spatial statistics – WBSS
Statistical comparison by Student's t-test was performed voxelwise to detect changes between the subject groups, i.e. FA values of one subject group were compared with the FA values of another subject group for each voxel separately – WBSS. FA values below 0.2 were not considered for calculation as cortical grey matter shows FA values up to 0.2 (Kunimatsu et al., 2004).
Statistical results were corrected for multiple comparisons using the false-discovery-rate (FDR) algorithm at p < 0.05 (Genovese et al., 2002). Further reduction of the alpha error was performed by a spatial correlation algorithm that eliminated isolated voxels or small isolated groups of voxels in the size range of the smoothing kernel leading to a threshold cluster size of 256 voxels.
2.2.4. Definition of tract systems according to the ALS-staging theory and tractwise fractional anisotropy statistics
From a cohort of 64 controls' DTI data sets, pathways for defined brain structures according to the ALS-staging system (Brettschneider et al., 2013) were identified with a seed-to-target approach as previously described (Kassubek et al., 2014, Rosenbohm et al., 2016). For this purpose, regions of interest (ROIs) were defined for the start and target regions. All potential fiber tracts (FTs) originating in the start ROI and ending in the target ROI of the respective pathway define the corresponding tract of interest (TOI). For the fiber tracking technique, a modified deterministic streamline tracking approach (FA threshold 0.2) was used that takes the directional information of neighboured FTs into account (Müller et al., 2007b). Representative TOIs for the definition of the four ALS stages are: the corticospinal tract (CST, representative for stage 1), the corticorubral and corticopontine tracts (representative for stage 2), the corticostriatal pathway (representative for stage 3), and the proximal perforant path (representative for stage 4) (Kassubek et al., 2014). As defined previously, a reference path was used originating from the corpus callosum (CC) area V where no involvement in ALS-associated neurodegeneration could be anticipated. TFAS was performed by statistically comparing the FA values in a respective tract system between two subject groups (Student's t-test).
2.2.5. Normalization of FA maps to disease duration
A correlation (Pearson correlation) could be observed for ALS patients between the revised ALS functional rating scale (ALS-FRS-R) (Cedarbaum et al., 1999) and the FA values along the CST (TFAS) (Fig. 2). Supplementary Fig. 1A shows the progression rate vs. the disease duration for LMND patients and ALS patients, respectively. One part of the LMND patients shows progression rates similar to ALS patients while the other part shows lower progression rates; details are provided in the Results section. Following this association between clinical scores and FA, it is safe to assume for LMND patients that FA values decrease in the respective disease related tract systems with increasing disease duration. That means that LMND patients with a long disease duration show decreased FA values although disease progression might be slow; on the other hand, LMND patients with a high disease progression rate (even ALS like) could present higher FA values due to a short disease duration. This scenario is mirrored in Fig. 1B for FA values in the CST of LMND patients:
Fig. 2.

Significant correlation for ALS patients between the ALS-FRS-R and the FA values along the CST (TFAS).
Supplementary Fig. 1.

(A) Progression rate vs. the disease duration for LMND patients and ALS patients. One part of the LMND patients shows progression rates similar to ALS patients where a smaller part shows lowered progression rates. (B) Correlation between LMND patients' FA values in the CST and disease duration. LMND patients with a long disease duration could show decreased FA values although they follow a slow disease progression; LMND patients with a high disease progression rate (even ALS like) could present comparatively high FA values due to a short disease duration. Mean FA values in the CST (± standard deviation) for controls are marked in blue, for ALS patients are marked in red.
Generally, there is a dependency in LMND between the progression rate of the disease and the disease duration, i.e. LMND patients with a fast progression rate usually show a short disease duration at the timepoint of MRI investigation and LMND patients with a long disease duration mostly show a lower progression rate. This means that, when the LMND group is simply separated according to disease duration or the LMND group as a whole is compared to controls, such an analysis will be blind to the fact that there is variability within the LMND group consisting of fast progressors (who are hypothesized to behave like ALS) and slow progressors (who are hypothesized to behave like controls). Thus, it is not only necessary to split the LMND group into fast and low progressors (what could be performed either by disease duration or by progression rate), but a normalization of FA values to the identical timepoint in the course of the disease has to be performed.
