

ABSTRACT
Molluscum contagiosum virus (MCV) is a dermatotropic poxvirus that causes benign skin lesions. MCV lesions persist because of virally encoded immune evasion molecules that inhibit antiviral responses. The MCV MC159 protein suppresses NF-κB activation, a powerful antiviral response, via interactions with the NF-κB essential modulator (NEMO) subunit of the IκB kinase (IKK) complex. Binding of MC159 to NEMO does not disrupt the IKK complex, implying that MC159 prevents IKK activation via an as-yet-unidentified strategy. Here, we demonstrated that MC159 inhibited NEMO polyubiquitination, a posttranslational modification required for IKK and downstream NF-κB activation. Because MCV cannot be propagated in cell culture, MC159 was expressed independent of infection or during a surrogate vaccinia virus infection to identify how MC159 prevented polyubiquitination. Cellular inhibitor of apoptosis protein 1 (cIAP1) is a cellular E3 ligase that ubiquitinates NEMO. Mutational analyses revealed that MC159 and cIAP1 each bind to the same NEMO region, suggesting that MC159 may competitively inhibit cIAP1-NEMO interactions. Indeed, MC159 prevented cIAP1-NEMO interactions. MC159 also diminished cIAP1-mediated NEMO polyubiquitination and cIAP1-induced NF-κB activation. These data suggest that MC159 competitively binds to NEMO to prevent cIAP1-induced NEMO polyubiquitination. To our knowledge, this is the first report of a viral protein disrupting NEMO-cIAP1 interactions to strategically suppress IKK activation. All viruses must antagonize antiviral signaling events for survival. We hypothesize that MC159 inhibits NEMO polyubiquitination as a clever strategy to manipulate the host cell environment to the benefit of the virus.
IMPORTANCE Molluscum contagiosum virus (MCV) is a human-specific poxvirus that causes persistent skin neoplasms. The persistence of MCV has been attributed to viral downregulation of host cell immune responses such as NF-κB activation. We show here that the MCV MC159 protein interacts with the NEMO subunit of the IKK complex to prevent NEMO interactions with the cIAP1 E3 ubiquitin ligase. This interaction correlates with a dampening of cIAP1 to polyubiquitinate NEMO and to activate NF-κB. This inhibition of cIAP1-NEMO interactions is a new viral strategy to minimize IKK activation and to control NEMO polyubiquitination. This research provides new insights into mechanisms that persistent viruses may use to cause long-term infection of host cells.
KEYWORDS: FLIP, IKK, MC159, NEMO, NF-κB, cIAP1, molluscum contagiosum virus, poxvirus, tumor necrosis factor, ubiquitination
INTRODUCTION
Molluscum contagiosum (MC) virus (MCV) is a member of the Poxviridae family and is the etiological agent of MC (1). MCV infections are common worldwide (2, 3); approximately 122 million MCV infections occurred in 2010 (4). MCV infects keratinocytes, resulting in small neoplasms that persist for months to years (1). There is little inflammation at the borders of these long-lasting lesions (1), suggesting that MCV encodes molecules that dampen the immune response (5–7). Unfortunately, the absence of cell culture or efficient animal models for MCV replication has greatly hampered MCV research. This has limited the field's understanding of how MCV evades the immune response to persist. To date, knowledge about MCV immune evasion strategies is derived from the expression of individual MCV proteins independent of infection or during infection with a surrogate virus, and these strategies include antagonism of apoptosis (8–11), NF-κB activation (12–14), interferon regulatory factor 3 (IRF3) activation (15), and chemokine (16) and interleukin-18 (IL-18) (17) actions.
Tumor necrosis factor (TNF) is upregulated at MC lesions (18). This has potential antiviral consequences because TNF-TNF receptor 1 (TNFR1) interactions trigger antiviral events, including NF-κB activation (19). The canonical IκB kinase (IKK) complex controls NF-κB activation and is comprised of NEMO (IKKγ) regulatory and IKKα and IKKβ kinase subunits (20–22). NEMO is instrumental in controlling NF-κB activation because it regulates the IKKα-IKKβ signaling axis during NF-κB activation.
NEMO has several important functions during TNF-induced NF-κB activation. First, the C terminus of NEMO facilitates IKK binding to polyubiquitinated recreceptor-interacting protein 1 (RIP1) (23, 24), which is part of the TNFR1-based signalsome. Second, NEMO itself is polyubiquitinated, allowing for subsequent IKK activation. K63-linked polyubiquitination of NEMO was reported first (23, 24), followed by M1-linked ubiquitination (25–27). Most recently, K63/M1 hybrid polyubiquitination of NEMO was reported (28). Although there is debate about the relative importance of each type of ubiquitination, it is likely that each type of ubiquitination is necessary for optimal NF-κB activation (29). Regardless, the covalent linkage of ubiquitin (Ub) to a protein substrate occurs via cellular E1, E2, and E3 ligases (30). cIAP1 is one such E3 ligase that polyubiquitinates NEMO in addition to other cellular proteins (31–33).
The MCV-encoded MC159 protein is perhaps the best-studied MCV molecule (6). MC159 expression neutralizes NF-κB activation, and MC159 interacts with the NEMO subunit of the IKK complex (14). Interestingly, MC159 does not disrupt the IKK complex (14), suggesting that MC159 antagonizes IKK activation in a manner that is distinct from those of other known viral NEMO inhibitors. We show here that MC159 prevented NEMO polyubiquitination, leading us to further examine how MC159 prohibited this posttranslational event. Here, the data indicated that MC159 competitively binds to NEMO in a manner that prevents cIAP1 from interacting with and polyubiquitinating NEMO. This is the first report of a viral protein that blocks cIAP1-NEMO interactions to block NEMO polyubiquitination. As such, this work extends the repertoire of mechanisms that viruses use to evade antiviral immune responses and illuminates one strategy that may allow the sustained presence of replicating MCV in skin neoplasms.
RESULTS
MC159 inhibits NEMO polyubiquitination.
We reported previously that MC159 inhibits NF-κB activation, and this inhibition correlated with MC159-NEMO interactions (14). MC159-NEMO interactions did not disrupt the IKK complex (14), and this implied that MC159 may prevent posttranslational modifications of NEMO or interactions with NEMO-binding partners that are critical for NF-κB activation.
NEMO polyubiquitination is one such posttranslational modification that is required for TNF-induced NF-κB activation (33). We used well-characterized immunoprecipitation (IP) assays to examine if MC159 altered NEMO polyubiquitination. For these assays, epitope-tagged versions of NEMO and ubiquitin were expressed in cells (Fig. 1A). Immunoprecipitated NEMO was then separated by SDS-PAGE, and polyubiquitinated NEMO was detected by immunoblotting (IB). In this system, NEMO polyubiquitination was detected only when cells expressed both NEMO-FLAG and Ub-Myc (Fig. 1A, lanes 1 to 3). Maximal NEMO ubiquitination was observed with 500 ng each of pNEMO-FLAG and pUb-Myc (Fig. 1A, lane 4). Figure 1A shows that the lower band in the immunoprecipitated samples represented monoubiquitinated NEMO, which is 56 kDa, while the remaining, higher-molecular-mass NEMO-containing bands represented polyubiquitinated NEMO. This same pattern is indicated for the remaining figures.
FIG 1.

MC159 inhibits NEMO polyubiquitination. (A) HEK 293T cells were transfected as follows: 1,000 ng pCI (lane 1), 500 ng pCI and 500 ng pUb-Myc (lane 2), 500 ng pCI and 500 ng pNEMO-FLAG (lane 3), 250 ng pNEMO-FLAG and 750 ng pUb-Myc (lane 4), 500 ng pNEMO-FLAG and 500 ng pUb-Myc (lane 5), and 750 ng pNEMO-FLAG and 250 ng pUb-Myc (lane 6). At 24 h posttransfection, cells were lysed in Ub lysis buffer. (B) HEK 293T cells were cotransfected with 500 ng pNEMO-FLAG, 500 ng pUb-Myc, and 1,000 ng of either pCI, pMC159, or pNEMOΔZnF. At 24 h posttransfection, cells were lysed in Ub lysis buffer. (C) HEK 293T cells were transfected with 250 ng pNEMO-FLAG, 250 ng pUb-Myc, and 1,000 ng of either pCI, pMC159, or pNEMOΔZnF. At 24 h posttransfection, cells were incubated in medium containing 10 ng/ml TNF for 15 min prior to lysis in Ub lysis buffer. For all experiments, a portion of each lysate prior to immunoprecipitation was set aside to detect protein expression by IB. The remaining cellular lysates were immunoprecipitated with anti-FLAG antibody or an IgG isotype control antibody conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect polyubiquitinated NEMO, MC159, or actin. Monoubiquitinated (Mono-Ub) and polyubiquitinated (Poly-Ub) forms of NEMO are indicated.
Next, the optimized conditions for Fig. 1A were used to examine the impact of MC159 on NEMO polyubiquitination. NEMO polyubiquitination was stimulated by either the overexpression of NEMO (Fig. 1B) or TNF stimulation of cells (Fig. 1C). For the latter case, we used a smaller amount of pNEMO-FLAG such that NEMO did not stimulate NF-κB activation but polyubiquitination of NEMO could still be detected. TNF was used as a stimulus because TNF is upregulated in MCV lesions (18), and the signal transduction events leading from TNF-TNFR1 interactions to NF-κB activation are well characterized (19). Cells were also cotransfected with an empty vector (pCI) or a plasmid containing the MC159L gene.
It was necessary to express MC159 independent of infection for two reasons. First, MCV cannot be propagated in cell culture (6). Second, there are two other MCV proteins that inhibit NF-κB activation (MC160 and MC132) (12, 13). Thus, the examination of MC159 function in cells infected with MCV isolated from patient lesions may be confounded by the presence of these or other as-yet-unidentified MCV NF-κB-inhibitory proteins. We chose to express MC159 via transient transfection. Of course, one weakness of the use of transient transfections is that MC159 ectopic expression may not necessarily reflect MC159 protein expression levels during infection. Indeed, natural MC159 expression levels are not known because there is no method to currently propagate MCV in cell culture. We attempted to create cell lines expressing MC159. However, the MC159 protein levels dramatically decrease each time after passaging of these cell lines, such that MC159 is no longer detectable by the fifth passage of cell lines. However, transient-transfection systems afford the opportunity to use reagents (e.g., TNF) that trigger NF-κB activation via well-defined signal transduction pathways.
Figures 1B and C show that the quantity and intensity of polyubiquitinated NEMO were decreased in MC159-expressing versus pCI-transfected cells, indicating that MC159 antagonized NEMO polyubiquitination. Namely, the intensities of bands representing polyubiquitinated NEMO were decreased when MC159 was present versus when MC159 was absent when comparing band intensities within each gel shown in Fig. 1B and C. NEMO protein levels were not decreased in transfected cells, indicating that this decrease in ubiquitination was not due to a decrease in NEMO protein levels. A separate set of cells was cotransfected with a plasmid encoding mutant NEMO that lacks its lysines, substrates for ubiquitin (NEMOΔZnF) (33, 34). Thus, NEMO polyubiquitination was also greatly reduced when cells expressed NEMOΔZnF, as would be expected. In Fig. 1B and C, parallel immunoprecipitations were performed, in which nonspecific IgG was used in place of an anti-FLAG antibody (Ab). NEMO was not immunoprecipitated under these conditions, showing the specificity of the immunoprecipitation conditions. MC159 was also coimmunoprecipitated with FLAG-NEMO, in agreement with data from previous reports (14, 35). Thus, these results suggest that MC159 antagonized NEMO polyubiquitination, likely by interacting with NEMO, and warranted further study into how MC159 prevented NEMO polyubiquitination.
