

ABSTRACT
Virus-like vesicles (VLVs) are membrane-enclosed vesicles that resemble native enveloped viruses in organization but lack the viral capsid and genome. During the productive infection of tumor-associated gammaherpesviruses, both virions and VLVs are produced and are released into the extracellular space. However, studies of gammaherpesvirus-associated VLVs have been largely restricted by the technical difficulty of separating VLVs from mature virions. Here we report a strategy of selectively isolating VLVs by using a Kaposi's sarcoma-associated herpesvirus (KSHV) mutant that is defective in small capsid protein and is unable to produce mature virions. Using mass spectrometry analysis, we found that VLVs contained viral glycoproteins required for cellular entry, as well as tegument proteins involved in regulating lytic replication, but lacked capsid proteins. Functional analysis showed that VLVs induced the expression of the viral lytic activator RTA, initiating KSHV lytic gene expression. Furthermore, employing RNA sequencing, we performed a genomewide analysis of cellular responses triggered by VLVs and found that PRDM1, a master regulator in cell differentiation, was significantly upregulated. In the context of KSHV replication, we demonstrated that VLV-induced upregulation of PRDM1 was necessary and sufficient to reactivate KSHV by activating its RTA promoter. In sum, our study systematically examined the composition of VLVs and demonstrated their biological roles in manipulating host cell responses and facilitating KSHV lytic replication.
IMPORTANCE Cells lytically infected with tumor-associated herpesviruses produce a high proportion of virus-like vesicles (VLVs). The composition and function of VLVs have not been well defined, largely due to the inability to efficiently isolate VLVs that are free of virions. Using a cell system capable of establishing latent KSHV infection and robust reactivation, we successfully isolated VLVs from a KSHV mutant defective in the small capsid protein. We quantitatively analyzed proteins and microRNAs in VLVs and characterized the roles of VLVs in manipulating host cells and facilitating viral infection. More importantly, we demonstrated that by upregulating PRDM1 expression, VLVs triggered differentiation signaling in targeted cells and facilitated viral lytic infection via activation of the RTA promoter. Our study not only demonstrates a new strategy for isolating VLVs but also shows the important roles of KSHV-associated VLVs in intercellular communication and the viral life cycle.
KEYWORDS: EBV, herpesvirus, KSHV, PRDM1, RTA, tegument protein, envelope protein, extracellular vesicle, microRNA, virus-like vesicle
INTRODUCTION
Cells productively infected with viruses release membrane-enclosed entities. Some of these membrane-enclosed entities are infectious progeny virions; others are extracellular vesicles (EVs), which contain proteins and RNAs important for cell-cell communication. However, EV is a broad term that describes vesicles heterogeneous in size, structure, and biogenesis, serving a wide variety of functions (1–3). There is a specific population of EVs that emerges only during virus infection and contains both viral proteins and nucleotide products. These vesicles resemble native enveloped virions in physical and chemical characteristics, so that it is hard to distinguish them from actual virions (4–6). As a result, we refer to this population of EVs as virus-like vesicles (VLVs). VLVs are noninfectious due to the absence of the viral capsid and genome (4, 5). These vesicles have been identified in a wide variety of virus infections, including infections with herpes simplex virus 1 (HSV-1) (7), human cytomegalovirus (HCMV) (8), hepatitis B virus (9), hepatitis C virus (10), human immunodeficiency virus (11), and vaccinia virus (12). Initially, VLVs were largely considered to be defective virion particles or debris from viral assembly, with no biological significance. However, it has been found that VLVs play roles in intercellular communication and viral infection. For example, VLVs from cells lytically infected with HSV-1 facilitate early stages of viral infection by delivering the virion host shutoff protein (vhs) into new host cells (13). In addition, VLVs (also termed “dense bodies”) of HCMV are able to induce the maturation and activation of dendritic cells (14). The VLVs attach to host cells via specific receptor binding and enter through endocytosis or fusion. Eventually, the proteins and/or RNAs contained within the VLVs interfere with cellular functions. The protein composition of VLVs is highly related to that of the corresponding virions, making VLVs a potentially useful tool in vaccinology (15–19).
Kaposi's sarcoma-associated herpesvirus (KSHV) is a member of the gammaherpesvirus subfamily. Like all herpesviruses, KSHV is a large, double-stranded DNA (dsDNA) virus with two distinct life cycle phases: latency and lytic (productive) replication. During latency, the viral genome persists as a nuclear episome, with minimal viral gene expression. During lytic replication, the viral genome replicates extensively, most viral genes are expressed, and virions are assembled and released from cells. KSHV infection is associated with several malignancies (20–23). Although KSHV-associated cancers are directly linked with latently infected cells, both latent and lytic infections contribute to tumorigenesis. Lytic replication contributes indirectly to tumorigenesis by providing a proinflammatory and proproliferative environment, as well as by spreading infection and expanding the population of latently infected cells, which subsequently develop into transformed cells. Previous studies have also shown that gammaherpesvirus lytic infection contributes to lymphoproliferative diseases in vivo (24). Moreover, clinical observations with AIDS patients at high risk of Kaposi's sarcoma (KS) showed that treatment with ganciclovir, an inhibitor of herpesvirus lytic DNA replication, significantly reduced the incidence of KS (25). This finding provides evidence for the significant role of gammaherpesvirus lytic replication in tumorigenesis (26–28).
During KSHV lytic replication, VLVs were generated in addition to mature virions (reference 29 and our unpublished results). Due to the technical difficulty of separating VLVs from virions, very little is known about the components and functions of KSHV-associated VLVs. Since KSHV is prevalent among AIDS patients and causes global cancer concern, understanding the composition and function of these VLVs may be an important key to alleviating these burdens. Here we report a strategy of isolating KSHV-associated VLVs from a mutant virus defective in the small capsid protein (SCP). Using mass spectrometry (MS) and high-throughput RNA sequencing (RNA-seq), we quantitatively characterized the protein and RNA compositions of these VLVs. Through a reporter assay, we showed that KSHV-associated VLVs were able to induce viral lytic replication by activating the RTA promoter. After evaluating the global cellular response by RNA-seq, we found that VLVs induced differentiation signaling in targeted cells by upregulating PRDM1. Through overexpression and knockdown by use of small interfering RNA (siRNA), we further confirmed that PRDM1 is critical for KSHV reactivation when induced by VLVs. Our study demonstrates the important roles of VLVs in intercellular communication and the facilitation of viral lytic infection.
RESULTS
Virus-like vesicles are released from cells during gammaherpesvirus lytic infection.
Due to the similarity in morphology between virions and VLVs, it is very challenging to separate these two populations. To examine the presence and quantity of KSHV-associated VLVs, we performed sucrose density gradient ultracentrifugation and collected the band containing viral particles. We then employed cryo-electron microscopy (cryo-EM) to visualize KSHV virions and VLVs. Under cryo-EM, VLVs are easily distinguishable from virions due to the lack of capsids (Fig. 1A, left). To determine whether VLVs were also produced by other gammaherpesviruses, we examined vesicles released from cells lytically infected with murine gammaherpesvirus 68 (MHV-68) or Epstein-Barr virus (EBV). Not surprisingly, VLVs were associated with both viruses (Fig. 1A, center and right). We noticed that the average diameters of both virions and VLVs were around 200 nm. However, while the diameters of virions have a small range of variance, the diameters of VLVs fluctuated significantly (Fig. 1B), indicating that the morphology of VLVs is more diverse than that of virions. In addition, the relatively large size of VLVs renders them distinguishable from exosomes, which normally fall into a range of 40 to 100 nm (3). We further determined the ratio of VLVs in cryo-EM images and found that VLVs made up 57.5% of total vesicles of related morphology in the KSHV samples. Interestingly, this percentage was not the same for the other gammaherpesvirus samples we tested. While fewer VLVs (49.6%) were present in the MHV-68 samples, there were dramatically more in the EBV samples (84%) (Fig. 1C).
FIG 1.