Since FA values could depend on the disease duration and the progression rate, FA values were normalized to the identical timepoint of disease duration. As such, a disease duration of 4 years was chosen which has already been suggested to be the timepoint to discriminate between LMND subtypes (van den Berg-Vos et al., 2003, Rosenbohm et al., 2016).
In a defined voxel or tract structure, the average FA decrease per year.
< ΔFA >ALS, year = ΔFA(controls ‐ ALS)/ < DD >ALS.for ALS patients was defined by the difference in FA between controls and ALS patients divided by the average disease duration of ALS patients.
The average progression rate in ALS patients was calculated by arithmetically averaging the individual progression rates.
< R >ALS£ ≤ (48 – ALS ‐ FRS ‐ R)/DD >ALS ‐ patients.
The individual FA loss per year was then estimated by ΔFAALS,year weighted by the ratio of individual progression rate and the average progression rate in ALS patients.
ΔFA,year ≤ ΔFA >ALS, year ∗ R/ < R >ALS.
The individual FA value for the defined timepoint 4 years can then be calculated by.
FA4years = FAscan + (DD ‐ 4years)ΔFA,year.
This normalization for disease duration was performed for averaged FA values of tract structures (TFAS) as well as on a voxelbasis prior to WBSS. NB: < ΔFA >ALS,year has to be calculated for each tract structure as well as for each voxel.
2.2.6. Separation of the LMND group into slow and fast progressors
The LMND sample was subdivided into two subgroups, i.e. fast progressors with the assumption of an ALS-like prognosis and slow progressive patients. In the literature, van den Berg-Vos et al. (2003) suggested a separation threshold for LMND fast and slow progressors by a disease duration of less or > 4 years. This threshold was optimised for the needs of this study (which is dependent on the time of scanning within the disease course) by a threshold that is based on the progression rate. The disease duration and the progression rate showed a linear dependency on a double logarithmic scaling (Supplementary Fig. 1). As a consequence, the threshold for the ALS-FRS-R-based progression rate to differentiate between slow and fast progressors was set to 2 ALS-FRS-R points per year. This separation resulted in 36 fast LMND progressors (22 scanned at 1.5 T and 14 scanned at 3.0 T) and 29 slow LMND progressors (15 scanned at 1.5 T and 14 scanned at 3.0 T). Average disease duration for fast progressors was 1.6 ± 1.5 years and average disease duration for slow progressors was 8.1 ± 7.9 years.
2.3. Statistics
For statistical comparison at the group level, WBSS and TFAS analysis used Student's t-test as the subject groups were large enough to show a Gaussian distribution of FA values. However, Mann-Whitney-U test shows almost identical statistical results. For correlation analysis, parametric Pearson correlation was used.
3. Results
3.1. Whole brain-based spatial statistics of FA maps
The comparison at the group level by WBSS for LMND patients (subdivided into fast and slow progressors), ALS patients, and controls demonstrated multiple clusters of regional FA reductions at p < 0.05 (corrected for multiple comparisons). Sagittal projectional views are depicted in Fig. 3A for the group comparisons. Comparing the LMND fast progressors to the LMND slow progressors, widespread FA reduction along the CST and also in frontal and prefrontal brain areas were observed. LMND fast progressors showed a widepread FA reduction pattern compared to controls (similar to ALS patients vs. controls), while LMND slow progressors showed no FA reduction compared to controls; the whole LMND group showed moderate FA reductions compared to controls along the CST and also in frontal and prefrontal brain areas (Fig. 3, upper panel). At contrast, ALS patients vs. the LMND group of fast progressors showed no significant FA reduction and ALS patients vs. the LMND group of slow progressors showed a widespread FA reduction along the CST and in frontal and prefrontal brain areas, i.e. a similar pattern as ALS patients compared to controls. ALS patients vs. the whole LMND group showed moderate FA reductions predominantly along the CST (Fig. 3, lower panel). ALS patients vs. controls show the well-known widespread FA reduction along the CST and also in frontal and prefrontal brain areas. A summary of the clusters is provided in Table 2. A centerwise WBSS analysis (in addition to Fig. 3 in order to demonstrate separate results for each center) is provided in Supplementary Fig. 2, indicating similar results at the single center level compared to the two-center level.