MC159 inhibits K63-linked NEMO ubiquitination.
Ubiquitination is an important posttranslational modification and can trigger events as diverse as protein degradation or protein activation (30, 36). For example, K48-linked polyubiquitination triggers the proteasomal degradation of a protein (30, 36). In contrast, K63-linked polyubiquitination is important for intracellular signaling (23, 24, 29, 37, 38). Indeed, K63-linked NEMO polyubiquitination was the first type of ubiquitination to be reported during TNF-induced NF-κB activation (23, 24, 29, 37, 38).
Given that MC159 blocks TNF-induced NF-κB activation (14), we queried whether MC159 could prevent the K63-linked polyubiquitination of NEMO. To answer this question, ubiquitin constructs were used, in which only lysine residue 48 (K48) or K63 remained intact (Fig. 2A), affording the examination of K48- or K63-linked polyubiquitination in isolation. All cells were treated with TNF to trigger NEMO polyubiquitination (Fig. 2B). As shown in Fig. 2B, TNF-stimulated NEMO polyubiquitination occurred when wild-type (WT) ubiquitin was present (lane 2). Polyubiquitinated NEMO was still detected when mutant ubiquitin expressing lysine only at residue 63 was present (Fig. 2B, lane 6). In this case, there was an increase in the molecular mobility of polyubiquitinated NEMO proteins compared to that of cells expressing wild-type ubiquitin. NEMO polyubiquitination was greatly reduced when pK48-Ub was expressed (lane 5). This was expected because TNF does not stimulate the K48-linked ubiquitination of NEMO (24). FLAG-tagged NEMO protein levels were similar regardless of wild-type or mutant ubiquitin. Thus, the diminished NEMO ubiquitination profile was not due to K48-mediated NEMO degradation.
FIG 2.
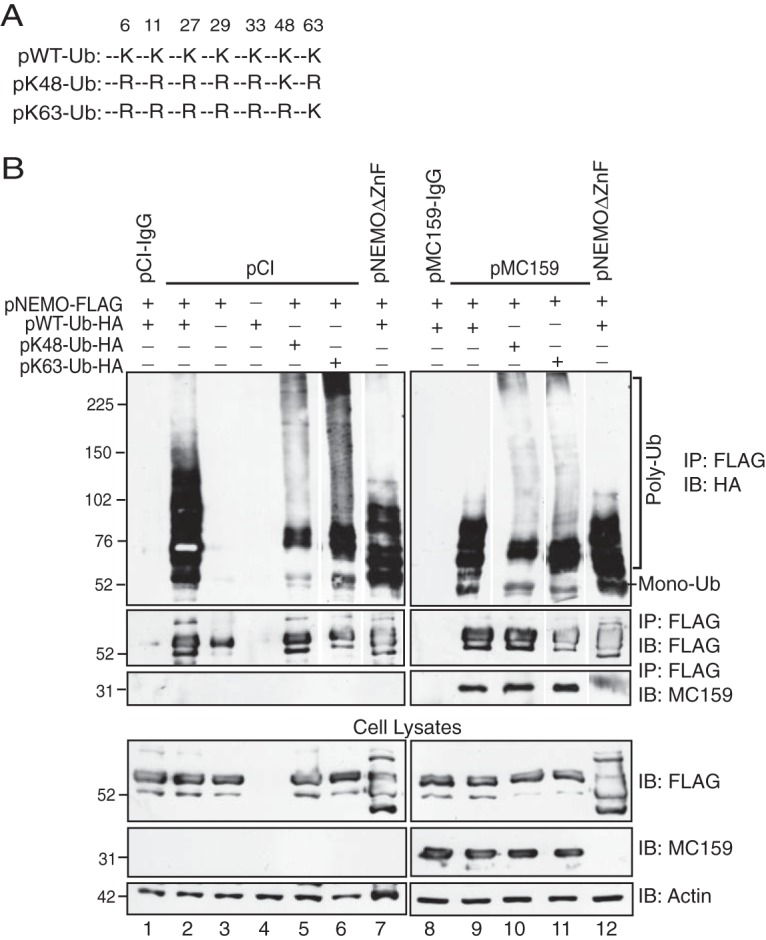
MC159 inhibits K63-linked NEMO polyubiquitination. (A) Schematic representation of wild-type or mutant Ub proteins that indicate lysine (K) or arginine (R) at a specific residue. (B) HEK 293T cells were cotransfected with 250 ng pNEMO-FLAG; 250 ng of pWT-Ub-HA, pK48-Ub-HA, or pK63-Ub-HA; and 1,000 ng of either pCI, pMC159, or pNEMOΔZnF for 24 h. Next, cells were incubated in medium containing 10 ng/ml TNF for 15 min. Cells were lysed in Ub lysis buffer. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IPs were performed by using either anti-FLAG or IgG antibodies conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect ubiquitinated forms of NEMO, MC159, or actin. For immunoprecipitated samples probed with anti-HA to detect ubiquitin, anti-FLAG, or anti-MC159 antibodies, lanes from the same gel were spliced to show the relevant samples for the experiment. The corresponding cellular lysates were probed by immunoblotting in a subsequent assay, avoiding the need for splicing. Monoubiquitinated (Mono-Ub) and polyubiquitinated (Poly-Ub) forms of NEMO are indicated.
MC159 inhibited TNF-induced NEMO polyubiquitination when either the wild-type or pK63-Ub proteins were expressed (Fig. 2B, lanes 9 and 11). Thus, MC159 inhibited the K63-linked ubiquitination of NEMO during TNF stimulation. MC159 coimmunoprecipitated with NEMO regardless of the expression of wild-type or mutant ubiquitin. As described above, the level of polyubiquitinated NEMO was greatly decreased when cells expressed NEMOΔZnF. These results suggest that MC159 blocked the K63-linked ubiquitination of NEMO during TNF stimulation.
MC159 does not inhibit RIP1 ubiquitination.
RIP1 polyubiquitination is essential for TNF-induced IKK activation (23). The K63-linked ubiquitin chains on RIP1 serve as a platform to recruit IKK (via the NEMO protein) and TAK binding protein (TAB)–transforming growth factor β (TGF-β)-activated kinase 1 (TAK1) (via the TAB2 protein) (19). Next, TAK1 phosphorylates and activates IKK (23). MC159 has been reported to coimmunoprecipitate with RIP1 (39, 40). It was of interest to ask if MC159 could prohibit the polyubiquitination of RIP1. Epitope-tagged versions of RIP1 and ubiquitin were expressed in cells. Immunoprecipitated RIP1 was then probed for polyubiquitination by immunoblotting. Ectopic RIP1 expression resulted in RIP1 polyubiquitination when Myc-tagged ubiquitin was coexpressed (Fig. 3A). A similar trend was observed during incubation of cells with TNF, which also increased RIP1 polyubiquitination (Fig. 3B). As shown in both Fig. 3A and B, RIP1 polyubiquitination was not decreased—in fact, it appeared to be enhanced—when MC159 was present when comparing ubiquitination patterns between pCI- and MC159-expressing cells within each gel shown in Fig. 3A and B. These data suggested that MC159 was not a global inhibitor of polyubiquitination. RIP1 overexpression is also known to activate apoptosis (41). While none of the cellular monolayers used in these experiments showed apoptotic morphologies (e.g., cellular blebbing or cellular detachment), we cannot rule out the possibility that this increase in polyubiquitination is due to an increase in cellular viability when the antiapoptotic MC159 protein is coexpressed.
FIG 3.
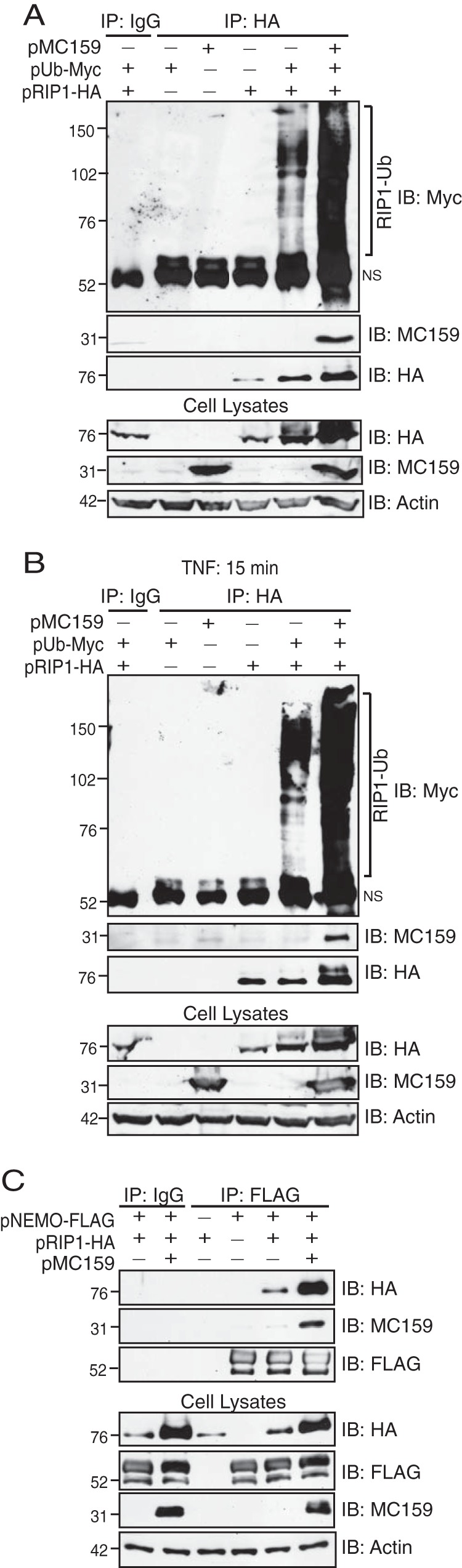
MC159 does not inhibit RIP1 polyubiquitination and still allows RIP1-NEMO interactions. (A and B) HEK 293T monolayers were cotransfected with 1,000 ng pCI or pMC159, 500 ng pUb-Myc, and 500 ng pRIP1-HA. At 24 h posttransfection, cells were incubated in medium lacking (A) or containing (B) 10 ng/ml TNF for 15 min to trigger RIP1 ubiquitination. Cells were lysed in Ub lysis buffer 24 h later. Some lysates were set aside to monitor protein expression. For the remaining clarified cellular lysates, IPs were performed by using IgG or anti-HA antibodies conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect RIP1, MC159, or actin. NS, nonspecific band. (C) HEK 293T monolayers were cotransfected with 1,000 ng pCI or pMC159, 500 ng pUb-Myc, and 500 ng pRIP1-HA. Cells were lysed in IP lysis buffer at 24 h posttransfection. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IP was performed by using IgG or anti-FLAG antibodies conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect either NEMO, RIP1, MC159, or actin.
We also observed that MC159 coimmunoprecipitated with RIP1, in agreement with data from previous reports (39, 40). Interestingly, hemagglutinin (HA)-tagged RIP1 protein levels were slightly higher in lysates and in immunoprecipitates when MC159 was expressed. The reason for this remains unclear, but this suggests that MC159 may enhance RIP1 stability. We also observed a higher-molecular-weight form of RIP1 in immunoprecipitates and lysates from TNF-stimulated cells (Fig. 3B). One possibility is that this band represents a phosphorylated form of RIP1 based on similar findings reported by others (42). Nevertheless, these data suggested that MC159 was not acting as a global inhibitor of polyubiquitination.
MC159 does not inhibit RIP1-NEMO interactions.
