

ABSTRACT
Nipah virus (NiV) is a zoonotic emerging paramyxovirus that can cause fatal respiratory illness or encephalitis in humans. Despite many efforts, the molecular mechanisms of NiV-induced acute lung injury (ALI) remain unclear. We previously showed that NiV replicates to high titers in human lung grafts in NOD-SCID/γ mice, resulting in a robust inflammatory response. Interestingly, these mice can undergo human immune system reconstitution by the bone marrow, liver, and thymus (BLT) reconstitution method, in addition to lung tissue engraftment, giving altogether a realistic model to study human respiratory viral infections. Here, we characterized NiV Bangladesh strain (NiV-B) infection of human lung grafts from human immune system-reconstituted mice in order to identify the overall effect of immune cells on NiV pathogenesis of the lung. We show that NiV-B replicated to high titers in human lung grafts and caused similar cytopathic effects irrespective of the presence of human leukocytes in mice. However, the human immune system interfered with virus spread across lung grafts, responded to infection by leukocyte migration to small airways and alveoli of the lung grafts, and accelerated oxidative stress in lung grafts. In addition, the presence of human leukocytes increased the expression of cytokines and chemokines that regulate inflammatory influx to sites of infection and tissue damage. These results advance our understanding of how the immune system limits NiV dissemination and contributes to ALI and inform efforts to identify therapeutic targets.
IMPORTANCE Nipah virus (NiV) is an emerging paramyxovirus that can cause a lethal respiratory and neurological disease in humans. Only limited data are available on NiV pathogenesis in the human lung, and the relative contribution of the innate immune response and NiV to acute lung injury (ALI) is still unknown. Using human lung grafts in a human immune system-reconstituted mouse model, we showed that the NiV Bangladesh strain induced cytopathic lesions in lung grafts similar to those described in patients irrespective of the donor origin or the presence of leukocytes. However, the human immune system interfered with virus spread, responded to infection by leukocyte infiltration in the small airways and alveolar area, induced oxidative stress, and triggered the production of cytokines and chemokines that regulate inflammatory influx by leukocytes in response to infection. Understanding how leukocytes interact with NiV and cause ALI in human lung xenografts is crucial for identifying therapeutic targets.
KEYWORDS: henipavirus, human lung grafts, virus replication, humanized mouse, inflammatory response, oxidative stress, antioxidant therapies
INTRODUCTION
Nipah virus (NiV) is a recently emerged zoonotic member of the family Paramyxoviridae (genus Henipavirus) that can cause both severe respiratory disease and encephalitis in humans (1, 2). NiV was discovered during an outbreak of acute encephalitis in 1998 to 1999 in Malaysia and Singapore (NiV strain Malaysia [NiV-M]) (3–5) and then reemerged on an almost yearly basis in Southeast Asia, primarily in Bangladesh (NiV strain Bangladesh [NiV-B]), where the case fatality rate (CFR) can reach up to 100%, depending on the size of the outbreak (6–8). Interestingly, the CFR and frequency of respiratory disease were higher during these outbreaks than during the initial outbreak in Malaysia and Singapore (8), ∼70% and 30%, respectively, and might originate from intrinsic differences between the two prototype virus strains, as previously suggested (9–13).
Lung examination of confirmed NiV-M cases revealed lesions, hemorrhages, and inflammation in the small airways (14). These features were also found in NiV-M-infected human lung xenografts (15) and in lungs of several animal models, such as Syrian hamsters, ferrets, and African Green monkeys (10, 13, 16–18), demonstrating the suitability of these models for the study of NiV pathogenesis. Previously, we demonstrated in vitro that NiV-B infection of primary human small airway epithelial cells caused a strong inflammatory response, in part induced by oxidative stress (19), that was accompanied by cell damage and chemotaxis of mononuclear cells (12, 20). We also showed that the tracheal bronchial epithelium could contribute to virus spread (12). However, there are still limited data on NiV pathogenesis in the human lung, including the respective contributions of the respiratory epithelium, vascular endothelium, and migrated leukocytes to lung inflammation and acute lung injury (ALI).
Humanized mouse models have been extensively used to study human diseases and virus pathogenesis (21–28). These models reconstitute the microenvironment of the human lung to evaluate the host response during virus infection in the human lung. First, our group recently developed a novel xenograft model of the human respiratory tract, allowing us to study in vivo NiV pathogenesis in human lung grafts lacking human immune cells (15). Here, we build on this model and characterized NiV-B pathogenesis in human lung grafts in the presence of the human immune system in mice using the bone marrow, liver, and thymus (BLT) reconstitution method (23), which allows peripheral and resident immune cells to respond to infection of lung grafts, as in NiV-infected patients. Specifically, we investigated whether human leukocytes impact virus replication and their role in oxidative stress, inflammation, and pathology of the lung. Our data show that NiV-B replicated efficiently to high titers in lung grafts and caused similar cytopathic effects regardless of the presence of human leukocytes in mice through reconstitution. However, the presence of the human immune system impacted infection of the lung grafts, as it interfered with virus dissemination, accelerated oxidative stress, as well as resulted in a high level of leukocyte migration, with a complex inflammatory response early in infection.
RESULTS
Generation of humanized mice.
Human lung grafts from 2 tissue donors were surgically implanted into 44 mice as previously described (nonreconstituted mice) (15) (Fig. 1, right), and reconstitution of the human immune system in half of them was established by using the BLT reconstitution method (23, 26) with the corresponding tissues from donor 1 or 2 (reconstituted mice) (Fig. 1, left). Groups of reconstituted and nonreconstituted mice with tissues from donor 1 or 2 were euthanized at either day 3 or 5 postinfection (p.i.) (Table 1). The levels of reconstitution, prior to challenge, in mice with tissues from donor 1 that were scheduled to be euthanized on days 3 and 5 averaged 30.2% and 24.0%, respectively (Table 1). Similarly, the reconstitution levels, prior to challenge, in mice with tissues from donor 2 that were scheduled to be euthanized on days 3 and 5 averaged 40.2% and 34.1%, respectively (Table 1). As previously shown (15), the lung grafts implanted on the 44 mice displayed similar structures of the lower respiratory tract and some of the typical differentiated cells, including ciliated and alveolar-like cells, as in the normal human lung.
FIG 1.
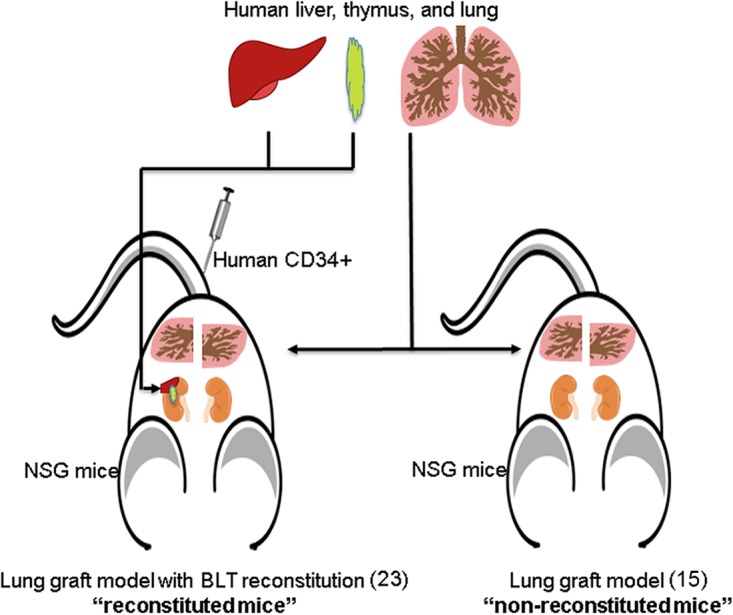
Generation of humanized mice. (Left) Human lung graft model in NSG mice with the human immune system using the BLT reconstitution method, as previously described (23). These mice are referred as reconstituted mice in this study. (Right) Human lung graft model in NSG mice, as previously described (15), that did not undergo human immune system reconstitution. These mice were named nonreconstituted mice in this study. Tissues were obtained from 2 donors.
