

Significance
Staphylococcus aureus is one of the most adaptable and prolific human pathogens, and it employs an arsenal of virulence factors to infect blood, bone, and soft tissues. The type VII secretion system (T7SS) is a dedicated virulence protein-secretion pathway that enables long-term survival of the bacteria in abscesses, where they are protected from host immune cells. Here we report that host-derived fatty acids are incorporated into the S. aureus membrane, altering bacterial membrane properties and activating the expression of the T7SS. Thus, this work identifies a mechanism by which an important human pathogen senses unique elements of the host environment and implements the expression of specific genes that enable bacterial survival and thereby promote human disease.
Keywords: type VII secretion, Staphylococcus aureus, fatty acids, virulence
Abstract
The type VII secretion system (T7SS) of Staphylococcus aureus is a multiprotein complex dedicated to the export of several virulence factors during host infection. This virulence pathway plays a key role in promoting bacterial survival and the long-term persistence of staphylococcal abscess communities. The expression of the T7SS is activated by bacterial interaction with host tissues including blood serum, nasal secretions, and pulmonary surfactant. In this work we identify the major stimulatory factors as host-specific cis-unsaturated fatty acids. Increased T7SS expression requires host fatty acid incorporation into bacterial biosynthetic pathways by the S. aureus fatty acid kinase (FAK) complex, and FakA is required for virulence. The incorporated cis-unsaturated fatty acids decrease S. aureus membrane fluidity, and these altered membrane dynamics are partially responsible for T7SS activation. These data define a molecular mechanism by which S. aureus cells sense the host environment and implement appropriate virulence pathways.
The bacterium Staphylococcus aureus is the leading cause of blood, skin, and soft tissue infections in the United States, and its treatment remains an important medical challenge worldwide. The increasingly common presence of the mecA gene, which confers resistance to the most frequently used antibiotic, methicillin, in clinically relevant strains of S. aureus has made these infections particularly challenging to treat. Despite its role as a pathogen, S. aureus is also a common component of the human skin and mucosal microbiota, colonizing the nares of nearly 30% of healthy individuals (1). One reason for this apparent paradox is the diverse arsenal of virulence factors that clinically relevant S. aureus strains, such as USA300, specifically deploy upon bacterial penetration of dermal and mucosal barriers. For example, transcription of a key virulence factor, Panton–Valentine leukocidin (LukF-PV), is decreased during nasal colonization but is substantially up-regulated during blood and cardiac infection (2). Some virulence genes enhance bacterial survival in blood through a variety of mechanisms, including the scavenging of scarce nutrients, as well as by evading and neutralizing the host immune response. Other virulence genes encode secreted and surface-anchored proteins that promote long-term survival via the formation of staphylococcal abscess communities that serve as replicative niches for the bacteria (3). One set of genes that promotes the long-term persistence of S. aureus in abscesses is the 12-gene operon encoding the type VII secretion system (T7SS), a multiprotein complex dedicated to the transport of several effector proteins into the extracellular milieu. Deciphering how the host environment affects expression of virulence pathways, such as the T7SS, is critical to understanding bacterial transition from mucosal colonizer to pathogen and thus is key to deciphering S. aureus-mediated diseases.
T7SSs are encoded in the genomes of diverse pathogenic and nonpathogenic bacteria across the Actinobacteria and Firmicutes phyla. The best studied of these is the Esx-1 secretion system of Mycobacteria, which is conserved among pathogenic Mycobacteria (as well as the commensal Mycobacterium smegmatis) and is essential for virulence in vivo (4). The T7SS of S. aureus similarly plays a key role in the virulence of staphylococcal infections in a variety of mouse infection models. At the molecular level, the S. aureus T7SS is composed of four membrane-associated proteins (EsaA, EssA, EssB, and EssC), three soluble cytosolic proteins (EsaB, EsaE, and EsaG), and five secreted virulence factors (EsxA, EsxC, EsxB, EsxD, and EsaD) (Fig. 1A). Through a poorly understood mechanism, the membrane and cytoplasmic proteins cooperate to export the effector proteins during host infection. Inactivation of the secretion apparatus or deletion of the secreted proteins results in decreased bacterial survival and a decrease in the number and size of abscesses in host kidneys, liver, and spleen in various mouse infection models (5–7).
Fig. 1.

Host fatty acids stimulate the expression of T7SS. (A) Diagram illustrating the genetic organization of genes encoding the T7SS. (B) USA300 WT and essB strains were grown in TSB alone or TSB supplemented with 0.25% human serum (HS). (Left) Whole-cell lysates and supernatants were assessed for EsxA protein by Western blot. (Right) Quantitation of the fold increase in EsxA protein stimulated by human serum. Data are from four independent experiments. (C) USA300 cultures were grown in TSB alone, TSB supplemented with 0.25% human serum, or TSB supplemented with 0.25% of the aqueous (lipid-free) or organic (org.) extract of human serum. EsxA and RpoB proteins in whole-cell lysates were measured by Western blot. (D) USA300 cultures were grown in TSB supplemented with various concentrations (20, 10, 5, 2.5, 1.25, or 0.625 µM, or zero) of LA or OA. EsxA protein in whole-cell lysates was measured by Western blot. (Upper) The dose-dependent stimulation of EsxA protein was fit to a sigmoid function and plotted. (Lower) The EsxA Western blot. Whole-cell lysates were also probed for RpoB by Western blot to demonstrate equal sample loading. (E) USA300 cultures were grown in TSB alone or in TSB supplemented with 10 µM LA, and total RNA was isolated. The relative mRNA levels of esxA, esaA, essC, esxC, and esaD were measured by RT-qPCR and normalized to the ribosomal gene rrsA. WT mRNA levels for each gene were normalized to 1 in each experiment. Relative mRNA levels are reported as the average of at least two independent experiments with three biological replicates in each experiment. Error bars represent the SEM; significance was evaluated using the Mann–Whitney u test: *P ≤0.05, **P ≤0.01, ****P ≤0.0001.
During in vitro growth in liquid culture, basal levels of T7SS proteins vary among different S. aureus strains (6, 8). However, we and others have found that S. aureus substantially increases expression of the T7SS upon exposure to host factors such as blood serum (9) and pulmonary surfactant (10). This is reminiscent of other better-studied environmental sensing pathways whereby pathogens use cues unique to the host environment to activate virulence mechanisms. For example, neutrophil-derived antimicrobial peptides are sensed by the SaeRS two-component system (TCS) and the Aps three-component sensor/regulator, and S. aureus gene expression is altered to protect against this insult (11, 12). To better understand regulation of this important pathway in S. aureus-mediated disease, we sought to identify the host factor(s) responsible for S. aureus T7SS activation, to elucidate its mechanism of action, and to ascertain whether this regulation represents a physiologically important pathway for S. aureus survival during host infection.
Results
Host Fatty Acids Stimulate Expression of the T7SS in S. aureus.
S. aureus cells grown in tryptic soy broth (TSB) supplemented with human serum or nasal secretions increased EsxA protein levels four- to eightfold in whole-cell lysates and resulted in EsxA secretion into the medium (Fig. 1B and Fig. S1A). Horse and mouse serum exhibited a similar effect, while FBS did not (Fig. S1B). The nasal secretion- and serum-stimulated EsxA was secreted into the extracellular medium in an essB-dependent manner, as expected for T7SS substrates. The expression of T7SS genes is dependent upon agr signaling (13); thus, higher concentrations of serum may have less stimulatory activity due to the presence of agr-inhibitory proteins such as hemoglobin (14) and apolipoprotein B (15). Horse serum proteins are reported to enhance the expression of the T7SS proteins including EsxC and EsaD (16). However, neither heat inactivation nor tryptic digestion of human serum diminished its activity, suggesting that the T7SS-activating factor is not a polypeptide (Fig. S1 C and D). In contrast, lipid-depleted serum, prepared by organic extraction, completely lacked T7SS-stimulating activity, while the organic extract retained the activity of untreated human serum (Fig. 1C). Thus, the serum factor responsible for EsxA activation is not a protein but is instead a lipophilic molecule found in human, mouse, and horse serum but not in FBS.
