

Abstract
Acute myeloid leukemia (AML) is an aggressive hematopoietic malignancy with an exceedingly poor prognosis, showing a 5-year overall survival rate of 40–45% in the young and a 5-year survival rate of below 10% in the elderly (>60y). Though a high percentage of patients enter complete remission following chemotherapeutic intervention, the majority of patients relapse within three years. Such stark prognostic outcomes highlight the need for additional clinical research, basic discovery, and molecular delineation of the etiologies and mechanisms behind responses to therapy that lead to relapse. Here we summarize recent discoveries in tumor heterogeneity at the genetic and epigenetic levels, and their independent molecular trajectories and dynamics in response to therapy. These new discoveries may have significant implications for understanding, monitoring, and treating leukemia and other cancers.
Keywords: heterogeneity, cancer evolution, epigenome, epiallele, prognosis
Introduction
The high relapse rates of leukemia may be due (in part) to intra-tumor genetic heterogeneity and chemo-resistance-enabling mutations, which allow many avenues of “escape” from treatment for AML. Indeed, it is often observed that subsets of malignant cells, through a combination of favorable genetic mutations (TET2, FLT3, IDH), are comparatively resistant to therapeutic intervention and thus persist in patients after treatment [1–5]. Yet, the mutation burden of AML at both diagnosis and relapse is one of the lowest relative to those of other cancers – suggesting that additional factors may contribute significantly to the aggressive nature of the disease [6, 7, 8]. Namely, epigenetic factors have been strongly implicated in aggressive AML pathologies [8], and there has been burgeoning interest in the interplay of the genetic and epigenetic interactions, as well as more recently the transcriptomic and epitranscriptomic (RNA modification) landscapes of cancer [9].
A systems-level understanding of the complex interactions between the biological levers and regulators of cancer progression, remission, and relapse may span many realms, including: genetic, epigenetic, transcriptomic, epitranscriptomic, proteomic, and metabolomic. As such, newly emerging molecular profiles of AML and the contribution of each “layer” to the evolutionary dynamics of the disease are necessary to advance the understanding of AML and other cancers. Here we describe several exciting new datasets that have performed innovative, longitudinal characterizations of AML and other cancers to reveal novel disease dynamics that open new avenues of understanding, research, and potentially treatment.
Genetics and Cytogenetics
Cytogenetic variations have long been established as diagnostically and clinically relevant markers of AML. Approximately 45% of AML cases feature a recurrent chromosomal alteration with 10–14% of cases featuring three or more alterations comprising a “complex karyotype” [10–12]. AML cases with translocations between chromosome 8 and 21 (t(8;21)) – the most frequently observed cytogenetic abnormality in AML – tend to have more favorable prognoses and feature comparatively high levels of remission, falling into a so-called “favorable” risk group [10, 13]. Intermediate-risk AML cases include those featuring the loss of the Y chromosome or those with t(9;11) resulting in a KMT2A-MLLT3 gene fusion [10, 14–15]. Adverse-risk AML cases associated with poor prognoses include those with t(6;9) resulting in a DEK-NUP214 gene fusion, those with inv(3)/t(3;3), and those with certain complex karyotypes [10–11, 16–19, 20]. Nearly half of all AML cases, however, feature a normal karyotype, indicating that a large part of AML malignancy arises not from structural chromosomal variations but from non-cytogenetic molecular factors, particularly nucleotide-level mutations and other molecular changes [10, 21]. There is strong evidence that many of the mutations present in AML genomes exist in progenitor hematopoietic stem cells just prior to the initiating leukemogenic event [22]. Welch et al. sequenced 24 AML genomes to characterize their genomic landscape, including: 12 AML FAB-M3 genomes with a known cytogenetic initiating event (PML/RARA gene fusion) and 12 AML FAB-M1 samples with no cytogenetic abnormalities and unknown initiating events. In both the M1 and M3 samples, the number of single nucleotide variants (SNVs) per genome increased proportionally with patient age, and the mutational spectra (types) of both AML stages were similar. Then, upon comparison with sequenced exomes of hematopoietic stem/progenitor cells (HSPCs) from healthy controls showed that the number of SNVs in HSPC samples also increased proportionally with age and that the mutational spectrum of the HSPC samples was similar to that of the AML samples. Such age-dependent mutational burden of TET2 was also observed in normal, elderly individuals with clonal hematopoiesis [23].
