

Abstract
Calcium (Ca2+) influx into the mitochondrial matrix stimulates ATP synthesis. Here, we investigate whether mitochondrial Ca2+ transport pathways are altered in the setting of deficient mitochondrial energy synthesis, as increased matrix Ca2+ may provide a stimulatory boost. We focused on mitochondrial cardiomyopathies, which feature such dysfunction of oxidative phosphorylation. We study a mouse model where the main transcription factor for mitochondrial DNA (transcription factor A, mitochondrial, Tfam) has been disrupted selectively in cardiomyocytes. By the second postnatal week (10–15 day old mice), these mice have developed a dilated cardiomyopathy associated with impaired oxidative phosphorylation. We find evidence of increased mitochondrial Ca2+ during this period using imaging, electrophysiology, and biochemistry. The mitochondrial Ca2+ uniporter, the main portal for Ca2+ entry, displays enhanced activity, whereas the mitochondrial sodium-calcium (Na+-Ca2+) exchanger, the main portal for Ca2+ efflux, is inhibited. These changes in activity reflect changes in protein expression of the corresponding transporter subunits. While decreased transcription of Nclx, the gene encoding the Na+-Ca2+ exchanger, explains diminished Na+-Ca2+ exchange, the mechanism for enhanced uniporter expression appears to be post-transcriptional. Notably, such changes allow cardiac mitochondria from Tfam knockout animals to be far more sensitive to Ca2+-induced increases in respiration. In the absence of Ca2+, oxygen consumption declines to less than half of control values in these animals, but rebounds to control levels when incubated with Ca2+. Thus, we demonstrate a phenotype of enhanced mitochondrial Ca2+ in a mitochondrial cardiomyopathy model, and show that such Ca2+ accumulation is capable of rescuing deficits in energy synthesis capacity in vitro.
Keywords: Mitochondrial calcium uniporter, mitochondrial sodium-calcium exchanger, whole-mitoplast electrophysiology, cardiac metabolism, respiratory-chain deficient cardiomyopathy, OXPHOS deficient cardiomyopathy
Graphical abstract

1. INTRODUCTION
Heart failure leads to a profound reprogramming of energetic production in cardiomyocytes[1]. As heart failure progresses, energetic supply fails to meet demand, and individuals with more severe supply-demand mismatch have reduced survival[2]. Amongst the pathways altered in heart failure, our interest focuses on calcium (Ca2+), which is central for excitation-contraction coupling, and is increasingly recognized as both an enhancer and inhibitor of mitochondrial function. Upon entering the mitochondrial matrix, Ca2+ can stimulate ATP production[3], but excess Ca2+ entry disrupts mitochondrial function. Because Ca2+ has bimodal effects, deciphering its contribution to energetic homeostasis during heart failure has remained difficult. Perhaps such difficulties arise from the relatively late occurrence of clinically-evident energetic depletion during heart failure[2], with mechanisms regulating mitochondrial Ca2+ becoming obscured by other homeostatic pathways triggered during heart failure progression. To identify clear examples of altered mitochondrial Ca2+ regulation, we sought a disease model that features energetic failure as an early phenotype. We expected that early onset of energetic dysfunction during heart failure would allow us to examine mitochondrial Ca2+ regulation in a relatively isolated manner.
A spectrum of diseases featuring energetic imbalance arise from a heterogeneous group of mutations in proteins encoded in either the nuclear or mitochondrial genome (mtDNA) that are involved in oxidative phosphorylation (OXPHOS) [4]. These disorders often feature cardiac involvement, termed the mitochondrial cardiomyopathies, and can be devastating, increasing the mortality rates of children suffering from these disorders three-fold[5]. For our studies, such deficient OXPHOS provides a unique opportunity to examine how the heart modulates mitochondrial Ca2+ fluxes when energy production is failing. One such mouse model was created by a cardiac-specific deletion of the main transcription factor for mtDNA (transcription factor A, mitochondrial, Tfam)[6], leading to depletion of electron transport chain (ETC) subunits encoded by mtDNA. In humans, two infants with TFAM mutations died within months of liver failure, had mitochondrial abnormalities on muscle biopsies, and in one case developed clinical heart failure[7].
In mice, cardiac Tfam deletion recapitulates the central problem caused by the heterogeneous group of mutations causing mitochondrial cardiomyopathies: mtDNA depletion and subsequent OXPHOS dysfunction. Multiple studies have revealed that these mice develop cardiac dysfunction with many of the same clinical, biochemical, and ultrastructural features found in human mitochondrial cardiomyopathies, such as an early decline in energetic function and abnormal cytoplasmic Ca2+ handling[8–10]. Although these mice have been otherwise characterized, regulation of their cardiac mitochondrial Ca2+ remains unexplored.