TABLE 1.
Experimental design and distribution of mice based on their human immune system reconstitution status and human tissue donor origin
| Donor | Group description | Day of euthanasia | Inoculum | Avg reconstitution (%) (range)a |
|---|---|---|---|---|
| 1 | Reconstituted | 3 | Virus | 30.2 (10.1–56.6) |
| 1 | Nonreconstituted | 3 | Virus | NA |
| 1 | Nonreconstituted | 3 | Mock | NA |
| 2 | Reconstituted | 3 | Virus | 36.0 (16.1–68.7) |
| 2 | Reconstituted | 3 | Mock | 44.3 (10.5–78.1) |
| 2 | Nonreconstituted | 3 | Virus | NA |
| 2 | Nonreconstituted | 3 | Mock | NA |
| 1 | Reconstituted | 5 | Virus | 23.6 (11.2–40.7) |
| 1 | Reconstituted | 5 | Mock | 24.4 (10.4–50.2) |
| 1 | Nonreconstituted | 5 | Virus | NA |
| 1 | Nonreconstituted | 5 | Mock | NA |
| 2 | Reconstituted | 5 | Virus | 35.6 (23.0–50.3) |
| 2 | Reconstituted | 5 | Mock | 32.6 (17.6–52.3) |
| 2 | Nonreconstituted | 5 | Virus | NA |
| 2 | Nonreconstituted | 5 | Mock | NA |
Percentage of human versus mouse CD45+ cells, with sorting from 100 μl mouse peripheral blood. NA, nonapplicable.
Nipah virus Bangladesh replicates efficiently in human lung grafts.
At 10 weeks postengraftment, human lung tissues were infected with NiV-B or mock infected by the direct injection of virus or phosphate-buffered saline (PBS) into the engrafted tissues. Animals were then monitored for the development of clinical signs of disease and body weight. None of the 24 mice with NiV-B-infected lung grafts had significant weight loss and did not exhibit any clinical signs throughout the experiment. Data obtained from lung grafts from donors 1 and 2 were pooled, as no significant differences in any of the parameters analyzed were observed throughout the study.
Assessment of NiV-B titers in lung grafts was done at days 3 and 5 p.i., based on results from our previous study (15) that validated this endpoint as being optimum. NiV-B titers increased by 2 log10 units over the course of infection in lung grafts regardless of the reconstitution status of the mice (Fig. 2a). Specifically, the virus titer reached 106.1 50% tissue culture infective doses (TCID50)/g tissue by day 3, the time point at which the peak titer was reached in our previous study (15), and remained as high as 107.0 TCID50/g tissue at day 5 p.i. The tropism and spread of virus in the human lung were then assessed by the expression of NiV-B nucleoprotein. Interestingly, the surface area and signal intensity of the NiV-B nucleoprotein antigen expressed in lung grafts from reconstituted mice were lower than those in lung grafts from nonreconstituted mice throughout infection (Fig. 2bto4). Virus antigen was also typically restricted to focal sites in the presence of large leukocytic infiltrates in lung grafts from reconstituted mice (Fig. 4a, b, e, and f), especially at day 3 p.i., which, taken together, suggests that leukocytes interfered with NiV dissemination.
FIG 2.
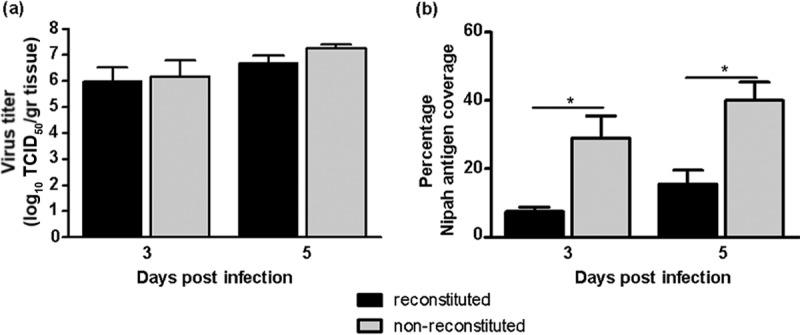
NiV-B replicates in human lung grafts from reconstituted and nonreconstituted mice. (a) Kinetics of NiV-B replication in lung grafts of donors 1 and 2 from reconstituted and nonreconstituted mice determined at days 3 and 5 postinfection. Results are expressed as averages of data from 3 repetitions per graft with standard deviations. (b) Percent coverage of NiV-B nucleoprotein antigen per section of lung graft, with 4 sections being observed per graft. *, P < 0.05 by a nonparametric Mann-Whitney test.
FIG 3.

Imaging of NiV-B replication in human lung grafts from nonreconstituted mice. Shown is H&E (left) and NiV-B nucleoprotein antigen (brown signal) (right) staining of human lung graft sections from nonreconstituted mice at day 3 (a and e) and day 5 (b and f) postinfection. Similar staining was performed on mock-infected lung graft sections from nonreconstituted mice at day 3 (c and g) and day 5 (d and h) postinfection. Images are representative of virus spread across lung grafts from nonreconstituted mice (a, b, e, and f). Original magnification, ×5. Bars, 200 μm.
FIG 4.
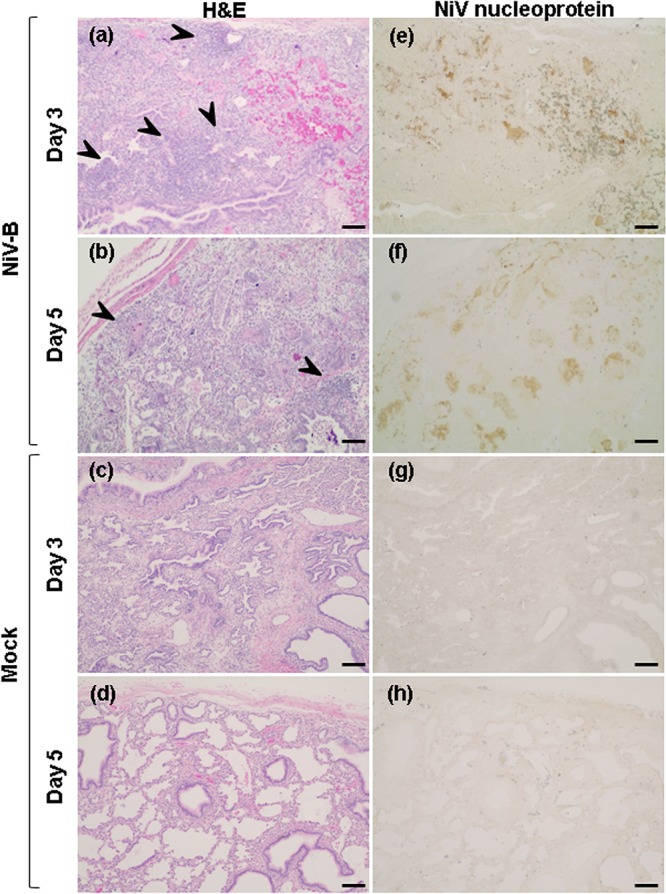
Imaging of NiV-B replication in human lung grafts from reconstituted mice. Shown is H&E (left) and NiV-B nucleoprotein antigen (brown signal) (right) staining of human lung grafts sections from reconstituted mice at day 3 (a and e) and day 5 (b and f) postinfection. Similar staining was performed on mock-infected lung graft sections from reconstituted mice at day 3 (c and g) and day 5 (d and h) postinfection. Images are representative of virus spread across lung grafts from reconstituted mice (a, b, e, and f). Large immune cell infiltrations are indicated by arrowheads. Original magnification, ×5. Bars, 200 μm.
Infection with Nipah virus Bangladesh causes lesions in human lung grafts.