These recent data suggest that many mutations found in AML genomes are simply age-related background mutations carried over into the AML genome when an aging progenitor cell undergoes a leukemogenetic event. These pre-existing background mutations, present in nearly all AML cells in the sample, are largely irrelevant in AML pathogenesis [22]. Welch et al. posit that as little as two somatic mutations are necessary to initiate AML pathogenesis, that only a tiny fraction of mutational events in each AML genome contributes to the majority of the disease while the rest are “background noise” [22]. Various genes (FLT3, NPM1, KIT, CEBPA, TET2, DNMT3A, IDH1, IDH2, U2AF1, EZH2, SMC1A, SMC3, etc.) and related mutations have been implicated as factors pertinent to AML.
However, relative to other adult cancers, AML genomes tend to be less mutated, with an average of only 13 cancer-specific mutations [6]. Thus, more than a quarter of AML genomes do not feature mutations in these known leukemogenic genes. Moreover, the observed genetic lesions can remain largely unchanged during AML progression, with some patients showing no new somatic mutations even at relapse [8, 24]. This suggests that in some cases the genetic landscape of AML is largely independent of disease progression, and that there is utility in understanding AML through other means, namely epigenetic and other “omics,” which can be tied more directly to disease progression [8].
Epigenetic Heterogeneity and Independence
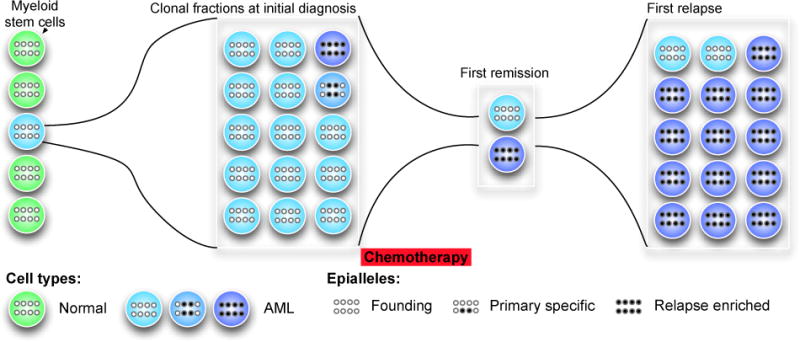
Li and Garrett-Bakelman et al. conducted one of the first large-scale analyses of epigenetic heterogeneity and of the interplay between genetic and epigenetics in acute myeloid leukemia [8]. The authors found that epiallele burden, or the degree of methylation variation and “epigenetic evolution” at the same loci between samples, can be used as a distinct and significant predictor of relapse risk among AML patients. Among patients divided into two groups based on the magnitude of their epiallele burden compared to normal bone marrow (median cutoff; highest and lowest 50%), those with a high epiallele burden at diagnosis relapsed more quickly than those with a low epiallele burden. Multivariate analysis showed that the magnitude of the epiallele burden was clinically relevant regardless of age, sex, cytogenetics, and white blood cell count at the time of diagnosis (P = 0.024, Cox proportional-hazards regression model). In an accompanying survival analysis based on mutational burden, no statistically significant difference was seen in time-to-relapse between the high-mutational burden and the low-mutational burden groups (P = 0.272, Mantel-Cox log-rank test) [8]. This indicates that, while epiallele burden is prognostically relevant, mutational burden was less so in this cohort of 139 AML longitudinally followed patients.
Also, epigenetic allele shifting (epiallele shifts) was shown to be a universal feature of AML, relative to normal bone marrow – 100% of AML patients showed significant epiallele shifting, both at diagnosis and at relapse. Epigenetic allele shifting, furthermore, is independent of age, subtype, mutation burden, and blast purity. Also, when the same loci were examined in the same patient at diagnosis and relapse, 92% of patients showed some degree of epiallele shifting during disease progression while 8% showed none. Thus, while epiallele shifting is universal in AML relative to normal bone marrow controls, the degree of epiallele shift during disease progression varies considerably from patient to patient [8].