Fig. 3.

(A) Whole brain-based spatial statistics (WBSS) of FA maps at the group level for ALS patients, LMND patients (subdivided into slow and fast progressors), and controls. WBSS of FA maps demonstrated multiple clusters of regional FA reductions at p < 0.05 (corrected for multiple comparisons), sagittal projectional views. (B) Comparison of FA maps at the group level for fast LMND progressors vs. slow LMND progressors. Whole brain-based spatial statistics (WBSS) of FA maps at p < 0.05, false discovery rate (FDR)-corrected.
Table 2.
Whole brain based spatial statistics (WBSS) for ALS patients vs. LMND patients vs. controls. Size of area of significant alterations in comparison at the group level (mm3).
| Size of area of significant alterations/mm3 | |
|---|---|
| ALS vs. controls | 289,928 |
| ALS vs. LMND fast | 0 |
| ALS vs. LMND slow | 150,405 |
| ALS vs. LMND | 66,660 |
| LMND fast vs LMND slow | 79,867 |
| LMND fast vs controls | 174,015 |
| LMND slow vs controls | 0 |
| LMND vs controls | 86,002 |
Supplementary Fig. 2.

Centerwise whole brain-based spatial statistics (WBSS) of FA maps at the group level for ALS patients, LMND patients (subdivided into slow and fast progressors), and controls. WBSS of FA maps demonstrated multiple clusters of regional FA reductions at p < 0.05 (corrected for multiple comparisons), sagittal projectional views.
A slicewise visualization of the FA alteration pattern for fast LMND progressors vs. slow LMND progressors including CST involvement is presented in Fig. 3B. For demonstration purposes, results without FA normalization to disease duration are provided in Supplementary Fig. 3.
Supplementary Fig. 3.

Whole brain-based spatial statistics (WBSS) and tractwise fractional anisotropy statistics (TFAS) of FA maps at the group level for ALS patients, LMND patients (subdivided into slow and fast progressors), and controls. (A) WBSS of FA maps demonstrated multiple clusters of regional FA reductions at p < 0.05 (corrected for multiple comparisons), sagittal projectional views. (B) TFAS demonstrated significant regional FA reductions in ALS-related tract systems and in the grand average predominantly between ALS patients and fast LMND progressors and slow LMND progressors and controls. No alterations between groups were observed in the reference tract. * p < 0.05, ** p < 0.001.