Studying mitochondrial Ca2+ regulation in this model of mitochondrial dysfunction may also provide broader clinical insight, as mitochondrial dysfunction is widespread in heart failure. OXPHOS dysfunction and mtDNA damage are prevalent in dilated cardiomyopathies, and indicate worse prognosis[11–14]. In animal models of ischemic[15], cardiotoxic[16], diabetic[17], tachycardia-induced[18], and pressure-overload[19] cardiomyopathies, reductions in TFAM, mtDNA, and OXPHOS are common. In a proteomic analysis, TFAM was the second-most downregulated protein in failing hearts[20]. Conversely, overexpressing TFAM improves function after cardiac injury[21], and is the subject of therapies targeted towards cardioprotection[22]. However, the late occurrence of clinically evident OXPHOS deficiency has made defining regulatory mechanisms difficult in common forms of heart failure, whereas it drives heart failure in mitochondrial cardiomyopathies.
Here, we study mitochondrial Ca2+ transport in the second postnatal week in mice with cardiac-specific Tfam deletion. As OXPHOS declines, these animals develop a dilated cardiomyopathy. In these failing hearts, we find that mitochondria become far more Ca2+ avid, taking up Ca2+ at twice the rate as controls. This occurs via increased activity of the mitochondrial Ca2+ uniporter, the main channel transporting Ca2+ from cytoplasm to mitochondrial matrix. In addition, mitochondria from these mice release matrix Ca2+ at half the rate as controls, indicating reduced activity of the sodium-calcium (Na+-Ca2+) exchanger. The increase in Ca2+ influx and reduction in Ca2+ efflux reflect corresponding changes in protein levels for the mitochondrial Ca2+ uniporter and Na+-Ca2+ exchanger. Though the decrease in Na+-Ca2+ exchanger levels occurs at the transcript level, the mechanism for increased uniporter function appears to be post-transcriptional, as mRNA levels are reduced despite higher protein amounts. Finally, we find substantial inhibition of mitochondrial respiration in these mice after Ca2+ depletion, whereas Ca2+ incubation boosts respiration to levels comparable to controls.
2. METHODS
Full description of methods is available in the Supplementary Materials.
2.1 Animal use
The floxed Tfam (a kind gift of Dr. Nils-Göran Larsson) and α-myosin heavy chain-promoter driven Cre recombinase (Myh6-Cre) mice (Jackson Labs, Bar Harbor, ME, strain #011038) have been backcrossed into a C57BL/6J background[6, 23]. All procedures were approved by the Institutional Animal Care and Use Committee in accordance with institutional guidelines. Animals used were 10–15 days old except where noted.
2.2 Mitochondrial isolation
For all assays, knockout and littermate controls were processed in parallel. Mice were euthanized with CO2 and hearts rapidly removed. A mitochondrial fraction was subsequently isolated by differential centrifugation[24].
2.3 Mitochondrial enzyme assays
We processed 20 μg of mitochondrial fractions using the Citrate Synthase Activity Kit (Sigma, St. Louis, MO). For subsequent assays of complex I–IV activity, we determined the citrate synthase activity in a sample of Tfam heterozygous mitochondria and adjusted amounts of wild-type and knockout littermate mitochondria to match that level. Complex I–IV were analyzed as detailed previously[25], using assay rates in the presence or absence of an inhibitor to determine the complex-specific activity.
2.4 Inductively coupled plasma-mass spectrometry (ICP-MS)
Mitochondria isolated by differential centrifugation were incubated in 40 μM digitonin to permeabilize compartments other than the mitochondrial matrix, prior to final pelleting for analysis. Mitochondria were processed by the ICP-MS metals lab at the University of Utah.
2.5 Fluorescent imaging of mitochondrial inner membrane voltage gradient (ΔΨ) and Ca2+ transport
We performed protocols as previously described using 50 μg of the mitochondrial fractions, except for Ca2+-induced tetramethylrhodamine methyl ester (TMRM) depolarization, where we used 20 μg[26].
2.6 Whole-mitoplast electrophysiology
We prepared mitoplasts from mitochondrial fractions and subsequently performed voltage-clamp analysis using the Kirichok protocol[24].
2.7 Oxygen consumption analysis
Oxygen consumption was determined with a Clark electrode (Hansatech Instruments, Norfolk, UK), or with the fluorescent oxygen probe MitoXpress Xtra (Luxcel Biosciences, Cork, Ireland). For MitoXpress Xtra, we used fluorescence lifetime imaging of mineral oil-sealed wells in a 96-well microplate [27].