VLVs are produced during gammaherpesvirus lytic infection. (A) Three gammaherpesviruses, KSHV, MHV-68, and EBV, were purified individually by sucrose density gradient ultracentrifugation. Then virions and VLVs were examined by cryo-EM. Red arrows indicate VLVs. Bar, 100 nm. (B) Sizes of virions and VLVs. (C) Quantification of virions and VLVs. More than 200 membrane particles each from KSHV, MHV-68, and EBV were counted. The percentages of VLVs and virions were then plotted.
Virus-like vesicles are generated in MVB-like compartments of lytically infected cells.
In order to explore the formation and release pathways of VLVs, we examined the localization of VLVs inside and outside infected cells. Since MHV-68 readily undergoes lytic replication, we used NIH 3T3 cells infected with MHV-68 as a model for this set of experiments. As shown in Fig. 2A, MHV-68 nucleocapsids underwent tegumentation and envelopment in cytoplasmic multivesicular body (MVB)-like compartments. VLVs, identifiable as empty vesicles with double membranes, were also enveloped in the same compartments. When we looked at the extracellular space of infected cells, we found that both virions and VLVs were present, and their ratios were similar to those observed in the cryo-EM images (Fig. 2B). These data suggest that VLVs form in the same cellular compartment as virions, and likely through a similar pathway.
FIG 2.
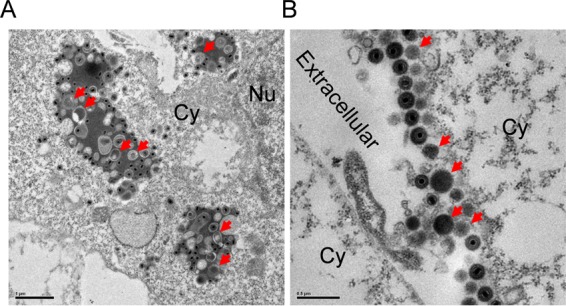
VLVs are generated in multivesicular bodies. (A) Formation of VLVs in cells lytically infected with MHV-68. NIH 3T3 cells were first infected with MHV-68 and then examined by plastic embedding, ultrathin sectioning, and transmission electron microscopy. Red arrows indicate VLVs. Cy, cytoplasm; Nu, nucleus. (B) Release of VLVs into extracellular space.
Purification of VLVs from a KSHV mutant defective in SCP.
One of the fundamental steps in the study of VLVs is to isolate VLVs that are free of virions. In our previous study of KSHV capsid assembly (30), we generated a KSHV mutant in which a stop codon was introduced into the coding region of the small capsid protein (SCP). The SCP-null virus (KSHV-SCPnull) is capable of DNA replication and the production of all viral proteins except SCP; as a result, the virus is defective in capsid maturation. KSHV-SCPnull produces 1,000- to 10,000-fold fewer infectious virions than the wild-type virus. Importantly, cells lytically infected with KSHV-SCPnull were still able to produce KSHV-associated VLVs (30), while in the absence of reactivation, iSLK cells latently infected with KSHV do not produce VLVs and barely release other extracellular vesicles (data not shown). By using the KSHV-SCPnull mutant and the iSLK cell system (31, 32), which allows for lytic KSHV replication, we efficiently purified KSHV-associated VLVs through sucrose density gradient ultracentrifugation. Upon cryo-EM examination, we found that VLVs derived from cells lytically infected with KSHV-SCPnull were similar to VLVs produced during wild-type virus infection: their average size was around 200 nm, and they did not contain capsids (Fig. 3A). To confirm that our VLV preparation was free of virions, we incubated 293T cells with either KSHV-SCPnull-derived VLVs or wild-type KSHV. Since our KSHV genome carries a green fluorescent protein (GFP) expression cassette driven by the EF-1α promoter, GFP can serve as an indicator for infection. We found that the cells incubated with VLVs were GFP negative, while cells infected with wild-type KSHV displayed a very strong GFP signal (Fig. 3B). This suggests that the KSHV-SCPnull-derived VLVs are free of virion contamination.
FIG 3.

The KSHV-SCPnull mutant serves as a model for the study of virus-like vesicles. (A) KSHV-associated VLVs were examined by cryo-EM. iSLK cells latently infected with KSHV-SCPnull were treated with doxycycline and sodium butyrate to induce lytic viral replication. Four days later, VLVs were purified from supernatants through a sucrose density gradient and were further examined by cryo-EM. Bar, 100 nm. (B) Detection of infectious virion particles by de novo infection. 293T cells were infected with a virion sample or a VLV sample at a final concentration of 2 μg/ml. Two days later, cells were examined under a fluorescence microscope. Bar, 20 μm. (C) Viral proteins in VLV and virion samples. One microgram of a virion sample, purified from iSLK cells infected with wild-type KSHV, or a VLV sample, purified from iSLK cells infected with KSHV-SCPnull, was first lysed in SDS buffer and then subjected to Western blotting.
Proteomic analysis of VLVs.
After successfully isolating VLVs, we examined their protein components. As shown in Fig. 2A, gammaherpesvirus-associated VLVs formed at MVB-like compartments, which are also the location for virion assembly. This result indicates that VLVs might contain virion proteins. Indeed, KSHV tegument proteins ORF52 and ORF45 were detected at comparable levels in virion and VLV samples by Western blotting, while capsid triplex protein 2 (ORF26) could be detected only in virions (Fig. 3C). Since triplex protein 2 is absolutely required for herpesvirus capsid assembly (33), its absence confirms that our KSHV-SCPnull-derived VLVs are free of virion contamination.
Next, we sought to systematically analyze the proteome of KSHV-SCPnull-derived VLVs. We performed mass spectrometry on the isolated VLVs and compared the results to those for KHSV virions. As shown in Table 1, VLVs contained envelope and tegument proteins at levels comparable to those in KSHV virions. In contrast to their detection in virions, however, capsid proteins were barely detected in VLVs, with a signal intensity significantly lower than that in the virion sample. This result is consistent with our EM observation that VLVs do not contain capsids (Fig. 3). Furthermore, two tegument proteins that we have previously shown to be directly associated with capsids, ORF19 and ORF32, were barely detectable in VLVs (34).
TABLE 1.