Fig. S1.

(A) USA300 WT or essB-mutant cells were grown in TSB alone or in TSB supplemented with 2.5% nasal secretions (NS). (Left) Whole-cell lysates and supernatants were assessed for EsxA protein by Western blot. (Right) Bar graphs represent the quantitation of EsxA stimulation in whole cells and supernatants from two independent experiments. (B) USA300 cells were grown in TSB alone or in TSB supplemented with the indicated concentrations and types of serum. Whole-cell lysates were assessed for EsxA protein by Western blot. (C) (Right) Human serum was digested with trypsin, and the degree of proteolysis was assessed by simplyBlue stain (Applied Biosystems). (Left) USA300 cells were grown in TSB alone or in TSB supplemented with the indicated concentration of human serum. Whole-cell lysates were analyzed for EsxA protein by Western blot. (D) Human serum either was left untreated or was heat-inactivated by incubation at 55 °C for 30 min. USA300 cells were grown in TSB alone or in TSB supplemented with varying concentrations (20, 4, 0.8, or 0.16%) of untreated human serum or heat-inactivated human serum. Whole-cell lysates were assessed for EsxA protein by Western blot. (E) USA300, USA400, and Newman cells were grown in TSB alone or in TSB supplemented with 10 µM LA. Whole-cell lysates (Left) and supernatants (Right) were assessed for EsxA and RpoB protein by Western blot. (F) Human serum and LA were preincubated with varying concentrations of BSA (50, 5, or 0.5 mg/mL, or zero). USA300 cells were grown in TSB alone or in TSB supplemented with the BSA-treated 0.25% human serum (Left) or 10 µM LA (Right). Whole-cell lysates were assessed for EsxA protein by Western blot.
A comparative lipidomics analysis of horse serum and FBS reported the unsaturated fatty acid linoleic acid (LA) is ninefold more abundant in the former, representing half of all the fatty acids present (17). Additionally, a study of the S. aureus transcriptional response to antimicrobial fatty acids found esxA mRNA levels increased upon growth in the presence of 10 μM LA (18). Thus, we hypothesized that LA is likely one of the serum factors responsible for enhanced EsxA protein expression. In accordance with this hypothesis, purified LA induced a dose-dependent increase in EsxA protein levels in USA300 S. aureus cells (Fig. 1D). The maximal stimulation occurred at a concentration of 10–20 μM LA and was similar in magnitude to maximal stimulation by human serum. The structurally related molecule oleic acid (OA) exhibited a similar but less potent stimulating activity (Fig. 1D and Table S1). Furthermore, 10 μM LA increased the mRNA transcript level of genes across the entire T7SS operon (Fig. 1E). LA (10 µM) also enhanced the expression of EsxA in S. aureus strains Newman and USA400, thereby demonstrating that the effect of LA is relevant to multiple S. aureus strains (Fig. S1E). Preincubation of human serum with the fatty acid-binding protein BSA depleted the serum of its EsxA-stimulating activity in a dose-dependent manner (Fig. S1F). We concluded that fatty acids are an important, if not the primary, EsxA-stimulating factor in human, horse, and mouse serum and that LA, because of its abundance, is likely the primary fatty acid responsible for this activity.
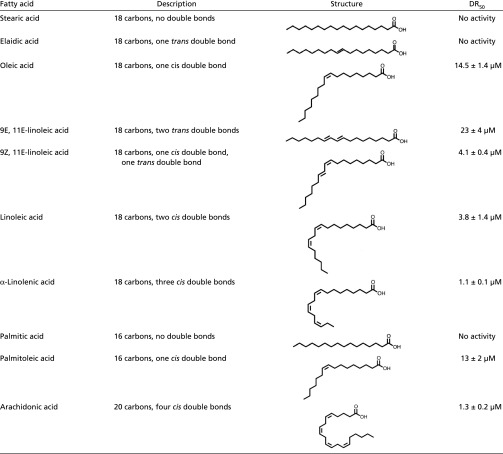
Table S1.
SARs of EsxA stimulation by strain-chain free fatty acids
 |
Unsaturated Fatty Acids Do Not Activate EsxA via Membrane Stress.
High concentrations of free fatty acids disrupt the bacterial cell membrane, leading to depolarization, leakage of small proteins, and ultimately cell death (19). This fact motivated us to investigate whether LA activation of EsxA might be the result of a general membrane stress response. Although 10 μM LA elicits maximal EsxA protein expression, there is no growth defect for S. aureus below 40 μM LA, thereby indicating a minimal impact on membrane integrity at LA concentrations relevant to our experiments (Fig. S2A). Additionally, EssB is required for secretion of the 11-kDa EsxA protein (Fig. 1B), implying an intact cell membrane that does not allow passive diffusion of small proteins into the extracellular space.
Fig. S2.

(A) USA300 cells were grown in TSB supplemented with the indicated concentration of LA in a 96-well plate at 37 °C with shaking. The optical density (600 nm) was measured at the indicated time points over the course of 7 h. The values reported are the average of three biological replicates; error bars represent the SE. (B, Left) USA300 cells were grown in TSB alone or in TSB supplemented with 10 µM LA or subinhibitory concentrations of daptomycin (1, 0.25, 0.063, or 0.016 µg/mL). (Right) USA300 cells were grown in TSB supplemented with the indicated chemical agents (twofold below the growth-inhibitory concentration). Whole-cell lysates were analyzed for EsxA protein by Western blot. (C) USA300 WT or fakA-mutant cells were grown in TSB alone or in TSB supplemented with the indicated concentrations of human serum. Whole-cell lysates were assessed for EsxA protein by Western blot using fluorescent secondary antibodies. The data represent the average values from three independent experiments; error bars represent the SE. (D) Overview of the fatty acid biosynthetic pathways in S. aureus. (E) The indicated USA300 strains were grown in TSB alone or in TSB supplemented with 10 µM LA, and whole-cell lysates were assessed for EsxA protein by Western blot. The ratio of EsxA protein (10 µM LA:TSB alone) was reported. Data represent the average of three biological replicates (minimum of two independent experiments); error bars represent the SE.
We also performed an RNA-sequencing (RNA-seq) experiment to measure global transcriptional changes in response to 10 μM LA and to determine whether membrane stress genes are up-regulated. The vast majority of genes were unaffected by 10 µM LA, as only 1.7% of genes are increased and 0.4% are decreased more than fourfold (Dataset S1). Expression of membrane stress-response genes, such as vraRS and asp23, as well as general stress-responsive genes, such as groEL/ES, clpP, and dnaK/J/grpE, was unaltered by the presence of 10 μM LA (Table S2 and Dataset S1). Finally, membrane-disrupting agents such as SDS, ethanol, or daptomycin had little effect on EsxA protein levels in USA300 cells (Fig. S2B) or in the N315 strain (20). In summary, USA300 cells grown in the presence of 10 μM LA do not resemble cells undergoing membrane stress, nor do membrane-damaging agents elicit EsxA expression. These data led us to conclude that LA-stimulated EsxA expression occurs via a specific signaling pathway and is not part of a general membrane stress response.
Table S2.