3.2. Differences of FA in tract systems
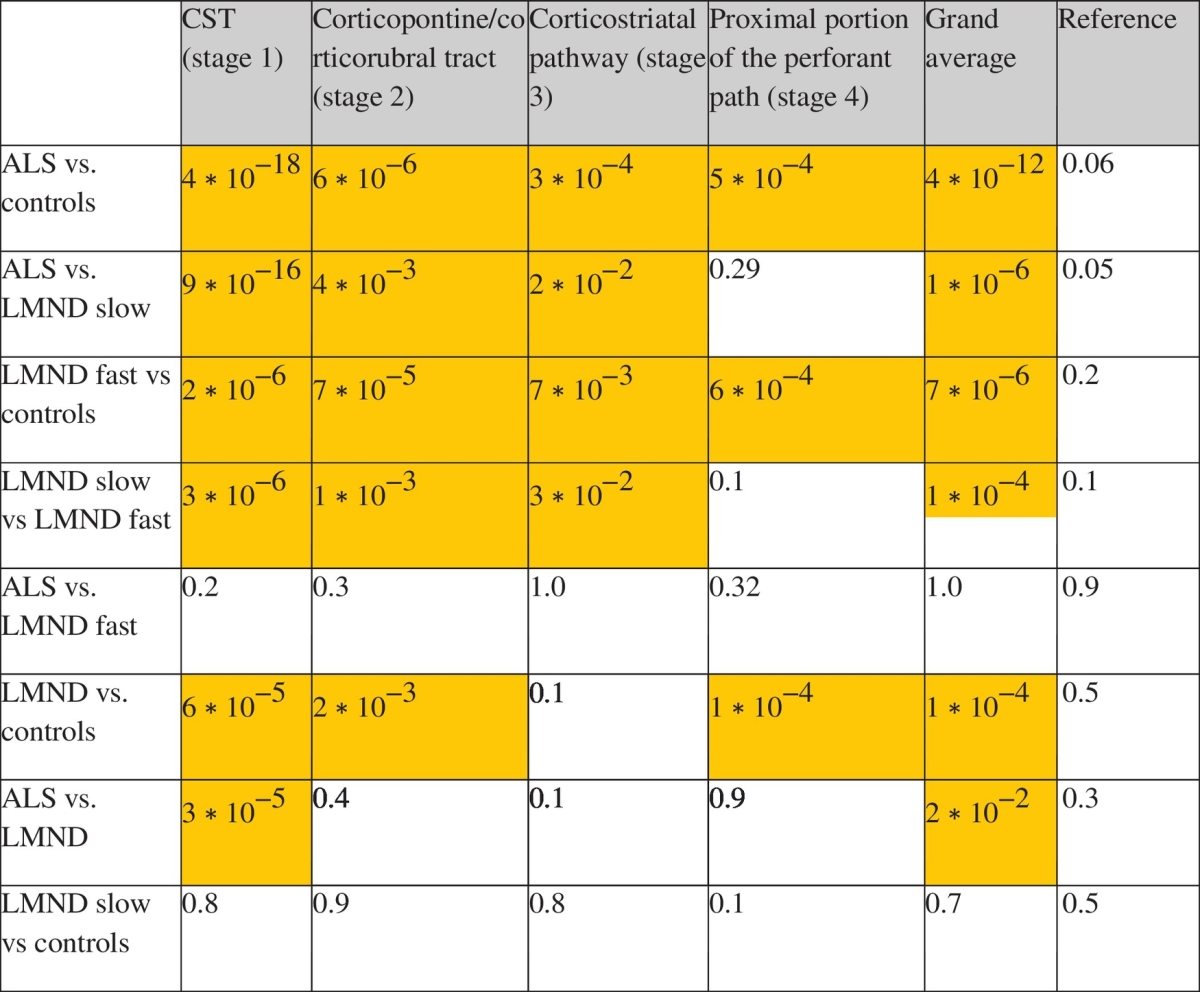
The hypothesis-guided analysis of the FA differences in the ALS-related tract systems (TFAS) showed differences of the averaged FA values between the ALS and LMND group; both, ALS and LMND group vs. the control group showed most prominent FA alterations in the CST, followed by FA reductions in the other ALS-staging-related tracts (Fig. 4). Here, significant FA reductions could be observed independently for the CST, the corticopontine and the corticorubral tract, and for the corticostriatal pathway (ALS stages 1–3) in ALS patients and fast LMND progressors each compared to controls and slow LMND progressors, respectively; less pronounced FA alterations were observed for the perforant path (ALS stage 4). For the grand average of the stage-related tract systems, significant FA reductions were observed for ALS patients and fast LMND progressors, both compared to controls and to slow LMND progressors. On the other hand, no significant FA alterations were found in the comparison between ALS patients and fast LMND progressors and between slow LMND progressors and controls, respectively. The whole LMND group showed moderate results compared to ALS patients and compared to controls – the averaged FA values of the whole LMND group were inbetween the averaged FA values of the fast and slow LMND progressors groups. No significant FA alterations could be observed for any group comparison in the reference tract system. A summary of all alterations in the tract systems at the group level is provided in Table 3. A centerwise TFAS analysis (corresponding to Fig. 4 with separate results for each center) is provided in Supplementary Fig. 4, indicating similar results also at the single-center level compared to the two-center level.
Fig. 4.