Proteomic analysis of viral proteins in KSHV virion and VLV samples using mass spectrometry
| Category and protein | Size (aa) | No. of peptides in: |
Intensity ratio (VLV/virion) | Function | |
|---|---|---|---|---|---|
| Virions | VLVs | ||||
| Capsid proteins | |||||
| ORF25 | 1,376 | 180 | 24 | 0.003 | Major capsid protein (MCP) |
| ORF26 | 305 | 23 | 6 | 0.021 | Triplex protein 2 (Tri2) |
| ORF17.5 | 288 | 22 | 4 | 0.031 | Scaffold protein |
| ORF65 | 170 | 20 | 2 | 0.005 | Small capsid protein (SCP) |
| ORF43 | 605 | 12 | 0 | 0.000 | Portal protein |
| ORF62 | 331 | 9 | 1 | 0.003 | Triplex protein 1 (Tri1) |
| Other virion proteins largely absent in EVs | |||||
| ORF32 | 454 | 11 | 1 | 0.009 | Binds triplex |
| ORF19 | 549 | 9 | 0 | 0.000 | Binds penton |
| Envelope proteins | |||||
| ORF8 | 845 | 55 | 60 | 0.763 | gB |
| ORF22 | 730 | 20 | 19 | 0.756 | gH |
| ORF68 | 545 | 13 | 14 | 1.129 | UL32 homolog |
| ORF39 | 400 | 7 | 9 | 1.169 | gM |
| K8.1 | 228 | 3 | 4 | 1.061 | gp35/37 |
| ORF47 | 167 | 4 | 3 | 0.624 | gL |
| ORF28 | 102 | 3 | 3 | 1.273 | EBV gp150 homolog |
| ORF4 | 550 | 12 | 11 | 0.610 | Complement control |
| Tegument and other viral proteins | |||||
| ORF75 | 1,296 | 72 | 75 | 0.740 | Tegument protein |
| ORF64 | 2,635 | 60 | 69 | 0.816 | Large tegument protein |
| ORF21 | 580 | 43 | 43 | 0.800 | Thymidine kinase |
| ORF63 | 928 | 35 | 32 | 0.490 | UL37 homolog |
| ORF33 | 334 | 28 | 29 | 0.844 | Virion egress |
| ORF45 | 407 | 29 | 27 | 0.857 | Inhibition of IRF-7 |
| ORF52 | 131 | 23 | 25 | 1.146 | Virion egress |
| ORF23 | 404 | 12 | 11 | 0.873 | UL21 homolog |
| ORF38 | 61 | 6 | 7 | 1.421 | Virion egress |
| ORF42 | 278 | 6 | 6 | 1.371 | Virion egress |
| ORF27 | 290 | 5 | 7 | 1.795 | Intercellular viral spread |
| ORF49 | 302 | 6 | 5 | 0.671 | Transcription regulation |
| ORF55 | 227 | 4 | 6 | 1.748 | Unknown |
In addition to viral proteins, cellular proteins were also identified in VLV and virion samples (Table 2). Many of these cellular proteins (including actin, HSP70, HSP90, pyruvate kinase, annexins, ezrin, and enolase) have been reported to be present in KSHV virion samples, which were purified from lytically infected B cells (35, 36). Interestingly, cellular proteins were more abundant in VLV samples than in virion samples. One possible explanation is that VLVs do not contain capsid, and thus, more space is allowed for packaging cellular proteins. Another explanation is that cellular proteins do not require the capsid to be packaged efficiently, a hypothesis supported by the observation that the VLV/virion signal ratios of tegument proteins are lower than those of cellular proteins in general. This observation may suggest that most tegument proteins need the presence of the capsid to be efficiently packaged. Taken together, these proteomics data suggest that KSHV-SCPnull-derived VLVs have a protein composition similar to that of KSHV virions.
TABLE 2.
Presence of cellular proteins in KSHV virions and VLVsa
| UniProtKB accession no. | Protein | Size (aa) | No. of peptides in: |
Intensity ratio (VLV/virion) | |
|---|---|---|---|---|---|
| Virions | VLVs | ||||
| P07355 | Annexin A2 | 339 | 29 | 29 | 1.30 |
| P60709 | Beta-actin | 376 | 23 | 30 | 1.65 |
| Q09666 | Neuroblast differentiation-associated protein | 5,890 | 80 | 110 | 1.63 |
| P14618 | Pyruvate kinase isozymes M1/M2 | 605 | 35 | 42 | 2.49 |
| P11142 | Heat shock cognate 71-kDa protein | 646 | 29 | 32 | 1.25 |
| P04406 | Glyceraldehyde-3-phosphate dehydrogenase | 293 | 22 | 29 | 1.54 |
| P08107 | Heat shock 70-kDa protein 1A/1B | 641 | 24 | 30 | 2.32 |
| P06733 | Alpha-enolase | 341 | 22 | 30 | 1.73 |
| P62937 | Peptidyl-prolyl cis-trans isomerase A | 165 | 14 | 17 | 1.39 |
| P11021 | 78-kDa glucose-regulated protein | 654 | 34 | 20 | 1.14 |
| P08758 | Annexin A5 | 320 | 23 | 22 | 0.86 |
| P08195 | 4F2 cell surface antigen heavy chain | 631 | 18 | 28 | 2.82 |
| P08133 | Annexin A6 | 673 | 25 | 30 | 1.26 |
| P07237 | Protein disulfide-isomerase | 508 | 28 | 12 | 1.12 |
| P21333 | Filamin-A | 2,647 | 29 | 58 | 3.77 |
| P63104 | 14-3-3 protein zeta/delta | 245 | 13 | 18 | 1.61 |
| P62158 | Calmodulin | 149 | 10 | 13 | 1.56 |
| P04075 | Fructose-bisphosphate aldolase A | 364 | 16 | 22 | 2.65 |
| P60174 | Triosephosphate isomerase | 286 | 17 | 23 | 2.16 |
| P23528 | Cofilin-1 | 166 | 9 | 13 | 1.64 |
| P08670 | Vimentin | 466 | 16 | 26 | 2.90 |
| P68104 | Elongation factor 1-alpha 1 | 462 | 10 | 17 | 2.29 |
| P15311 | Ezrin | 586 | 16 | 24 | 2.10 |
| P08238 | Heat shock protein HSP 90-beta | 724 | 10 | 19 | 3.09 |
| P60903 | Protein S100-A10 | 97 | 9 | 11 | 0.90 |
| P05556 | Integrin beta-1 | 798 | 18 | 22 | 1.46 |
| P30101 | Protein disulfide-isomerase A3 | 505 | 22 | 13 | 1.25 |
| P04083 | Annexin A1 | 346 | 21 | 20 | 1.15 |
| P07900 | Heat shock protein HSP 90-alpha | 732 | 11 | 22 | 3.14 |
| P37802 | Transgelin-2 | 199 | 13 | 16 | 1.64 |
The cellular proteins encoded by the top 30 genes are listed in order of decreasing signal intensity of the gene in VLVs.
Quantification of microRNA in VLVs.
It has been shown previously that microRNAs (miRNAs) are packaged into gammaherpesvirus virions (37, 38) and also into circulating exosomes from KSHV patients (39). To test whether microRNAs are also present in KSHV-associated VLVs, we first purified total RNA from the VLV sample and then analyzed the size and abundance of the purified RNA. We found that large amounts of microRNAs were packaged into KSHV-associated VLVs (Fig. 4A). Using microRNA deep sequencing, we further identified and quantified individual microRNAs. We found that 69 host microRNAs and 14 KSHV microRNAs showed >100 reads (see Table S1 in the supplemental material). In terms of microRNA quantity, 18.0% of total microRNAs were encoded by KSHV, while 82.0% were derived from host cells (Fig. 4B). Many microRNAs previously identified in KSHV virion samples also showed high abundance in VLVs (37) (Table S1). In addition, pathway union analysis of the gene targets of the top 24 cellular microRNAs revealed significant overrepresentation of signaling pathways, including the extracellular matrix (ECM)-receptor pathway, proteoglycan-related pathways in cancer, and cell adhesion molecules (Fig. 4C). Thus, VLVs indeed package microRNAs, which may be important for regulating cellular signaling pathways.
FIG 4.

VLVs contain significant amounts of microRNA. (A) MicroRNA in VLVs. Total RNA was purified from VLVs and was then subjected to sizing and quantitation analysis using an Agilent 2100 Bioanalyzer. LM, Agilent lower marker (15 bp). (B) Percentages of microRNA reads from the host cell genome and the KSHV genome. (C) Pathway analysis of the top 24 human miRNAs (reads, >1,000) by the DIANA miRPath server (v3.0) (65). Shown are P values, corrected for the false discovery rate by the Benjamini-Hochberg procedure, for miRNAs associated with KEGG pathways.
VLVs promote KSHV lytic replication.
In our mass spectrometry analysis, we found that viral envelope glycoproteins were present in VLVs at levels comparable to those in the virion sample (Table 1). Since envelope proteins facilitate viral attachment and entry, we speculated that VLVs could also attach to and enter host cells. To test our hypothesis, we first incubated cells with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD)-labeled VLVs and then examined the DiD signal by fluorescence microscopy. As shown in Fig. 5A, host cells showed strong dot-like signals in the cytoplasm, indicating that VLVs were indeed capable of entering cells.
FIG 5.