The epigenetic and genetic landscapes of AML are also largely independent. Li and Garrett-Bakelman et al. defined epigenetically-shifted loci (eloci) based on their presence at specific time-points in AML progression: eloci present only at diagnosis (cluster 1; diagnosis-specific); eloci present only at relapse (cluster 2; relapse-specific); and eloci present at both diagnosis and relapse (cluster 3; shared). Somatic mutations are also classified as diagnosis-specific or relapse-specific depending on when they were present during AML progression. The proportions of mutations in the diagnosis-specific and relapse-specific categories did not match the distributions of the corresponding eloci patient categories (cluster 1/diagnosis-specific mutations, P = 0.89; cluster 2/relapse-specific mutations, P = 0.40; Wilcoxon-signed rank tests), and there was no significant association between specific somatic mutations in genes recurrent in AML and the presence of eloci (P > 0.05 for all relevant genes, chi-squared test with Monte Carlo simulation) [8]. This strengthens the claim of independence between epiallele burden and mutation burden [8], and there was also not a significant increase in genes that regulate DNA methylation (DNMT1, DNMT3a, TET1,2,3, IDH1,2) in those samples with higher levels of epiallele burden.
These results buttress earlier claims that the genetic landscape of AML – at least for about one-third of patients – is comprised of inherited background mutations that are largely irrelevant in the pathophysiology of the disease, while the epigenetic landscape of AML is acting on and shaping the disease at diagnosis and especially at relapse. This newly proposed independence between the genetics and epigenetics of AML has interesting ramifications, chief of which is the development of tumor clonal selection and heterogeneity simultaneously at genetic and epigenetic levels [25]. In the diagnosis stage of AML progression, there is relative genetic heterogeneity and epigenetic homogeneity. Yet, patients at subsequent relapse feature greater epigenetic heterogeneity due to epiallele shift and greater genetic homogeneity, as the subset of cells surviving treatment may be genetically selected. Thus, as it progresses to relapse, AML in many patients becomes simultaneously more heterogeneous (epigenome) and more homogenous (genome).
Transcriptomic and Epigenetic Linkage
In contrast with the independent interplay between genetic and epigenetic changes, the transcriptional landscape of acute myeloid leukemia seems is strongly tied to epigenetic regulation at specific genomic loci. Landau et al. showed that loci with increased epigenetic heterogeneity in chronic lymphocytic leukemia (CLL) lead to increased dysregulation of those genes [26], which manifested as increased gene expression coefficient of variation (CV). Similarly, genes with higher epiallelle heterogeneity in their promoter regions in AML patients also have high levels of transcriptional heterogeneity (P < 2.2×10−6), and the presence of epigenetically-altered loci (eloci) correlated with deregulated expression in the associated genes [8]. Moreover, these loci shifted depending on the progression of the disease, with 114 genes differentially expressed between the diagnosis-specific cluster and the relapse-specific cluster, with genes encoding protein kinases being upregulated in the diagnosis cluster and genes encoding inflammation and immune response being upregulated in the relapse cluster [8].
Excitingly, such links between epigenetic evolution and heterogeneity are no longer just observed as a function of global gene expression differences, but can now be delineated at the single-cell level. In particular, Li and Garrett-Bakelman et al. showed that epigenetic heterogeneity associated with increased CV in global RNA-sequencing (RNA-seq) transcriptome-wide, and then used single-cell RNA-sequencing (scRNA-seq) to characterize five serial samples from a patient who relapsed four times. Their data showed that the cell-to-cell variation in gene expression was highest in the genes that showed the fastest and most significant levels of epiallele burden. Also, their data showed that this variation was established as the first stage of disease and persisted across all five time points, indicating that such heterogeneity might represent a consistent and persistent feature of leukemia’s dysregulation.