The formation of syncytia is a hallmark of NiV infection, and indeed, similar NiV-B-induced syncytium formation was observed predominantly in the epithelium of the small airways and alveolar areas of all infected lung grafts at day 3 p.i. (Fig. 5a to d) and day 5 p.i. The syncytia were overall larger at day 5 than at day 3 p.i. and were often accompanied by a large amount of necrotic cellular debris or tissue necrosis (data not shown), as previously reported (15). In addition, there was a marked increase in the amount of mononuclear cells such as macrophages in the alveolar space of NiV-B-infected lung grafts from reconstituted mice compared to those from nonreconstituted mice at day 3 p.i. (Fig. 5a to c) or day 5 p.i. (data not shown). The presence of a few immune cells in lung grafts from nonreconstituted mice could originate from progenitor cells circulating in the human lung at the time of engraftment. Two specific observations for NiV-B-infected lung grafts from reconstituted mice were an abundance of myeloperoxidase (MPO)-positive neutrophils in the lumen of the small airways and alveolar area near syncytia (Fig. 5e and 6) and, along with an influx of CD8+ T cells, in large leukocytic perivascular infiltrations around these infected areas, as shown at day 3 p.i. (Fig. 7). Mock-infected lung grafts also had a detectable basal population of CD8+ T cells, while similar numbers of CD8+ T cells were observed in the peripheral blood of reconstituted mice with mock- and NiV-B-infected lung grafts (data not shown). These results suggest the potential for immune contributions by tissue-resident, as well as recruited, CD8+ T cell populations. Potentially, tissue-resident CD8+ T cells could respond during earlier stages of infection independent of antigen specificity. Bystander activation is likely to occur following exposure to cytokines (e.g., interleukin-15 [IL-15]) produced in the lung grafts as part of the antiviral innate immune response seen at days 3 and 5 p.i. The increase in the number CD8+ T cells above the basal level is expected here to reflect the recruitment of antigen-specific cells from the periphery following antigen presentation and activation in lymphoid tissues. Altogether, these observations suggest that macrophages, neutrophils, and CD8+ T cells are leukocyte subpopulations recruited to the site of infection.
FIG 5.

NiV-B-induced histopathological changes in respiratory epithelium and alveoli of human lung grafts. Shown is H&E staining of NiV-B-infected (a, c, and e) and mock-infected (b and d) lung graft sections from nonreconstituted (a and b) or reconstituted (c to e) mice at day 3 postinfection. Images are representative of virus-induced lesions regardless of the donor. Syncytium formation (arrows), the presence of macrophages (asterisks), and large leukocyte infiltrations (arrowheads) were observed in small airways/alveoli. Neutrophils (circles) were detected in the lumen of airways. A, alveolar area; V, vasculature. Original magnifications, ×10 (a to d) and ×40 (e). Bars, 80 μm (a to d) and 10 μm (e).
FIG 6.
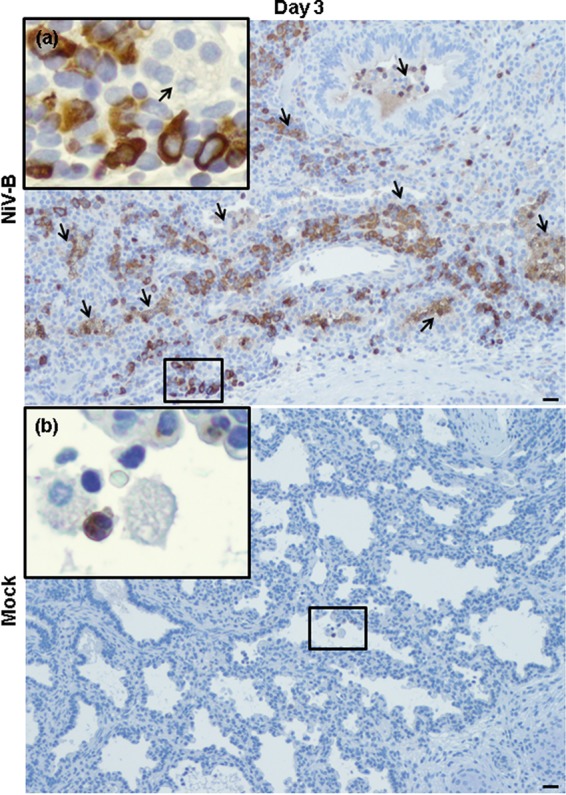
NiV-B-induced human neutrophil migration to human lung interstitium from reconstituted mice. Shown is myeloperoxidase (brown signal) in NiV-B-infected (a) and mock-infected (b) human lung graft sections from reconstituted mice at day 3 postinfection. Neutrophils (a and b) in lumen of the small airways/alveolar area and in large perivascular infiltrations (a) are shown. Syncytium formation is indicated by arrows. This was observed regardless of the donor. Original magnifications, ×10. Original magnifications of windows, ×40. Bars, 100 μm.
FIG 7.
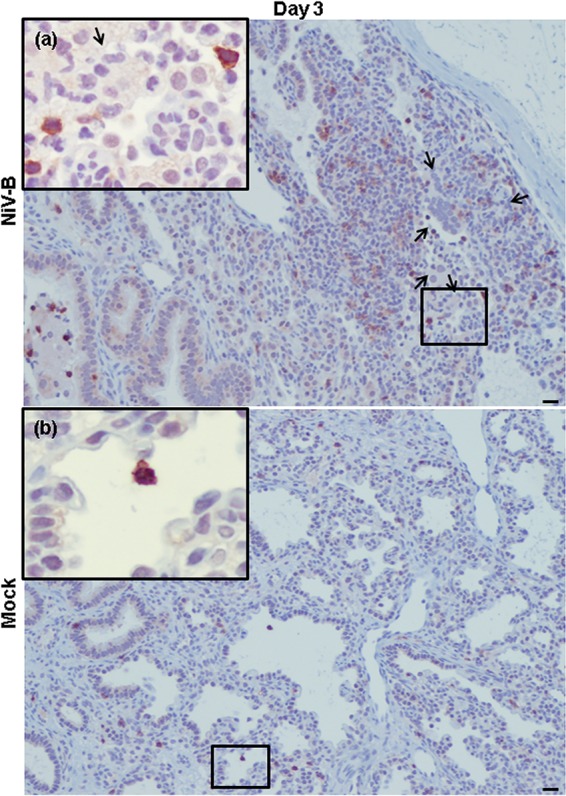
NiV-B-induced human T CD8+ cell migration to human lung interstitium from reconstituted mice. Shown are T CD8+ cells (red signal) in NiV-B-infected (a) and mock-infected (b) human lung graft sections from reconstituted mice at day 3 postinfection. T CD8+ cells (a and b) are located in the lumen of the small airways/alveolar area and in large perivascular infiltrations (a). Syncytium formation is indicated by arrows. This was observed regardless of the donor. Original magnifications, ×10. Original magnifications of windows, ×40. Bars, 100 μm.
Next, the level of human pulmonary surfactant-associated protein D (SP-D) in mouse serum was quantified in order to determine whether infection of lung grafts causes lung function impairment via damage of lung parenchyma. The SP-D level was equally increased, by as early as day 3 p.i., in sera of reconstituted (P < 0.01) and nonreconstituted (P < 0.05) mice that had NiV-B-infected lung grafts compared to the levels in sera of mice with mock-infected lung grafts (Fig. 8). This was also true at day 5 p.i., suggesting that NiV-B damages alveoli, which in turn increases alveolar-capillary barrier permeability and causes hemorrhages.
FIG 8.