VLVs promote lytic replication of KSHV. (A) VLVs can attach to and enter host cells. Vero cells were treated with DiD-labeled VLVs or PBS, used as a control (Ctl), for 1 h at 37°C. Then the cells were fixed and were examined under a fluorescence microscope. Bars, 20 μm. (B) VLVs reactivate KSHV from latency. HOK.219 cells were treated with VLVs at a final concentration of 1 μg/ml or with PBS as a control. Two days later, the cells were examined under a fluorescence microscope. Bar, 20 μm. (C) VLVs enhance de novo lytic replication of KSHV. HOK16B cells were infected with KSHV.219 at an MOI of 3. At the same time, VLVs were added to the viral inoculate at a concentration of 1 μg/ml, while the same volume of PBS was used as a control. Two days later, cells were examined under a fluorescence microscope. (D) VLVs upregulate KSHV lytic gene transcription. RNA was purified from the same cells as those shown in panel B, and viral transcripts were quantified by qRT-PCR. Viral gene expression relative to that in PBS-treated cells was plotted as means ± SD from triplicate experiments. **, P < 0.01. (E) VLVs induce infectious virion production. (F) Genomewide analysis of viral gene expression after VLV treatment. HOK16B.219 cells were treated with PBS or VLVs. Total RNAs were purified 1 or 2 days after VLV treatment (1d or 2d) and were sequenced. Transcriptional levels of viral genes were quantified in reads per kilobase of coding region per million total reads (RPKM) in the sample. Viral genes are listed in decreasing order of fold induction, and their RPKMs are plotted as means ± SD from two experiments. (G) Activation of the RTA promoter (RTAp) by VLVs. 293T cells were transfected with an RTA promoter plasmid and were split into new 24-well plates 12 h later. Cells were further treated with PBS or increasing amounts of VLVs for 24 h and were then lysed for luciferase activity analysis.
VLVs were shown to contain viral tegument and envelope proteins (Table 1). Since many of these proteins play important roles in facilitating KSHV infection (40), we speculated that VLVs may regulate KSHV lytic replication. We tested the hypothesis by determining if VLVs could reactivate KSHV from latency. For this experiment, we used immortalized human keratinocytes (HOK16B) latently infected with KSHV.219, a virus that expresses GFP from the EF-1α promoter during both lytic and latent infection and also expresses red fluorescent protein (RFP) from the KSHV PAN promoter during lytic infection only. Therefore, KSHV.219 facilitates the identification of cells undergoing lytic KSHV replication (41). First, we generated a HOK.219 cell line by latently infecting HOK16B cells with KSHV.219. Then we incubated the HOK.219 cells with VLVs and examined lytic viral replication by microscopy. As shown in Fig. 5B, exposure to VLVs efficiently induced RFP expression, indicating viral reactivation.
In addition, we tested the impact of VLVs on de novo lytic infection by mixing VLVs with wild-type virions during virus inoculation. We observed that VLVs were able to enhance de novo KSHV lytic infection (Fig. 5C), further supporting the notion that VLVs have a stimulating impact on KSHV lytic replication. To quantify the lytic replication induced by VLVs, we further analyzed viral gene expression and virion production. During the herpesvirus lytic cycle, viral gene expression is highly coordinated and occurs in a sequential cascade divided into immediate early (IE), early (E), and late (L) genes. Treatment of latently KSHV infected HOK.219 cells with VLVs induced the expression of all three classes of viral genes, including RTA (IE), ORF59 (E), ORF52 (L), and K8.1A (L) (Fig. 5D), and resulted in the production of infectious progeny viruses (Fig. 5E).
To systematically quantify the impact of VLVs on the expression of each viral gene, we used RNA-seq to examine the viral transcriptome during VLV treatment (Fig. 5F; see also Table S2 in the supplemental material). We found that all viral genes were upregulated and that the most upregulated genes were those responsive to RTA, including ORF59, PAN, and K12 (kaposin) (42–44) (Fig. 5F, top). This observation led us to test the hypothesis that VLVs might induce lytic replication by activating RTA. We set up a luciferase reporter assay to detect activation of the RTA promoter and found that the RTA promoter was activated by VLVs in a dose-dependent manner (Fig. 5G). Since VLVs do not contain the viral genome, the results from this reporter assay also indicate that activation of the RTA promoter by VLVs does not require new expression of viral proteins. In conclusion, these results suggest that VLVs promote KSHV lytic replication by inducing RTA expression.
VLVs induce differentiation signaling in host cells.
In order to explore cellular responses to VLVs and investigate the potential mechanism of activation of viral lytic replication, we performed RNA-seq to globally analyze cellular gene expression with and without VLV treatment. In brief, HOK16B cells were treated with phosphate-buffered saline (PBS) or VLVs for 1 day, and then mRNA was purified and subjected to DNA library construction and sequencing. Raw reads were aligned to a human genome assembly, and the expression levels of individual genes were expressed as reads per million total reads (RPM) (see Table S3 in the supplemental material). Fifty-five genes were upregulated >2-fold in VLV-treated cells (P < 0.05; difference in RPM, >2), and 40 genes were downregulated >2-fold (P < 0.05; difference in RPM, >2) (Fig. 6A; see also Table S4 in the supplemental material). We then performed Gene Ontology enrichment analysis on the 55 upregulated genes and found that they are significantly enriched for the following biological processes: cell differentiation, protein dephosphorylation, and antiapoptosis (Fig. 6B; see also Table S5 in the supplemental material). With regard to the 40 downregulated genes, we did not identify significant enrichment for specific biological processes (data not shown), but we did find that transcription of the tumor suppressor gene TP53 was downregulated 65%.
FIG 6.

VLVs induce cell differentiation. (A) Heat map diagram of differential gene expression after VLV treatment. HOK16B cells were treated with PBS or VLVs in quadruplicate for 1 day. Then RNA was purified and sequenced. The cluster diagram represents 55 upregulated genes and 40 downregulated genes with P values of <0.05 and >2-fold changes in expression. Expression levels are color-coded blue for low intensities and red for high intensities (see the scale at the bottom). The arrow outlined in green indicates PRDM1; the arrow outlined in brown indicates p53. (B) Gene Ontology enrichment analysis of the 55 upregulated genes using DAVID bioinformatics resources. (C) Expression of PRDM1 and other epithelial cell differentiation-related genes after knockdown of PRDM1. HOK16B cells were transfected with either a control siRNA, siPRDM1#8, or siPRDM1#10 and were then treated with PBS or VLVs for 1 day. Total RNA was purified and subjected to qRT-PCR analysis. Gene expression levels relative to those with the control siRNA (siCtl) and PBS-treated cells were plotted as means ± SD from triplicate experiments. For all genes, the P values for the differences in expression between siCtl- and siPRMD1-transfected cells in the presence of VLVs are <0.01. (D) Kinetics of PRDM1 and SPRR3 expression after VLV treatment. HOK16B cells were treated with VLVs for 1 h at 4°C with gentle agitation, washed with culture medium three times, and then transferred to a 37°C incubator. RNA was purified at the indicated times, and the transcription levels of PRDM1 and SPRR3 were quantified.
VLVs induced the expression of a panel of epithelial differentiation genes, especially genes belonging to the epidermal differentiation complex (SPRR2C, SPRR2D, SPRR3, LCE3D, and SCEL) (45). Notably, PRDM1 was found to be upregulated >5-fold. PRDM1, also known as Blimp-1, functions as a transcriptional repressor and plays a critical role in cell differentiation (46, 47). Since epithelial cell differentiation is a process reported to induce KSHV lytic replication (48), we examined the role of PRDM1 in VLV-treated cells. We knocked down PRDM1 expression in HOK16B cells by siRNA, treated those cells with VLVs, and then tested the expression of genes related to epithelial cell differentiation. As shown in Fig. 6C, VLVs significantly induced the expression of SPRR genes and KRT34 in control siRNA-transfected cells. However, this induction was largely impaired in PRDM1 siRNA-transfected cells. This result suggests that VLVs induce a host cell differentiation response through the upregulation of PRDM1 expression.
To characterize the host cell differentiation response, we further examined the kinetics of PRDM1 and SPRR3 expression after VLV treatment. We found that the expression of PRDM1 reached its highest level at 2 h after VLV treatment and then decreased gradually over time. In contrast, SPRR3 expression increased continuously between 4 h and 24 h after treatment with VLVs (Fig. 6D). This result showed that PRDM1 expression was transiently activated after VLV treatment, suggesting that a brief event—e.g., VLV attachment and/or entry—might be responsible for signaling PRDM1 upregulation. This result also showed that SPRR3 was expressed later than PRDM1, further supporting our conclusion that VLVs induced the host cell differentiation response through upregulation of PRDM1 expression.