Conclusion
When some of the first studies on cancer and heterogeneity were first completed, there were no methods to simultaneously quantify RNA levels and DNA methylation from the same cell. However, recent proof-of-principle experiments have helped realize the promise of single-cell epigenetic and integrated genomic methods, with protocols like single-cell reduced representation bisulfite sequencing (scRRBS), the assay for transposase-accessible chromatin (scATAC) (Figure 1), and single-cell triple-omics (scTrio-seq) [27]. As such, it is now feasible to precisely discern the intra-cellular and inter-cellular variance of states of epigenetic regulation (e.g. methylation) and the concomitant gene expression changes within each cell, using sequence-based measurements of genetic, epigenetic, and transcriptome states at once. These technological advances will be instrumental in teasing out the regulation and dynamics of cellular heterogeneity and their changes during therapy.
Figure 1. Single-cell reduced representation bisulfite sequencing (scRRBS).
New single-cell and clonality methods can be controlled and combined into one channel or tube. (a) The 10 steps of RRBS can be centralized into one plate of reactions and automated for improved reproducibility. These data or inference from deeply sequenced tumor samples can show evolution (left to right), selection after chemotherapy (red), and emergence of chemo-resistant clones that are driven by epigenetic changes called epialleles and shifted epialleles per million loci (EPM).
The rapid development of these single-cell technological methods will eventually lead to a more comprehensive view of the biology of each cancer cell, which can help understand heterogeneity states of leukemia and many other cancers. Moreover, new single-base resolution methods are also emerging, which can leverage regional, phased, and long-range information from differentially methylated regions, single molecule sequencing, and new nanopore-based methods [28–30]. Notabl, y these techniques are already leading to new combined therapies and measure in leukemia patients with specifical mutations, like DNMT3A and TET mutations [31–32]. But beyond improving the reliability and accuracy of these low-input methods (which often involve extensive PCR or signal amplification), there is an ongoing debate about the applicability of these methods for improving clinical care. While the papers published to date have shown novel prognostic abilities for separating out high- and low-risk patients, the challenge is that often they actually separate “bad” from “very bad” patient cohorts. Thus, to improve their utility for a broader population, these methods and measures need to be applied to more cancers and rigorously examined in terms of their applicability as a function of treatment type, cancer stage, and microenvironment, which also will be relevant factors.
Prior work has shown that genetic and epigenetic “hotspots” can reveal new therapeutic targets, thus providing novel means to eliminate aberrant cells. But these robust measures of heterogeneity have yet to be fully applied as part of standard, ongoing care in a clinical context. The whole-genome, pan-methylome, five-timepoint patient evolutionary landscape from Li and Garrett-Bakelman et al. is one of the deepest longitudinal, and multi–omic characterizations of a cancer patient to date, showing the first clues about the dynamics of heterogeneity.
These studies serve as a guide for future therapeutic directions and additional technological innovation, which ideally would measure the exact proportion of cells that have responded to a therapy as well as the proportions of loci or eloci across all cells that have been altered or evolutionarily selected. Once found, they could then be targeted by CRISPR-based systems that leverage dCas9 and TET-fusion proteins, and “re-programmed” back to the original state of normal cells, such as normal bone marrow for leukemia. Such measures and technologies could enable patients to fix their cancers with their own cells, which would also reduce treatment rejection for cell transplants. While not yet available for clinicians’ hands as monitoring/treatment tools, the sites and states of heterogeneity are finally emerging, and with them, clues as to where we might strike first against such functional inter-cellular variation.
Highlights.
Genetic and epigenetic evolution can follow distinct and independent trajectories.
New epigenetic technologies improve diagnostic accuracy and clinical applications.
Epigenetic CRISPR editing can reprogram the aberrant sites of DNA methylation.
Acknowledgments
We would like to thank the Starr Cancer Consortium grants (I7-A765, I9-A9-071) and funding from the Irma T. Hirschl and Monique Weill-Caulier Charitable Trusts, Bert L. and N Kuggie Vallee Foundation, the WorldQuant Foundation, The Pershing Square Sohn Cancer Research Alliance, NASA (NNX14AH50G, 15-15Omni2-0063), the Bill and Melinda Gates Foundation (OPP1151054).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts: The authors have no conflicts of interest.