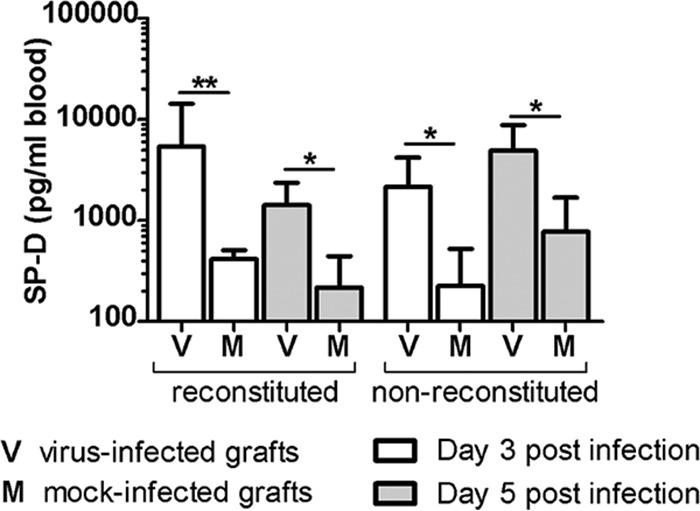
NiV-B increases alveolar-capillary permeability of human lung grafts. The level of human pulmonary SP-D in sera of reconstituted and nonreconstituted mice was measured at days 3 and 5 postinfection. *, P < 0.05; **, P < 0.01 (by a nonparametric Mann-Whitney test).
Fibrinoid necrosis and syncytia in the lining of the human lung graft vasculature were sometimes observed and accompanied by edema at day 3 p.i. (Fig. 9a and b) and day 5 p.i. (data not shown), regardless of the reconstitution status of the mice. High frequencies of fibrin deposition and fibrin thrombi were also found in large and small arterioles (Fig. 9c and d) and, to a lesser extent, in alveolar capillaries (Fig. 9e and f).
FIG 9.
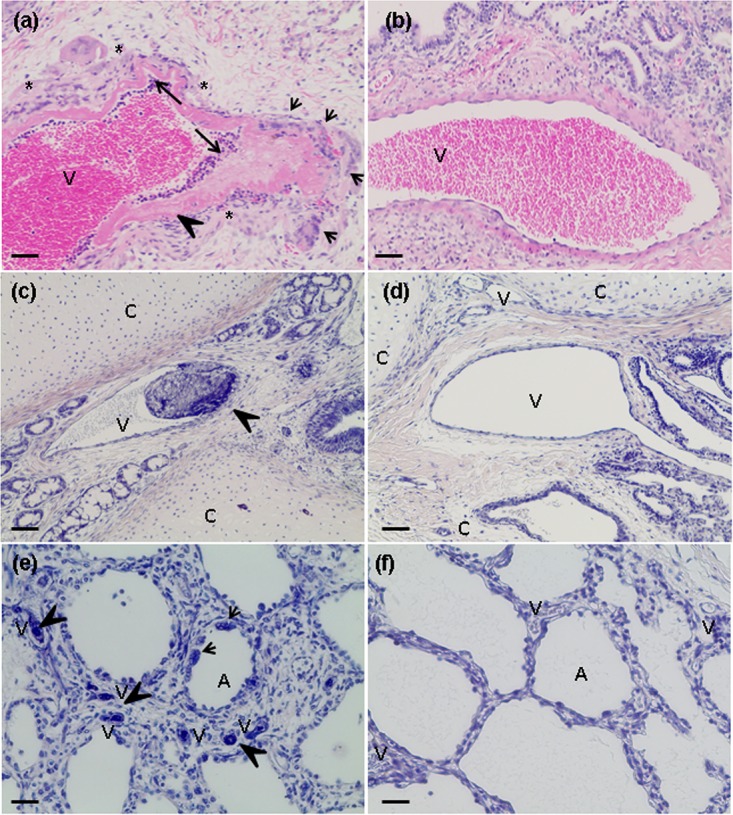
NiV-B-induced histopathological changes in the vasculature of human lung grafts. (a, c, and e) H&E (a) and PTAH (c and e) staining of NiV-B-infected lung graft sections from reconstituted mice showing fibrin accumulation/thrombus formation (arrowhead), fibrinoid necrosis (long arrow), syncytium formation (short arrow), and edema formation (asterisk), which were observed in multisized vessels. These lesions were noticed irrespective of the donor and reconstitution status of the mice. (b, d, and f) H&E (b) and PTAH (d and f) staining of mock-infected lung grafts from reconstituted mice. A, alveolar area; V, vessel; C, cartilage. Magnifications, ×10 (a to d) and ×20 (e and f). Bars, 100 μm (a to d) and 50 μm (e and f).
Nipah virus Bangladesh infection induces oxidative stress in human lung grafts.
NiV-B-induced oxidative stress was previously shown to play a role in inflammation in infected human respiratory epithelium (19), suggesting that oxidative stress contributes to NiV-induced ALI. Here, the formation of malondialdehyde (MDA)-protein adducts was quantified in lung grafts and used as a marker of oxidative stress. While the highest levels of MDA detected in NiV-B-infected lung grafts did not always correlate with the highest reconstitution level in mice (data not shown), the level of MDA adducts was higher in NiV-B- than in mock-infected lung grafts from both reconstituted mice at day 3 p.i. (P < 0.01) and day 5 p.i. (P < 0.05) and nonreconstituted mice at day 5 p.i. (P < 0.05) (Fig. 10a). The MDA concentration was also higher in NiV-B-infected lung grafts from reconstituted mice than in those from nonreconstituted mice at day 3 p.i. (P < 0.05). This suggests altogether that the NiV-induced recruitment of circulating leukocytes causes an early induction of oxidative stress in lung grafts that otherwise occurs at a later stage of infection in the absence of the human immune system. In an attempt to determine whether NiV-B-induced oxidative stress in the lung grafts resulted from impaired antioxidant cellular defenses, the antioxidant enzyme activities of catalase (CAT), glutathione peroxidase (GPx), glutathione S-transferase (GST), and total superoxide dismutase (SOD) were assessed in lung homogenates (Fig. 10b to e). CAT, GPx, and GST activities remained unaffected by NiV-B infection at any time point. However, total SOD activity was reduced at day 5 p.i. in NiV-B- compared to mock-infected lung grafts from both reconstituted (P < 0.05) and nonreconstituted (P < 0.01) mice, suggesting that a lack of antioxidant enzymes contributes to oxidative stress only at a late stage of infection.
FIG 10.

NiV-B induces oxidative stress in human lung grafts. Shown is quantification of MDA-protein adducts as a marker of oxidative stress (a) as well as measurement of CAT, GPx, GST, and total SOD antioxidant enzyme activities (b to e) in lung grafts from reconstituted and nonreconstituted mice throughout infection. *, P < 0.05; **, P < 0.01 (by a nonparametric Mann-Whitney test).
Nipah virus Bangladesh induces a differential inflammatory response in human lung grafts depending on the reconstitution status of mice.
A multiplex approach was then used to investigate the role of the human innate response in lung graft injury during NiV-B infection and to differentiate inflammatory mediators secreted by the lung tissue from those resulting from the effect of leukocyte recruitment to the lung tissue. Out of a total of 31 cytokines and chemokines analyzed, the levels of the following 14 were significantly increased by NiV-B infection: granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-6, IL-8, RANTES, vascular endothelial growth factor (VEGF), eotaxin, alpha 2 interferon (IFN-α2), IL-1 receptor A (IL-1RA), IL-15, macrophage inflammatory protein 1α (MIP-1α), MIP-1β, IFN-γ-inducible protein 10 (IP-10 or CXCL10), and IL-10 (Fig. 11). The secretion levels of leukocyte chemoattractants, including G-CSF, GM-CSF, IL-6, IL-8, RANTES, as well as VEGF, were similar in NiV-B-infected lung grafts irrespective of the reconstitution status of the mice at day 3 or 5 p.i., suggesting that the secretion of these inflammatory mediators stems mostly from the lung tissue in response to infection. However, significant increases in eotaxin, IFN-α2, IL-1RA, IL-15, MIP-1α, and MIP-1β secretion levels were detected earlier (day 3 p.i.) in NiV-B-infected lung grafts from reconstituted mice, whereas their secretion levels were no longer significantly different from those in NiV-B-infected lung grafts from nonreconstituted mice by day 5 p.i. Therefore, this suggests that the earlier secretion of the same mediators in lung grafts from reconstituted mice originates in part from the response of the lung tissue to infection following its interaction with activated circulating, resident, or recruited immune cells. IP-10 levels were also higher in NiV-B-infected lung grafts from reconstituted mice than in those from nonreconstituted mice at days 3 and 5 p.i. (P < 0.01), indicating here a combined response of the lung tissue and multiple activated leukocyte subpopulations, including neutrophils, macrophages, and CD8+ T cells. Finally, a specific observation for the NiV-induced inflammatory response was that the level of the regulatory cytokine IL-10, which often dampens excessive inflammatory responses, was increased only in infected lung grafts from reconstituted mice throughout infection (P < 0.01). This result suggests that the presence of leukocytes, and notably of T cells and possibly monocyte/macrophage populations, in NiV-infected lungs also mitigates the immune response.
