

Abstract
Using an iridium catalyst modified by PhanePhos, CF3-allenes react with methanol to form branched products of hydrohydroxymethylation as single regioisomers with excellent levels of enantiomeric enrichment. This hydrogen auto-transfer process enables catalytic enantioselective formation of acyclic CF3-bearing all-carbon quaternary stereocenters in the absence of stoichiometric metals or byproducts.
Graphical abstract
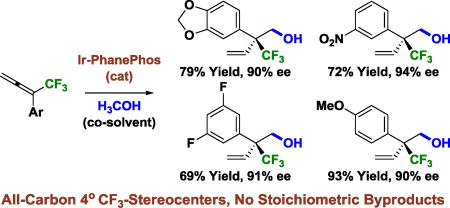
The catalytic enantioselective formation of all-carbon quaternary stereocenters remains a formidable challenge in chemical synthesis.1,2,3 In this context, the enantioselective formation of acyclic CF3-bearing all-carbon quaternary centers is especially daunting.4,5,6,7 As established in seminal work by Shibata,4a methods capable of delivering this motif are restricted to conjugate additions of β,β-disubstituted enones4a,b,e,g,h or nitroolefins4c,d,f,i,j,k and two isolated reports of asymmetric allyl5a and propargyl5b substitution. In connection with ongoing studies of alcohol mediated C-C coupling via hydrogen auto-transfer,8 we recently developed catalytic enantioselective methods for the formation of acyclic all-carbon quaternary stereocenters that utilize vinyl epoxides3a,b,d and 1,3-dienes3c as pronucleophiles. Attempts to adapt this technology to the formation of acyclic CF3-bearing stereocenters via ruthenium catalyzed CF3-allene-paraformaldehyde reductive coupling mediated by 2-propanol failed to deliver highly enantiomerically enriched adducts.9
The unique efficacy of iridium(I)-PhanePhos10 complexes in recently developed methanol mediated hydrohydroxymethylations of 2-substituted-1,3-dienes,3c along with the availability of improved protocols for the preparation of CF3-allenes,11 motivated our continued efforts toward this elusive bond formation. Here, expanding upon the use of methanol as a C1-feedstock in metal catalyzed C-C coupling,12,13,14 we report that iridium-PhanePhos complexes promote the methanol-mediated hydrohydroxymethylation of CF3-allenes to form highly enantiomerically enriched adducts with complete branched-regioselectivity. Thus, catalytic enantioselective formation of acyclic CF3-bearing all-carbon quaternary stereocenters is achieved in the absence of stoichiometric metals or byproducts.
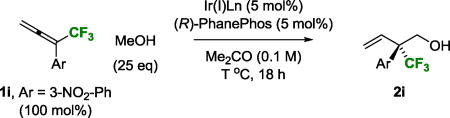
Given the singular effectiveness of iridium(I)-PhanePhos in asymmetric diene hydrohydroxymethylation,3c these conditions were applied to the coupling of methanol with 1,1-disubstituted CF3-allenes 1a–1o. Although the desired coupling products 2a–2o were generated with complete branched regioselectivity, the reaction was highly substrate dependent with significant variation in isolated yield and enantioselectivity. Furthermore, all other chiral ligands that were evaluated failed to deliver products of C-C coupling. These circumstances led us to explore the influence of alternate reaction parameters. As illustrated in the optimization of the methanol-mediated hydrohydroxymethylation of CF3-allene 1i to form 2i, several interesting trends emerged (Table 1). For CF3-allene 1i and other allenes bearing electron deficient aryl moieties, changing the precatalyst from [Ir(cod)Cl]2 to Ir(cod)(acac) resulted in higher levels of enantiomeric enrichment, but with lower conversion (Table 1, entries 1 and 2). A modest increase in reaction temperature improved the isolated yield of 2i without compromising enantioselectivity (Table 1, entry 3). The addition of water (500 mol%) increased enantioselectivity, but led to competing transfer hydrogenation of 1i (Table 1, entry 4). Tetrabutylammonium iodide (TBAI) (10 mol%) suppresses competing transfer hydrogenation and increases enantioselectivity to a small extent (Table 1, entry 5). Using water (500 mol%) and TBAI (10 mol%) in concert, the neopentyl alcohol 2i could be obtained in 81% yield and 90% ee (Table 1, entry 6). Exchanging acetone for ethyl acetate as solvent improved enantioselectivity but diminished yield (Table 1, entry 7).