Our and data and those reported previously by others showed that MC159 coimmunoprecipitates with RIP1 (39, 40) and NEMO (14, 35). IKK must interact with RIP1 for NF-κB activation (23). Thus, an alternative explanation for the MC159-mediated inhibition of NF-κB activation could be that MC159 inhibited RIP1-NEMO interactions (23). To test this possibility, epitope-tagged versions of NEMO and RIP1 were coexpressed in the presence or absence of MC159. FLAG-tagged NEMO was immunoprecipitated, and the presence of RIP1 was evaluated by using immunoblotting (Fig. 3C). NEMO-RIP1 interactions were detected in cells lacking or expressing MC159, implying that MC159 did not prevent IKK from interacting with RIP1. This interaction was not due to the nonspecific binding of proteins to protein G-Sepharose beads because RIP1 was not detected when IgG was used in place of anti-FLAG for coimmunoprecipitations. Similar to the data shown in Fig. 3B, MC159 protein expression correlated with an increased level of RIP1 proteins present in immunoprecipitates and cellular lysates (Fig. 3C).
MC159 is not ubiquitinated by the cell but associates with ubiquitin.
Ubiquitination involves the covalent attachment of an ∼8-kDa ubiquitin to a lysine on a recipient protein (20). MC159 has five lysine residues. Thus, one possible reason for the observed decrease in NEMO polyubiquitination would be that MC159 outcompeted cellular proteins for host cell ubiquitin machinery. To test this possibility, MC159 was coexpressed with Myc-tagged ubiquitin in cells. In this case, Myc-tagged ubiquitin was immunoprecipitated, and this immunoprecipitate was probed for the presence of MC159 by immunoblotting (Fig. 4). MC159 was present in immunoprecipitates collected from either unstimulated or TNF-stimulated cells, as indicated by a 31-kDa band (43), and this is the same size as the that of MC159-containing band observed in cellular lysates. We conclude that it is unlikely that MC159 itself received covalent ubiquitin linkages because covalent ubiquitin linkages would be expected to increase the molecular mass of MC159 to at least 39 kDa. Because MC159 coimmunoprecipitated with Myc-tagged ubiquitin under these conditions (Fig. 4A and B), one possibility is that MC159 may interact with unanchored, noncovalently linked forms of ubiquitin. However, it is not clear if this modification to MC159 is beneficial.
FIG 4.

MC159 coimmunoprecipitates with ubiquitin but is not covalently linked to ubiquitin. Subconfluent HEK 293T cellular monolayers were cotransfected with 500 ng pUb-Myc and 500 ng pMC159. At 24 h posttransfection, cells were incubated in medium either lacking (A) or containing (B) 10 ng/ml TNF. After 15 min, cells were lysed in Ub lysis buffer. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IP was performed by using IgG or anti-Myc antibodies conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect either MC159, Myc-tagged ubiquitinated proteins, or actin.
MC159 binds to the N-terminal region of NEMO.
NEMO has two C-terminal regions that contain lysines that are targeted for covalently linked ubiquitination: ubiquitin binding domain (UBAN) (residues 249 to 339) and zinc finger (ZnF) (residues 389 to 419) (20). Since MC159 inhibited NEMO polyubiquitination, one hypothesis was that MC159 binds to the C terminus of NEMO to physically cloak lysine substrates. This possibility was examined by mapping NEMO-MC159 interactions using the wild-type or mutant NEMO constructs shown in Fig. 5A. As shown in Fig. 5B, HEK 293T cells were transfected with plasmids expressing NEMO, MC159, and ubiquitin to duplicate the conditions used for ubiquitination assays shown in Fig. 1 and 2. All transfected cells were treated with TNF to trigger NEMO polyubiquitination. These conditions were conducive for NEMO-MC159 interactions because MC159 coimmunoprecipitated with FLAG-tagged wild-type NEMO as expected (Fig. 5B). Interestingly, MC159 did not interact with NEMO residues 250 to 419, the NEMO region that contains the lysines that receive ubiquitin linkages. Instead, MC159 interacted with the N-terminal region of NEMO (residues 1 to 250), a region that does not possess known ubiquitination sites (Fig. 5B).
FIG 5.
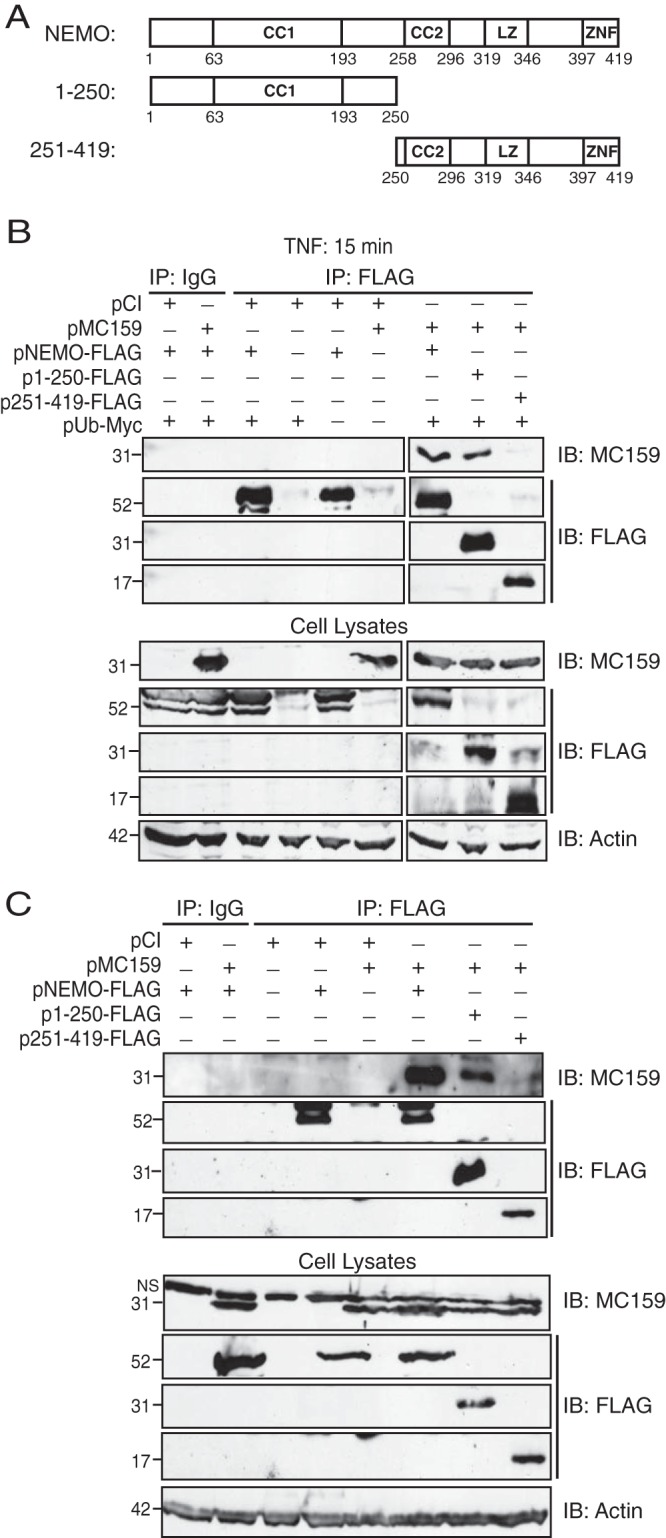
MC159 binds to the N-terminal region of NEMO. (A) Schematic representation of wild-type and mutant NEMO constructs used here. NEMO domains, including the two coiled-coil regions (CC1 and CC2), a leucine zipper (LZ), and a zinc finger (ZnF) domain, are shown here. (B) Subconfluent HEK 293T cellular monolayers were cotransfected with the indicated plasmids (1,000 ng pMC159, 500 ng of pNEMO-based plasmids, and 500 ng pUb-Myc). Where necessary, pCI was present in transfection reaction mixtures to bring the amount of total DNA in each transfection reaction mixture up to 2,000 ng. At 24 h posttransfection, cells were incubated with medium containing TNF (10 ng/ml) for 15 min. (C) NEMO−/− MEFs were cotransfected with the indicated plasmids (1,000 ng pMC159 and 500 ng of pNEMO-based plasmids). Where necessary, pCI was present in transfection reaction mixtures to bring the amount of total DNA in each transfection reaction mixture up to 1,500 ng. At 24 h posttransfection, HEK 293T cells or MEFs were lysed in 150 μl of IP lysis buffer. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IP was performed by using IgG or anti-FLAG antibodies conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect either NEMO, MC159, or actin.
Additional experiments were performed by using NEMO−/− mouse embryonic fibroblasts (MEFs) (Fig. 5C) that were reconstituted with the NEMO constructs shown in Fig. 5A. This second experimental approach was used to rule out contributions of endogenous NEMO. Once again, MC159 coimmunoprecipitated with wild-type NEMO and the NEMO construct consisting of residues 1 to 250. No MC159-NEMO interactions were detected when a NEMO construct expressing NEMO residues 251 to 419 was used. These data suggested that MC159 interacted with the N-terminal region of NEMO and not with the NEMO regions that are known to contain lysines that are targeted for ubiquitination.
MC159 reduces cIAP1-induced NF-κB activation and NEMO ubiquitination.
cIAP1 and cIAP2 are similar proteins, and both proteins are required to activate NF-κB during TNF stimulation (44). Each protein acts as an E3 ligase to covalently link ubiquitin to cellular proteins, including RIP1 and NEMO (33, 44, 45). cIAP1 interacts with the N terminus of NEMO (amino acids 64 to 106) (32). Moreover, cIAP1 adds K63 ubiquitin linkages to cellular substrates under certain conditions (31). Since MC159 and cIAP1 each interact with the N terminus of NEMO, one possibility was that MC159 prevented cIAP1-NEMO interactions as a mechanism to inhibit NEMO ubiquitination.
To test our hypothesis, we chose to overexpress an epitope-tagged version of cIAP1 instead of small interfering RNA (siRNA) silencing of cIAP1. This is because both cIAP1 and cIAP2 must be siRNA silenced to completely block TNF-induced NF-κB activation. Unfortunately, the siRNA silencing of both cIAP1 and cIAP2 also triggers apoptosis (44). Thus, the overexpression of cIAP1 (instead of siRNA silencing of cIAP1) avoids confounding effects.
As an initial test of this hypothesis, we capitalized on previous reports that cIAP1 overexpression stimulates NF-κB activation (33). Luciferase activity assays were developed under conditions similar to those for examining NEMO ubiquitination (Fig. 1B). Figure 6A shows that cIAP1-induced luciferase activity was highest when cells were transfected with 750 or 1,000 ng of a cIAP1-containing plasmid. MC159 expression greatly reduced cIAP1-mediated NF-κB activation (Fig. 6A). The Kaposi's sarcoma herpesvirus K13 protein contains two tandem death effector domains, similar to MC159 (46). K13 also binds to NEMO but is known to activate NF-κB (47, 48). Thus, K13 expression would be expected to enhance cIAP1-induced NF-κB activation, and this trend was observed (Fig. 6A). Examination of lysates used for luciferase activity assays showed that MC159 and FLAG-NEMO protein expression levels were similar under all conditions examined. K13-FLAG protein levels were also similar in cells receiving 750 or 1,000 ng cIAP1. cIAP1 levels appeared slightly lower in K13-expressing cells when smaller amounts of the cIAP1-containing plasmid were present, but this trend did not continue when larger amounts of the cIAP1-containing plasmid were transfected into cells.
FIG 6.