FIG 11.

NiV-B-induced inflammatory response in human lung grafts. Shown are cytokine/chemokine levels in NiV-B- and mock-infected lung grafts from reconstituted and nonreconstituted mice at both days 3 and 5 postinfection. Results are expressed as averages of data from triplicates, and bars represent standard deviations. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (by a nonparametric Mann-Whitney test).
DISCUSSION
Nipah virus is a recently emerged bat-borne paramyxovirus that can induce severe and often lethal respiratory disease and encephalitis in humans. The mechanisms involved in NiV-induced ALI remain unclear, and only a limited number of experimental studies using in vitro (11, 12, 19, 29) and in vivo (15) models of infection of the human airway have addressed this question. We previously showed that a human lung xenograft model supports high levels of NiV replication and can be used for studying viral pathogenesis (15). Here, we build on this model and characterize NiV-B pathogenesis in human lung xenografts in the presence of the human immune system using the BLT reconstitution method in mice (23). Notably, we focused on whether leukocytes impact virus replication, oxidative stress, inflammation, and pathology of the lung. NiV-B was used in this study, as it is the most recent circulating strain responsible for sporadic outbreaks in Southeast Asia, with evidence of human-to-human transmission and a higher incidence of respiratory disease than for the original outbreak in Malaysia and Singapore in 1998 to 1999 (6–8, 30).
BLT-humanized mice, or reconstituted mice, exhibit a nearly complete human immune system, and their levels of reconstitution in the present study were similar to those reported in previous studies (23, 26). All human lung grafts also resembled human lungs, as demonstrated in our previous study (15), therefore validating that lung grafts from reconstituted and nonreconstituted (no BLT reconstitution) mice are relevant models to study NiV pathogenesis in the human lung. However, circulating human leukocytes from reconstituted mice migrated to NiV-B-infected lung grafts and slowed virus dissemination without affecting the virus titer. However, it is possible that NiV-B in lung grafts from reconstituted mice might be less virulent than its counterpart in lung grafts from nonreconstituted mice because of a difference in the amount of a particular viral protein, as shown previously for rabies virus-infected mice (31). The biological function and origin of the differential expression of nucleoprotein in lung grafts from reconstituted compared to nonreconstituted mice will require further investigations.
While alveolar macrophages were previously reported to be infected in a hamster model of NiV infection (32), both neutrophils and CD8+ T cells contributed to limiting NiV-B dissemination and facilitating virus clearance in our model. This was previously shown for the former with influenza virus (33–35) and for the latter with pneumonia virus of mice and measles virus, 2 other paramyxoviruses (36, 37). The perivascular infiltrations of CD8+ T cells, although detected early compared to the typical immune response pattern during acute infection in humans (38), likely result from the selection and differentiation of naive lymphocytes into effector T cells specific for NiV-B antigens. This response could supplement the immediate activation of resident CD8+ T cells with heterologous specificity due to the secretion of IL-15 by surrounding infected cells (38–41), as shown at day 3 in NiV-B-infected lung grafts from reconstituted mice only. The relative contribution of CD8+ T cells, neutrophils, or both compared to those of other immune cells in NiV-B pathogenesis and virus clearance in the human lung will require additional studies. NiV-B-induced multinucleated cells were found in the bronchiolar interstitium, as previously shown for human lung grafts (11, 15). The 10-fold increase in the level of human pulmonary SP-D in sera of mice also suggested increases in alveolar-capillary permeability and alveolar damage of the human lung grafts and was previously considered, when detected in the human bloodstream, a hallmark of ALI (42, 43). In addition, NiV-B infected the endothelial lining of the lung vasculature, which is one of the main cellular targets for henipaviruses (14, 44) and where edema, fibrinoid necrosis, and thrombus formation were observed in the vicinity, suggesting vasculitis. The presence of large perivascular infiltrates and extensive macrophage accumulations seen in NiV-B-infected small airways and alveolar spaces, respectively, of lung grafts from reconstituted mice is in agreement with immune cell infiltrations seen in the lung parenchyma of human NiV cases (14, 45). Overall, these pathological findings are consistent with data from in vitro and in vivo studies of NiV infection (2, 10, 12, 13, 29, 46) as well as histopathological reports of NiV-M-infected patients (4, 14). Based on all these observations, NiV infection of human lung grafts results in an outcome reminiscent of ALI, suggesting that human lung grafts in the reconstituted mouse model constitute a robust model that recapitulates typical pulmonary histopathological lesions observed in NiV-infected patients.
We previously reported that NiV-B induces oxidative stress in epithelial cells from human small airways (19). Here, we demonstrated that oxidative stress also occurs in NiV-B-infected human lung grafts by quantifying MDA-protein adducts, a well-accepted oxidative stress marker (47–49). Interestingly, the total activity of SOD, as measured for three subtypes (SOD1, -2, and -3) involved in cellular defenses that control oxidative stress, was decreased late during infection. This is consistent with our previous findings showing gene downregulation of SOD2 and SOD3 in respiratory epithelial cells (19), suggesting that reduced SOD activity contributes to an increased level of reactive oxygen species (ROS), the actors of oxidative stress that cause cellular oxidative damage across the lung at a late stage of infection. However, the earlier induction of oxidative stress in NiV-B-infected lung grafts from reconstituted mice suggests that human leukocytes also contribute to oxidative stress when recruited to the site of infection. Here, neutrophils in NiV-B-infected lung grafts from reconstituted mice were found, in agreement with data from previous studies (2, 10, 18, 50–52), and were detected concomitant with secreted mediators that can recruit them, including G-CSF, GM-CSF, IL-6, IL-8, and IP-10 (53, 54). Interestingly, neutrophils can generate oxidative stress by an oxidative burst that releases ROS and MPO (55, 56), the levels of which in the lung have been shown to be positively correlated with the level of MDA, which was increased in our study as well. The conclusive role of neutrophils in oxidative stress induction in NiV-B-infected lung grafts from reconstituted mice will require additional studies.
The earlier secretion of the anti-inflammatory mediator IL-1RA and the proinflammatory mediators eotaxin, IFN-α2, MIP-1α, MIP-1β, and IL-15 in NiV-B-infected lung grafts from reconstituted mice could indicate an effect of activated circulating or resident immune cells, such as alveolar macrophages, on the lung tissue to recruit more circulating leukocytes to the site of infection. The secretion of the anti-inflammatory mediator IL-10, a unique feature of the NiV-B-induced inflammatory response in lung grafts from reconstituted mice, could originate here from several leukocyte subpopulations (57, 58) and has been shown to regulate excessive inflammatory T cell responses to limit tissue damage (58–61). This includes the regulation of IL-6, IL-8, and IP-10 (58), which were also detected in NiV-B-infected lung grafts from reconstituted mice and whose levels could be restrained to balance protection and immune system-mediated pathology, as the blockage of IL-10 during influenza virus infection in the mouse lung resulted in higher levels of IL-6 (62). Altogether, these findings suggest that the human immune system attempted to temper NiV-B-induced inflammation in human lung grafts while limiting virus dissemination.
In summary, this study presents a human immune system-reconstituted mouse model as a complementary model to nonreconstituted mice that can be jointly used to implant human lung tissue and study NiV-induced lung pathogenesis. The lesions and leukocytic infiltrations observed in the lung grafts were in line with results reported previously for NiV patients. While no differences in virus replication in lung grafts were seen between the two models, the presence of multiple subpopulations of circulating or resident leukocytes in the lung grafts interfered with virus spread and caused an earlier induction of oxidative stress and a more elaborated innate immune response to infection than the one from the lung tissue itself. This included IL-10 secretion, which regulates excessive inflammatory responses to limit tissue damage. This reconstituted model provides the setting to investigate the mechanisms of how human leukocytes aim to fight infection across the lung and will help identify targets for drug development for the treatment of NiV-induced ALI.