Table 1.
Influence of various reaction parameters in the optimization of the enantioselective iridium-catalyzed C-C coupling of CF3-allene 1i with methanol to form alcohol 2i.a
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Ir(I)Ln | Additive (mol%) | T °C | Yield (%) | ee (%) |
| 1 | [Ir(cod)Cl]2 | --- | 70 | 56 | 64 |
| 2 | Ir(cod)(acac) | --- | 70 | 14 | 79 |
| 3 | Ir(cod)(acac) | --- | 80 | 79 | 79 |
| 4 | Ir(cod)(acac) | H2O (500 mol%) | 80 | 56 | 89 |
| 5 | Ir(cod)(acac) | TBAI (10 mol%) | 80 | 79 | 84 |
| 6 | Ir(cod)(acac) | H2O/TBAI | 80 | 81 | 90 |
| 7b | Ir(cod)(acac) | H2O/TBAI | 80 | 72 | 94 |
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis.
EtOAc solvent.
Taking into account the aforesaid influence of the indicated reaction parameters, the conversion of 1,1-disubstituted CF3-allenes 1a–1o to neopentyl alcohols 2a–2o was explored (Table 2). Two general sets of conditions emerged. In the coupling of CF3-allenes 1a–1f and 1k, which incorporate electron rich, electron neutral or slightly electron deficient aryl moieties, use of the iridium catalyst derived from [Ir(cod)Cl]2 and (R)-PhanePhos in acetone solvent at 70 °C was optimal. For CF3-allenes bearing highly electron deficient aryl moieties, the precatalyst Ir(cod)(acac) in ethyl acetate solvent at 80 °C was preferred. Under both sets of conditions, TBAI and H2O were frequently required to enhance enantioselectivity. By tailoring reaction conditions in this manner, alcohols 2a–2o with CF3-bearing all-carbon quaternary stereocenters could be formed with uniformly high levels of enantioselectivity and as single regioisomers. Alkyl substituted CF3-allenes engage in efficient methanol-mediated hydrohydroxymethylation, but enantioselectivities were lower (<50% ee). The absolute stereochemical assignment of adducts 2a–2o is based on single crystal X-ray diffraction analysis of 2b and 2f. Attempted use of higher alcohols led to regio- and enantioselective transfer hydrogenation of the internal allene π-bond (ca. 40% ee).
Table 2.
Enantioselective iridium-catalyzed coupling of methanol with CF3-allenes 1a–1o to form higher alcohols 2a–2o.a

|
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis.
[Ir(cod)Cl]2, 70 °C, Me2CO;
Ir(cod)(acac), TBAI (10 mol%), 80 °C, EtOAc;
TBAI (10 mol%);
H2O (200 mol%);
H2O (500 mol%). See Supporting Information for further experimental details.
To demonstrate how adducts 2a–2o can be used in chemical synthesis, alcohol 2a was subjected to a series of functional group manipulations. Conversion of alcohol 2a to the corresponding p-toluenesulfonate followed by exposure to sodium cyanide in DMSO at 150 °C provided the nitrile 3, representing a remarkable example of an SN2 reaction at a highly congested neopentyl center (eq. 1). Jones oxidation of alcohol 2a followed by Fischer esterification provides the β,γ-unsaturated methyl ester 4 (eq. 2). Finally, conversion of alcohol 2a to the benzoate followed by oxidative cleavage15 of the vinyl moiety delivers the chiral α-stereogenic carboxylic acid 5 (eq. 3).