Tractwise fractional anisotropy statistics (TFAS) of FA maps at the group level for ALS patients, LMND patients (subdivided into slow and fast progressors), and controls. TFAS demonstrated significant regional FA reductions in ALS-related tract systems and in the grand average predominantly between ALS patients and fast LMND progressors and slow LMND progressors and controls. No alterations between groups were observed in the reference tract. * p < 0.05, ** p < 0.001.
Table 3.
p-values for differences between groups for different ALS-related tract systems. Significant FA alterations (p < 0.05) are coloured.
Supplementary Fig. 4.

Centerwise tractwise fractional anisotropy statistics (TFAS) of FA maps at the group level for ALS patients, LMND patients (subdivided into slow and fast progressors), and controls. TFAS demonstrated significant regional FA reductions in ALS-related tract systems and in the grand average predominantly between ALS patients and fast LMND progressors and slow LMND progressors and controls. No alterations were observed in the reference tract. * p < 0.05, ** p < 0.001.
4. Discussion
This DTI data analysis study supports the hypothesis that LMND is an ALS variant by demonstrating central nervous system involvement of the corticofugal tracts in fast progressive LMND. The data of this two-centre study strongly suggest that progressive LMND is probably one end of a clinicopathologic continuum of MND spanning primary lateral sclerosis, ALS, and LMND. The FA based affectation pattern for the group comparisons of ALS patients and LMND patients (subdivided into slow and fast progressors) could be demonstrated in an unbiased approach by WBSS as well as by the application of the hypothesis-guided tract of interest-based in vivo transfer of the stages according to the proposed staging scheme for ALS (Braak et al., 2013). With respect to the hypotheses, this two-center MRI study provides novelty in the following respects:
1) The LMND affectation pattern of our previous study (Rosenbohm et al., 2016), i.e. alterations of FA along the CST, was reproducible in a larger subject sample independent of the scanner and the DTI scanning protocol. In order to show the consistency with previous studies, WBSS of LMND vs. controls was calculated without normalization to disease duration, both for the combined samples as well as for a centerwise analysis (results in Supplementary Fig. 5). WBSS of the Milan data showed no differences between LMND and controls, in accordance with the previous analysis (Spinelli et al., 2016), while the Ulm data showed the alterations as previously described (Rosenbohm et al., 2016). We therefore conclude that the normalization to disease duration and the discrimination between fast and slow processors is essential in the analysis of MRI-based LMND data.
Supplementary Fig. 5.

Comparison of FA maps at the group level for LMND vs. controls without correction for disease duration. Whole brain-based spatial statistics (WBSS) of FA maps at p < 0.05, false discovery rate (FDR)-corrected. Left: combined data set. Center: Milan data only. Right: Ulm data only.
2) By dividing the LMND group into fast and slow progressors, it could be demonstrated that fast progressors show a similar pattern to ALS and slow progressors show a similar pattern to controls. It is important to note in the analysis of differences at the group level in LMND that, due to the inhomogeneous disease durations, FA values in the CST could be low due to a long disease duration (despite a low progression rate) and also FA values could be high due to a short disease duration (despite a high progression rate). Thus, a normalization procedure of FA values to an identical time-point was suggested. It has to be noted that this estimation is based on the assumption of a linear progression of FA values and also that a basic progression rate (i.e., the average progression of FA in ALS) has to be taken into account. By this approach, it was possible to show the differences of slow and fast progressors in the LMND group and to relate these resulting differences (at least for the fast progressors) to the staging based ALS affectation pattern. Therefore, one might assume that results of previous studies at the group level might differ due the possible inhomogeneity of the LMND-associated affectation pattern. The normalization to disease duration might help here to understand the differences in results of previous PMA/LMND based studies (Cosottini et al., 2005, van der Graaff et al., 2011, Spinelli et al., 2016, Rosenbohm et al., 2016). DTI-analysis based methodological differences may also explain inconsistent findings between the two previous reports (Rosenbohm et al., 2016, Spinelli et al., 2016). For WBSS (Rosenbohm et al., 2016), spatial normalization is required to make anatomically different subjects comparable in each individual voxel. Here, residual anatomical alterations were cleared by spatial smoothing. Tract-based spatial statistics (Spinelli et al., 2016) matches each voxel in different subjects projecting DTI data on a common anatomical skeleton and therefore does not require smoothing. Moreover, given the different statistical methods used by WBSS (Student's t-test with FDR correction) and TBSS (non-parametric permutation-based inference with cluster-level error correction), a direct comparison of findings can only be performed at the gross anatomical level. However, the main difference to previous studies is the normalization to disease duration, which helps to provide a differentiation of FA values between slow and fast LMND progressors. In order to show the consistency to previous studies, WBSS of LMND vs. controls without normalization to disease duration was calculated for the combined samples as well as for a centerwise analysis (Supplementary Fig. 3). WBSS of the Milan data showed no differences between LMND and controls, in accordance with the previous analysis (Spinelli et al., 2016), whereas the Ulm data show the alterations as previously described (Rosenbohm et al., 2016). We therefore conclude that the normalization to disease duration and the discrimination between fast and slow processors is essential in the analysis of MRI-based LMND data.