MATERIALS AND METHODS
Ethics statement.
Approval for animal experiments was obtained from the Institutional Animal Care and Use Committee, University of Texas Medical Branch (UTMB) (protocol number 0905041), as previously described (15). Discarded tissue from deceased human fetuses (15 to 19 weeks of age) was obtained via a nonprofit partner (Advanced Bioscience Resources, Alameda, CA), as approved under exemption 4 of HHS regulations (45 CFR part 46) (63).
Lung grafting and BLT reconstitution in the mouse model.
Forty-four NOD-SCID/γc(null) (NSG) mice (Jackson Laboratories), 6 months of age, were each engrafted with two fragments of human fetal lung from the same donor under general anesthesia and under aseptic conditions, as previously described (15) (Fig. 1). Specifically, 21 and 23 mice received lung tissue from donors 1 and 2, respectively, as previously described (15). Next, 9 and 12 mice among the groups of 21 and 23 mice, respectively, also underwent human immune system reconstitution, as previously described, with the BLT reconstitution method (23, 26) by receiving fetal liver and thymus tissue as well as CD34+ fetal liver cells from the corresponding donor 1 or 2. These mice with human immune system reconstitution are referred to in this study as reconstituted mice. The surgical procedure did not successfully reconstitute the immune system in all animals, limiting both the number of groups and the number animals allocated per group in the present study. Note that the data obtained from lung grafts from donors 1 and 2 were pooled, as no significant differences in any of the parameters analyzed were observed throughout the study.
Flow cytometry.
The level of human immune system reconstitution in BLT mice was assessed before infection by flow cytometry and is expressed as the percentage of human leukocyte CD45+ compared to murine CD45+ populations in 100 μl mouse peripheral blood, as previously described (23, 26). The human leukocyte CD45+ subpopulations were further analyzed and showed the presence of T cells (human CD3+, CD4+, and CD8+ markers) and monocytes/macrophages (CD68+ marker).
Viruses and cells.
NiV-B was kindly provided by the Special Pathogens Branch (Centers for Disease Control and Prevention, Atlanta, GA, USA). NiV-B was isolated from a throat swab of a patient exhibiting respiratory involvement during the 2004 outbreak in Rajbari district, Bangladesh. The virus was propagated and titrated on Vero cells (ATCC CCL-81) as previously described (11). All work involving infectious NiV was performed under biosafety level 4 (BSL4) conditions at the Robert E. Shope BSL4 Laboratory and the BSL4 Galveston National Laboratory, UTMB.
Infection process and sample preparation.
Animals were transferred to the BSL4 laboratory at 10 weeks postengraftment. Under isoflurane anesthesia, human lung tissues were directly injected with a 50-μl volume of either 105 TCID50 NiV-B (virus) or PBS (mock), as previously described (15). Animals were then monitored daily for weight loss and clinical signs of disease, including ataxia, scruffy coat, hunched posture, lethargy, irregular breathing, seizure, and paralysis. Fourteen groups of 3 mice and one group of 2 mice were euthanized on day 3 or 5 postinfection (Table 1), and human lung grafts were harvested and processed for histology or weighed and homogenized in a buffer containing 20 mM HEPES, 1 mM EGTA, 90 mM mannitol, and 70 mM sucrose (neutral pH) for subsequent analyses. Mouse blood was also collected in EDTA vacutainers.
Virus titration.
The virus titer in lung homogenates was assessed by a TCID50 assay in 96-well plates as previously described (15) and is expressed as TCID50 per gram of lung tissue.
Immunoassays and biochemical assays.
Measurements of MDA-protein adducts in lung graft homogenates and of human pulmonary SP-D levels in mouse serum were performed by using competitive enzyme immunoassays from Cell Biolabs (San Diego, CA) and Novatein Biosciences (Woburn, MA), respectively, according to the manufacturers' instructions. Note that SP-D is specific to humans and does not cross-react with mouse samples. SOD, CAT, GST, and GPx enzymatic activities in lung graft homogenates were measured by using Cayman Chemicals (Ann Arbor, MI) kits, according to the manufacturer's instructions.
Histology and immunohistochemistry.
Pieces of lung grafts were fixed by using 10% neutral buffered formalin for 7 days under BSL4 conditions. Specimens were then processed for paraffin embedding, sectioning at a 6-μm thickness, and staining with hematoxylin and eosin (H&E) or phosphotungstic acid-hematoxylin (PTAH) for histopathological analyses. Other tissue sections were stained by immunohistochemistry for NiV nucleoprotein by using a rabbit anti-NiV N antibody (kindly provided by C. Broder, Uniformed Services University, Bethesda, MD), for neutrophils by using a rabbit anti-MPO antibody from Abcam (Cambridge, MA), or for CD8+ T cells by using a rabbit CD8 antibody from Dako (Santa Barbara, CA). Observations were made with an Evos XL core microscope at a ×5, ×10, ×20, or ×40 magnification. Grading of the distribution of NiV nucleoprotein in lung grafts was expressed as percent coverage per section in 5 and 10% increments for grafts fixed at days 3 and 5, respectively, with 10 to 12 sections being observed under each condition. Grading was performed by blind slide reading. Note that the pictures of histopathological lesions, virus antigen staining, and immune cell staining are also representative of observations of human lung grafts from both donors.
Cytokine/chemokine analysis.
Cytokine/chemokine concentrations in lung homogenates were determined by using a Milliplex Human Cytokine 31 Plex Immunoassay custom kit (Millipore). Prior to analysis, samples were inactivated on dry ice by gamma irradiation (5 Mrad). The assays were performed according to the manufacturer's instructions. The concentrations of 31 cytokines [epidermal growth factor (EGF), eotaxin, G-CSF, GM-CSF, fractalkine (CX3CL1), IFN-α2, IFN-γ, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-15, IL-17A, IL-1RA, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, chemokine ligand 10 (IP-10 or CXCL10), monocyte chemotactic protein (MCP-1), MIP-1α, MIP-1β, RANTES, tumor necrosis factor alpha (TNF-α), TNF-β, and VEGF] were quantified.
Statistical analyses.
Comparisons of virus replication levels, abundances of NiV nucleoprotein antigen across sections, levels of MDA-protein adducts, SP-D concentrations, levels of secreted inflammatory mediators, as well as enzymatic activities were performed by using the nonparametric Mann-Whitney test. All data are presented in figures as means ± standard deviations (SD). Each condition was tested with biological duplicates (1 group of 2 mice) or triplicates (15 groups of 3 mice).
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant 1R21AI111042-01 to B.R. and A.N.F). Fellowship support was provided for R.J.N. through the UTMB McLaughlin Endowment and for M.B.H. through the UTMB Kempner Scholar Foundation and the American Society for Microbiology Robert D. Watkins Graduate Research Fellowship.