MC159 reduces cIAP1-mediated NF-κB activation and NEMO polyubiquitination. (A) HEK 293T cells were cotransfected for 24 h with 225 ng pNF-κBluc; 25 ng pRL-TK; 250 ng pUb-myc; 250 ng pNEMO-FLAG; 750 ng of pcDNA3.1 (vector control [VC]), pMC159, or pK13-FLAG; and increasing amounts of pcIAP1. Cells were lysed, and luciferase activities were quantified. Results are shown as the fold induction of luciferase activity relative to those of cells transfected with the empty vector. *, P < 0.05; **, P < 0.001 (compared to cells transfected with the empty vector). Immunoblot analysis of lysates was also performed. (B) HEK 293T cells were cotransfected with 250 ng pNEMO-HA, 250 ng pUb-Myc, 250 or 500 ng pcIAP1, and an amount of pCI sufficient to make 1,500 ng present in all reaction mixtures. (C) HEK 293T cells were cotransfected with 250 ng pNEMO-HA, 250 ng pUb-Myc, 250 ng pcIAP1, and 1,000 ng of either pCI or pMC159. (B and C) At 24 h posttransfection, cells were lysed in 150 μl of Ub lysis buffer. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IP was performed by using the indicated antibodies conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect cIAP1, NEMO, MC159, K13, or actin. Monoubiquitinated (Mono-Ub) and polyubiquitinated (Poly-Ub) forms of NEMO are indicated.
A second strategy to examine if MC159 negatively regulated cIAP1 was to determine cIAP1-induced NEMO polyubiquitination in the presence or absence of MC159. In this case, cells coexpressed epitope-tagged NEMO, cIAP1, and ubiquitin. HA-tagged NEMO was immunoprecipitated and probed for polyubiquitination by immunoblotting (Fig. 6B and C). Cells overexpressing cIAP1 showed more polyubiquitinated forms of NEMO than did cells lacking cIAP1, and 250 ng pcIAP1 was the best inducer of NEMO polyubiquitination (Fig. 6B). This finding suggests that cIAP1 facilitates the polyubiquitination of NEMO. While these reactions each showed higher-molecular-weight bands indicative of polyubiquitinated NEMO, these higher-molecular-weight bands were less intense when MC159 was present, suggesting that MC159 partially inhibited cIAP1 ubiquitination of NEMO (Fig. 6C).
MC159 inhibits NEMO-cIAP1 interactions.
When examining the immunoprecipitates shown in Fig. 6B, we observed that cIAP1 was absent from NEMO-based immunoprecipitations in MC159-expressing cells. This suggested that MC159 prevented cIAP1-NEMO interactions. This seemed likely because cIAP1 binds to residues 64 to 106 of NEMO (32), while MC159 binds to a region in the first 250 residues of NEMO.
Additional immunoprecipitations were performed (in which the plasmid expressing ubiquitin was absent) to further investigate if MC159 could interfere with cIAP1-NEMO interactions (Fig. 7). Figure 7A shows immunoprecipitations of an HA-tagged version of NEMO. FLAG-tagged cIAP1 coimmunoprecipitated with NEMO only when MC159 was absent (Fig. 7A). MC159 is a viral FLICE inhibitory protein (FLIP), and there are other viral FLIPs expressed by herpesviruses. The expression of Kaposi's sarcoma-associated herpesvirus (KSHV) K13 did not perturb cIAP1-NEMO interactions (Fig. 7B), indicating that MC159 inhibition of cIAP1-NEMO interactions was a unique property of this MCV vFLIP. We also asked if cIAP1 was an MC159 binding partner (Fig. 7C). In this case, FLAG-tagged cIAP1 was immunoprecipitated, and immunoprecipitates were probed for the presence of HA-tagged NEMO or HA-tagged MC159. While cIAP1-NEMO interactions were detectable, cAIP1-MC159 interactions were not (Fig. 7C). This finding suggested that MC159 specifically targets NEMO.
FIG 7.

MC159, but not the K13 homolog, inhibits NEMO-cIAP1 interactions. (A and B) HEK 293T cells were cotransfected with 500 ng pcIAP1-FLAG, 500 ng pNEMO-HA, and 1,000 ng of pCI, pMC159 (A), or pK13-FLAG (B). At 24 h posttransfection, cells were collected and lysed in IP lysis buffer. A portion of each lysate was set aside to monitor protein expression. The remaining clarified cellular lysates were incubated with IgG or anti-HA antibody conjugated to protein G-Sepharose beads. (C) HEK 293T cells were cotransfected with 500 ng pcIAP1-FLAG and 500 ng pCI, pNEMO-HA, or pMC159-HA. At 24 h posttransfection, cells were collected and lysed in IP lysis buffer. Clarified cellular lysates were incubated with IgG or anti-HA antibodies conjugated to protein G-Sepharose beads. (D) Dishes (10 cm2) of subconfluent monolayers of HeLa cells were transfected with 5,000 ng of pCI or pMC159-HA. At 24 h posttransfection, cells were incubated in medium containing TNF (10 ng/ml) for 10 min. Cells were collected and lysed in IP lysis buffer. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IPs were performed by using IgG or anti-cIAP1 antibodies conjugated to protein G-Sepharose beads. (E) HEK 293T cells were cotransfected with 250 ng pcIAP1-Myc; 250 ng pNEMO-FLAG; 250 ng pWT-Ub-HA, pK48-HA, or pK63-Ub-HA; and 1,000 ng pCI or pMC159, as indicated. At 24 h posttransfection, cells were collected and lysed in IP lysis buffer. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IPs were performed by using IgG or anti-HA antibodies conjugated to protein G-Sepharose beads. For all experiments, immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect cIAP1, NEMO, Ub, MC159, K13, or actin.
The above-described experiments detected interactions of epitope-tagged proteins that were overexpressed in HEK 293T cells. Additional experiments were performed to show that MC159 blocked the interactions of endogenous cIAP1 and NEMO (Fig. 7D). In this case, cells were transfected with only pCI or MC159/pCI. While TNF was added to these cells, note that cIAP-NEMO interactions can be detected in the absence or presence of TNF (33). cIAP1 was immunoprecipitated by using anti-cIAP1 antibodies, and immunoprecipitates were probed with antibodies against NEMO or HA-tagged MC159. While cIAP1-NEMO interactions were detected in pCI-transfected cells, these interactions were reduced when MC159 was present.
Figure 7E shows a reverse approach to examine the effect of MC159 on the cIAP1-mediated ubiquitination of NEMO. In this case, cells were transfected with the HA-tagged Ub constructs shown in Fig. 2 (WT, K48, or K63), and ubiquitinated proteins were immunoprecipitated by using anti-HA antibodies. Immunoprecipitates were probed with antibodies specific for NEMO. When NEMO and ubiquitin were overexpressed (Fig. 7E, lane 3), a 56-kDa NEMO protein was detected, representing the monoubiquitinated form of NEMO. This was expected because NEMO has a UBAN (ubiquitin binding region) domain. Also, the diubiquitinated form of NEMO (64 kDa) was also observed. When cIAP1 was overexpressed, a third, higher-migrating band of NEMO was detected (approximately 72 kDa), indicating that cIAP1 facilitates NEMO polyubiquitination (Fig. 7E, lane 4). This banding pattern changed when K48 or K63 mutant ubiquitins were expressed. When K48 was expressed, monoubiquitinated NEMO was observed, but there was a dramatic reduction in diubiquitinated NEMO levels. This was expected because K48-linked ubiquitination is usually associated with proteosomal degradation. When K63 ubiquitin was expressed, we instead observed a doublet NEMO band with a slightly higher molecular weight than that of the diubiquitin NEMO observed in Fig. 7E, lanes 3 and 4. We speculate that this doublet may represent a mixed population of NEMO that has ubiquitin binding to the UBAN region versus the zinc finger domain region, but we have no means to prove this. It was most interesting that when MC159 was expressed, only monoubiquitinated NEMO was detected in Fig. 7E. In summary, these data suggest that MC159 greatly reduced cIAP-NEMO interactions and thus cIAP1-mediated NEMO polyubiquitination.
MC159 inhibits NEMO-cIAP1 interactions during surrogate virus infection.
Vaccinia virus and other poxviruses are genetically tractable because these viruses can be propagated in vitro. However, MCV cannot be propagated in cell culture (6, 7), making it difficult to assess if MC159 inhibits NEMO-cIAP1 interactions during MCV infection. As an alternative strategy to study MC159 function during infection, the MCV MC159L gene was stably inserted into a vaccinia virus that lacked the vaccinia virus A49R gene, a gene that encodes an NF-κB-inhibitory protein (49). MC159 protein expression was detected in infected cells at both 2 and 6 h postinfection (p.i.), and this was expected because MC159 was under the control of a synthetic early/late promoter (Fig. 8A). The parental virus (vΔA49) and the revertant virus, in which the A49R gene was reintroduced into vΔA49, did not express MC159 (Fig. 8A).
FIG 8.
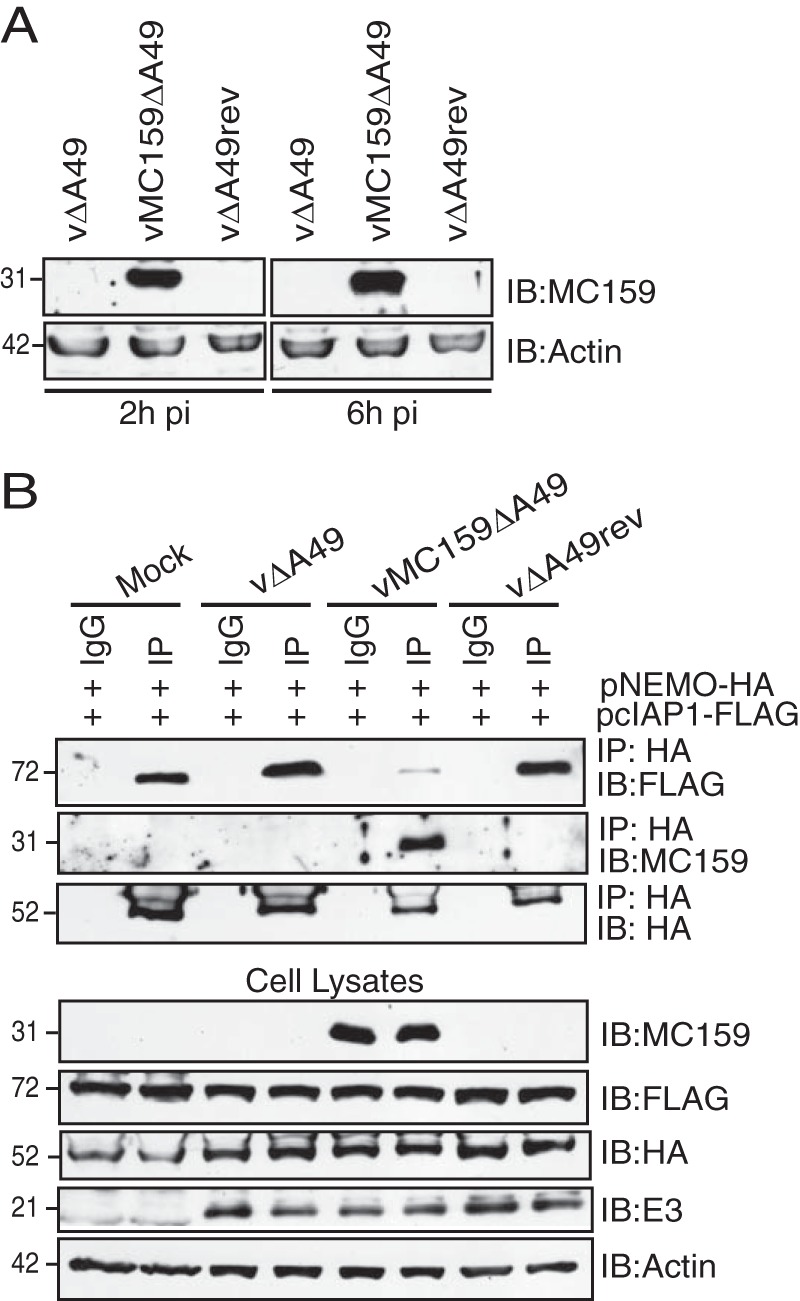
MC159 inhibits NEMO-cIAP1 interactions during surrogate virus infection. (A) RK13 cellular monolayers were infected with vΔA49, vΔA49rev, or vMC159ΔA49 (MOI = 10). At 2 or 6 h p.i., cells were collected and lysed in CE buffer. A portion of each lysate was electrophoretically separated by using SDS–12% PAGE, and proteins were transferred onto PVDF membranes. Membranes were probed with antibodies to detect MC159 or actin. (B) HEK 293T cellular monolayers were cotransfected with 500 ng pNEMO-HA and 500 ng pcIAP1-FLAG. At 24 h posttransfection, cells were either mock infected or infected with vΔA49, vΔA49rev, or vMC159ΔA49 (MOI = 10). At 24 h postinfection, cells were collected and lysed in IP lysis buffer. A portion of each lysate was set aside to monitor protein expression. For the remaining clarified cellular lysates, IPs were performed by using IgG or anti-HA antibodies conjugated to protein G-Sepharose beads. Immunoprecipitated samples or cellular lysates were separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB. Membranes were probed with the indicated antibodies to detect NEMO, MC159, cIAP1, E3, or actin.