VLVs induce KSHV lytic replication by activating PRDM1.
Differentiation signaling has been reported previously to play a role in gammaherpesvirus reactivation (48–51). Since PRDM1 has been reported to activate EBV lytic replication (49) and is also upregulated in the presence of VLVs, we reasoned that VLVs induced KSHV lytic replication as a consequence of PRDM1 upregulation. To test this hypothesis, we first investigated whether PRDM1 overexpression was able to induce KSHV lytic replication in latently infected cells (iSLK-KSHV). As shown in Fig. 7A, the expression levels of KSHV lytic transcripts (ORF57, ORF59, ORF52, and ORF26) in cells transfected with PRDM1 were similar to those in RTA-transfected cells. These results suggest that PRDM1 expression is sufficient to induce KSHV lytic gene expression at levels comparable to those with RTA, the master regulator of gammaherpesvirus lytic replication.
FIG 7.

PRDM1 induces lytic replication of KSHV after VLV treatment. (A) Overexpression of PRDM1 activates lytic replication of KSHV. iSLK-KSHV cells were transfected either with an empty vector or with an RTA or PRDM1 expression plasmid. Two days later, RNA was purified, and the transcription levels of viral genes were quantified. Gene expression levels relative to those in vector-transfected cells are plotted as means ± SD for triplicate experiments. (B) Knocking down PRDM1 expression reduced VLV-induced lytic replication of KSHV. HOK16B.KSHV cells were transfected with either a control siRNA (siCtl), siPRDM1#8, or siPRDM1#10 and were then treated with PBS or VLVs for 2 days. Total RNA was purified, and the transcription levels of viral genes were quantified. (Top) Gene expression levels relative to those with the control siRNA and PBS-treated cells are plotted as means ± SD for triplicate experiments. For all genes, the P values for the differences in expression between siCtl- and siPRMD1-transfected cells in the presence of VLVs are <0.01. (Bottom) The cell lysates were harvested for Western blotting with an anti-PRDM1 antibody. (C) Knocking down PRDM1 expression reduced VLV-induced production of infectious virions. Supernatants from the same cells as those for which results are shown in panel B were collected to determine viral titers. Infectious virion production relative to that with the control siRNA and VLV-treated cells is plotted as means ± SD from triplicate experiments. **, P < 0.01. (D) (Top) Results of luciferase reporter assays showing the activation of the RTA promoter (RTAp) by PRDM1. (Bottom) The expression of the HA-PRDM1 protein was analyzed by Western blotting with an anti-HA antibody.
Next, we examined the function of PRDM1 during KSHV reactivation after treatment with VLVs. As shown above, treatment of latently infected cells with VLVs induced the expression of KSHV lytic genes (Fig. 3G and 5B). This induction was significantly impaired when PRDM1 expression was knocked down by siRNA (Fig. 7B), leading to a reduction in virion production (Fig. 7C). In the reporter assay, we showed that VLVs were able to activate the RTA promoter (Fig. 5G). We further tested whether PRDM1 was able to activate the RTA promoter. As shown in Fig. 7D, PRDM1 expression robustly activated the RTA promoter in a dose-dependent manner. Taking these data together, we propose a model in which VLVs facilitate KSHV lytic replication by upregulating PRDM1 expression, which, in turn, induces RTA expression (Fig. 8).
FIG 8.

Model illustrating the mechanism by which VLVs reactivate KSHV from latency.
DISCUSSION
After centuries of coevolution with their hosts, viruses have acquired many strategies for modulating the cellular machinery to facilitate their own replication. During the productive phase of viral infection, cells are manipulated to produce virions for transmission. Infection also generates noninfectious vesicles, which, in turn, enhance virus replication and pathogenesis (4, 13, 52). In this study, we expanded our understanding of VLVs by using KSHV-derived VLVs as a model. Infection with KSHV, a human tumor-associated virus, produces VLVs that contain viral tegument and envelope proteins in addition to various cellular proteins. These VLVs were also found to be rich in viral and host microRNAs. In characterizing the function of KSHV-associated VLVs during viral replication, we found that VLVs could efficiently reactivate KSHV from latently infected cells as well as enhancing de novo lytic infection of native cells. In an attempt to explore the functional mechanism, we showed that VLVs activated the RTA promoter, the master regulator of gammaherpesvirus replication. Using RNA-seq, we found that VLVs upregulated a group of cellular genes involved in cellular differentiation signaling. A functional analysis confirmed that VLVs triggered a differentiation response in target cells by inducing PRDM1, the master transcriptional regulator during cell differentiation (46, 47). Interestingly, PRDM1 was also found to be necessary and sufficient for KSHV reactivation induced by VLVs.
Previous studies have identified VLVs, also termed L-particles or dense bodies, from alpha- and betaherpesviruses (7, 8, 53). HSV-1 L-particles are distinguishable from mature virions by use of a density gradient; the former appear as a broad, low-density band above the narrow, high-density virion band. L-particles contain viral envelope and tegument proteins and can deliver these proteins into target cells. Functional studies have also shown that L-particles are able to facilitate HSV-1 infection (13, 53) by enhancing virus replication and impairing the host immune response (52, 54). However, no systematic analysis of the protein and RNA compositions of HSV-1 L-particles, or the cellular response from targeted cells, has been carried out. Given that an alphaherpesvirus, a betaherpesvirus, and three gammaherpesviruses have been shown to produce VLVs (Fig. 1A), it seems likely that VLV production is a general feature of herpesviruses. Additionally, virion samples from the three gammaherpesviruses tested in this study contain large proportions of VLVs. Especially, EBV-associated VLVs constituted 84% of the total population (Fig. 1C). Therefore, some viral and cellular proteins, which were previously classified as virion components, may need to be reexamined, since they may be mostly or only present in VLVs. From the cryo-EM images, we quantified the ratios of VLVs to virions (Fig. 1C) and found that the percentage of VLVs in gammaherpesviruses is higher than those in HSV-1 and HCMV (data not shown). This finding indicates that VLVs may have a stronger impact on infection and/or pathogenesis for gammaherpesviruses than for alpha- and betaherpesviruses.
In this report, we also observed that VLVs were assembled in the same MVB-like compartments as virions (Fig. 2A). Taking this result together with the finding that VLVs and virions contain comparable amounts of envelope proteins and most tegument proteins, we conclude that VLVs may be generated in the same pathway as virions. Moreover, we propose that VLVs do not simply represent a by-product of virus infection, since the following evidence supports the intrinsic biological function of VLVs: (i) all gammaherpesviruses tested produce VLVs; (ii) production of VLVs is not restricted to a particular cell type or animal species; (iii) production of VLVs is not correlated to the yield of virions.
PRDM1, also known as Blimp-1, plays a critical role in controlling cell differentiation (46, 47). By using RNA-seq and functional assays, we found that PRDM1 upregulation is central to cellular differentiation responses induced by VLVs and KSHV reactivation. From studies of the regulation of PRDM1 expression, it is known that several signaling pathways, including extracellular signal-related kinases (ERKs) and NF-κB, can activate PRDM1 transcription (55, 56). Studies from Bala Chandran's group showed that NF-κB and ERK signaling were activated by the binding of KSHV envelope proteins to cell surface receptors (40, 57, 58). Based on the facts that abundant KSHV envelope proteins are present on VLVs (Table 1) and that VLVs are able to enter target cells (Fig. 5A), it is very likely that VLVs could activate NF-κB and ERK signaling. In addition, the hypothesis that VLVs trigger PRDM1 expression during their binding and/or entry is further supported by our finding that PRDM1 was activated very early but not continuously (Fig. 6D). However, the questions of whether and how ERK and NF-κB signaling pathways play a role in activating PRDM1 in the presence of VLVs still need to be examined. Future detailed signaling and transcriptional analysis is necessary to clarify the role of PRDM1 and its mechanism of function.