REFERENCES
- 1.Eaton BT, Broder CC, Wang LF. 2005. Hendra and Nipah viruses: pathogenesis and therapeutics. Curr Mol Med 5:805–816. doi: 10.2174/156652405774962308. [DOI] [PubMed] [Google Scholar]
- 2.Hooper P, Zaki S, Daniels P, Middleton D. 2001. Comparative pathology of the diseases caused by Hendra and Nipah viruses. Microbes Infect 3:315–322. doi: 10.1016/S1286-4579(01)01385-5. [DOI] [PubMed] [Google Scholar]
- 3.Chua KB, Lam SK, Goh KJ, Hooi PS, Ksiazek TG, Kamarulzaman A, Olson J, Tan CT. 2001. The presence of Nipah virus in respiratory secretions and urine of patients during an outbreak of Nipah virus encephalitis in Malaysia. J Infect 42:40–43. doi: 10.1053/jinf.2000.0782. [DOI] [PubMed] [Google Scholar]
- 4.Goh KJ, Tan CT, Chew NK, Tan PS, Kamarulzaman A, Sarji SA, Wong KT, Abdullah BJ, Chua KB, Lam SK. 2000. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N Engl J Med 342:1229–1235. doi: 10.1056/NEJM200004273421701. [DOI] [PubMed] [Google Scholar]
- 5.Mounts AW, Kaur H, Parashar UD, Ksiazek TG, Cannon D, Arokiasamy JT, Anderson LJ, Lye MS. 2001. A cohort study of health care workers to assess nosocomial transmissibility of Nipah virus, Malaysia, 1999. J Infect Dis 183:810–813. doi: 10.1086/318822. [DOI] [PubMed] [Google Scholar]
- 6.Harit AK, Ichhpujani RL, Gupta S, Gill KS, Lal S, Ganguly NK, Agarwal SP. 2006. Nipah/Hendra virus outbreak in Siliguri, West Bengal, India in 2001. Indian J Med Res 123:553–560. [PubMed] [Google Scholar]
- 7.Hossain MJ, Gurley ES, Montgomery JM, Bell M, Carroll DS, Hsu VP, Formenty P, Croisier A, Bertherat E, Faiz MA, Azad AK, Islam R, Molla MA, Ksiazek TG, Rota PA, Comer JA, Rollin PE, Luby SP, Breiman RF. 2008. Clinical presentation of Nipah virus infection in Bangladesh. Clin Infect Dis 46:977–984. doi: 10.1086/529147. [DOI] [PubMed] [Google Scholar]
- 8.Lo MK, Rota PA. 2008. The emergence of Nipah virus, a highly pathogenic paramyxovirus. J Clin Virol 43:396–400. doi: 10.1016/j.jcv.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Clayton BA, Middleton D, Bergfeld J, Haining J, Arkinstall R, Wang L, Marsh GA. 2012. Transmission routes for Nipah virus from Malaysia and Bangladesh. Emerg Infect Dis 18:1983–1993. doi: 10.3201/eid1812.120875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeBuysscher BL, de Wit E, Munster VJ, Scott D, Feldmann H, Prescott J. 2013. Comparison of the pathogenicity of Nipah virus isolates from Bangladesh and Malaysia in the Syrian hamster. PLoS Negl Trop Dis 7:e2024. doi: 10.1371/journal.pntd.0002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Escaffre O, Borisevich V, Carmical JR, Prusak D, Prescott J, Feldmann H, Rockx B. 2013. Henipavirus pathogenesis in human respiratory epithelial cells. J Virol 87:3284–3294. doi: 10.1128/JVI.02576-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escaffre O, Borisevich V, Vergara LA, Wen JW, Long D, Rockx B. 2016. Characterization of Nipah virus infection in a model of human airway epithelial cells cultured at an air-liquid interface. J Gen Virol 97:1077–1086. doi: 10.1099/jgv.0.000441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mire CE, Satterfield BA, Geisbert JB, Agans KN, Borisevich V, Yan L, Chan YP, Cross RW, Fenton KA, Broder CC, Geisbert TW. 2016. Pathogenic differences between Nipah virus Bangladesh and Malaysia strains in primates: implications for antibody therapy. Sci Rep 6:30916. doi: 10.1038/srep30916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, Guarner J, Goldsmith CS, Chua KB, Lam SK, Tan CT, Goh KJ, Chong HT, Jusoh R, Rollin PE, Ksiazek TG, Zaki SR. 2002. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am J Pathol 161:2153–2167. doi: 10.1016/S0002-9440(10)64493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valbuena G, Halliday H, Borisevich V, Goez Y, Rockx B. 2014. A human lung xenograft mouse model of Nipah virus infection. PLoS Pathog 10:e1004063. doi: 10.1371/journal.ppat.1004063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baseler L, de Wit E, Scott DP, Munster VJ, Feldmann H. 2015. Syrian hamsters (Mesocricetus auratus) oronasally inoculated with a Nipah virus isolate from Bangladesh or Malaysia develop similar respiratory tract lesions. Vet Pathol 52:38–45. doi: 10.1177/0300985814556189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clayton BA, Middleton D, Arkinstall R, Frazer L, Wang LF, Marsh GA. 2016. The nature of exposure drives transmission of Nipah viruses from Malaysia and Bangladesh in ferrets. PLoS Negl Trop Dis 10:e0004775. doi: 10.1371/journal.pntd.0004775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rockx B, Brining D, Kramer J, Callison J, Ebihara H, Mansfield K, Feldmann H. 2011. Clinical outcome of henipavirus infection in hamsters is determined by the route and dose of infection. J Virol 85:7658–7671. doi: 10.1128/JVI.00473-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Escaffre O, Halliday H, Borisevich V, Casola A, Rockx B. 2015. Oxidative stress in Nipah virus-infected human small airway epithelial cells. J Gen Virol 96:2961–2970. doi: 10.1099/jgv.0.000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Escaffre O, Borisevich V, Rockx B. 2013. Pathogenesis of Hendra and Nipah virus infection in humans. J Infect Dev Ctries 7:308–311. doi: 10.3855/jidc.3648. [DOI] [PubMed] [Google Scholar]
- 21.Berges BK, Rowan MR. 2011. The utility of the new generation of humanized mice to study HIV-1 infection: transmission, prevention, pathogenesis, and treatment. Retrovirology 8:65. doi: 10.1186/1742-4690-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bird BH, Spengler JR, Chakrabarti AK, Khristova ML, Sealy TK, Coleman-McCray JD, Martin BE, Dodd KA, Goldsmith CS, Sanders J, Zaki SR, Nichol ST, Spiropoulou CF. 2016. Humanized mouse model of Ebola virus disease mimics the immune responses in human disease. J Infect Dis 213:703–711. doi: 10.1093/infdis/jiv538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calderon VE, Valbuena G, Goez Y, Judy BM, Huante MB, Sutjita P, Johnston RK, Estes DM, Hunter RL, Actor JK, Cirillo JD, Endsley JJ. 2013. A humanized mouse model of tuberculosis. PLoS One 8:e63331. doi: 10.1371/journal.pone.0063331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J, Brehm MA, Greiner D, Shultz LD, Kornfeld H. 2013. Engrafted human cells generate adaptive immune responses to Mycobacterium bovis BCG infection in humanized mice. BMC Immunol 14:53. doi: 10.1186/1471-2172-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morton JJ, Bird G, Refaeli Y, Jimeno A. 2016. Humanized mouse xenograft models: narrowing the tumor-microenvironment gap. Cancer Res 76:6153–6158. doi: 10.1158/0008-5472.CAN-16-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nusbaum RJ, Calderon VE, Huante MB, Sutjita P, Vijayakumar S, Lancaster KL, Hunter RL, Actor JK, Cirillo JD, Aronson J, Gelman BB, Lisinicchia JG, Valbuena G, Endsley JJ. 2016. Pulmonary tuberculosis in humanized mice infected with HIV-1. Sci Rep 6:21522. doi: 10.1038/srep21522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith DJ, Lin LJ, Moon H, Pham AT, Wang X, Liu S, Ji S, Rezek V, Shimizu S, Ruiz M, Lam J, Janzen DM, Memarzadeh S, Kohn DB, Zack J, Kitchen SG, An DS, Yang L. 2016. Propagating humanized BLT mice for the study of human immunology and immunotherapy. Stem Cells Dev 25:1863–1873. doi: 10.1089/scd.2016.0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spengler JR, Lavender KJ, Martellaro C, Carmody A, Kurth A, Keck JG, Saturday G, Scott DP, Nichol ST, Hasenkrug KJ, Spiropoulou CF, Feldmann H, Prescott J. 2016. Ebola virus replication and disease without immunopathology in mice expressing transgenes to support human myeloid and lymphoid cell engraftment. J Infect Dis 214(Suppl 3):S308–S318. doi: 10.1093/infdis/jiw248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo MK, Miller D, Aljofan M, Mungall BA, Rollin PE, Bellini WJ, Rota PA. 2010. Characterization of the antiviral and inflammatory responses against Nipah virus in endothelial cells and neurons. Virology 404:78–88. doi: 10.1016/j.virol.