Next, viruses were used to infect cells expressing epitope-tagged versions of NEMO and cIAP1 (Fig. 8B). In this case, NF-κB activation was triggered by the overexpression of cIAP1 and NEMO. cIAP1 coimmunoprecipitated with NEMO in mock-infected cells or in cells infected vΔA49 or vΔA49rev. However, these interactions were greatly reduced when cells were infected with vMC159ΔA49. E3, a vaccinia virus protein expressed early in infection, was present in similar amount in lysates of all infected cells, demonstrating similar extents of virus infection (Fig. 8B). An identical set of lysates was incubated with IgG antibodies. Under these conditions, neither NEMO, cIAP1, nor MC159 was detected in immunoprecipitation reactions, indicating that these proteins did not nonspecifically bind to protein G-Sepharose beads. In summary, these data indicate that MC159 interferes with cIAP1-NEMO interactions during virus infection.
DISCUSSION
There is no reliable FDA-approved treatment for MCV infections (6, 7). Understanding how MC159 represses NEMO activation may provide a rationale for the design of compounds that may have therapeutic value for MCV infections. We show here that MC159 inhibited NEMO polyubiquitination, piquing curiosity as to how MC159 might interact with NEMO to interfere with its posttranslational modification. MC159 probably is not a competitive substrate for the host cell ubiquitination machinery because MC159 is not covalently linked to ubiquitin. MC159 also does not physically cloak the C-terminal lysine residues of NEMO that are known substrates of the ubiquitin machinery because MC159 did not interact with the C-terminal region of NEMO.
A potential mechanism for the MC159 antagonist function became apparent when we observed that MC159 interacted with the N terminus of NEMO, the same region to which cIAP1 binds (32). Here, several lines of evidence support the model that MC159 binds to NEMO to prevent cIAP1 from interacting with and polyubiquitinating NEMO. First, cIAP1 no longer coimmunoprecipitated with NEMO when MC159 was present. Second, cIAP1 had a greatly reduced capacity to polyubiquitinate NEMO when MC159 was present. Third, cIAP1-induced NF-κB activation was greatly reduced by MC159. This is the first report of a viral protein disrupting cIAP1-NEMO interactions as a strategy to antagonize NF-κB activation. The current model is that by competitively inhibiting cIAP1-NEMO interactions, MC159 prevents the cIAP1-induced polyubiquitination of NEMO. Thus, MC159 represents a novel viral strategy to antagonize IKK activation. One caveat is that our coimmunoprecipitation data do not rule out the possibility that MC159 indirectly binds to NEMO. We think that this possibility is less likely because a viral homolog of MC159 (K13) binds directly to NEMO in a cell-free system (47).
We used TNF to trigger signal transduction events in many experiments here because TNF is expressed at MC lesions (18). Also, many of the signal transduction events that occur from TNFR1 to NF-κB transcriptional activation are well known (19), which would aid in determining how MC159 may alter IKK activation. For example, the K63-linked polyubiquitination of NEMO was the first type of ubiquitination event shown to be important for TNF-induced NF-κB activation (23, 24). TNF also stimulates the formation of M1-linked and K63/M1-ubiquitin hybrid chains on NEMO (28, 50). K63-linked chains are substrates for M1-linked polyubiquitination (28, 50). Since MC159 inhibited K63-linked ubiquitination (Fig. 2), it is predicted that MC159 would also block the formation of K63/M1 hybrid chains and, as such, may also inhibit NF-κB activation triggered via other innate immune signaling pathways such as those triggered by the IL-1 receptor (IL-1R) (28, 50).
RIP1 polyubiquitination is critical for IKK and NF-κB activation, and cIAP1 ubiquitinates RIP1 (23, 25, 26, 51–53). Thus, it was quite surprising to observe that MC159 did not prevent RIP1 polyubiquitination despite RIP1-MC159 coimmunoprecipitations. Since these are coimmunoprecipitations, it cannot be discerned if MC159 binds directly to RIP1 or indirectly to RIP1 via NEMO. Nevertheless, our results raise the question of why the inhibition of NEMO but not RIP1 polyubiquitination may be beneficial to MCV. TNF-TNFR1 interactions possess the capacity to trigger either NF-κB activation, apoptosis, or necroptosis (19, 54). The ubiquitination status of RIP1 can control this outcome (23, 51, 55, 56). The general consensus is that polyubiquitinated RIP1 allows NF-κB activation (23). However, if RIP1 is unmodified, apoptosis (57–59) or necroptosis (56, 60–62) signaling events may occur. Thus, a strategic allowance of RIP1, polyubiquitination would decrease signaling events associated with cell death and increase events associated with virus survival. There are reports that show that MC159 inhibits TNF-induced necroptosis (35) and programmed necrosis (39, 63), which support this hypothesis. MC159 is already known to bind to and inhibit procaspase-8 to inhibit apoptosis (8, 9, 43). Thus, the allowance of RIP1 ubiquitination for the purposes of cell survival would represent a second (albeit indirect) antiapoptosis mechanism of MC159.
cIAP1 is an E3 ligase that polyubiquitinates RIP1 (44, 45). Other reports have shown that cIAP1 also ubiquitinates NEMO (32, 33). For example, using recombinant proteins, Tang et al. showed that cIAP1 conjugates K6-linked ubiquitin to NEMO (33). cIAP1 has also been reported to add K63-linked ubiquitin to BCL10 to stimulate IKK activation in B cell lymphoma cell lines (31). No reports show that cIAP1 ubiquitinates NEMO during Toll-like receptor 2 (TLR2)- or IL-1R-induced NF-κB activation. Instead, TRAF6 ubiquitinated NEMO for TLR2-induced and IL-1R-induced NF-κB activation (64). Thus, it is difficult to predict if MC159 would inhibit TNF receptor-associated factor 6 (TRAF6)-induced NEMO ubiquitination. While there is no reported evidence to show that cIAP1 adds K63-linked ubiquitin chains to NEMO, cIAP1 has been reported to K63 link ubiquitinated Bcl-10 (31) and RIP1 (65). Thus, we became interested in the possibility that cIAP1 may act as an E3 ligase in our system of study because of the coincidence that MC159 interacted with the N terminus of NEMO, where cIAP1 was reported to bind (32). We show here that MC159 inhibited NEMO-cIAP1 interactions and cIAP1-induced NF-κB activation and NEMO polyubiquitination. From these data, our current model is that MC159 prevents cIAP1-induced NEMO polyubiquitination. We observed that MC159 did not completely abolish NEMO polyubiquitination under any conditions examined. It has been proposed that there are multiple E3 ligases that ubiquitinate NEMO (20). We speculate that perhaps MC159 does not prevent these other ligases from interacting with and modifying NEMO. The biological relevance of this remains unclear. We also observed that MC159 was a stronger inhibitor of NEMO polyubiquitination when cells were stimulated with TNF than with the overexpression of cIAP1. Perhaps the latter result is due to competition between cIAP1 and MC159 for NEMO in the transfection system used here.
MCV cannot be propagated in cell culture, making it difficult to examine the biochemical function of MC159 during infection. Here, we expressed the MCV MC159L gene in vaccinia virus as a strategy to explore MC159 functions during poxvirus infections. We show that MC159 prevented cIAP1-NEMO interactions during virus infection, implying that MC159-NEMO interactions are biologically relevant during MCV infections. We purposefully did not examine NEMO ubiquitination or NF-κB activation in virus-infected cells because the parental vaccinia virus (vΔA49) expresses at least nine other NF-κB-inhibitory proteins that inhibit NF-κB activation, including some that act upstream of IKK activation (e.g., A52, A46, and K7) (66). Thus, NEMO is expected to remain in its nonubiquitinated state during vΔA49 infection due to these other vaccinia virus proteins. Because of this, it would be technically impossible to detect MC159's effects on NEMO polyubiquitination in these virus-infected cells.
To date, MCV encodes three proteins that target the NF-κB activation pathway: MC159, MC160, and MC132 (6, 12). MC159 and MC160 each target the IKK complex (13, 14, 67). MC159 binds to the NEMO subunit of the IKK complex (14). In contrast, MC160 triggers IKKα degradation (67). MC132 acts at a step downstream of MC159 and MC160; MC132 degrades the p65 subunit of the NF-κB complex (12). The mechanism of each MCV protein described above was determined by expressing each protein independent of infection because MCV cannot be propagated in cell culture. Of course, all three proteins are predicted to be expressed simultaneously during MCV infection. It is not clear why MCV codes for proteins with seemingly redundant functions. Similar questions are raised with vaccinia virus, a virus that expresses at least 10 different NF-κB inhibitors (66). One prevailing theory for vaccinia virus is that each NF-κB-inhibitory protein is important to block NF-κB activation triggered at different stages of infection or by different signal transduction pathways. Perhaps MCV encodes multiple proteins for similar reasons. For example, MC159 may target canonical IKK activation, while MC132 may be a means to neutralize NF-κB activation that occurs through the noncanonical (IKK-independent NF-κB activation) pathway.
This report focuses on MC159 inhibition of NF-κB activation (14, 35, 40, 68). However, there are several reports showing that MC159 activates the NF-κB pathway when MC159 is expressed at low levels (14, 35, 68, 69). This begs the question of which phenotype has biological relevance. Interestingly, the murine cytomegalovirus (MCMV) M45 protein induces NF-κB early after infection (70) and then inhibits NF-κB during later times postinfection (71). For M45, the former function is due to virion-associated M45 molecules, and it has been proposed that NF-κB activation is important for initial virus infection (70). In contrast, de novo-synthesized M45 proteins inhibit NF-κB, and this may be important for MCMV persistence (71). An attractive hypothesis is that reports of MC159 activating and inhibiting NF-κB activation may reflect MC159 up- or downregulating NF-κB activation temporally during infection to reorganize the cell landscape in a manner that is beneficial for persistent infection. Unfortunately, this hypothesis cannot be tested currently because MCV cannot be propagated in cell culture or in animals. In an elegant approach to further examine MC159 within the context of virus infection, Huttmann et al. examined MC159 properties within the context of M45-less MCMV infection (35). MC159 did not inhibit MCMV-induced NF-κB activation at late times postinfection (35), which implies that the NF-κB-inhibitory properties of MC159 are important during the initial phases of infection.