As exemplified in methanol-mediated transfer hydrogenations of C-C π-bonds,16 the reversible and highly endothermic nature of methanol dehydrogenation (ΔH(MeOH) = +20 kcal/mol vs ΔH(EtOH) = +16 kcal/mol)17 can be overcome by linking dehydrogenation to an exothermic process. Methanol dehydrogenation also poses a significant challenge in the hydrohydroxymethylation of π-unsaturated reactants and invariably represents the rate-limiting step.3c,14 However, as the present allene-methanol C-C couplings involve formation of a highly congested CF3-bearing all-carbon quaternary stereocenter, it was unclear whether methanol dehydrogenation or carbonyl addition would be rate-limiting. To gain further insight into the catalytic mechanism, CF3-allene 1a was subjected to d4-methanol under otherwise standard conditions (Scheme 2, eq. 4). The product, deuterio-2a, completely retains deuterium at the carbinol position (Ha, Hb = >95% 2H), suggesting deuterio-2a is kinetically inert with respect to dehydrogenation due to coordination of the homoallylic olefin to iridium, blocking the adjacent coordination site required for β-hydride elimination. While deuterium is not incorporated at the vinylic terminus of deuterio-2a (Hd, He = <5% 2H), a significant quantity of deuterium appears at the internal vinylic position (Hc = 58% 2H). Unlike related diene-methanol C-C couplings,3c this data suggests the hydrometalation event is a completely regioselective process. Incomplete incorporation of deuterium at the internal vinylic position may be due to adventitious water.17 In a related competition kinetics experiment, CF3-allene 1a was exposed to equimolar quantities of methanol and d4-methanol under otherwise standard conditions (Scheme 2, eq. 5). Deuterium incorporation at the carbinol methylene (Ha, Hb = 27% 2H) of the product deuterio-2a constitutes a normal primary kinetic isotope effect (kH/kD ≈ 3.0) consistent with turnover-limiting methanol dehydrogenation. The influence of the precatalyst and additives (TBAI, H2O) on enantioselectivity suggest the counterion (and water) are present during the enantiodetermining carbonyl addition event. To accommodate this observation, we propose that carbonyl addition occurs from an allyliridium(III) intermediate (Scheme 2, bottom).
Scheme 2.

General catalytic mechanism and deuterium labelling studies.a
aYields of material isolated by silica gel chromatography. The extent deuterium incorporation was determined by HRMS and 1H and 2H NMR analysis. See Supporting Information for further experimental details. Haptomeric equilibria and equilibria involving alkoxy-bridged dimers are not depicted.
To summarize, CF3-bearing all-carbon quaternary stereocenters are exceptionally difficult to prepare in enantiomerically enriched form, with existing protocols for their construction largely restricted to conjugate additions to β,β-disubstituted CF3-enones and nitroolefins.4 Here, we demonstrate that iridium complexes modified by PhanePhos catalyze the methanol-mediated hydrohydroxymethylation of CF3-allenes to generate this structural motif in a completely regioselective and highly enantioselective fashion in the absence of stoichiometric metals or byproducts. Future studies will focus on the development of related C-C bond forming transfer hydrogenations for the conversion of abundant feedstocks to value-added products.
Supplementary Material
Scheme 1.

Catalytic enantioselective formation of acyclic CF3-bearing all-carbon quaternary stereocenters.
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research.