The role of RTA in regulating gammaherpesvirus lytic replication has been studied extensively. Expression of KSHV RTA is necessary and sufficient to switch on the lytic gene expression program from latency (59, 60). In this study, we investigated the mechanism of KSHV reactivation by VLVs and found that VLVs triggered the expression of KSHV RTA by activating its transcription (Fig. 5D, F, and G). Furthermore, we showed that RTA activation by VLVs depends on PRDM1 (Fig. 7B) and its ability to activate the KSHV RTA promoter (Fig. 7D). In a recent study, PRDM1 was found to induce EBV lytic reactivation by activating R (EBV RTA) and Z promoters (49). These findings support a common gammaherpesvirus strategy of control of lytic reactivation by PRDM1, especially through the control of RTA promoters. However, the mechanism by which PRDM1, normally considered a transcription suppressor, activates RTA promoters is not fully understood. Future studies of the interactions between RTA promoters and PRDM1 downstream factors may help answer this question.
In addition, VLVs generated from capsid assembly-deficient mutants offer obvious advantages for the development of a noninfectious subunit vaccine. Previous studies with HSV-1 L-particles were severely limited due to the inability to produce large amounts of L-particles free of contaminating wild-type virus. Although very few infectious particles are produced from KSHV-SCPnull, the risk of contamination could be further reduced by combining this mutation with mutations of other capsid proteins. In addition, an in vitro cell system enabling KSHV latent infection and robust reactivation has been established successfully (31, 32), and this system shows a significant advantage for producing VLVs on a large scale.
MATERIALS AND METHODS
Cells.
Human papillomavirus 16-immortalized keratinocytes, (HOK16B cells) were maintained in keratinocyte growth medium (Lonza) (61). HOK16B.219 cells were established by infection with KSHV.219 and were further selected and maintained with 0.5 μg/ml puromycin. HEK293T, iSLK-KSHV BAC16 (32), and iSLK-SCPnull (30) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. iSLK-KSHV and iSLK-SCPnull cells were maintained in the presence of 1 μg/ml puromycin, 250 μg/ml G418, and 1,200 μg/ml hygromycin B. NIH 3T3 cells were cultured in DMEM supplemented with 10% newborn calf serum. EBV-producing marmoset B cells (B95-8) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum.
Production and purification of virions (KSHV, MHV-68, and EBV) and KSHV-associated VLVs.
To produce wild-type KSHV or VLVs, iSLK-KSHV cells or iSLK-SCPnull cells were expanded to 30 15-cm plates and were then induced with 1 μg/ml doxycycline plus 1 mM sodium butyrate for 3 to 4 days, when 90% of the cells were round and detached from the plates. To produce MHV-68, 30 15-cm plates of NIH 3T3 cells were infected with MHV-68 at a multiplicity of infection (MOI) of 0.05 for 4 days. To produce EBV, 15 T175 flasks of B95-8 cells were induced with 25 ng/μl tetradecanoyl phorbol acetate (TPA) plus 0.5 mM sodium butyrate for 5 days. Virions and KSHV-associated VLVs were then purified from supernatants by a procedure described previously (30). Briefly, the supernatant was collected and was centrifuged at 10,000 × g for 10 min at 4°C to clear it of cells and debris. Then viral particles or VLVs were pelleted by ultracentrifugation (21,000 rpm for 1 h at 4°C; SW27 rotor), followed by resuspension in PBS. Viral particles or VLVs were further purified by ultracentrifugation using a 10-to-50% sucrose gradient; sharp viral particle bands or the corresponding section of VLVs was collected and then pelleted by ultracentrifugation. Pellets of virions or VLVs were resuspended in PBS, aliquoted, flash frozen in liquid nitrogen, and further stored at −80°C. The number of PFU of MHV-68 virions was determined by infecting Vero cells (62). Infectious units (IU) of KSHV virions were quantified by counting GFP-positive clusters after infecting 293T cells (61). The concentration of KSHV VLVs was measured using a Bradford protein assay.
Cryo-electron microscopy (cryo-EM).
Two microliters of a purified sample was applied to holey carbon-coated grids, plunge-frozen in liquid ethane, and imaged at liquid nitrogen temperature with an FEI Tecnai F20 transmission electron microscope.
Plastic embedding and transmission electron microscopy.
NIH 3T3 cells were infected with MHV-68 at an MOI of 0.1 for 3 days. Then the cells were collected and were subjected to plastic embedding, ultrathin sectioning, and transmission electron microscopy examination. Briefly, cells were washed twice with PBS, fixed with 2% glutaraldehyde, postfixed in 1% OsO4, en bloc stained in 2% uranyl acetate, dehydrated in an ascending ethanol series, and then embedded using Spurr resin (Electron Microscopy Sciences). Sections (∼75 nm) were stained with saturated aqueous uranyl acetate and lead citrate and were examined with an FEI Tecnai 12 transmission electron microscope.
Quantitative MS.
KSHV virion and VLV samples were prepared in duplicate, digested with trypsin, and concentrated for liquid chromatography (LC)-tandem mass spectrometry (MS-MS). Digested peptide mixtures were analyzed by a Thermo Scientific LTQ Orbitrap Elite mass spectrometry system equipped with a Proxeon Easy-nLC 1000 ultrahigh-pressure liquid chromatography and autosampler system.
KSHV titration.
To determine the concentration of infectious KSHV virions released from HOK16B.219 cells, the cells were treated with VLVs or PBS for 3 days. Then supernatants were collected, centrifuged at 10,000 × g for 10 min to remove cells and debris, serially diluted, and then used to infect 293T cells by spinoculation (at 3,000 × g for 1 h at 30°C). Three days postinfection, GFP-positive cell clusters containing two or more cells were counted under a fluorescence microscope to determine the titers of infectious KSHV viruses. Infectious units are expressed as the number of GFP-positive cell clusters in each well at the time of analysis (61).
Western blotting.
KSHV virions, VLVs, or cells were lysed in 1× Western blotting loading buffer, resolved by SDS-PAGE, and transferred to a polyvinylidene difluoride (PVDF) membrane. Viral proteins were detected with antibodies against K8.1A (Advanced Biotechnologies), ORF26 (Novus), ORF52 (monoclonal antibody produced from mouse hybridoma cells), or ORF45 (Abcam). PRDM1 expression was detected with antibodies against either the native protein (Cell Signaling) or the hemagglutinin (HA) epitope (Sigma).
Luciferase reporter assay.
A dual-luciferase reporter assay was performed according to a previously described method with some modifications (62). Briefly, 293T cells in a 12-well plate were transfected with 50 ng RTA promoter and 50 ng pRL-PGK, with or without pCMVHA-PRDM1 (100 ng, 300 ng, or 900 ng) and/or an empty vector. Two days later, the cells were lysed, and luciferase activity was examined using a dual-luciferase reporter assay system (Promega).
Reverse transcription and real-time PCR.