Expression of stress-response genes upon growth in 10 µM LA
| Gene | Fold change | P value | Gene function |
| esxA | 7.57 | 3 × 10−13 | Type VII secretion (5) |
| esaA | 3.86 | 4 × 10−12 | |
| essA | 4.32 | 9 × 10−11 | |
| esaB | 3.53 | 2 × 10−6 | |
| essB | 3.84 | 5 × 10−9 | |
| essC | 6.96 | 3 × 10−13 | |
| esxC | 6.73 | 2 × 10−10 | |
| esxB | 7.01 | 2 × 10−11 | |
| esaE | 7.62 | 5 × 10−12 | |
| esxD | 8.46 | 1 × 10−11 | |
| esaD | 4.14 | 2 × 10−11 | |
| esaG | 2.22 | 1 × 10−8 | |
| vraR | 1.10 | 0.19 | Two-component system, cell wall stress response (20) |
| vraS | 1.07 | 0.46 | |
| groEL | 0.68 | 1 × 10−5 | Chaperonins, cell wall stress response (20) |
| groES | 0.48 | 2 × 10−7 | |
| asp23 | 1.07 | 0.24 | Alkaline/cell wall stress response (39) |
| dnaJ | 0.98 | 0.60 | Chaperonins, cell wall stress response (20) |
| dnaK | 0.92 | 0.09 | |
| grpE | 0.77 | 2 × 10−4 | |
| clpP | 0.99 | 0.9 | Proteostasis, stress tolerance (40) |
Stimulation of EsxA Requires Incorporation of LA into Biosynthetic Pathways by Fak.
S. aureus are unable to produce unsaturated fatty acids, such as LA, as they lack the oxidative enzymes required for fatty acid olefination; thus, the LA that leads to expression of the T7SS must come from an exogenous source. S. aureus cells readily incorporate environmental fatty acids into their phospholipids and lipoproteins, and fatty acid kinase (FAK) is a cytoplasmic enzyme complex required for the incorporation of exogenous fatty acids into S. aureus biosynthetic pathways (21, 22). Free fatty acids in the extracellular environment are thought to passively insert into the outer leaflet of the S. aureus cell membrane and spontaneously flip to the inner leaflet in a pH-dependent manner. FakA works in conjunction with two semiredundant proteins, FakB1 and FakB2, to extract both saturated and unsaturated free fatty acids from the membrane and to phosphorylate the carboxyl oxygen, activating them for transfer to glycerol 3-phosphate by the enzyme PlsY (23). The fatty-acyl glycerol 3-phosphate intermediate is a key substrate for the synthesis of phosphatidic acid and other fatty acyl-containing biomolecules such as LPs, lipoteichoic acid (LTA), and various components of the phospholipid membrane.
USA300 cells lacking a functional fakA gene were insensitive to stimulation by LA or human serum (Fig. 2A and Fig. S2C). The EsxA-stimulating activity of LA in fakA-mutant cells could be rescued by expression of full-length FakA from a plasmid. Inactivation of individual fakB genes had no effect on LA stimulation of EsxA due to their redundant function in cells; the double mutant fakB1fakB2, however, did not up-regulate EsxA in response to incubation with 10 µM LA (Fig. 2A). Linoleamide (LAm) is a LA derivative that has a structure identical to that of LA but cannot be phosphorylated by FakA due to the amide substitution of the phosphorylatable carboxyl group. LAm had no effect on EsxA protein expression at the highest concentration tested (10 μM), indicating that FakA phosphorylation of the exogenous LA is required, and passive insertion of the cis-unsaturated lipid into the bacterial membrane is not sufficient for increased EsxA expression (Fig. 2B). Similarly, while OA stimulated EsxA expression, the nonphosphorylatable OA analog oleyl sulfate did not. These data demonstrate that LA-dependent stimulation of EsxA requires its incorporation into S. aureus biosynthetic pathways via phosphorylation by the bacterial fatty acid kinase complex FAK.
Fig. 2.

Fatty acid stimulation of EsxA requires Fak activity. (A) USA300 cultures (WT, fakA-mutant, complemented fakA, fakB1-mutant, fakB2-mutant, or fakB1fakB2-mutant) were grown in TSB alone or in TSB supplemented with the indicated concentration of LA. Whole-cell lysates were assessed for EsxA protein by Western blot, and the fold-stimulation (relative to TSB alone) was reported. Each measurement is the average of a minimum of three independent experiments; error bars represent the SEM. Significance was evaluated using the Mann–Whitney u test: ***P ≤ 0.001; n.s., not significant, >0.05. (B) USA300 cultures were grown in TSB or in TSB supplemented with 10 µM OA, 10 µM LA, 10 µM oleyl sulfate, or 10 µM LAm. EsxA protein was measured by Western blot and reported as described in A. Significance was evaluated using the Mann–Whitney u test: ***P ≤ 0.001, ****P ≤ 0.0001. (C) C57BL/6 mice were infected via tail vein injection with 1 × 107 cfu of USA300 WT, fakA-mutant, or essB-mutant strains, and the mice were monitored for survival over 8 d. The reported data represent three independent experiments (n = 14 or 15 for each strain for each experiment). Significance between mutants and WT bacteria were determined by using the Mann–Whitney u test. (D) Surviving mice were killed after 8 d, livers were homogenized, and S. aureus was enumerated. Each data point represents cfus from one liver. Significance was evaluated using the Mann–Whitney u test: *P ≤ 0.05, **P ≤ 0.01.
Next, we sought to determine which pathway(s) downstream of FAK might be involved in LA-dependent stimulation of EsxA. Fatty acids are primarily incorporated into six molecules in S. aureus: phosphatidic acid (PA), phosphatidyl glycerol (PG), LTA, LPs, lysyl-phosphatidyl glycerol (L-PG), and cardiolipin (CL) (Fig. S2D) (23). PG is a core component of the cell membrane, and both PG and PA are precursors to LTA, LP, L-PG, and CL; thus, their central roles preclude inactivation of the essential genes responsible for their production. However, the genes required for synthesis of LTA, LP, L-PG, and CL are dispensable, enabling us to test whether these pathways are required for LA stimulation of T7SS. We found that USA300 strains lacking any of the individual genes required for the synthesis of LTA (ltaS, ypfP), CL (cls1, cls2), LP (lgt), or L-PG (fmtC) remain sensitive to LA stimulation of EsxA protein (Fig. S2E). Thus, FAK-mediated phosphorylation of exogenous LA is required for induction of EsxA expression, likely through incorporation into phospholipid membranes as PA, PG, or a combination of fatty acyl molecules.
Inactivation of fakA Recapitulates the essB Phenotype in a Mouse Infection Model.
Our findings raised the possibility that enhancement of T7SS gene expression by host-derived fatty acids is an important virulence signal in S. aureus infection. The T7SS has been shown to play an important role in virulence for several different S. aureus strains, including the common laboratory strains Newman, COL, SA1113, and RN6890 (5, 6, 9, 16) as well as ST398, an emerging cause of community-acquired infections in humans that was originally found in livestock (7). However, whether regulation of T7SS gene expression occurs in vivo and whether host-derived fatty acids may contribute is unknown. We hypothesized that, if host-derived fatty acids have a role in the virulence function of T7SS, the fakA mutant should phenocopy the virulence defect of a T7SS-deficient strain. To test this, we infected C57BL/6 mice with USA300 WT or essB- or fakA-mutant strains i.v. via tail vein injection. Mice infected with WT USA300 had a threefold higher mortality rate than mice infected with either essB or fakA mutants, indicating a virulence defect for both mutants (Fig. 2C). Bacterial cfu recovered from liver were also significantly greater in mice infected with WT USA300 than in mice infected with essB or fakA (Fig. 2D). Interestingly, there was no difference in cfu recovered from kidneys and no defect in the severity of kidney abscesses for USA300 strains lacking either essB or fakA (Fig. S3A). Thus, the USA300 fakA mutant exhibits a tissue-specific virulence defect very similar to that of the USA300 essB mutant in a mouse model of S. aureus bacteremia.
Fig. S3.