2.8 Statistical analysis
Microsoft Excel and R were used for data analysis. We rejected the null hypothesis for P values less than 0.05. For multiple comparison tests on samples < 15, we established an overall P-value using a Kruskal-Wallis test, and for P < 0.05 proceeded to a Dunn’s multiple comparison test. For multiple comparisons on samples > 15, we established an overall P-value using a one-way ANOVA, and for P < 0.05 proceeded to two-tailed, unequal variance, Student’s t-tests. For multiple comparison tests, final P-values between groups were Bonferroni-corrected.
3. RESULTS
3.1 The dilated cardiomyopathy produced by cardiac-specific deletion of Tfam is evident in the second postnatal week
For our studies, we crossed the previously-developed Tfam loxP mice with mice bearing a Cre recombinase gene under the control of the α-myosin heavy chain promoter (Myh6-Cre), which becomes active during embryogenesis in a cardiomyocyte-specific manner[28]. For our studies, we compared knockout animals (TfamloxP/loxP; +/Myh6-Cre, [Tfam KO]) to littermate Tfam heterozygotes (TfamloxP/+; +/Myh6-Cre, [Tfam HET]) and wild-type animals (TfamloxP/loxP; +/+ or TfamloxP/+; +/+. [Tfam WT]).
We conducted our experiments on P10–P15 animals, an age range in which most Tfam KO mice were alive, yet hearts were large enough to yield sufficient mitochondria for functional characterization of individual animals. By this age, Tfam deletion was essentially complete, reflected in the near absence of mRNA and protein in knockout hearts (Fig. 1A–C; numbers are average ± standard error fold changes compared to controls here and throughout: 0.12 ± 0.02 mRNA, 0.05 ± 0.02 protein). The Tfam KO mice experienced early mortality, with animals dying mostly from postnatal week 2 onwards (Fig. 1D). This is consistent with prior reports of cardiac Tfam deletion[6]. In one study using TfamloxP/loxP; +/Myh6-Cre animals, >75% of knockout animals died in the first postnatal week[29]. However, those animals were in a mixed background, and the Myh6-Cre transgene construct differed from the one used here.
Figure 1. Myh6-Cre deletes Tfam in cardiac tissue and leads to a dilated cardiomyopathy in 10–15 day old animals.
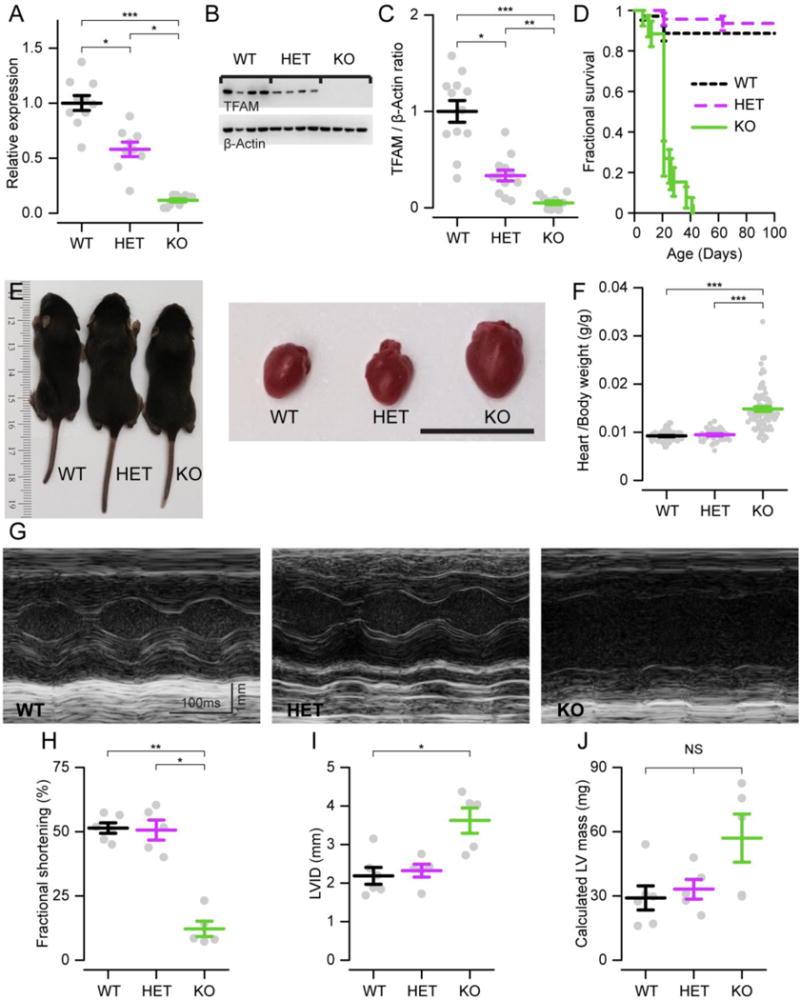
A. qPCR analysis of Tfam expression, displayed as fold change relative to wild-type animals. Myh6-Cre produces mice that are wild-type (WT), heterozygous (HET), or knockout (KO) for cardiomyocyte Tfam. Here and throughout grey points represent samples from individual mice, summary bars show mean ± standard error, and numbers listed are animals used per genotype, unless otherwise noted. (n = 9–10) B. Exemplar Western blot with each lane showing cardiac TFAM expression (top) and β-actin (loading control, bottom) from a mouse with genotype listed above. C. Summary data showing ratio of TFAM to β-actin chemiluminescence on Western, normalized to WT (n = 12). D. Mouse survival data (n = 26–71). E. Left, photograph of mice on postnatal day 10. Right, hearts extracted from the corresponding animal. Scale, 1 cm. F. Ratio of heart to body weight for individuals of listed genotype (n = 45–86). G. Exemplar transthoracic echocardiograph M-mode recordings in left ventricular short axis of mouse hearts. Calculated fractional shortening (H), left ventricular dimension in diastole (LVID, I), and calculated left ventricular (LV) mass (J) on transthoracic echocardiography (n = 5–6). P < 0.05 (*), <0.01 (**), <0.001 (***), > 0.05 (NS) here and throughout.