Footnotes
Supporting Information Available: Experimental procedures and spectral data. HPLC traces corresponding to racemic and enantiomerically enriched samples. Single crystal X-ray diffraction data for 2b and 2f. This material is available free of charge via the internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.For selected reviews on the enantioselective formation of all-carbon quaternary centers, see: Fuji K. Chem. Rev. 1993;93:2037.Corey EJ, Guzman-Perez A. Angew. Chem. Int. Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.Bella M, Gasperi T. Synthesis. 2009:1583.Shimizu M. Angew. Chem. Int. Ed. 2011;50:5998. doi: 10.1002/anie.201101720.Hong AY, Stoltz BM. Eur. J. Org. Chem. 2013:2745. doi: 10.1002/ejoc.201201761.Minko Y, Marek I. Chem. Comm. 2014:12597. doi: 10.1039/c4cc04391j.Marek I, Minko Y, Pasco M, Mejuch T, Gilboa N, Chechik H, Das JP. J. Am. Chem. Soc. 2014;136:2682. doi: 10.1021/ja410424g.Quasdorf KW, Overman LE. Nature. 2014;516:181. doi: 10.1038/nature14007.Liu Y, Han S-J, Liu W-B, Stoltz BM. Acc. Chem. Res. 2015;48:740. doi: 10.1021/ar5004658.Zeng X-P, Cao Z-Y, Wang Y-H, Zhou F, Zhou J. Chem. Rev. 2016;116:7330. doi: 10.1021/acs.chemrev.6b00094.
- 2.For recent examples of the catalytic enantioselective formation of acyclic all-carbon quaternary centers, see: Dabrowski JA, Gao F, Hoveyda AH. J. Am. Chem. Soc. 2011;133:4778. doi: 10.1021/ja2010829.Zhang P, Le H, Kyne RE, Morken JP. J. Am. Chem. Soc. 2011;133:9716. doi: 10.1021/ja2039248.Jung B, Hoveyda AH. J. Am. Chem. Soc. 2012;134:1490. doi: 10.1021/ja211269w.Gao L, Kang BC, Ryu DH. J. Am. Chem. Soc. 2013;135:14556. doi: 10.1021/ja408196g.Liu W-B, Reeves CM, Stoltz BM. J. Am. Chem. Soc. 2013;135:17298. doi: 10.1021/ja4097829.Mei T-S, Patel HH, Sigman MS. Nature. 2014;508:340. doi: 10.1038/nature13231.Zhang C, Santiago CB, Crawford JM, Sigman MS. J. Am. Chem. Soc. 2015;137:15668. doi: 10.1021/jacs.5b11335.Zhou Q, Cobb KM, Tan T, Watson MP. J. Am. Chem. Soc. 2016;138:12057. doi: 10.1021/jacs.6b08075.Patel HH, Sigman MS. J. Am. Chem. Soc. 2016;138:14226. doi: 10.1021/jacs.6b09649.
- 3.For catalytic enantioselective formation of acyclic all-carbon quaternary centers via C-C bond forming transfer hydrogenation, see: Feng J, Garza VJ, Krische MJ. J. Am. Chem. Soc. 2014;136:8911. doi: 10.1021/ja504625m.Feng J, Noack F, Krische MJ. J. Am. Chem. Soc. 2016;138:12364. doi: 10.1021/jacs.6b08902.Nguyen KD, Herkommer D, Krische MJ. J. Am. Chem. Soc. 2016;138:14210. doi: 10.1021/jacs.6b09333.Guo Y-A, Lee W, Krische MJ. Chem. Eur. J. 2017;23:2557. doi: 10.1002/chem.201606046.