References
- 1.Craddock Charles, et al. Biology and management of relapsed acute myeloid leukaemia. British journal of haematology. 2005;129(1):18–34. doi: 10.1111/j.1365-2141.2004.05318.x. [DOI] [PubMed] [Google Scholar]
- 2.Schlenk Richard F, Hartmut Döhner. Genomic applications in the clinic: use in treatment paradigm of acute myeloid leukemia. ASH Education Program Book. 2013;2013(1):324–330. doi: 10.1182/asheducation-2013.1.324. [DOI] [PubMed] [Google Scholar]
- 3.Verma Dushyant, et al. Late relapses in acute myeloid leukemia: analysis of characteristics and outcome. Leukemia & lymphoma. 2010;51(5):778–782. doi: 10.3109/10428191003661852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Preisler HD, et al. The frequency of long‐ term remission in patients with acute myelogenous leukaemia treated with conventional maintenance chemotherapy: a study of 760 patients with a minimal follow-up time of 6 years. British journal of haematology. 1989;71(2):189–194. doi: 10.1111/j.1365-2141.1989.tb04253.x. [DOI] [PubMed] [Google Scholar]
- 5.Ishikawa Fumihiko, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nature biotechnology. 2007;25(11):1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;2013(368):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grove Carolyn S, Vassiliou George S. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Disease Models and Mechanisms. 2014;7(8):941–951. doi: 10.1242/dmm.015974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Sheng, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nature Medicine. 2016;22(7):792–799. doi: 10.1038/nm.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, CE Mason. The Pivotal Regulatory Landscape of RNA Modifications. Annual Review of Genomics and Human Genetics. 2014;15:127–50. doi: 10.1146/annurev-genom-090413-025405. [DOI] [PubMed] [Google Scholar]
- 10.Betz Bryan L, Hess Jay L. Acute myeloid leukemia diagnosis in the 21st century. Archives of pathology & laboratory medicine. 2010;134(10):1427–1433. doi: 10.5858/2010-0245-RA.1. [DOI] [PubMed] [Google Scholar]
- 11.Stölzel F, et al. Karyotype complexity and prognosis in acute myeloid leukemia. Blood cancer journal. 2016;6(1):e386. doi: 10.1038/bcj.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mrózek Krzysztof, Heerema Nyla A, Bloomfield Clara D. Cytogenetics in acute leukemia. Blood reviews. 2004;18(2):115–136. doi: 10.1016/S0268-960X(03)00040-7. [DOI] [PubMed] [Google Scholar]
- 13.Nishii K, et al. Characteristics of t (8; 21) acute myeloid leukemia (AML) with additional chromosomal abnormality: concomitant trisomy 4 may constitute a distinctive subtype of t (8; 21) AML. Leukemia. 2003;17(4):731–737. doi: 10.1038/sj.leu.2402871. [DOI] [PubMed] [Google Scholar]
- 14.Holmes Romayne I, et al. Loss of the Y chromosome in acute myelogenous leukemia: a report of 13 patients. Cancer genetics and cytogenetics. 1985;17(3):269–278. doi: 10.1016/0165-4608(85)90018-4. [DOI] [PubMed] [Google Scholar]
- 15.Walter Claudia Ulrike, et al. The t (9; 11) Confers Good Prognosis in AML Patients Treated with Stem Cell Transplantation As Compared to Non-t (9; 11) and Other Adverse-Risk Abnormalities. Blood. 2011;118(21):2527–2527. doi: 10.5144/1658-3876.2012.169. [DOI] [PubMed] [Google Scholar]
- 16.Gröschel Stefan, et al. Mutational spectrum of myeloid malignancies with inv (3)/t (3; 3) reveals a predominant involvement of RAS/RTK signaling pathways. Blood. 2015;125(1):133–139. doi: 10.1182/blood-2014-07-591461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers Heesun J, et al. Complex or monosomal karyotype and not blast percentage is associated with poor survival in acute myeloid leukemia and myelodysplastic syndrome patients with inv (3)(q21q26. 2)/t (3; 3)(q21; q26. 2): a Bone Marrow Pathology Group study. Haematologica. 2014;99(5):821–829. doi: 10.3324/haematol.2013.096420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Jianlan, et al. De novo acute myeloid leukemia with inv (3)(q21q26. 2) or t (3; 3)(q21; q26. 2): a clinicopathologic and cytogenetic study of an entity recently added to the WHO classification. Modern Pathology. 2011;24(3):384–389. doi: 10.1038/modpathol.2010.210. [DOI] [PubMed] [Google Scholar]