Quantitative reverse transcription-PCR (qRT-PCR) was performed as described previously (63). Briefly, total RNA was extracted from cells with a PureLink RNA minikit (Ambion), treated with DNase I, and reverse transcribed with SuperScript III reverse transcriptase (Thermo Fisher). The sequences of primers used to quantify DNA and RNA are as follows: for actin, 5′-GGA CTT CGA GCA AGA GAT GG-3′ and 5′-AGC ACT GTG TTG GCG TAC AG-3′; for RTA, 5′-CAC AAA AAT GGC GC AAG ATG A-3′ and 5′-TGG TAG AGT TGG GCC TTC AGT T-3′; for ORF59, 5′-TTG GCA CTC CAA CGA AAT ATT AGA A-3′ and 5′-CGG GAA CCT TTT GCG AAG A-3′; for ORF57, 5′-TGG ACA TTA TGA AGG GCA TCC TA-3′ and 5′-CGG GTT CGG ACA ATT GCT-3′; for ORF52, 5′-CTT ACG ATG GAA GAC CTA ACC G-3′ and 5′-ATC CCA GTG CTT TCC GAA G-3′; for ORF26, 5′-AGC CGA AAG GAT TCC ACC AT-3′ and 5′-TCC GTG TTG TCT ACG TCC AG-3′; for PRDM1, 5′-TGT GGT ATT GTC GGG ACT TTG-3′ and 5′-CTT TGG GAC ATT CTT TGG GC-3′; for SPRR2C, 5′-GAC TCC TAA ACT CCT GGT ACT TG-3′ and 5′-TGG CTC TGG ACG CTT TG-3′; for SPRR3, 5′-AAA GTT CCT GAG CAA GGA TAC AC-3′ and 5′-GAC ATG GCT CTG GTA GCT TTG-3′; for KRT34, 5′-AGG AGT CAG TAT GAG GCT CTG-3′ and 5′-GTG CGT CTC AGC TCG ATG-3′.
Directional RNA-seq library preparation and sequencing.
For poly(A) RNA deep-sequencing analysis, HOK16B or HOK16B.219 cells were either mock treated or treated with VLVs for 1 or 2 days; then total RNA was purified with TRIzol, and RNA-seq libraries were prepared by a method described previously (61). Multiplex sequencing was performed on 50-bp single-end reads with an Illumina HiSeq 2000 machine at the UCLA Clinical Microarray Core. Raw reads were aligned to a human genome assembly (hg38) or KSHV RNA transcripts using TopHat with default parameters (64). Results were quantified by reads per kilobase of coding region per million total reads (RPKM). For microRNA deep-sequencing analysis, total RNA was purified from KSHV-associated VLVs with TRIzol, and a miRNA-seq library was constructed using a TruSeq Small RNA Library Prep kit (Illumina). Deep sequencing was performed on 50-bp single-end reads with an Illumina HiSeq 2000 machine at the UCLA Clinical Microarray Core. Raw reads were aligned to both KSHV and human microRNAs using TopHat with default parameters.
MicroRNA deep sequencing.
Two independent batches of KSHV virion and VLV samples were purified, mixed in a 1:1 ratio (determined by protein concentration), and then subjected to total-RNA purification with TRIzol. MicroRNAs were further isolated by gel purification and were subjected to library construction using the TruSeq Small RNA Library Prep kit (Illumina) and deep sequencing with an Illumina HiSeq 2000 machine at the UCLA Clinical Microarray Core.
Statistical analysis.
All numerical data were calculated and plotted as means ± standard deviations (SD). Results were analyzed by an unpaired Student t test. Differences were considered statistically significant when the P value was <0.05.
Accession number(s).
Raw read data are available from NCBI Gene Expression Omnibus under accession number GSE91386.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Ren Sun and Ting-Ting Wu labs for helpful suggestions and discussions.
This work was supported by NIH grants CA091791, DE023591, and CA177322 to Ren Sun. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00362-17.
REFERENCES
- 1.Meckes DG Jr, Gunawardena HP, Dekroon RM, Heaton PR, Edwards RH, Ozgur S, Griffith JD, Damania B, Raab-Traub N. 2013. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc Natl Acad Sci U S A 110:E2925–E2933. doi: 10.1073/pnas.1303906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yáñez-Mó M, Siljander PRM, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, Colas E, Cordeiro-da Silva A, Fais S, Falcon-Perez JM, Ghobrial IM, Giebel B, Gimona M, Graner M, Gursel I, Gursel M, Heegaard NHH, Hendrix A, Kierulf P, Kokubun K, Kosanovic M, Kralj-Iglic V, Kramer-Albers EM, Laitinen S, Lasser C, Lener T, Ligeti E, Line A, Lipps G, Llorente A, Lotvall J, Mancek-Keber M, Marcilla A, Mittelbrunn M, Nazarenko I, Nolte-'t Hoen ENM, Nyman TA, O'Driscoll L, Olivan M, Oliveira C, Pallinger E, del Portillo HA, Reventos J, Rigau M, Rohde E, Sammar M, et al. . 2015. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4:27066. doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raposo G, Stoorvogel W. 2013. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nolte-'t Hoen E, Cremer T, Gallo RC, Margolis LB. 2016. Extracellular vesicles and viruses: are they close relatives? Proc Natl Acad Sci U S A 113:9155–9161. doi: 10.1073/pnas.1605146113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meckes DG Jr, Raab-Traub N. 2011. Microvesicles and viral infection. J Virol 85:12844–12854. doi: 10.1128/JVI.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalamvoki M, Deschamps T. 2016. Extracellular vesicles during herpes simplex virus type 1 infection: an inquire. Virol J 13:63. doi: 10.1186/s12985-016-0518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szilágyi JF, Cunningham C. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J Gen Virol 72(Part 3):661–668. doi: 10.1099/0022-1317-72-3-661. [DOI] [PubMed] [Google Scholar]
- 8.Craighead JE, Kanich RE, Almeida JD. 1972. Nonviral microbodies with viral antigenicity produced in cytomegalovirus-infected cells. J Virol 10:766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chai N, Chang HE, Nicolas E, Han Z, Jarnik M, Taylor J. 2008. Properties of subviral particles of hepatitis B virus. J Virol 82:7812–7817. doi: 10.1128/JVI.00561-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, Chisari FV. 2010. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol 84:10999–11009. doi: 10.1128/JVI.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantin R, Diou J, Belanger D, Tremblay AM, Gilbert C. 2008. Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J Immunol Methods 338:21–30. doi: 10.1016/j.jim.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Spehner D, Drillien R. 2008. Extracellular vesicles containing virus-encoded membrane proteins are a byproduct of infection with modified vaccinia virus Ankara. Virus Res 137:129–136. doi: 10.1016/j.virusres.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 13.McLauchlan J, Addison C, Craigie MC, Rixon FJ. 1992. Noninfectious L-particles supply functions which can facilitate infection by HSV-1. Virology 190:682–688. doi: 10.1016/0042-6822(92)90906-6. [DOI] [PubMed] [Google Scholar]
- 14.Sauer C, Klobuch S, Herr W, Thomas S, Plachter B. 2013. Subviral dense bodies of human cytomegalovirus stimulate maturation and activation of monocyte-derived immature dendritic cells. J Virol 87:11287–11291. doi: 10.1128/JVI.01429-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynolds TD, Buonocore L, Rose NF, Rose JK, Robek MD. 2015. Virus-like vesicle-based therapeutic vaccine vectors for chronic hepatitis B virus infection. J Virol 89:10407–10415. doi: 10.1128/JVI.01184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rose NF, Buonocore L, Schell JB, Chattopadhyay A, Bahl K, Liu X, Rose JK. 2014. In vitro evolution of high-titer, virus-like vesicles containing a single structural protein. Proc Natl Acad Sci U S A 111:16866–16871. doi: 10.1073/pnas.1414991111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becke S, Aue S, Thomas D, Schader S, Podlech J, Bopp T, Sedmak T, Wolfrum U, Plachter B, Reyda S. 2010. Optimized recombinant dense bodies of human cytomegalovirus efficiently prime virus specific lymphocytes and neutralizing antibodies without the addition of adjuvant. Vaccine 28:6191–6198. doi: 10.1016/j.vaccine.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Cayatte C, Schneider-Ohrum K, Wang ZT, Irrinki A, Nguyen N, Lu J, Nelson C, Servat E, Gemmell L, Citkowicz A, Liu Y, Hayes G, Woo J, Van Nest G, Jin H, Duke G, McCormick AL. 2013. Cytomegalovirus vaccine strain Towne-derived dense bodies induce broad cellular immune responses and neutralizing antibodies that prevent infection of fibroblasts and epithelial cells. J Virol 87:11107–11120. doi: 10.1128/JVI.01554-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pepperl S, Munster J, Mach M, Harris JR, Plachter B. 2000. Dense bodies of human cytomegalovirus induce both humoral and cellular immune responses in the absence of viral gene expression. J Virol 74:6132–6146. doi: 10.1128/JVI.74.13.6132-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boshoff C, Weiss R. 2002. AIDS-related malignancies. Nat Rev Cancer 2:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- 21.Sunil M, Reid E, Lechowicz MJ. 2010. Update on HHV-8-associated malignancies. Curr Infect Dis Rep 12:147–154. doi: 10.1007/s11908-010-0092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neparidze N, Lacy J. 2014. Malignancies associated with Epstein-Barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol 12:358–371. [PubMed] [Google Scholar]