- 4.For catalytic enantioselective conjugate addition to form acyclic CF3-bearing quaternary stereocenters, see: Kawai H, Okusu S, To-kunaga E, Sato H, Shiro M, Shibata N. Angew. Chem. Int. Ed. 2012;51:4959. doi: 10.1002/anie.201201278.Kawai H, Yuan Z, Kitayama T, Tokunaga E, Shibata N. Angew. Chem. Int. Ed. 2013;52:5575. doi: 10.1002/anie.201301123.Gao J-R, Wu H, Xiang B, Yu W-B, Han L, Jia Y-X. J. Am. Chem. Soc. 2013;135:2983. doi: 10.1021/ja400650m.Chen Q, Wang G, Jiang X, Xu Z, Lin L, Wang R. Org. Lett. 2014;16:1394. doi: 10.1021/ol500157b.Kwiatkowski P, Cholewiak A, Kasztelan A. Org. Lett. 2014;16:5930. doi: 10.1021/ol502941d.Ma C-H, Kang T-R, He L, Liu Q-Z. Eur. J. Org. Chem. 2014:3981.Sanz-Marco A, Blay G, Vila C, Pedro JR. Org. Lett. 2016;18:3538. doi: 10.1021/acs.orglett.6b01494.Zhu Y, Dong Z, Cheng X, Zhong X, Liu X, Lin Li, Shen Z, Yang P, Li Y, Wang H, Yan W, Wang K, Wang R. Org. Lett. 2016;18:3546. doi: 10.1021/acs.orglett.6b01498.Lai X, Zha G, Liu W, Xu Y, Sun P, Xia T, Shen Y. Synlett. 2016;27:1983. doi: 10.1039/c6ob00119j.Hou X, Ma H, Zhang Z, Xie L, Qin Z, Fu B. Chem. Commun. 2016:1470. doi: 10.1039/c5cc08480f.Hao X-Q, Wang C, Liu S-L, Wang X, Wang L, Gong J-F, Song M-P. Org. Chem. Front. 2017;4:308.
- 5.(a) Kimura M, Yamazaki T, Kitazume T, Kubota T. Org. Lett. 2004;6:4651. doi: 10.1021/ol0481941. [DOI] [PubMed] [Google Scholar]; (b) Tsuchida K, Senda Y, Nakajima K, Nishi-bayashi Y. Angew. Chem. Int. Ed. 2016;55:9728. doi: 10.1002/anie.201604182. [DOI] [PubMed] [Google Scholar]
- 6.For selected reviews on methods for the preparation of organofluorine compounds, see: Ma J-A, Cahard D. Chem. Rev. 2004;104:6119. doi: 10.1021/cr030143e.Brunet VA, O'Hagan D. Angew. Chem. Int. Ed. 2008;47:1179. doi: 10.1002/anie.200704700.Bobbio C, Gouverneur V. Org. Biomol. Chem. 2006;4:2065. doi: 10.1039/b603163c.Audouard C, Ma J-A, Cahard D. Adv. Org. Synth. 2006;2:431.Ma J-A, Cahard D. Chem. Rev. 2008;108:PR1–PR43. doi: 10.1021/cr800221v.Cao L-L, Gao BL, Ma S-T, Liu Z-P. Curr. Org. Chem. 2010;14:888.Kang YK, Kim DY. Curr. Org. Chem. 2010;14:917.Cahard D, Xu X, Couve-Bonnaire S, Pannecoucke X. Chem. Soc. Rev. 2010;39:558. doi: 10.1039/b909566g.Nie J, Guo H-C, Cahard D, Ma J-A. Chem. Rev. 2011;111:455. doi: 10.1021/cr100166a.Furuya T, Kamlet AS, Ritter T. Nature. 2011;473:470. doi: 10.1038/nature10108.Besset T, Schneider C, Cahard D. Angew. Chem. Int. Ed. 2012;51:5048. doi: 10.1002/anie.201201012.Studer A. Angew. Chem. Int. Ed. 2012;51:8950. doi: 10.1002/anie.201202624.Chen P, Liu G. Synthesis. 2013;45:2919.Liang T, Neumann CN, Ritter T. Angew. Chem. Int. Ed. 2013;52:8214. doi: 10.1002/anie.201206566.Barata-Vallejo S, Lantano B, Postigo A. Chem. Eur. J. 2014;20:16806. doi: 10.1002/chem.201404005.Camp-bell MG, Ritter T. Chem. Rev. 2015;115:612. doi: 10.1021/cr500366b.Xu X-H, Matsuzaki K, Shibata N. Chem. Rev. 2015;115:731. doi: 10.1021/cr500193b.Yang X, Wu T, Phipps RJ, Toste FD. Chem. Rev. 2015;115:826. doi: 10.1021/cr500277b.Alonso C, Martinez de Marigorta E, Rubiales G, Palacios F. Chem. Rev. 2015;115:1847. doi: 10.1021/cr500368h.