C57BL/6 mice were infected via tail vein injection with 1 × 107 cfu of USA300 WT, fakA-, or essB-mutant strains. (Upper Left) After 8 d, the mice were killed, and bacterial survival in one kidney from each surviving mouse was enumerated. (Upper Right) The remaining kidney was formalin fixed, processed by routine methods, and H&E stained. Histologic sections were manually scored for pyelonephritis severity on a four-point scale based on the microscopically estimated fraction of tissue involved by inflammation and/or necrosis: 1, <1% involvement; 2, 1–5% involvement; 3, 5–25% involvement; 4, >25% involvement. (Lower) Gram staining (images) confirms renal Gram-positive bacterial microabscesses associated with features of microfilm (amorphous matrix and branching channels), regardless of genotype.
cis-Unsaturated Fatty Acids, but Not Their Saturated or Trans-Unsaturated Counterparts, Activate T7SS Expression.
To gain insight into the molecular mechanism of LA-dependent EsxA stimulation, we examined the structure–activity relationships (SAR) of a panel of fatty acids (Table S1). Specifically, we measured the dose-dependent increase in EsxA expression of USA300 cells grown in the presence of various C18 fatty acids and determined the half-maximal stimulating concentration (DR50) of each. LA, a straight-chain fatty acid with two cis double bonds, enhanced EsxA protein levels fivefold, with a calculated DR50 of 3.8 ± 1.4 μM. OA, which has one cis double bond, was nearly fourfold less potent than LA (DR50 = 14.5 ± 1.4 μM). In contrast, α-linolenic acid (α-LA), which has a total of three cis double bonds, was more than threefold more potent than LA (DR50 = 1.1 ± 0.1 μM). Strikingly, the saturated analog stearic acid (SA), and the trans-unsaturated elaidic acid (EA) had no stimulatory effect, even at the highest concentration tested (20 μM). Conjugated C18 fatty acids such as 9Z, 11E-linoleic acid, and 9E, 11E-linoleic acid similarly showed greater activity when cis double bonds were present in the molecule. Fatty acids of different lengths, such as palmitic acid, palmitoleic acid, and arachidonic acid, also displayed the same correlation between increasing cis double bonds and more potent EsxA stimulation. Thus, fatty acids with more cis double bonds are more potent EsxA activators, while saturated and trans-unsaturated fatty acids have little or no effect. Importantly, S. aureus lacks the ability to olefinate fatty acids, so these activators of T7SS gene expression must come from an exogenous source such as host tissues.
LA Alters S. aureus Membrane Fluidity in a FAK-Dependent Manner.
The viscosity of cell membranes is a fundamental property that governs cell integrity, limits lateral diffusion of molecules within the lipid bilayer, and affects the activity of membrane-associated signaling enzymes. Incorporation of nonnative fatty acids into membrane-associated molecules is likely to affect these properties. Thus, we examined the effects of LA on the biophysical properties of the S. aureus cytosolic membrane by measuring membrane fluidity of USA300 cells grown in TSB alone or supplemented with 10 μM SA, 10 μM OA, or 10 μM LA, using the excimer-forming lipid technique (24). USA300 cells grown in medium supplemented with 10 µM LA or 10 µM OA, which activate EsxA production, exhibited more rigid membranes than cells grown with 10 μM SA or without supplemented fatty acids, conditions in which EsxA production is low (Fig. 3A). S. aureus bacteria with more rigid cell membranes are better able to survive incubation with the membrane-disrupting agent SDS (25). As expected from our membrane fluidity experiment, LA-treated USA300 cells were better able to survive SDS treatment than cells grown in TSB alone or with 10 μM PA (Fig. S3B). USA300 fakA-mutant cells have more rigid membranes overall, but their membrane fluidity is not altered upon LA treatment (Fig. 3B). In summary, exogenous unsaturated fatty acids that stimulated EsxA expression also decreased the fluidity of S. aureus membranes, whereas saturated fatty acids had no effect on either EsxA levels or membrane fluidity.
Fig. 3.

Unsaturated fatty acids decrease S. aureus membrane fluidity, resulting in enhanced EsxA expression. (A) USA300 cultures were grown in TSB alone or in TSB supplemented with the indicated fatty acids. The membrane fluidity of the bacteria was determined at various temperatures using the pyrene decanoic acid fluorescence method. (B) USA300 WT or fakA-mutant strains were grown in TSB alone or in TSB supplemented with 10 µM LA, and the membrane fluidity of the bacteria at 37 °C was determined as in A. The percent change in membrane fluidity with or without LA is plotted. (C) USA300 WT or fakA-mutant strains were grown at the indicated temperatures in TSB alone or in TSB supplemented with 10 µM LA. Whole-cell lysates were assessed for EsxA protein by Western blot. The fold increase in EsxA protein for each condition was plotted relative to the average EsxA protein for the indicated strain grown in TSB alone at 37 °C. (D) USA300 WT or pCrtMN strains were grown in TSB alone or in TSB supplemented with 10 µM LA. Whole-cell lysates were assessed for EsxA protein by Western blot. The fold increase in EsxA protein was plotted relative to the average EsxA protein level in the indicated strain grown in TSB alone. Significance was evaluated using the Mann–Whitney u test: **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
To determine whether changes in membrane fluidity have a role in the mechanism of LA stimulation of T7SS, we performed studies in which we altered this property without the addition of exogenous fatty acids. When cells are shifted to higher temperatures, their membranes become more fluid (26), and when the temperature is decreased, their membranes become more rigid (27). Growth of S. aureus at 30 °C increased basal EsxA protein abundance two- to threefold, while growth at 42 °C suppressed the basal EsxA protein level fivefold (Fig. 3C). We also conducted an RNA-seq experiment comparing mRNA levels in USA300 cells grown at 30 °C and 37 °C and found that T7SS transcripts are increased two- to threefold at 30 °C (Fig. S4 and Dataset S1). Indeed, other than urease genes, T7SS were the only genes up-regulated by both 10 µM LA and deceased temperature, two conditions which decrease membrane fluidity. The fakA mutant strain exhibited a similar response to temperature, suggesting that changes in membrane viscosity are downstream of FakA but upstream of EsxA expression. LA stimulation of EsxA was enhanced at 30 °C and suppressed at 42 °C (Fig. S5A) in USA300 WT, while there was no LA-dependent effect in the fakA-mutant strain.
Fig. S4.

(Top) USA300 cells were grown in TSB alone or in TSB supplemented with 10 µM LA, and global transcript levels were measured by RNA-seq. (Middle) The fold-change (+LA/−LA) is plotted on the y axis, and the chromosomal position of each gene is plotted on the x axis. (Bottom) USA300 cells were grown in TSB at 37 °C or 30 °C, and global transcript levels were measured by RNA-seq. The fold change (30 °C/37 °C) is plotted on the y axis, and the chromosomal position of each gene is plotted on the x axis.
Fig. S5.

(A) USA300 cells were grown at the temperatures shown (30 °C, 37 °C, and 42 °C) in TSB alone or in TSB supplemented with the indicated LA concentration. Whole-cell lysates were analyzed for EsxA protein by Western blot. (B) USA300 WT or USA300 cells expressing crtMN from the pMK4 plasmid were grown in TSB alone or in TSB supplemented with 10 µM LA. Membrane fluidity was measure using the pyrenedecanoic acid fluorescence method. The increasing ratio of fluorescence at 460 nm and 405 nm reflects increased membrane fluidity. (C) USA300 WT, arlS-, agrB-, and saeRS-mutant strains were grown in TSB alone or in TSB supplemented with 10 µM LA. Whole-cell lysates were assessed for EsxA protein by Western blot. The data represent the average values of three independent experiments; error bars represent the SE.