As a limitation, the study design was cross-sectional, while, in order to finally confirm the sequential spreading model (Kassubek et al., 2014) for fast LMND progressors, longitudinal data from different time points during the course of the disease are necessary. Thus, a longitudinal design would be more appropriate to assess the question of interest and the proposed goal of this work, investigating the relationship between the presence of tract involvement and the clinical development of UMN signs at the individual patient level. Future prospective studies could help to evaluate the changes in quantitative DTI measures over time and to correlate them with progression in clinical pathology. The current retrospective study was set up to combine data of LMND patients of different studies so that the differences of field strengths (1.5 T and 3.0 T) and different scanning protocols had to be dealt with. In the future, prospective studies with the identical protocol would be preferred. In general, with the growing number of multicenter studies in large-scale patient samples, the challenge of improved normalization of different scanning protocols will also be of rising importance (Müller et al., 2016). A further limitation is that genetic testing was not available for the ALS and LMND patients.
In summary, the neuroimaging results of this two-centre study confirm the clinical approach to the phenotype of fast progressive LMND as an ALS variant, in accordance with the latest revision of the El Escorial criteria for ALS (Ludolph et al., 2015, Agosta et al., 2015), in favour of the consequence to treat these patients like ALS and also to include them into clinical trials of ALS.
The following are the supplementary data related to this article.
Author contributions
Hans-Peter Müller: study concept and design, data analysis and interpretation of data, critical revision of manuscript for intellectual content.
Federica Agosta: Data collection, interpretation of data, critical revision of manuscript for intellectual content.
Nilo Riva: Data collection, interpretation of data, critical revision of manuscript for intellectual content.
Edoardo Gioele Spinelli: Data collection, interpretation of data, critical revision of manuscript for intellectual content.
Giancarlo Comi: Interpretation of data, critical revision of manuscript for intellectual content.
Albert Ludolph: Interpretation of data, study supervision, critical revision of manuscript for intellectual content.
Massimo Filippi: study concept and design, interpretation of data, study supervision, drafting of manuscript for intellectual content.
Jan Kassubek: study concept and design, interpretation of data, study supervision, drafting of manuscript.
Author disclosures
F. Agosta is Section Editor of NeuroImage: Clinical; has received speaker honoraria from ExceMED – Excellence in Medical Education and Biogen Idec; and receives or has received research supports from the Italian Ministry of Health, AriSLA (Fondazione Italiana di Ricerca per la SLA), and the European Research Council.
G. Comi has received consulting fees for participating on advisory boards from Novartis, Teva Pharmaceutical Ind. Ltd., Sanofi, Genzyme, Merck Serono, Bayer, Actelion and honorarium for speaking activities for Novartis, Teva Pharmaceutical Ind. Ltd., Sanofi, Genzyme, Merck Serono, Bayer, Biogen, ExceMED.