All Tfam KO animals assayed had a dilated cardiomyopathy within this period (Fig. 1E–F). Cardiac function on echocardiography was impaired, with severely reduced fractional shortening and dilated left ventricles (Fig. 1G–J). Tfam HET animals, despite having an intermediate reduction in TFAM, had normal cardiac function at this age.
3.2 Tfam KO cardiac mitochondria have impaired OXPHOS activity but preserved numbers and ΔΨ in the second postnatal week
Next, we examined mitochondrial function in Tfam KO animals in this timespan. First, we studied the TFAM-mediated transcription of mtDNA. Quantitative reverse transcription polymerase chain reaction (qPCR) analysis of a subset of mtDNA genes revealed minimal mRNA levels in Tfam KO compared to controls (Fig. 2A). Such disruption was replicated in enzymatic assays of ETC function. We found marked inhibition of complex I, III, and IV but not complex II activity (Fig. 2B–E; complex I: 0.23 ± 0.06, complex II: 1.18 ± 0.12, complex III: 0.72 ± 0.07, complex IV: 0.64 ± 0.07). This pattern reflects the encoding of ETC subunits in nuclear versus mtDNA, as, unlike the other complexes, complex II subunits are wholly nuclear-encoded. Additionally, the absence of heart failure in Tfam HET animals at this age, despite intermediate reductions in ETC activity, is consistent with the significant reserve capacity of cardiac mitochondria. Next, we measured respiration on complex I (glutamate/malate) or complex II substrates (rotenone/succinate), using a Clark electrode (Fig. S1A). Consistent with the qPCR and isolated complex measurements, complex I respiration was reduced in the Tfam KO (Fig. S1B; 0.6±0.1), whereas respiration on complex II was unchanged compared to controls (Fig. S1C; 1.3±0.2). Finally, we assayed ATP production. For the F1-FO ATP synthase, beyond the mtDNA-encoded mt-Atp6 and mt-Atp8 genes (Fig. 2A), we quantified the expression of the nuclear-encoded ATP5A1, ATP5F1, ATP5G1/2/3, and ATP5O subunits, and found that several of these were reduced in Tfam KO hearts (Fig. S1D–F, Fig. 5). We then measured ATP production using a luciferin-luciferase assay (Fig. S1G–I), integrating OXPHOS and ATP transport. Using this measure, we found that, as with respiration, Tfam KO mice had a lower rate of ATP production (0.73±0.07). Subsequent to blocking the F1-FO-ATP synthase with oligomycin, we found no difference between genotypes. Such deficiencies in electron transport chain activity and ATP synthesis are notable in the development of heart failure, including in the Tfam KO animals [6, 9, 30].
Figure 2. Disruption of Tfam inhibits ETC activity in KO hearts.