Because changes in temperature may affect numerous cellular pathways, we sought an additional strategy for perturbing membrane fluidity independently of LA. Overexpression of proteins involved in the initial steps of the conversion of farnesyl pyrophosphate into staphyloxanthin, CrtM and CrtN, leads to an increase in carotenoid levels and a concomitant decrease in membrane fluidity (25). We overexpressed crtMN using the sarA promoter and confirmed that crtMN overexpression reduces membrane fluidity in USA300 cells (Fig. S5B). Consistent with a role for membrane viscosity in EsxA production, there was twofold more LA-induced EsxA in cells overexpressing crtMN (Fig. 3D). Taken together, our data indicate that LA induces esxA transcription and EsxA protein expression, at least in part through effects on membrane fluidity itself.
Membrane-associated signaling enzymes have been reported to alter their activity in response to changes in membrane fluidity (28). Since TCSs are a key class of bacterial enzymes involved in environmental sensing and cellular signaling, we measured basal and LA-induced EsxA expression in USA300 strains lacking each known TCS and found that inactivation of agr and arl decreased basal EsxA protein levels, while the sae strain exhibited increased basal EsxA protein (Fig. S5C), as previously described (13, 29). However, the stimulation of EsxA by 10 µM LA was unaffected by these gene disruptions, indicating that agr, arl, and sae play a role in the regulation of basal but not LA-induced T7SS gene expression.
Discussion
Understanding how bacterial pathogens sense the host environment and utilize these cues to orchestrate virulence gene expression is key to understanding the molecular mechanisms of infectious disease. While there has been a great deal of interest in understanding how mammalian hosts sense bacterial pathogens, there has been less focus on deciphering how potential pathogens survey the host environment and respond to these signals. This is particularly interesting for organisms such as S. aureus, which are capable of colonizing diverse niches throughout the host. Although S. aureus is a common member of the normal human microflora, it is also a frequent cause of infection in both healthy and immunocompromised individuals. Identification of the pathways by which S. aureus responds to human cues may aid in the development of novel therapeutics that block sensing of the host environment and thereby reduce pathogen virulence (30).
We discovered that S. aureus detects specific cis-unsaturated fatty acids, such as LA and arachidonic acid, which cannot be produced by the bacteria but are abundant in host tissues. The bacteria respond to these host-derived fatty acids by enhancing the expression of the T7SS operon, a dedicated virulence pathway that promotes bacterial survival and pathogenesis in vivo. Furthermore, we found this pathway requires that the host-derived fatty acids be incorporated into bacterial membranes through a metabolic pathway involving the FAK enzyme complex. Incorporation of the cis-unsaturated fatty acids affects bacterial membrane fluidity, which is at least part of the signal for the transcriptional changes induced by the host environment. We also found that this process is required for virulence, and fakA and essB mutant strains exhibit similar defects in host mortality and bacterial survival in a mouse model of bacteremia, suggesting that type VII secretion may be the major virulence mechanism downstream of sensing host fatty acids. Thus, our work yields insight into S. aureus host–pathogen interactions and identifies FakA as a potential therapeutic drug target.
Fatty acid stimulation of EsxA protein exhibits remarkably precise SARs: Fatty acids with more cis double bonds are more potent EsxA activators, while saturated and trans-unsaturated fatty acids exhibit little or no activity. Straight-chain fatty acids with trans double bonds have a 3D structure quite similar to saturated fatty acids such as SA. In contrast, the presence of a cis double bond leads to a sharp bend in the structure of fatty acids (31). Fatty acids with additional cis double bonds, such as LA and α-LA, display a sharper angle of bend. Thus, the observed SAR suggests that fatty acids with a more linear structure do not stimulate EsxA protein levels, while more bent fatty acids are more potent EsxA activators. Importantly, S. aureus cells synthesize only straight-chain and branched saturated fatty acids, as they do not possess the oxidative enzymes required for fatty acid olefination (23). In contrast, LA is an essential fatty acid for humans, is a key component of mammalian cell membranes, and is the initial substrate for the biosynthesis of signaling lipids such as arachidonic acid (itself a potent EsxA stimulator) as well as some prostaglandins and leukotrienes.
FakA is a gateway enzyme that facilitates the incorporation of exogenous fatty acids into a variety of S. aureus biomolecules including phospholipids, lipoproteins, CL, and LTAs. Since inactivation of fakA or fakB1/B2 abrogated fatty acid-dependent EsxA expression, we hypothesized the mechanism of T7SS stimulation involves incorporation of cis-unsaturated fatty acids into S. aureus lipid molecules. We have been unable to pinpoint a single class of lipids that accounts for the signaling downstream of fakA; this may imply that more than one class of lipids, glycolipids, or LPs is capable of inducing the change in membrane lipid fluidity when host-derived cis-unsaturated fatty acids are incorporated. Whatever the responsible lipids, our data suggest that a decrease in membrane fluidity is itself a signal that stimulates EsxA expression. There is precedent in bacteria for membrane fluidity serving as a signal of the environment as well as directly affecting the activity of membrane-associated enzymes (26, 28).
Clinically, S. aureus is a relatively common cause of liver abscesses (32, 33). Thus, a particularly interesting observation from this study is that the in vivo survival defect for USA300 fakA- and essB-mutant strains is much stronger in the liver than in the kidneys in the i.v. infection mouse model. This is consistent with the known critical role of the liver in uptake of free fatty acids, fatty acid metabolism, and lipid trafficking including the assembly of very low density lipoprotein (34). Furthermore, pulmonary surfactant, which also contains an array of host-derived fatty acids, may play a role in tissue-specific activation of T7SS genes in host lungs (10), suggesting that this mechanism may be relevant in S. aureus pneumonia as well. Taken together, these observations suggest that the S. aureus T7SS may be regulated by tissue-specific differences in fatty acid composition and abundance. This may provide an approach to novel therapeutics that limit S. aureus disease by disabling its sensing of the host environment and thereby reducing pathogen virulence.
Materials and Methods
Bacterial Plasmids, Strains, and Media.
Bacterial strains, plasmids, primers, and RT-qPCR oligonucleotides are summarized in Tables S3–S5. To briefly summarize, “USA300 WT strain” refers to NRS384 ∆hsdR∆mcr (35), and all mutants were constructed in this background. In-frame gene deletions were made by homologous recombination using pIMAY vector (36). Transposon mutants were obtained from the Nebraska Transposon Library and were transduced into the NRS384 ∆hsdR∆mcr strain by bacteriophage 85 (ATCC 27708-B1). Complementation of fakA and overexpression of crtMN were carried out by expressing fakA or crtMN from the pMK4 vector (37) under control of the sarA promoter.
Table S3.
Bacterial strains used in this study
| Designation | Strain | Genotype, description | Source |
| Parent | NRS384 | USA300 S. aureus | NARSA |
| USA300 WT | RC07 | NRS384 ΔhsdRΔmcr | M. Tan laboratory* (35) |
| USA300 essB | RC249 | NRS384 ΔhsdRΔmcrΔessB | Koushik Paul† |
| USA400 WT | MW2 | USA400 S. aureus | NARSA |
| Newman | Newman | Newman S. aureus | NARSA |
| USA300 fakA | RC780 | NRS384 ΔhsdRΔmcr fakA::ermR | This study |
| USA300 fakB1 | RC784 | NRS384 ΔhsdRΔmcr fakB1::ermR | This study |
| USA300 fakB2 | RC785 | NRS384 ΔhsdRΔmcr fakB2::ermR | This study |
| USA300 fakB1 fakB2 | RC792 | NRS384 ΔhsdRΔmcr∆fakB1 fakB2::ermR | This study |
| USA300 ypfP | RC789 | NRS384 ΔhsdRΔmcr ypfP::ermR | This study |
| USA300 cls1 | RC788 | NRS384 ΔhsdRΔmcr cls1::ermR | This study |
| USA300 cls2 | RC790 | NRS384 ΔhsdRΔmcr cls2::ermR | This study |
| USA300 lgt | RC793 | NRS384 ΔhsdRΔmcr ∆lgt | This study |
| USA300 fmtC | RC791 | NRS384 ΔhsdRΔmcr fmtC::ermR | This study |
| USA300 gdpP | NRS384 ΔhsdRΔmcr gdpP::ermR | M. Tan laboratory | |
| USA300 gdpP ltaS | NRS384 ΔhsdRΔmcr ∆ltaS gdpP::ermR | M. Tan laboratory | |
| Escherichia coli DC10B | DH10B ∆dcm | M. Tan laboratory (35) | |
| E. coli XL10-Gold | Stratagene |
NARSA, Network on Antimicrobial Resistance in Staphylococcus aureus (https://www.niaid.nih.gov/research/network-antimicrobial-resistance-staphylococcus-aureus).