M. Filippi is Editor-in-Chief of the Journal of Neurology; serves on a scientific advisory board for Teva Pharmaceutical Industries; has received compensation for consulting services and/or speaking activities from Biogen Idec, ExceMED, Novartis, and Teva Pharmaceutical Industries; and receives research support from Biogen Idec, Teva Pharmaceutical Industries, Novartis, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, Cure PSP, Alzheimer's Drug Discovery Foundation (ADDF), the Jacques and Gloria Gossweiler Foundation (Switzerland), and ARiSLA (Fondazione Italiana di Ricerca per la SLA).
Jan Kassubek has received consulting fees as an advisory board member and honoraria as a speaker from UCB Pharma, Teva Pharmaceuticals, Zambon, Medtronic, Desitin, AbbVie, Boehringer Ingelheim, GlaxoSmithKline, Merz Pharmaceuticals, and Hoffmann-La Roche. Albert C. Ludolph has received consultant fees and honoraria as a speaker from Desitin, GSK and Biogen.
Statement
All human studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
References
- Agosta F., Al-Chalabi A., Filippi M. The El Escorial criteria: strengths and weaknesses. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:1–7. doi: 10.3109/21678421.2014.964258. [DOI] [PubMed] [Google Scholar]
- Braak H., Brettschneider J., Ludolph A.C. Amyotrophic lateral sclerosis - a model of corticofugal axonal spread. Nat. Rev. Neurol. 2013;9:708–714. doi: 10.1038/nrneurol.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J., Del Tredici K., Toledo J.B. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013;74:20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J., Del Tredici K., Lee V.M. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 2015;16:109–120. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B.R., Miller R.G., Swash M. World federation of neurology research group on motor neuron diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amytroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- Cedarbaum J.M., Stambler N., Malta E. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS study group (phase III) J. Neurol. Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- Chiò A., Calvo A., Moglia C. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J. Neurol. Neurosurg. Psychiatry. 2011;82:740–746. doi: 10.1136/jnnp.2010.235952. [DOI] [PubMed] [Google Scholar]
- Cosottini M., Giannelli M., Siciliano G. Diffusion-tensor MR imaging of corticospinal tract in amyotrophic lateral sclerosis and progressive muscular atrophy. Radiology. 2005;237:258–264. doi: 10.1148/radiol.2371041506. [DOI] [PubMed] [Google Scholar]
- Filippi M., Agosta F., Grosskreutz J. Progress towards a neuroimaging biomarker for amyotrophic lateral sclerosis. Lancet Neurol. 2015;14:786–788. doi: 10.1016/S1474-4422(15)00134-9. [DOI] [PubMed] [Google Scholar]
- Genovese C.R., Lazar N.A., Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. NeuroImage. 2002;15:870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- Ince P.G., Lowe J., Shaw P.J. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol. Appl. Neurobiol. 1998;24:104–117. doi: 10.1046/j.1365-2990.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Jones D.K., Symms M.R., Cercignani M. The effect of filter size on VBM analyses of DT-MRI data. NeuroImage. 2005;26:546–554. doi: 10.1016/j.neuroimage.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Kassubek J., Müller H.P. Computer-based magnetic resonance imaging as a tool in clinical diagnosis in neurodegenerative diseases. Expert. Rev. Neurother. 2016;16:295–306. doi: 10.1586/14737175.2016.1146590. [DOI] [PubMed] [Google Scholar]
- Kassubek J., Müller H.P., Del Tredici K. Diffusion tensor imaging analysis of sequential spreading of disease in amyotrophic lateral sclerosis confirms patterns of TDP-43 pathology. Brain. 2014;137:1733–1740. doi: 10.1093/brain/awu090. [DOI] [PubMed] [Google Scholar]