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Gurley ES, Montgomery JM, Hossain MJ, Bell M, Azad AK, Islam MR, Molla MA, Carroll DS, Ksiazek TG, Rota PA, Lowe L, Comer JA, Rollin P, Czub M, Grolla A, Feldmann H, Luby SP, Woodward JL, Breiman RF. 2007. Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg Infect Dis 13:1031–1037. doi: 10.3201/eid1307.061128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wirblich C, Schnell MJ. 2011. Rabies virus (RV) glycoprotein expression levels are not critical for pathogenicity of RV. J Virol 85:697–704. doi: 10.1128/JVI.01309-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baseler L, Scott DP, Saturday G, Horne E, Rosenke R, Thomas T, Meade-White K, Haddock E, Feldmann H, de Wit E. 2016. Identifying early target cells of Nipah virus infection in Syrian hamsters. PLoS Negl Trop Dis 10:e0005120. doi: 10.1371/journal.pntd.0005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujisawa H. 2001. Inhibitory role of neutrophils on influenza virus multiplication in the lungs of mice. Microbiol Immunol 45:679–688. doi: 10.1111/j.1348-0421.2001.tb01302.x. [DOI] [PubMed] [Google Scholar]
- 34.Fujisawa H. 2008. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J Virol 82:2772–2783. doi: 10.1128/JVI.01210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. 2011. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179:199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Vries RD, Yuksel S, Osterhaus AD, de Swart RL. 2010. Specific CD8(+) T-lymphocytes control dissemination of measles virus. Eur J Immunol 40:388–395. doi: 10.1002/eji.200939949. [DOI] [PubMed] [Google Scholar]
- 37.Frey S, Krempl CD, Schmitt-Graff A, Ehl S. 2008. Role of T cells in virus control and disease after infection with pneumonia virus of mice. J Virol 82:11619–11627. doi: 10.1128/JVI.00375-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janeway C, Travers P, Walport M, Shlomchik M. 2004. Immunobiology: the immune system in health and disease, 6th ed Garland Science, New York, NY. [Google Scholar]
- 39.Bangs SC, McMichael AJ, Xu XN. 2006. Bystander T cell activation—implications for HIV infection and other diseases. Trends Immunol 27:518–524. doi: 10.1016/j.it.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Unutmaz D, Pileri P, Abrignani S. 1994. Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J Exp Med 180:1159–1164. doi: 10.1084/jem.180.3.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 8:591–599. doi: 10.1016/S1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- 42.Doyle IR, Nicholas TE, Bersten AD. 1999. Partitioning lung and plasma proteins: circulating surfactant proteins as biomarkers of alveolocapillary permeability. Clin Exp Pharmacol Physiol 26:185–197. doi: 10.1046/j.1440-1681.1999.03015.x. [DOI] [PubMed] [Google Scholar]
- 43.Hermans C, Bernard A. 1999. Lung epithelium-specific proteins: characteristics and potential applications as markers. Am J Respir Crit Care Med 159:646–678. doi: 10.1164/ajrccm.159.2.9806064. [DOI] [PubMed] [Google Scholar]
- 44.Maisner A, Neufeld J, Weingartl H. 2009. Organ- and endotheliotropism of Nipah virus infections in vivo and in vitro. Thromb Haemost 102:1014–1023. doi: 10.1160/TH09-05-0310. [DOI] [PubMed] [Google Scholar]
- 45.Wong KT, Shieh WJ, Zaki SR, Tan CT. 2002. Nipah virus infection, an emerging paramyxoviral zoonosis. Springer Semin Immunopathol 24:215–228. doi: 10.1007/s00281-002-0106-y. [DOI] [PubMed] [Google Scholar]
- 46.Bossart KN, Rockx B, Feldmann F, Brining D, Scott D, LaCasse R, Geisbert JB, Feng YR, Chan YP, Hickey AC, Broder CC, Feldmann H, Geisbert TW. 2012. A Hendra virus G glycoprotein subunit vaccine protects African green monkeys from Nipah virus challenge. Sci Transl Med 4:146ra107. doi: 10.1126/scitranslmed.3004241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ayala A, Munoz MF, Arguelles S. 2014. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA, Colasurdo GN, Khan AM, Garofalo RP, Casola A. 2006. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respir Crit Care Med 174:1361–1369. doi: 10.1164/rccm.200603-319OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giera M, Lingeman H, Niessen WM. 2012. Recent advancements in the LC- and GC-based analysis of malondialdehyde (MDA): a brief overview. Chromatographia 75:433–440. doi: 10.1007/s10337-012-2237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeBuysscher BL, Scott D, Marzi A, Prescott J, Feldmann H. 2014. Single-dose live-attenuated Nipah virus vaccines confer complete protection by eliciting antibodies directed against surface glycoproteins. Vaccine 32:2637–2644. doi: 10.1016/j.vaccine.2014.02.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mohd Nor MN, Gan CH, Ong BL. 2000. Nipah virus infection of pigs in peninsular Malaysia. Rev Sci Tech 19:160–165. doi: 10.20506/rst.19.1.1202. [DOI] [PubMed] [Google Scholar]
- 52.Yoneda M, Georges-Courbot MC, Ikeda F, Ishii M, Nagata N, Jacquot F, Raoul H, Sato H, Kai C. 2013. Recombinant measles virus vaccine expressing the Nipah virus glycoprotein protects against lethal Nipah virus challenge. PLoS One 8:e58414. doi: 10.1371/journal.pone.0058414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, Sasaki T, Ohteki T, Ranieri VM, dos Santos CC, Kawaoka Y, Akira S, Luster AD, Lu B, Penninger JM, Uhlig S, Slutsky AS, Imai Y. 2013. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am J Respir Crit Care Med 187:65–77. doi: 10.1164/rccm.201203-0508OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wright HL, Moots RJ, Bucknall RC, Edwards SW. 2010. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 49:1618–1631. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- 55.McGovern TK, Chen M, Allard B, Larsson K, Martin JG, Adner M. 2016. Neutrophilic oxidative stress mediates organic dust-induced pulmonary inflammation and airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol 310:L155–L165. doi: 10.1152/ajplung.00172.2015. [DOI] [PubMed] [Google Scholar]
- 56.McGovern TK, Goldberger M, Allard B, Farahnak S, Hamamoto Y, O'Sullivan M, Hirota N, Martel G, Rousseau S, Martin JG. 2015. Neutrophils mediate airway hyperresponsiveness after chlorine-induced airway injury in the mouse. Am J Respir Cell Mol Biol 52:513–522. doi: 10.1165/rcmb.2013-0430OC. [DOI] [PubMed] [Google Scholar]
- 57.Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galan JE, Harhaj E, Flavell RA. 2006. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 25:941–952. doi: 10.1016/j.immuni.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 58.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. 2001. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 59.Couper KN, Blount DG, Riley EM. 2008. IL-10: the master regulator of immunity to infection. J Immunol 180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 60.Joss A, Akdis M, Faith A, Blaser K, Akdis CA. 2000. IL-10 directly acts on T cells by specifically altering the CD28 co-stimulation pathway. Eur J Immunol 30:1683–1690. doi:. [DOI] [PubMed] [Google Scholar]
- 61.Schandene L, Cogan E, Crusiaux A, Goldman M. 1997. Interferon-alpha upregulates both interleukin-10 and interferon-gamma production by human CD4+ T cells. Blood 89:1110–1111. [PubMed] [Google Scholar]
- 62.Sun J, Madan R, Karp CL, Braciale TJ. 2009. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med 15:277–284. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Code of Federal Regulations. 2009. Title 45. Public welfare. Part 46. Protection of human subjects. https://www.hhs.gov/ohrp/regulations-and-policy/regulations/45-cfr-46/. [PubMed]