Man Wah Tan Lab, Department of Infectious Disease, Genentech Inc.
Koushik Paul, Department of Infectious Disease, Genentech Inc.
Table S5.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′–3′) | Purpose |
| ML072 | TTATTGCAAACCGAAATTATTAGAAAGTTGTTGGTC | esxA RT primer |
| ML073 | CTATTTAAACCATCTAATCTTTTGATAAGCTTGGTTTGCG | essC RT primer |
| ML074 | CTACTTATTTAATATTCTTCTAATATTTCTTTCACCATTAATTTC | esaD RT primer |
| ML075 | TTAGATTAATCTCTCTTTCTTAAAGTGTTTTATAAACATGTTCAG | esaA RT primer |
| ML092 | TTAATTCATTGCTTTATTAAAATATTCACTTGCC | esxC RT primer |
| ML135 | ATGCATCGGATCCCCTTAGGAGGATGATTATTTATGATTAGCAAAATTAATGGTAAATTATTTGCCGATATGATTATACAAGGGG | fakA SmaI pMK4 forward |
| ML136 | GACGGCCAGTGAATTTTATTCTACTGAAAAGAAATATTG | fakA EcoRI pMK4 reverse |
| ML169 | ATATGTCGACAGCACCGCTATAGGCGCG | ΔfakA pIMAY F1 |
| ML170 | GACTCTTTTATTAAAATTTTATAAAGTCGTCCTCCTAAA ATATACTCATAGCAATG | ΔfakA pIMAY R1 |
| ML171 | GTATATTTTAGGAGGACGACTTTATAAAATTTTAATAAAAGAGTCTATATTGTAATTGGAAATTATCTCTCG | ΔfakA pIMAY F2 |
| ML172 | ATATGAATTCTGTCTCTTTCTAATGGCTCGAACAATATG | ΔfakB1 pIMAY R2 |
| ML203 | TACGGTTGGATCCCCATGACAATGATGGATATGAATT | crtMN SmaI pMK4 forward |
| ML204 | GACGGCCAGTGAATTTTATACGCCCCGCTCAATA | crtMN EcoRI pMK4 reverse |
| ML205 | ATATGTCGACGTACTAATTACCGGTGATTCAGGAATAGGT | Δlgt pIMAY F1 |
| ML206 | CCTCAAACTATCATCAACCTACTCCTCACTCTTATG | Δlgt pIMAY R1 |
| ML207 | GTGAGGAGTAGGTTGATGATAGTTTGAGGAAATTTTTATCAAAAACAC | Δlgt pIMAY F2 |
| ML208 | ATATGAATTCAAAATTGTCACCTCCTAATCATTTTTATATAC | Δlgt pIMAY R2 |
| MLqPCR08 | ACACGTGCACAAGGTGAAAT | esxA qPCR forward |
| MLqPCR09 | GTTGTTGGAATTGCTCTTCG | esxA qPCR reverse |
| MLqPCR10 | FAM-TGACCTTCCCAGTTCGCTGCA-BHQ1 | esxA qPCR probe |
| MLqPCR17 | ATCGGAAGTCCAGGATATGG | essC qPCR forward |
| MLqPCR18 | TTGGTACCGAAATCGAACAA | essC qPCR reverse |
| MLqPCR19 | FAM-TGTTGCAAGACACCATCGTCCTGA-BHQ1 | essC qPCR probe |
| MLqPCR20 | GCCCCTTAGTGCTGCAGCTA | rrsA qPCR forward |
| MLqPCR21 | AGTTTCAACCTTGCGGTCGTA | rrsA qPCR reverse |
| MLqPCR22 | VIC-CGCATTAAGCACTCCGCCTGGG-BHQ1 | rrsA qPCR probe |
| MLqPCR26 | AATCATGGAAGGACTTTCGC | esaD qPCR forward |
| MLqPCR27 | CGCCTTTGAAGCCTAATTTC | esaD qPCR reverse |
| MLqPCR28 | FAM-TCCCAGCACCTTTGAGGAAGCC-BHQ1 | esaD qPCR probe |
| MLqPCR38 | GAAGTCAATGGCGTTGCTAA | esaA qPCR forward |
| MLqPCR39 | CCACAACACTCGTTGCTTTC | esaA qPCR reverse |
| MLqPCR40 | FAM-TTCATCTTGACGATTTGCTTGTCCAAA-BHQ1 | esaA qPCR probe |
| MLqPCR47 | TTGAAAGCACAGCAAAGGAC | esxC qPCR forward |
| MLqPCR48 | GCTTGAAATGTTGGGTGAAG | esxC qPCR reverse |
| MLqPCR49 | FAM-AAGGGAAAGGCGCCCAATACG-BHQ1 | esxC qPCR probe |
Table S4.
Plasmids used in this study
| Plasmid | Parent | Description | Source |
| pSarA.MK4 | pMK4 | S. aureus complementation vector, driven by SarA promoter and a ribosomal-binding site of the SodA gene, ampicillin-resistance in E. coli, chloramphenicol-resistance in S. aureus | M. Tan laboratory (38) |
| pIMAY | Vector that facilitates allelic replacement in S. aureus, chloramphenicol-resistance | M. Tan laboratory (35) | |
| pML01 | pSarA.MK4 | S. aureus expression vector, expresses FakA under SarA promoter | This study |
| pML02 | pSarA.MK4 | S. aureus expression vector, expresses CrtMN under SarA promoter | This study |
| pML03 | pIMAY | S. aureus allelic replacement vector targeting fakB1 | This study |
| pML04 | pIMAY | S. aureus allelic replacement vector targeting lgt | This study |
Reagents.
All chemical reagents were purchased from the indicated commercial vendors and used without further purification (SI Materials and Methods for product numbers). Anti-EsxA and anti-SrtA polyclonal antibodies were produced in rabbits in-house. The anti-RpoB antibody was purchased from Invitrogen (MA25425). Serum samples were obtained from Sigma Aldrich (SI Materials and Methods for product numbers).
T7SS Protein Secretion Assay.
Saturated overnight S. aureus cultures were diluted to an OD600 of 0.05 in 30 mL fresh TSB or RPMI medium and were grown at the indicated temperature in a shaking incubator. At an OD600 of 0.9–1.0 (TSB) or 0.8–0.9 (RPMI) the cultures were harvested by centrifugation at 3,000 × g for 5 min. Culture supernatants were passed through 0.22-μm filters, combined with 7.5 mL of 100% TCA (final concentration of 20%), and incubated on ice for 20 min. TCA-precipitated proteins were centrifuged at 3,000 × g and 4 °C for 10 min, washed twice with 2 mL acetone, and allowed to air-dry for 20 min at room temperature. The TCA-precipitated proteins were suspended in 400 μL of 1× sample-loading buffer and incubated at ambient temperature overnight or at 55 °C for 1–2 h. Cell pellets were washed once with PBS, suspended in 400 μL of 1× sample-loading buffer, incubated at 80° for 10 min with shaking, and lysed by sonication (20% amplitude, seven 2-s pulses). For Western blot and Coomassie analysis, 20 μL of each sample was loaded onto 4–20% Tris-Gly gels, and proteins were separated at 200 V over 40 min. For Western blots, the proteins were transferred to nitrocellulose using the iBlot transfer system. Proteins were probed using appropriate primary antibodies and fluorescent secondary antibodies. Proteins levels were quantitated using LI-COR imaging system and software.