The IKK complex is a powerful means to activate NF-κB (20). Several viruses manipulate NEMO to antagonize IKK. For example, the Merkel cell polyomavirus (McPyV) small T antigen (ST) and the MCMV M45 protein each bind to NEMO to prevent IKK activation (71, 72). Each protein uses a different mechanism to antagonize NEMO, and this likely reflects the different needs of a virus during infection. McPyV ST draws cellular molecules serine/threonine-protein phosphatase 4 (PP4C) and protein phosphatase 2 beta (PP2aβ) to NEMO to presumably dephosphorylate and inactivate IKK (72). In contrast, M45-NEMO interactions target NEMO to the autophagosome for subsequent NEMO degradation (71). Here, MC159 inhibits NEMO polyubiquitination. Other viral proteins target other IKK subunits. The vaccinia virus B14 protein binds to IKKβ to prevent IKKβ phosphorylation by upstream kinases (73). The MCV MC160 protein degrades IKKα to destroy the IKK complex (13, 67). These viral proteins exemplify the importance of IKK for the antiviral response and that multiple viruses antagonize IKK activation using multiple strategies.
On the other side of the coin, some oncogenic viruses encode NEMO-binding proteins to stimulate IKK activation and induce the transformation of infected cells. These proteins include the KSHV K13 protein (47, 48). MC159 and K13 are members of the FLIP family of proteins (46). These proteins are also present in cells in two forms, cFLIP-long and cFLIP-short (cFLIPL and cFLIPS), and these proteins were characterized originally by two tandem death effector domains and their ability to block extrinsic apoptosis (46). It is rather striking that MC159 and K13 each bind to the N terminus of NEMO, but MC159-NEMO interactions inhibit IKK activation, while K13-NEMO interactions trigger IKK activation (14, 47, 48). How do these seemingly similar proteins interact with NEMO but cause such disparate biological outcomes? The solved crystal structures of MC159 (74, 75) and a K13-NEMO complex (76) have not shed light on potential mechanisms. It was originally proposed that K13 would induce major conformational changes in NEMO that favor IKK activation. However, Bagneris et al. recently reported that K13 binding causes only subtle conformational changes in NEMO, suggesting that K13 may rely on other binding partners to stimulate IKK (77). In our hands, MC159 prevented cIAP1-NEMO interactions, while K13 did not (Fig. 7B). One hypothesis for future research is that K13 may enhance cIAP1-NEMO interactions to activate IKK, while MC159 prevents such interactions to inhibit IKK. Although much is known about IKK activation, there are still many questions remaining about NEMO and IKK, including their temporal regulation by different stimulants and different cell types. It is proposed that parallel studies of structural homologs like MC159 and K13 provide a unique platform to learn how to precisely manipulate IKK activation to the benefit of human health.
MATERIALS AND METHODS
Cell lines and plasmids.
Human embryonic kidney HEK 293T cells and human cervical carcinoma (HeLa) cells were obtained from the American Type Culture Collection. NEMO−/− MEFs were obtained from Tak Mak (University of Toronto). Rabbit kidney cells (RK13) were a kind gift from Grant McFadden (University of Florida). All cells were cultured in Eagle's minimal essential medium (MEM) supplemented with 2 mM l-glutamine and 10% fetal bovine serum (FBS; Fisher).
Plasmids pCI and pcDNA3.1 were obtained from Promega and Invitrogen, respectively. Plasmids harboring the MCV MC159 gene (pMC159) or an HA epitope-tagged MC159 gene (pMC159-HA) were described previously (14, 15, 68). pK13-FLAG harbors a FLAG-tagged KSHV K13 gene and was provided by Jeffery Cohen (National Institutes of Health). The RIP1 plasmid (pRIP1-HA) was a kind gift from Preet Chaudary (University of Texas Southwestern Medical Center) (40). pNEMO-FLAG and pNEMO-HA express an epitope-tagged version of NEMO. The p1-250-FLAG and p251-419-FLAG plasmids were provided by Kunitada Shimotohno (Chiba Institute of Technology). p1-250-FLAG encodes residues 1 to 250 of NEMO, while p251-419-FLAG encodes residues 251 to 419 of NEMO. pIKKγΔZnF (called pNEMOΔZnF here) encodes a mutant NEMO protein in which the ZnF domain of NEMO is absent and cannot be ubiquitinated (33). Plasmids pWT-Ub-HA (Yossi Yarden, Weizmann Institute of Science) and pUb-Myc (Kunitada Shimotohno) encode HA and Myc epitope-tagged ubiquitins, respectively (78). The constructs pK48-Ub-HA and pK63-Ub-HA were kind gifts from Derek Abbott (Case Western Reserve University). Plasmids pcIAP1-FLAG and pcIAP1-Myc, which produce FLAG and Myc epitope-tagged cIAP1 proteins, respectively, were purchased from Addgene. Plasmid pNF-κBluc carries the firefly luciferase gene under the control of a synthetic NF-κB promoter (Promega). Plasmid pRL-TK (Promega) expresses constitutively low levels of sea pansy luciferase and is used to determine transfection efficiency.
For all experiments involving plasmids, DNA was transfected into cells by using FuGene 6 transfection reagent (Promega), according to the manufacturer's protocols.
Viruses.
vΔA49 and vΔA49rev were described previously (49). A recombinant virus was created, in which the MC159L gene, under the control of the synthetic p7.5 promoter, was inserted into a mutant vaccinia virus (vΔA49) with a deletion of nucleotides (nt) 113 to 473 of the A49R gene (49). The first step in this process was to create a pΔA49MCS plasmid that had multiple cloning sites for the insertion of the MC159L gene.
A plasmid lacking nt 113 to 473 of the A49R gene (pΔA49Z11) was used as a source of DNA. From this plasmid, two regions were PCR amplified: the A48 flank and the A50 flank. A48 begins at A48R nt 457 and continues through to A49R nt 112 for a 338-bp product. The primers used to PCR amplify this product are A48R forward primer 5′-ATCGGGATCCAACAAAAGGTATTACAAGAATAT-3′ and A48R reverse primer 5′-CGCGGATACTAGTATCGATATAGTTTCTATCTTGGCAATAACTAATTG-3′. The underlined regions are regions of vaccinia virus DNA. The forward primer introduces a BamHI restriction enzyme site (in italics). The reverse primer introduces the ClaI and SpeI restriction enzyme sites (in italics). The A50-flanking region was PCR amplified from pΔA49Z11. This PCR product begins at A49R nt 474 and ends at A50R nt 293 to PCR amplify a 339-bp product. The A50 forward primer is 5′-GAAACTATATCGATACTAGTATCCGCGAACGATATTTGTGA-3′. The A50 reverse primer is 5′-ATATGAATTCGATTTCTGTGTTCTCTTTGAAGAAAGTC-3′. The underlined nucleotides are located in the vaccinia virus genome. The forward primer introduces the ClaI and SpeI restriction enzyme sites (in italics). The reverse primer introduces an EcoRI restriction enzyme site. Next, 50 to 100 ng of each gel-purified PCR product was stitching-of-ends (SOE) joined (the DNA regions that overlapped for SOE are in boldface type) and PCR amplified by using A48R forward and A50R reverse primers. The resultant PCR product was restriction enzyme digested with BamHI and EcoRI and inserted into pUC13/gpt/EGFP that was digested with BamHI and EcoRI and shrimp alkaline phosphatase (SAP) modified. This plasmid is referred to as pΔA49MCS.
To create an MC159L-containing plasmid for the insertion of MC159L into the vaccinia virus genome, the MCV MC159L gene was PCR amplified from MC159/pCI by using forward primer 5′-CGGATCTATAATCATGTCCGACTCCAAGGAGG-3′ and reverse primer 5′-GAAAACTAGTTCAAGTCGTTTGCTCGGGG-3′ to yield a 726-bp PCR product. The MC159 nucleotides are underlined. The reverse primer engineered in the SpeI restriction enzyme digestion site is in italic type. The p7.5 promoter was PCR amplified from pUC13/gpt/EGFP. The forward primer is 5′-TTTTATCGATTAAATAATAAATACAATAATTAATTTCTCGT-3′, and the reverse primer is 5′-GGAGTCGGACATGATTATAGATCCGTCACTG-3′, which yielded a 121-bp PCR product. The forward primer introduced a ClaI restriction enzyme site (italic type). Next, 50 to 100 ng of each gel-purified PCR product was SOE joined and PCR amplified by using A48R forward and A50R reverse primers. The DNA regions that overlapped for SOE are in boldface type. The resultant PCR product of 847 bp was restriction enzyme digested with ClaI and SpeI and inserted into pΔA49MCS that was digested with ClaI and SpeI and SAP modified. This plasmid was called MC159/pΔA49MCS.
To create vMC159ΔA49R, BSC-1 cells were infected with vΔA49R and transfected with MC159/pΔA49R. Recombinant viruses were collected 24 h later and selected in the presence of mycophenolic acid, xanthine, and hypoxanthine (79). This process was repeated three times to isolate recombinant viruses away from parental viruses. Intermediate viruses expressing the Escherichia coli gpt gene product were resolved into vΔA49 or vMC159ΔA49 by growing viruses in BSC-1 cells in the absence of drugs. The genotype of each virus was confirmed by using PCR to amplify MC159L and the flanking A48R and A50R regions.
MC159 expression in vMC159ΔA49-infected cells was assessed by immunoblotting. RK13 cellular monolayers were infected with vΔA49, vΔA49rev, or vMC159vΔA49 (multiplicity of infection [MOI] of 10 PFU/cell). At 2 or 6 h p.i., cells were removed from the plate by scraping and collected by centrifugation (18,000 × g for 5 min). Cellular pellets were resuspended and lysed in 150 μl CE buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA [pH 8], 0.1 mM EGTA [pH 8], 0.05% Nonidet P-40) that was supplemented with Halt protease inhibitors (Fisher) for 30 min at 4°C. Cellular lysates were centrifuged at 18,000 × g for 5 min. Clarified supernatants were removed to new tubes, and 15 μg of protein from each supernatant was used for immunoblotting.
Luciferase reporter assay.
A dual-reporter luciferase assay was performed to quantify NF-κB activation according to previously reported protocols (14, 15, 68). Briefly, subconfluent cellular monolayers of HEK 293T cells in 12-well plates were cotransfected with 225 ng pNF-κBluc; 12.5 ng pRL-TK; and 750 ng of pcDNA3.1, pMC159, or pK13. Cells were additionally cotransfected with 250 ng, 500 ng, 750 ng, or 1,000 ng pcIAP1. All transfections were performed in triplicate. Cells were lysed in 1× passive lysis buffer (PLB; Promega), and lysates were analyzed for firefly and sea pansy luciferase activities. For each lysate, the ratio of firefly luciferase activity to sea pansy luciferase activity was calculated to correct for differences in transfection efficiencies. The data from triplicates were averaged. The resultant values were used to compare the expression of the firefly luciferase gene in stimulated cells to that in unstimulated, pcDNA3.1-transfected cells, whose value was set to 1. Values are shown as means ± standard deviations (SD). The Student t test was used to determine statistical significance. Statistically significant inhibition of luciferase activity is indicated by asterisks.
Lysates from luciferase assays were also analyzed for the presence of relevant proteins by immunoblotting. In this case, 20 μg of protein from each lysate was separated by using SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were probed with polyclonal rabbit anti-MC159 (1:1,000) (43), rabbit anti-FLAG (1:2,000) (catalog number F7425; Sigma-Aldrich), and goat anti-cIAP1 (1:400) (catalog number AF8181; R&D Systems) and then with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:10,000) (CalBiotech) or HRP-conjugated rabbit anti-goat IgG (1:5,000) (R&D Systems) to detect MC159, K13, or cIAP1 expression levels. Antibody-antigen reactions were detected by using chemiluminescence reagents (Thermo Scientific) and autoradiography.