- 19.Chi Yiqing, et al. Acute myelogenous leukemia with t (6; 9)(p23; q34) and marrow basophilia: an overview. Archives of pathology & laboratory medicine. 2008;132(11):1835–1837. doi: 10.5858/132.11.1835. [DOI] [PubMed] [Google Scholar]
- 20.Mrózek Krzysztof. Seminars in oncology. 4. Vol. 35. WB Saunders; 2008. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaidi Syed Z, et al. The challenge of risk stratification in acute myeloid leukemia with normal karyotype. Hematology/oncology and stem cell therapy. 2008;1(3):141–158. doi: 10.1016/s1658-3876(08)50023-9. [DOI] [PubMed] [Google Scholar]
- 22.Welch John S, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Busque L, Patel J, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, Hassimi M, Socci N, Bhatt P, Gonen M, Mason CE, Melnick A, Godley L, Brennan C, Abdel-Wahab O, Levine R. Recurrent Somatic TET2 Mutations in Normal Elderly Individuals With Clonal Hematopoiesis. Nature Genetics. 2012 Nov;44(11):1179–81. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Yang, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118(20):5593–5603. doi: 10.1182/blood-2011-03-343988. [DOI] [PubMed] [Google Scholar]
- 25.Sasca Daniel, Huntly BJ. Independence of epigenetic and genetic diversity in AML. Nature medicine. 2016;22(7):708. doi: 10.1038/nm.4136. [DOI] [PubMed] [Google Scholar]
- 26.Landau DA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou Y, et al. Cell Res. 2016;26:304–19. doi: 10.1038/cr.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Garrett-Bakelman FE, Akalin A, Zumbo P, Levine R, Melnick A, Mason CE. eDMR: An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 2013 Apr;10(14)(S5):S10. doi: 10.1186/1471-2105-14-S5-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mason CE, Afshinnekoo E, Tighe S, Wu S, Levy S. International Standards for Genomes, Transcriptomes, and Metagenomes. Journal of Biomolecular Techniques. 2017 Apr;28(1):8–18. doi: 10.7171/jbt.17-2801-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McIntyre ABR, Rizzardi L, Yu AM, Alexander N, Rosen GL, Botkin DJ, Stahl SS, John KK, Castro-Wallace SL, McGrath K, Burton AS, Feinberg AP, Mason CE. Nanopore Sequencing in Microgravity. Nature Partner Journals (npj) Microgravity. 2016;2:16035. doi: 10.1038/npjmgrav.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, Ganzel C, Durham BH, Mohanty A, Hoermann G, Rivera SA, Chramiec AG, Pronier E, Bastian L, Keller MD, Tovbin D, Loizou E, Weinstein AR, Gonzalez AR, Lieu Y, Rowe JM, Pastore F, McKenney AS, Krivtsov AV, Sperr WR, Cross JR, Mason CE, Tallman MS, Arcila ME, Abdel-Wahab O, Armstrong SA, Kubicek S, Staber PB, Gönen M, Paietta EM, Melnick AM, Nimer SD, Mukherjee S, Levine RL. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nature Medicine. 2016 Dec;22(12):1488–1495. doi: 10.1038/nm.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih A, Meydan C, Shank K, Garrett-Bakelman F, Ward F, Intlekofer A, Nazir A, Stein E, Knapp K, Glass J, Travins J, Straley K, Gliser C, Mason CE, Yen K, Thompson C, Melnick A, Levine R. Combination targeted therapy to disrupt aberrant oncogenic signaling and reverse epigenetic dysfunction in IDH2- and TET2-mutant acute myeloid leukemia. Cancer Discovery. 2017 Feb 13; doi: 10.1158/2159-8290.CD-16-1049. pii: CD-16-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]