- 7.For examples of the catalytic enantioselective formation of cyclic CF3-bearing quaternary stereocenters, see: Denton JR, Sukumaran D, Davies HML. Org. Lett. 2007;9:2625. doi: 10.1021/ol070714f.Deng Q-H, Wadepohl H, Gade LH. J. Am. Chem. Soc. 2012;134:10769. doi: 10.1021/ja3039773.Woźniak L, Murphy JJ, Melchiorre P. J. Am. Chem. Soc. 2015;137:5678. doi: 10.1021/jacs.5b03243.Zhang Z-M, Xu B, Xu S, Wu H-H, Zhang J. Angew. Chem. Int. Ed. 2016;55:6324. doi: 10.1002/anie.201602542.
- 8.For selected reviews on C-C bond forming transfer hydrogenation, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew. Chem., Int. Ed. 2014;53:9142. doi: 10.1002/anie.201403873.Feng J, Kasun ZA, Krische MJ. J. Am. Chem. Soc. 2016;138:5467. doi: 10.1021/jacs.6b02019.
- 9.Sam B, Montgomery TP, Krische MJ. Org. Lett. 2013;15:3790. doi: 10.1021/ol401771a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pye PJ, Rossen K, Reamer RA, Tsou NN, Volante RP, Reider PJ. J. Am. Chem. Soc. 1997;119:6207. [Google Scholar]
- 11.For synthesis of CF3-bearing allenes, see: Ambler BR, Peddi S, Altman RA. Org. Lett. 2015;17:2506. doi: 10.1021/acs.orglett.5b01027.Boreux A, Lonca GH, Riant O, Gagosz F. Org. Lett. 2016;18:5162. doi: 10.1021/acs.orglett.6b02636.
- 12.For a review on the use of methanol and paraformaldehyde as C1-feedstocks in metal catalyzed C-C coupling, see: Sam B, Breit B, Krische MJ. Angew. Chem. Int. Ed. 2015;54:3267. doi: 10.1002/anie.201407888.
- 13.For selected examples of the use of methanol as a methyl donor in catalytic C-C coupling, see: Chan LKM, Poole DL, Shen D, Healy MP, Donohoe TJ. Angew. Chem. Int. Ed. 2014;53:761. doi: 10.1002/anie.201307950.Ogawa S, Obora Y. Chem. Comm. 2014:2491. doi: 10.1039/c3cc49626k.Li Y, Li H, Junge H, Beller M. Chem. Comm. 2014:14991. doi: 10.1039/c4cc06933a.Shen D, Poole DL, Shotton CC, Kornahrens AF, Healy MP, Donohoe TJ. Angew. Chem. Int. Ed. 2015;54:1642. doi: 10.1002/anie.201410391.Quan X, Kerdphon S, Andersson PG. Chem. Eur. J. 2015;21:3576. doi: 10.1002/chem.201405990.
- 14.Moran J, Preetz A, Mesch RA, Krische MJ. Nature Chem. 2011;3:287. doi: 10.1038/nchem.1001. [DOI] [PubMed] [Google Scholar]
- 15.Lemieux RU, von Rudloff E. Can. J. Chem. 1955;33:1701. [Google Scholar]
- 16.For methanol-mediated transfer hydrogenation catalyzed by iridium, see: Yamagata T, Iseki A, Tani K. Chem. Lett. 1997;26:1215.Tani K, Iseki A, Yamagata T. Chem. Commun. 1999:1821. doi: 10.1039/b102395k.
- 17.(a) Qian M, Liauw MA, Emig G. Appl. Catal. A. Gen. 2003;238:211. [Google Scholar]; (b) Lin W-H, Chang H-F. Catal. Today. 2004;97:181. [Google Scholar]
- 18.Ruthenium hydrides engage in H-D exchange with water: Tse SKS, Xue P, Lin Z, Jia G. Adv. Synth. Catal. 2010;352:1512.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.