Preparation of Lipid-Free Human Serum.
Twenty milliliters of human serum was combined with 16 mL diisopropyl ether (or methyl tert-butyl ether) and 8 mL l-butanol in a 50-mL Erlenmeyer flask, and the mixture was stirred vigorously for 30 min at ambient temperature. The mixture was transferred to a 50-mL falcon tube and centrifuged at 3,500 × g for 15 min. The organic layer was removed to a fresh vial, and the aqueous portion was extracted with an additional portion of diisopropyl ether (or methyl tert-butyl ether) and n-butanol as before. The combined organic portions were evaporated in vacuo and dissolved in 20 mL DMSO. The aqueous portion was left in an open falcon tube for 1 h to evaporate residual organics before being dialyzed against a 1,000× volume of PBS three times. The dialyzed aqueous portion is the delipidated serum, and the organic DMSO solution is the serum organic extract.
RT-qPCR.
Saturated overnight cultures of USA300 cells were diluted to OD600 of 0.05 and incubated in a 37 °C shaking incubator until the cultures reached OD600 of 1.0 in 10 mL of TSB supplemented with various fatty acids. The cultures were centrifuged at 8,000 × g for 5 min, and the supernatant was removed by aspiration. RNA was isolated from the bacteria using the RNAeasy Mini Kit (Qiagen) as previously described (37). RT-qPCR probes and primers were designed using GenScript software (https://www.genscript.com).
Membrane Fluidity Assay.
Saturated overnight cultures of USA300 cells were diluted to an OD600 of 0.05 and were incubated in a 37 °C shaking incubator until the cultures reached an OD600 of 1.0 in 10 mL of TSB supplemented with various fatty acids. The cultures were centrifuged at 8,000 × g for 5 min, and the supernatant was removed by aspiration. The cell pellets were then suspended in PBS containing 20% sucrose and 75 μg/mL lysostaphin and were incubated in a 37 °C water bath for 30 min. After digestion, the spheroplasts were centrifuged at 8,000 × g for 10 min, and the supernatant was removed by aspiration. The spheroplasts were then suspended in 400 μL of osmotically stabilized labeling solution (20% sucrose, 0.01% F-127, 10 µM pyrenedecanoic acid) and incubated at room temperature in the dark for 1 h. After washing in PBS with 20% sucrose, the spheroplasts were transferred to black, clear-bottomed 96-well plates and were incubated at the indicated temperatures. The cell suspensions were analyzed for fluorescence by excitation at 360 nm and emission at 405 nm and 460 nm.
Mouse Infections.
Mouse experiments were reviewed and approved by Genentech's Institutional Animal Care and Use Committee. Bacteria (1 × 107 cfu) suspended in 100 μL sterile PBS were injected i.v. into C57BL/6 mice. Surviving mice were killed at day 8, and liver and kidneys were resected and homogenized in 5 mL sterile PBS (0.05% Tween). The bacteria were enumerated by plating serial dilutions on blood agar plates. For the in vivo esxA expression experiment, RNA was isolated from surviving bacteria in kidneys and liver of infected mice using the RNAeasy Mini Kit (Qiagen) as previously described (37).
SI Materials and Methods
Reagents.
Sodium stearate (sc-215884), sodium oleate (sc-215879), sodium palmitate (sc-215881), elaidic acid (sc-205309), sodium linoleate (sc-215247), linoleic acid (sc-200788), α-linolenic acid (sc-205545), palmitoleic acid (sc-205424), linoleamide (sc-221852A), and arachidonic acid (sc-221261) were obtained from Santa Cruz Biotechnology. Conjugated (9E, 11E)-linoleic acid (90983) and sodium oleyl sulfate (CDS003399) were purchased from Sigma Aldrich. Horse serum (H1270) and human serum (male AB, S2145) were obtained from Sigma Aldrich. FBS (10082139) was obtained from Thermo Fisher Scientific. Human nasal fluid (MBS170293) was purchased from MyBioSource. The membrane fluidity kit (ab189819) was purchased from Abcam. Goat anti-mouse IRDye 680RD (925-68070) and goat anti-rabbit IRDye 800CW (925-32211) were purchased from LI-COR.
Fatty Acid Stimulation of EsxA Protein.
USA300 cultures were grown in TSB alone or in TSB supplemented with individual fatty acids (20, 10, 5, 2.5, 1.25, or 0.625 µM), and whole-cell lysates were assessed for EsxA protein levels by Western blot. The fold stimulation of EsxA (relative to TSB alone) was plotted against the negative logarithm of the fatty acid concentration. The data were fit to a sigmoid function using GraphPad Prism software, and the half-maximal EsxA-stimulating concentration was calculated and is reported as the DR50 value (average value ± SEM).
RNA-Seq.
RNA-seq was performed using USA300 S. aureus in the presence and absence of 10 μM LA. For each condition we included three replicates, thereby yielding a total of six samples. Quality control of RNA samples was done to determine RNA quantity and quality before their processing by RNA-seq. The concentration of RNA samples was determined using a NanoDrop 8000 spectrophotometer (Thermo Scientific), and the integrity of RNA was determined by a 2100 Bioanalyzer system (Agilent Technologies). Ribosomal RNA depletion was performed using a Ribo-Zero rRNA Removal Kit (Bacteria) (Illumina) according to the manufacturer’s instructions. RNA-seq library generation was continued using the TruSeq RNA protocol (Illumina) starting at the “Elute, Prime, Fragment” step and was completed according to the manufacturer’s instructions.
Bioinformatic analyses were performed using R (https://www.r-project.org/) and packages from the Bioconductor project (bioconductor.org). RNA-seq reads were processed using the HTSeqGenie package (Bioconductor). Reads were aligned to the Staphylococcus aureus USA300 NRS384 genome sequence using the GSNAP algorithm. Uniquely aligned read pairs were tallied to estimate gene-expression levels.
To determine differential expression of genes, we used the DESeq and DESeq2 algorithms with default parameters. To control for multiple testing, P values were adjusted using the Benjamini–Hochberg method. Reads per kilobase of transcript per million reads mapped (RPKMs) were normalized using the library size normalization defined by Anders and Huber (41). Genes were considered differentially expressed if they differed between comparison groups by at least twofold, with an adjusted P < 0.05.
RNA-seq data and analyses were deposited in the Gene Expression Omnibus (GEO) public data repository. The accession number for the RNA-seq data for the with/without LA comparison is GSE101580, and the accession number for the comparison of growth at 37 °C and 30 °C is GSE102279.
Supplementary Material
Acknowledgments
We thank Man Wah Tan and Nicholas Nickerson for editing this manuscript, Man Wah Tan, Paolo Manzanillo, and Koushik Paul for helpful discussions and for providing reagents, and Charles Jones III for histologic processing and staining.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: RNA-seq data and analyses have been deposited in the Gene Expression Omnibus (GEO) public data repository (accession nos. GSE101580 for the RNA-seq data for the with/without linoleic acid comparison and GSE102279 for the comparison of growth at 37 °C and 30 °C).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1700627114/-/DCSupplemental.