- 23.Thompson MP, Kurzrock R. 2004. Epstein-Barr virus and cancer. Clin Cancer Res 10:803–821. doi: 10.1158/1078-0432.CCR-0670-3. [DOI] [PubMed] [Google Scholar]
- 24.Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. 2005. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol 79:13993–14003. doi: 10.1128/JVI.79.22.13993-14003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med 340:1063–1070. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- 26.Ganem D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu Rev Pathol 1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- 27.Chatterjee M, Osborne J, Bestetti G, Chang Y, Moore PS. 2002. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298:1432–1435. doi: 10.1126/science.1074883. [DOI] [PubMed] [Google Scholar]
- 28.Cesarman E, Nador RG, Bai F, Bohenzky RA, Russo JJ, Moore PS, Chang Y, Knowles DM. 1996. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J Virol 70:8218–8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu YM, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. 1999. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol 80:83–90. doi: 10.1099/0022-1317-80-1-83. [DOI] [PubMed] [Google Scholar]
- 30.Dai X, Gong D, Xiao Y, Wu TT, Sun R, Zhou ZH. 2015. CryoEM and mutagenesis reveal that the smallest capsid protein cements and stabilizes Kaposi's sarcoma-associated herpesvirus capsid. Proc Natl Acad Sci U S A 112:E649–E656. doi: 10.1073/pnas.1420317112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pellett PE, Roizman B. 2007. The family Herpesviridae: a brief introduction, p 2479–2500. In Knipe DM, Howley PM (ed), Fields virology, 5th ed Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 34.Dai X, Gong D, Wu TT, Sun R, Zhou ZH. 2014. Organization of capsid-associated tegument components in Kaposi's sarcoma-associated herpesvirus. J Virol 88:12694–12702. doi: 10.1128/JVI.01509-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79:4952–4964. doi: 10.1128/JVI.79.8.4952-4964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol 79:800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin X, Li X, Liang D, Lan K. 2012. MicroRNAs and unusual small RNAs discovered in Kaposi's sarcoma-associated herpesvirus virions. J Virol 86:12717–12730. doi: 10.1128/JVI.01473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jochum S, Ruiss R, Moosmann A, Hammerschmidt W, Zeidler R. 2012. RNAs in Epstein-Barr virions control early steps of infection. Proc Natl Acad Sci U S A 109:E1396–E1404. doi: 10.1073/pnas.1115906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chugh PE, Sin SH, Ozgur S, Henry DH, Menezes P, Griffith J, Eron JJ, Damania B, Dittmer DP. 2013. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog 9:e1003484. doi: 10.1371/journal.ppat.1003484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandran B. 2010. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J Virol 84:2188–2199. doi: 10.1128/JVI.01334-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Cao Y, Liang D, Gao Y, Xia T, Robertson ES, Lan K. 2008. Kaposi's sarcoma-associated herpesvirus RTA activates the processivity factor ORF59 through interaction with RBP-Jκ and a cis-acting RTA responsive element. Virology 380:264–275. doi: 10.1016/j.virol.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song MJ, Li XD, Brown HJ, Sun R. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J Virol 76:5000–5013. doi: 10.1128/JVI.76.10.5000-5013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song MJ, Deng H, Sun R. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J Virol 77:9451–9462. doi: 10.1128/JVI.77.17.9451-9462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kypriotou M, Huber M, Hohl D. 2012. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp Dermatol 21:643–649. doi: 10.1111/j.1600-0625.2012.01472.x. [DOI] [PubMed] [Google Scholar]
- 46.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. 2002. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17:51–62. doi: 10.1016/S1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 47.Bikoff EK, Morgan MA, Robertson EJ. 2009. An expanding job description for Blimp-1/PRDM1. Curr Opin Genet Dev 19:379–385. doi: 10.1016/j.gde.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Johnson AS, Maronian N, Vieira J. 2005. Activation of Kaposi's sarcoma-associated herpesvirus lytic gene expression during epithelial differentiation. J Virol 79:13769–13777. doi: 10.1128/JVI.79.21.13769-13777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reusch JA, Nawandar DM, Wright KL, Kenney SC, Mertz JE. 2015. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J Virol 89:1731–1743. doi: 10.1128/JVI.02781-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson SJ, Tsao EH, Webb BLJ, Ye HT, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol 81:13578–13586. doi: 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu FQ, Feng JY, Harada JN, Chanda SK, Kenney SC, Sun R. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated herpesvirus. FEBS Lett 581:3485–3488. doi: 10.1016/j.febslet.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 52.Temme S, Eis-Hubinger AM, McLellan AD, Koch N. 2010. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J Immunol 184:236–243. doi: 10.4049/jimmunol.0902192. [DOI] [PubMed] [Google Scholar]
- 53.Dargan DJ, Subak-Sharpe JH. 1997. The effect of herpes simplex virus type 1 L-particles on virus entry, replication, and the infectivity of naked herpesvirus DNA. Virology 239:378–388. doi: 10.1006/viro.1997.8893. [DOI] [PubMed] [Google Scholar]
- 54.Heilingloh CS, Kummer M, Muhl-Zurbes P, Drassner C, Daniel C, Klewer M, Steinkasserer A. 2015. L particles transmit viral proteins from herpes simplex virus 1-infected mature dendritic cells to uninfected bystander cells, inducing CD83 downmodulation. J Virol 89:11046–11055. doi: 10.1128/JVI.01517-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yasuda T, Kometani K, Takahashi N, Imai Y, Aiba Y, Kurosaki T. 2011. ERKs induce expression of the transcriptional repressor Blimp-1 and subsequent plasma cell differentiation. Sci Signal 4:ra25. doi: 10.1126/scisignal.2001592. [DOI] [PubMed] [Google Scholar]
- 56.Calame K. 2008. Activation-dependent induction of Blimp-1. Curr Opin Immunol 20:259–264. doi: 10.1016/j.coi.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 57.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol 79:10308–10329. doi: 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J Virol 81:3949–3968. doi: 10.1128/JVI.02333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deng H, Liang Y, Sun R. 2007. Regulation of KSHV lytic gene expression. Curr Top Microbiol Immunol 312:157–183. [DOI] [PubMed] [Google Scholar]
- 60.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gong D, Wu NC, Xie Y, Feng J, Tong L, Brulois KF, Luan H, Du Y, Jung JU, Wang CY, Kang MK, Park NH, Sun R, Wu TT. 2014. Kaposi's sarcoma-associated herpesvirus ORF18 and ORF30 are essential for late gene expression during lytic replication. J Virol 88:11369–11382. doi: 10.1128/JVI.00793-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qi J, Han C, Gong D, Liu P, Zhou S, Deng H. 2015. Murine gammaherpesvirus 68 ORF48 is an RTA-responsive gene product and functions in both viral lytic replication and latency during in vivo infection. J Virol 89:5788–5800. doi: 10.1128/JVI.00406-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gong D, Kim YH, Xiao Y, Du Y, Xie Y, Lee KK, Feng J, Farhat N, Zhao D, Shu S, Dai X, Chanda SK, Rana TM, Krogan NJ, Sun R, Wu TT. 2016. A herpesvirus protein selectively inhibits cellular mRNA nuclear export. Cell Host Microbe 20:642–653. doi: 10.1016/j.chom.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T, Hatzigeorgiou AG. 2015. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res 43:W460–W466. doi: 10.1093/nar/gkv403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.