Immunoblotting and antibodies.
For all immunoblots, the protein concentration of lysates was determined by using the 660-nm protein assay (Pierce). Twenty micrograms of clarified cellular lysates was subjected to SDS-PAGE, and proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked in 5% (wt/vol) milk or bovine serum albumin (BSA) in Tris-buffered saline and Tween 20 (TBST) (150 mM NaCl, 50 mM Tris base, 0.05% Tween 20) for at least 30 min at room temperature. Membranes were incubated with the indicated primary antibodies overnight at 4°C. Next, membranes were washed three times in large volumes of TBST and incubated with the appropriate HRP-conjugated secondary antibodies. Immunoblots were developed by using chemiluminescence reagents (Thermo Scientific) and autoradiography. Primary antibodies used in these experiments were polyclonal rabbit anti-MC159 (1:1,000) (43), anti-FLAG (1:2,000) (catalog numbers F3165 and F7425; Sigma-Aldrich), anti-HA (1:2,000) (catalog number 3724; Cell Signaling Technology), anti-Myc (1:2,000) (catalog numbers 2272 and 2276; Cell Signaling Technology), anti-NEMO (1:1,000) (catalog number sc8330; Santa Cruz Biotechnology), and anti-cIAP1 (1:400) (catalog number AF8181; R&D Systems). Monoclonal mouse anti-E3 (1:5,000) was a kind gift from Stuart Isaacs (University of Pennsylvania) (80). Secondary HRP-conjugated antibodies were obtained from either Thermo Scientific (goat anti-mouse IgG; 1:10,000), Calbiochem (goat anti-rabbit IgG;1:10,000), or R&D Systems (rabbit anti-goat IgG; 1:5,000).
Ubiquitination assay.
There were two approaches used to stimulate NEMO polyubiquitination. The first approach was to overexpress NEMO in sufficient amounts to ensure NF-κB activation. For the first approach, HEK 293T cells were seeded into 6-well plates at 5 × 105 cells/well and transfected with 500 ng pUb-Myc, 500 ng pNEMO-FLAG, and 1,000 ng pCI or pMC159. In some cases, plasmids encoding HA epitope-tagged wild-type (pWT-Ub-HA) or mutant (pK63-Ub-HA or pK48-Ub-HA) ubiquitin were used instead of pUb-Myc. The second approach provided the opportunity to examine TNF-induced NEMO polyubiquitination. In this case, HEK 293T cells were transfected with 250 ng pUb-Myc, 250 ng pNEMO-FLAG, and 1,000 ng pCI or pMC159. At 24 h posttransfection, cells were incubated with TNF (10 ng/ml; Roche) for 15 min.
To examine RIP1 polyubiquitination, cells were instead transfected with 500 ng pRIP1-HA and 500 ng pUb-Myc. At 24 h posttransfection, cells were incubated in medium lacking or containing TNF. To examine MC159 ubiquitination, cells were instead transfected with 500 ng pMC159 and 500 ng pUb-Myc. At 24 h posttransfection, cells were incubated in medium lacking or containing TNF (10 ng/ml) for 15 min. For reverse immunoprecipitation, cells were transfected with 250 ng pNEMO-HA; 250 ng pcIAP1-Myc; 250 ng either pWT-Ub-HA, pK48-Ub-HA, or pK63-Ub-HA; and 1,000 ng pCI or MC159.
In all cases, cells were removed from the plate by scraping and collected by centrifugation (18,000 × g for 5 min). Cellular pellets were resuspended in 150 μl Ub lysis buffer (50 mM Tris [pH 7.6], 250 mM NaCl, 1% Triton X-100, 0.5% Nonidet P-40, 3 mM EGTA, 3 mM EDTA, 10% glycerol, 0.1 mM Na3VO4, 10 μM N-ethylmaleimide) supplemented with a mixture of protease inhibitors (Halt; Fisher-Pierce) for 30 min at 4°C as previously described (33). Cellular lysates were centrifuged at 18,000 × g for 5 min. Clarified supernatants were collected. Fifty microliters of the supernatants was set aside and used to detect protein expression levels. The remaining 100 μl of clarified lysates was used for IPs to detect the polyubiquitination of the protein of interest. In this case, lysates were precleared for no more than 2 h with protein G-Sepharose beads (Invitrogen) at 4°C with rotation. Beads were collected by centrifugation. Clarified lysates were removed to new tubes and incubated with 1 μg mouse anti-FLAG (catalog number F3165; Sigma-Aldrich), anti-HA (catalog number H9658; Sigma-Aldrich), or anti-Myc (catalog number 2276; Cell Signaling Technology) antibodies or mouse IgG isotype control antibodies (catalog number I5381; Sigma-Aldrich) for 2 h at 4°C with constant rotation. After 2 h, 50 μl protein G-Sepharose beads was added to each sample, and the samples were incubated overnight with constant rotation at 4°C. Beads were collected by centrifugation (18,000 × g for 5 min) and washed three times with 750 μl Ub lysis buffer and once with Ub lysis buffer supplemented with 3 M urea.
Coimmunoprecipitation assays.
To detect NEMO-MC159 interactions, subconfluent HEK 293T or NEMO−/− MEF cellular monolayers were cotransfected with 500 ng pUb-Myc; 500 ng of either pNEMO-FLAG, p1-250-FLAG, or p251-419-FLAG; and 1,000 ng pCI or pMC159. At 24 h posttransfection, cells were incubated in medium lacking or containing TNF (10 ng/ml) for 15 min. Cells were lysed in 150 μl of IP lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 0.5% Nonidet P-40, 50 mM NaF, 0.1 mM Na3VO4, 1 mM dithiothreitol [DTT]) supplemented with a mixture of protease inhibitors (Halt; Fisher-Pierce) as previously described (33). Fifty microliters of the supernatants was set aside and used to detect protein expression levels. The remaining 100 μl of clarified lysates was used for IPs to detect protein-protein interactions. Clarified lysates were precleared for 1 to 2 h with protein G-Sepharose beads (Invitrogen) at 4°C with rotation. Next, clarified lysates were incubated with 1 μg anti-FLAG or IgG overnight at 4°C. Fifty microliters of protein G-Sepharose beads was added to each tube, and reaction mixtures were incubated for an additional 4 h at 4°C with constant rotation.
To detect endogenous NEMO-cIAP1 interactions, 10-cm2 dishes of subconfluent HeLa cell monolayers were transfected with 5,000 ng of pCI or pMC159-HA. At 24 h posttransfection, cells were incubated in medium containing TNF (10 ng/ml) for 10 min. Next, cells were dislodged from plates by scraping into 1 ml chilled PBS and collected by centrifugation (1,000 × g for 10 min). Cellular pellets were incubated in 150 μl IP lysis buffer for 30 min at 4°C. Cellular lysates were centrifuged (18,000 × g for 5 min). Fifty microliters of the supernatants was set aside and used to detect protein expression levels. The remaining 100 μl of clarified lysates was used for IPs to detect protein-protein interactions. For IPs, lysates were precleared for 2 h with protein G-Sepharose beads (Invitrogen) at 4°C with rotation. Beads were removed by centrifugation, and clarified lysates were incubated with 2 μg mouse monoclonal anti-cIAP1 (catalog number sc-271419; Santa Cruz) or IgG isotype control (catalog number I5381; Sigma-Aldrich) antibodies for 2 h at 4°C with constant rotation. After incubation with anti-cIAP1 Ab, 50 μl protein G-Sepharose beads was added, and supernatants were incubated for an additional 1 h with constant rotation at 4°C.
For all IPs, pelleted bead-protein complexes were suspended in 30 μl of 2× Laemmli buffer containing 5% 2-mercaptoethanol (2-ME) and boiled for 5 min. Immunoprecipitated samples or a portion of the remaining clarified lysates was electrophoretically separated by SDS-PAGE, and proteins were transferred onto PVDF membranes for IB.
ACKNOWLEDGMENTS
We thank Geoff Smith at Cambridge University for providing laboratory space to create the recombinant vaccinia viruses used here. We thank Chris Brooke, Brian Nichols, Ariana Bravo Cruz, Melissa Ryerson, and Lauren Gates for helpful discussion.
This work was supported by NIH grant AI117105 and the Department of Microbiology, University of Illinois, Urbana-Champaign.