References
- 1.Gorwitz RJ, et al. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001-2004. J Infect Dis. 2008;197:1226–1234. doi: 10.1086/533494. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins A, et al. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. MBio. 2015;6:e0227214-14. doi: 10.1128/mBio.02272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng AG, DeDent AC, Schneewind O, Missiakas D. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol. 2011;19:225–232. doi: 10.1016/j.tim.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA. 2003;100:13001–13006. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burts ML, Williams WA, DeBord K, Missiakas DM. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci USA. 2005;102:1169–1174. doi: 10.1073/pnas.0405620102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kneuper H, et al. Heterogeneity in ess transcriptional organization and variable contribution of the Ess/Type VII protein secretion system to virulence across closely related Staphylocccus aureus strains. Mol Microbiol. 2014;93:928–943. doi: 10.1111/mmi.12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, et al. Role of the ESAT-6 secretion system in virulence of the emerging community-associated Staphylococcus aureus lineage ST398. Sci Rep. 2016;6:25163. doi: 10.1038/srep25163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warne B, et al. The Ess/Type VII secretion system of Staphylococcus aureus shows unexpected genetic diversity. BMC Genomics. 2016;17:222. doi: 10.1186/s12864-016-2426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burts ML, DeDent AC, Missiakas DM. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol Microbiol. 2008;69:736–746. doi: 10.1111/j.1365-2958.2008.06324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishii K, et al. Induction of virulence gene expression in Staphylococcus aureus by pulmonary surfactant. Infect Immun. 2014;82:1500–1510. doi: 10.1128/IAI.01635-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zurek OW, et al. The role of innate immunity in promoting SaeR/S-mediated virulence in Staphylococcus aureus. J Innate Immun. 2014;6:21–30. doi: 10.1159/000351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kraus D, et al. The GraRS regulatory system controls Staphylococcus aureus susceptibility to antimicrobial host defenses. BMC Microbiol. 2008;8:85. doi: 10.1186/1471-2180-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulthess B, Bloes DA, Berger-Bächi B. Opposing roles of σB and σB-controlled SpoVG in the global regulation of esxA in Staphylococcus aureus. BMC Microbiol. 2012;12:17. doi: 10.1186/1471-2180-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pynnonen M, Stephenson RE, Schwartz K, Hernandez M, Boles BR. Hemoglobin promotes Staphylococcus aureus nasal colonization. PLoS Pathog. 2011;7:e1002104. doi: 10.1371/journal.ppat.1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson MM, et al. Apolipoprotein B is an innate barrier against invasive Staphylococcus aureus infection. Cell Host Microbe. 2008;4:555–566. doi: 10.1016/j.chom.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson M, Chen Y-H, Butler EK, Missiakas DM. EsaD, a secretion factor for the Ess pathway in Staphylococcus aureus. J Bacteriol. 2011;193:1583–1589. doi: 10.1128/JB.01096-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson C, et al. Cell culture models demonstrate that CFTR dysfunction leads to defective fatty acid composition and metabolism. J Lipid Res. 2008;49:1692–1700. doi: 10.1194/jlr.M700388-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kenny JG, et al. The Staphylococcus aureus response to unsaturated long chain free fatty acids: Survival mechanisms and virulence implications. PLoS One. 2009;4:e4344. doi: 10.1371/journal.pone.0004344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons JB, Yao J, Frank MW, Jackson P, Rock CO. Membrane disruption by antimicrobial fatty acids releases low-molecular-weight proteins from Staphylococcus aureus. J Bacteriol. 2012;194:5294–5304. doi: 10.1128/JB.00743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muthaiyan A, Silverman JA, Jayaswal RK, Wilkinson BJ. Transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob Agents Chemother. 2008;52:980–990. doi: 10.1128/AAC.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parsons JB, Frank MW, Jackson P, Subramanian C, Rock CO. Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol Microbiol. 2014;92:234–245. doi: 10.1111/mmi.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parsons JB, et al. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc Natl Acad Sci USA. 2014;111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhn S, Slavetinsky CJ, Peschel A. Synthesis and function of phospholipids in Staphylococcus aureus. Int J Med Microbiol. 2015;305:196–202. doi: 10.1016/j.ijmm.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 24.Galla HJ, Hartmann W. Excimer-forming lipids in membrane research. Chem Phys Lipids. 1980;27:199–219. doi: 10.1016/0009-3084(80)90036-5. [DOI] [PubMed] [Google Scholar]
- 25.Mishra NN, et al. Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob Agents Chemother. 2011;55:526–531. doi: 10.1128/AAC.00680-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botello E, Jiménez-Sánchez A. A temperature upshift induces initiation of replication at oriC on the Escherichia coli chromosome. Mol Microbiol. 1997;26:133–144. doi: 10.1046/j.1365-2958.1997.5621924.x. [DOI] [PubMed] [Google Scholar]
- 27.Alonso A, Queiroz CS, Magalhães AC. Chilling stress leads to increased cell membrane rigidity in roots of coffee (Coffea arabica L.) seedlings. Biochim Biophys Acta. 1997;1323:75–84. doi: 10.1016/s0005-2736(96)00177-0. [DOI] [PubMed] [Google Scholar]
- 28.Mansilla MC, de Mendoza D. The Bacillus subtilis desaturase: A model to understand phospholipid modification and temperature sensing. Arch Microbiol. 2005;183:229–235. doi: 10.1007/s00203-005-0759-8. [DOI] [PubMed] [Google Scholar]
- 29.Anderson M, Aly KA, Chen Y-H, Missiakas D. Secretion of atypical protein substrates by the ESAT-6 secretion system of Staphylococcus aureus. Mol Microbiol. 2013;90:734–743. doi: 10.1111/mmi.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maura D, Ballok AE, Rahme LG. Considerations and caveats in anti-virulence drug development. Curr Opin Microbiol. 2016;33:41–46. doi: 10.1016/j.mib.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nile AH, Hannoush RN. Fatty acylation of Wnt proteins. Nat Chem Biol. 2016;12:60–69. doi: 10.1038/nchembio.2005. [DOI] [PubMed] [Google Scholar]
- 32.Muorah M, et al. Liver abscesses in children: A single center experience in the developed world. J Pediatr Gastroenterol Nutr. 2006;42:201–206. doi: 10.1097/01.mpg.0000189344.23387.26. [DOI] [PubMed] [Google Scholar]
- 33.Mishra K, Basu S, Roychoudhury S, Kumar P. Liver abscess in children: An overview. World J Pediatr. 2010;6:210–216. doi: 10.1007/s12519-010-0220-1. [DOI] [PubMed] [Google Scholar]
- 34.Mashek DG. Hepatic fatty acid trafficking: Multiple forks in the road. Adv Nutr. 2013;4:697–710. doi: 10.3945/an.113.004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. Transforming the untransformable: Application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio. 2012;3:e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kofoed EM, et al. De novo guanine biosynthesis but not the riboswitch-regulated purine salvage pathway is required for Staphylococcus aureus infection in vivo. J Bacteriol. 2016;198:2001–2015. doi: 10.1128/JB.00051-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Date SV, et al. Global gene expression of methicillin-resistant Staphylococcus aureus USA300 during human and mouse infection. J Infect Dis. 2014;209:1542–1550. doi: 10.1093/infdis/jit668. [DOI] [PubMed] [Google Scholar]
- 38.Hazenbos WLW, et al. Novel staphylococcal glycosyltransferases SdgA and SdgB mediate immunogenicity and protection of virulence-associated cell wall proteins. PLoS Pathog. 2013;9:e1003653. doi: 10.1371/journal.ppat.1003653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller M, et al. Deletion of membrane-associated Asp23 leads to upregulation of cell wall stress genes in Staphylococcus aureus. Mol Microbiol. 2014;93:1259–1268. doi: 10.1111/mmi.12733. [DOI] [PubMed] [Google Scholar]
- 40.Frees D, Gerth U, Ingmer H. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int J Med Microbiol. 2014;304:142–149. doi: 10.1016/j.ijmm.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.