- Kunimatsu A., Aoki S., Masutani Y. The optimal trackability threshold of fractional anisotropy for diffusion tensor tractography of the corticospinal tract. Magn. Reson. Med. Sci. 2004;3:11–17. doi: 10.2463/mrms.3.11. [DOI] [PubMed] [Google Scholar]
- Le Bihan D., Mangin J.F., Poupon C. Diffusion tensor imaging: concepts and applications. J. Magn. Reson. Imaging. 2001;13:534–546. doi: 10.1002/jmri.1076. [DOI] [PubMed] [Google Scholar]
- Ludolph A., Drory V., Hardiman O. A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015;29:1–2. doi: 10.3109/21678421.2015.1049183. [DOI] [PubMed] [Google Scholar]
- Müller H.P., Kassubek J. Diffusion tensor magnetic resonance imaging in the analysis of neurodegenerative diseases. J. Vis. Exp. 2013;77 doi: 10.3791/50427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H.P., Unrath A., Ludolph A.C. Preservation of diffusion tensor properties during spatial normalization by use of tensor imaging and fibre tracking on a normal brain database. Phys. Med. Biol. 2007;52:N99–109. doi: 10.1088/0031-9155/52/6/N01. [DOI] [PubMed] [Google Scholar]
- Müller H.P., Unrath A., Sperfeld A.D. Diffusion tensor imaging and tractwise fractional anisotropy statistics: quantitative analysis in white matter pathology. Biomed. Eng. Online. 2007;6:42. doi: 10.1186/1475-925X-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H.P., Turner M.R., Grosskreutz J. A large-scale multicentre cerebral diffusion tensor imaging study in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 2016;87:570–579. doi: 10.1136/jnnp-2015-311952. [DOI] [PubMed] [Google Scholar]
- Norris F., Shepherd R., Denys E. Onset, natural history and outcome in idiopathic adult motor neuron disease. J. Neurol. Sci. 1993;118:48–55. doi: 10.1016/0022-510x(93)90245-t. [DOI] [PubMed] [Google Scholar]
- Rascovsky K., Hodges J.R., Knopman D. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbohm A., Müller H.P., Hübers A. Corticoefferent pathways in pure lower motor neuron disease: a diffusion tensor imaging study. J. Neurol. 2016;263:2430–2437. doi: 10.1007/s00415-016-8281-2. [DOI] [PubMed] [Google Scholar]
- Rosenfeld A., Kak A.C. 2nd ed. Vol. 349. Academic Press Inc.; Orlando: 1982. Digital Picture Processing. [Google Scholar]
- Rosskopf J., Müller H.P., Dreyhaupt J. Ex post facto assessment of diffusion tensor imaging metrics from different MRI protocols: preparing for multicentre studies in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:92–101. doi: 10.3109/21678421.2014.977297. [DOI] [PubMed] [Google Scholar]
- Smith S.M., Jenkinson M., Johansen-Berg H. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. NeuroImage. 2006;31:1487–1505. doi: 10.1016/j.neuroimage.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Spinelli E.G., Agosta F., Ferraro P.M. Brain MR imaging in patients with lower motor neuron-predominant disease. Radiology. 2016;280:545–556. doi: 10.1148/radiol.2016151846. [DOI] [PubMed] [Google Scholar]
- Traynor B.J., Codd M.B., Corr B. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie house diagnostic criteria. Arch. Neurol. 2000;57:1171–1176. doi: 10.1001/archneur.57.8.1171. [DOI] [PubMed] [Google Scholar]
- Turner M.R., Grosskreutz J., Kassubek J. Towards a neuroimaging biomarker for amyotrophic lateral sclerosis. Lancet Neurol. 2011;10:400–403. doi: 10.1016/S1474-4422(11)70049-7. [DOI] [PubMed] [Google Scholar]
- Unrath A., Müller H.P., Riecker A. Whole brain-based analysis of regional white matter tract alterations in rare motor neuron diseases by diffusion tensor imaging. Hum. Brain Mapp. 2010;31:1727–1740. doi: 10.1002/hbm.20971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg-Vos R.M., Visser J., Franssen H. Sporadic lower motor neuron disease with adult onset: classification of subtypes. Brain. 2003;126:1036–1047. doi: 10.1093/brain/awg117. [DOI] [PubMed] [Google Scholar]
- van der Graaff M.M., Sage C.A., Caan M.W. Upper and extra-motoneuron involvement in early motoneuron disease: a diffusion tensor imaging study. Brain. 2011;134:1211–1228. doi: 10.1093/brain/awr016. [DOI] [PubMed] [Google Scholar]