REFERENCES
- 1.Damon IK. 2013. Poxviruses, p 2160–2184. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Reynolds MG, Holman RC, Yorita Christensen KL, Cheek JE, Damon IK. 2009. The incidence of molluscum contagiosum among American Indians and Alaska natives. PLoS One 4:e5255. doi: 10.1371/journal.pone.0005255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherwani S, Farleigh L, Agarwal N, Loveless S, Robertson N, Hadaschik E, Schnitzler P, Bugert JJ. 2014. Seroprevalence of molluscum contagiosum virus in German and UK populations. PLoS One 9:e88734. doi: 10.1371/journal.pone.0088734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, Abraham J, Ackerman I, Aggarwal R, Ahn SY, Ali MK, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Bahalim AN, Barker-Collo S, Barrero LH, Bartels DH, Basanez MG, Baxter A, Bell ML, Benjamin EJ, Bennett D, Bernabe E, Bhalla K, Bhandari B, Bikbov B, Bin Abdulhak A, Birbeck G, Black JA, Blencowe H, Blore JD, Blyth F, Bolliger I, Bonaventure A, Boufous S, Bourne R, Boussinesq M, Braithwaite T, Brayne C, Bridgett L, Brooker S, Brooks P, et al. 2012. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senkevich TG, Bugert JJ, Sisler JR, Koonin EV, Darai G, Moss B. 1996. Genome sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science 273:813–816. doi: 10.1126/science.273.5276.813. [DOI] [PubMed] [Google Scholar]
- 6.Shisler JL. 2015. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res 92:201–252. doi: 10.1016/bs.aivir.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Anstey AV, Bugert JJ. 2013. Molluscum contagiosum virus infection. Lancet Infect Dis 13:877–888. doi: 10.1016/S1473-3099(13)70109-9. [DOI] [PubMed] [Google Scholar]
- 8.Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, Moss B, Lenardo MJ, Tomaselli KJ, Cohen JI. 1997. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci U S A 94:1172–1176. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu S, Vincenz C, Buller M, Dixit VM. 1997. A novel family of viral death effector domain-containing molecules that inhibit both CD-95- and tumor necrosis factor receptor-1-induced apoptosis. J Biol Chem 272:9621–9624. doi: 10.1074/jbc.272.15.9621. [DOI] [PubMed] [Google Scholar]
- 10.Shisler JL, Senkevich TG, Berry MJ, Moss B. 1998. Ultraviolet-induced cell death blocked by a selenoprotein from a human dermatotropic poxvirus. Science 279:102–105. doi: 10.1126/science.279.5347.102. [DOI] [PubMed] [Google Scholar]
- 11.Coutu J, Ryerson MR, Bugert JJ, Nichols DB. 2017. The molluscum contagiosum virus protein MC163 localizes to the mitochondria and dampens mitochondrial mediated apoptotic responses. Virology 505:91–101. doi: 10.1016/j.virol.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 12.Brady G, Haas DA, Farrell PJ, Pichlmair A, Bowie AG. 2015. Poxvirus protein MC132 from molluscum contagiosum virus inhibits NF-κB activation by targeting p65 for degradation. J Virol 89:8406–8415. doi: 10.1128/JVI.00799-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols DB, Shisler JL. 2006. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha-induced NF-kappaB activation via inhibition of I kappa kinase complex formation. J Virol 80:578–586. doi: 10.1128/JVI.80.2.578-586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randall CM, Jokela JA, Shisler JL. 2012. The MC159 protein from the molluscum contagiosum poxvirus inhibits NF-kappaB activation by interacting with the IkappaB kinase complex. J Immunol 188:2371–2379. doi: 10.4049/jimmunol.1100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Randall CM, Biswas S, Selen CV, Shisler JL. 2014. Inhibition of interferon gene activation by death-effector domain-containing proteins from the molluscum contagiosum virus. Proc Natl Acad Sci U S A 111:E265–E272. doi: 10.1073/pnas.1314569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damon I, Murphy PM, Moss B. 1998. Broad spectrum chemokine antagonistic activity of a human poxvirus chemokine homolog. Proc Natl Acad Sci U S A 95:6403–6407. doi: 10.1073/pnas.95.11.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang Y, Moss B. 1999. IL-18 binding and inhibition of interferon gamma induction by human poxvirus-encoded proteins. Proc Natl Acad Sci U S A 96:11537–11542. doi: 10.1073/pnas.96.20.11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ku JK, Kwon HJ, Kim MY, Kang H, Song PI, Armstrong CA, Ansel JC, Kim HO, Park YM. 2008. Expression of Toll-like receptors in verruca and molluscum contagiosum. J Korean Med Sci 23:307–314. doi: 10.3346/jkms.2008.23.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brenner D, Blaser H, Mak TW. 2015. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15:362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 20.Clark K, Nanda S, Cohen P. 2013. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat Rev Mol Cell Biol 14:673–685. doi: 10.1038/nrm3644. [DOI] [PubMed] [Google Scholar]
- 21.Rothwarf DM, Zandi E, Natoli G, Karin M. 1998. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 22.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. 1998. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 93:1231–1240. doi: 10.1016/S0092-8674(00)81466-X. [DOI] [PubMed] [Google Scholar]
- 23.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. 2006. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 24.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. 2006. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation. Nat Cell Biol 8:398–406. doi: 10.1038/ncb1384 (Author Correction, 8:424, .) [DOI] [PubMed] [Google Scholar]
- 25.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H. 2009. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 26.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, Walczak H. 2011. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 27.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K. 2009. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol 11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 28.Emmerich CH, Bakshi S, Kelsall IR, Ortiz-Guerrero J, Shpiro N, Cohen P. 2016. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem Biophys Res Commun 474:452–461. doi: 10.1016/j.bbrc.2016.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadian K, Griesbach RA, Dornauer S, Wanger TM, Nagel D, Metlitzky M, Beisker W, Schmidt-Supprian M, Krappmann D. 2011. NF-kappaB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-kappaB activation. J Biol Chem 286:26107–26117. doi: 10.1074/jbc.M111.233163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komander D, Rape M. 2012. The ubiquitin code. Annu Rev Biochem 81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Kelly P, Shaffer AL III, Schmitz R, Yoo HM, Liu X, Huang DW, Webster D, Young RM, Nakagawa M, Ceribelli M, Wright GW, Yang Y, Zhao H, Yu X, Xu W, Chan WC, Jaffe ES, Gascoyne RD, Campo E, Rosenwald A, Ott G, Delabie J, Rimsza L, Staudt LM. 2016. Targeting non-proteolytic protein ubiquitination for the treatment of diffuse large B cell lymphoma. Cancer Cell 29:494–507. doi: 10.1016/j.ccell.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin HS, Lee DH, Kim DH, Chung JH, Lee SJ, Lee TH. 2009. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res 69:1782–1791. doi: 10.1158/0008-5472.CAN-08-2256. [DOI] [PubMed] [Google Scholar]
- 33.Tang ED, Wang CY, Xiong Y, Guan KL. 2003. A role for NF-kappaB essential modifier/IkappaB kinase-gamma (NEMO/IKKgamma) ubiquitination in the activation of the IkappaB kinase complex by tumor necrosis factor-alpha. J Biol Chem 278:37297–37305. doi: 10.1074/jbc.M303389200. [DOI] [PubMed] [Google Scholar]
- 34.Cordier F, Grubisha O, Traincard F, Veron M, Delepierre M, Agou F. 2009. The zinc finger of NEMO is a functional ubiquitin-binding domain. J Biol Chem 284:2902–2907. doi: 10.1074/jbc.M806655200. [DOI] [PubMed] [Google Scholar]
- 35.Huttmann J, Krause E, Schommartz T, Brune W. 2015. Functional comparison of molluscum contagiosum virus vFLIP MC159 with murine cytomegalovirus M36/vICA and M45/vIRA proteins. J Virol 90:2895–2905. doi: 10.1128/JVI.02729-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komander D. 2009. The emerging complexity of protein ubiquitination. Biochem Soc Trans 37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 37.Lo Y-C, Lin S-C, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, Eliezer D, Wu H. 2009. Structural basis for recognition of diubiquitins by NEMO. Mol Cell 33:602–615. doi: 10.1016/j.molcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Veron M, Agou F, Israel A. 2009. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J 28:2885–2895. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. 2003. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 40.Chaudhary PM, Jasmin A, Eby MT, Hood L. 1999. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18:5738–5746. doi: 10.1038/sj.onc.1202976. [DOI] [PubMed] [Google Scholar]
- 41.Grimm S, Stanger BZ, Leder P. 1996. RIP and FADD: two “death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc Natl Acad Sci U S A 93:10923–10927. doi: 10.1073/pnas.93.20.10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee TH, Shank J, Cusson N, Kelliher MA. 2004. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem 279:33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- 43.Shisler JL, Moss B. 2001. Molluscum contagiosum virus inhibitors of apoptosis: the MC159 v-FLIP protein blocks Fas-induced activation of procaspases and degradation of the related MC160 protein. Virology 282:14–25. doi: 10.1006/viro.2001.0834. [DOI] [PubMed] [Google Scholar]
- 44.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG. 2008. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A 105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertrand MJ, Lippens S, Staes A, Gilbert B, Roelandt R, De Medts J, Gevaert K, Declercq W, Vandenabeele P. 2011. cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1-4). PLoS One 6:e22356. doi: 10.1371/journal.pone.0022356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu JW, Shi Y. 2008. FLIP and the death effector domain family. Oncogene 27:6216–6227. doi: 10.1038/onc.2008.299. [DOI] [PubMed] [Google Scholar]
- 47.Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. 2003. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci 116:3721–3728. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- 48.Matta H, Sun Q, Moses G, Chaudhary PM. 2003. Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-kappaB activation. J Biol Chem 278:52406–52411. doi: 10.1074/jbc.M307308200. [DOI] [PubMed] [Google Scholar]
- 49.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, Ren H, Strnadova P, Bowie AG, Smith GL. 2013. Poxvirus targeting of E3 ligase beta-TrCP by molecular mimicry: a mechanism to inhibit NF-kappaB activation and promote immune evasion and virulence. PLoS Pathog 9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, Cohen P. 2013. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A 110:15247–15252. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H, Kobayashi M, Blonska M, You Y, Lin X. 2006. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem 281:13636–13643. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 52.Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael-Tomasevic A, Phu L, Helgason E, Fairbrother WJ, Deshayes K, Kirkpatrick DS, Vucic D. 2010. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J 29:4198–4209. doi: 10.1038/emboj.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu M, Skaug B, Zeng W, Chen ZJ. 2009. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol Cell 36:302–314. doi: 10.1016/j.molcel.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ting AT, Bertrand MJ. 2016. More to life than NF-kappaB in TNFR1 signaling. Trends Immunol 37:535–545. doi: 10.1016/j.it.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. 2009. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. 2009. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 57.Micheau O, Tschopp J. 2003. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114:181–190. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- 58.Wang L, Du F, Wang X. 2008. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 59.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. 2011. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. 1998. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 62.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. 2009. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 63.Thurau M, Everett H, Tapernoux M, Tschopp J, Thome M. 2006. The TRAF3-binding site of human molluscipox virus FLIP molecule MC159 is critical for its capacity to inhibit Fas-induced apoptosis. Cell Death Differ 13:1577–1585. doi: 10.1038/sj.cdd.4401847. [DOI] [PubMed] [Google Scholar]
- 64.Hayden MS, Ghosh S. 2012. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. 2008. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 66.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 67.Nichols DB, Shisler JL. 2009. Poxvirus MC160 protein utilizes multiple mechanisms to inhibit NF-kappaB activation mediated via components of the tumor necrosis factor receptor 1 signal transduction pathway. J Virol 83:3162–3174. doi: 10.1128/JVI.02009-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murao LE, Shisler JL. 2005. The MCV MC159 protein inhibits late, but not early, events of TNF-alpha-induced NF-kappaB activation. Virology 340:255–264. doi: 10.1016/j.virol.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 69.Challa S, Woelfel M, Guildford M, Moquin D, Chan FK. 2010. Viral cell death inhibitor MC159 enhances innate immunity against vaccinia virus infection. J Virol 84:10467–10476. doi: 10.1128/JVI.00983-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krause E, de Graaf M, Fliss PM, Dolken L, Brune W. 2014. Murine cytomegalovirus virion-associated protein M45 mediates rapid NF-kappaB activation after infection. J Virol 88:9963–9975. doi: 10.1128/JVI.00684-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fliss PM, Jowers TP, Brinkmann MM, Holstermann B, Mack C, Dickinson P, Hohenberg H, Ghazal P, Brune W. 2012. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog 8:e1002517. doi: 10.1371/journal.ppat.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Griffiths DA, Abdul-Sada H, Knight LM, Jackson BR, Richards K, Prescott EL, Peach AH, Blair GE, Macdonald A, Whitehouse A. 2013. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J Virol 87:13853–13867. doi: 10.1128/JVI.02159-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen RA, Ryzhakov G, Cooray S, Randow F, Smith GL. 2008. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog 4:e22. doi: 10.1371/journal.ppat.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li FY, Jeffrey PD, Yu JW, Shi Y. 2006. Crystal structure of a viral FLIP: insights into FLIP-mediated inhibition of death receptor signaling. J Biol Chem 281:2960–2968. doi: 10.1074/jbc.M511074200. [DOI] [PubMed] [Google Scholar]
- 75.Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ, Wu H. 2005. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell 20:939–949. doi: 10.1016/j.molcel.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bagneris C, Ageichik AV, Cronin N, Wallace B, Collins M, Boshoff C, Waksman G, Barrett T. 2008. Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol Cell 30:620–631. doi: 10.1016/j.molcel.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 77.Bagneris C, Rogala KB, Baratchian M, Zamfir V, Kunze MB, Dagless S, Pirker KF, Collins MK, Hall BA, Barrett TE, Kay CW. 2015. Probing the solution structure of IkappaB kinase (IKK) subunit gamma and its interaction with Kaposi sarcoma-associated herpes virus Flice-interacting protein and IKK subunit beta by EPR spectroscopy. J Biol Chem 290:16539–16549. doi: 10.1074/jbc.M114.622928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, Shimotohno K. 2010. Polyubiquitin conjugation to NEMO by triparite [sic] motif protein 23 (TRIM23) is critical in antiviral defense. Proc Natl Acad Sci U S A 107:15856–15861. doi: 10.1073/pnas.1004621107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Falkner FG, Moss B. 1990. Transient dominant selection of recombinant vaccinia viruses. J Virol 64:3108–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weaver JR, Shamim M, Alexander E, Davies DH, Felgner PL, Isaacs SN. 2007. The identification and characterization of a monoclonal antibody to the vaccinia virus E3 protein. Virus Res 130:269–274. doi: 10.1016/j.virusres.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]