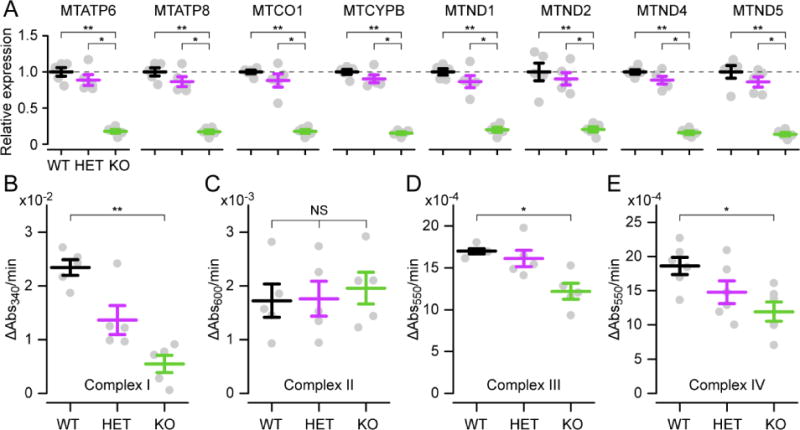
A. qPCR analysis of mtDNA gene expression. (n = 5). B. Spectrophotometric assay of complex I activity measured via decline in absorbance at 340 nm (ΔAbs340/min) produced by NADH oxidation (n = 5). C. Complex II activity measured via decline in absorbance at 600 nm (ΔAbs600/min) produced by 2,6-dichlorophenolindophenol reduction (n = 5). D. Complex III activity measured via increase in absorbance at 550 nm (ΔAbs550/min) produced by cytochrome c reduction (n = 5). E. Complex IV activity measured via decrease in absorbance at 550 nm (ΔAbs550/min) produced by cytochrome c oxidation (n = 5).
Figure 5. Changes in Ca2+ transporter protein expression underlie altered mitochondrial Ca2+ flux.
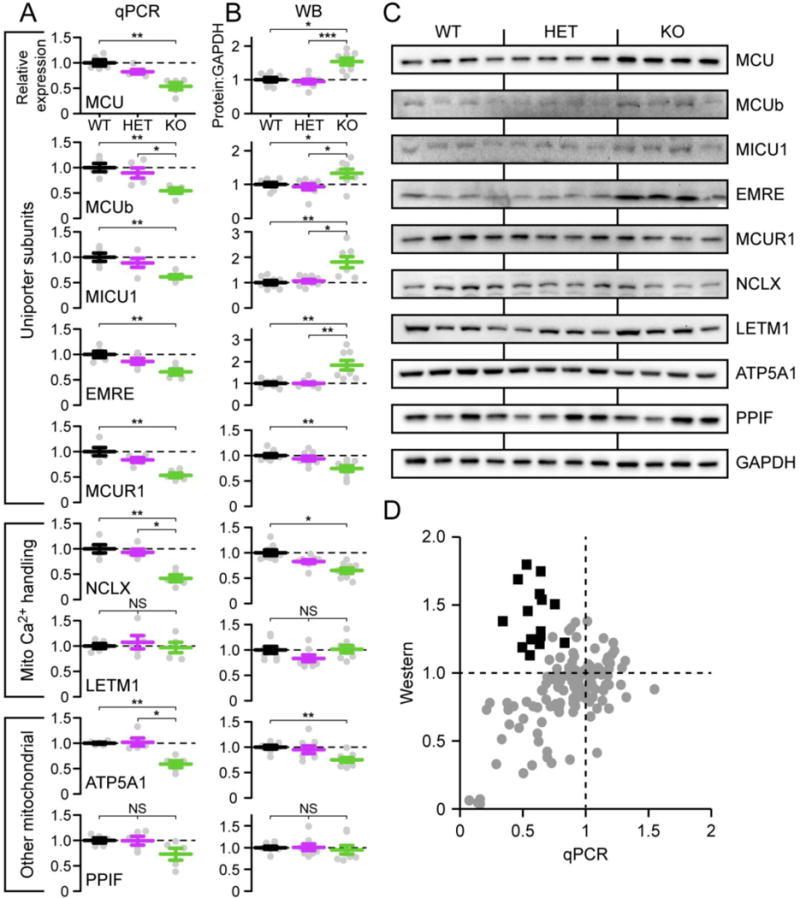
A. qPCR analysis of expression of mitochondrial Ca2+ uniporter subunits, other Ca2+ handling proteins, including the Na+-Ca2+ exchanger (Nclx) and Ca2+-H+ exchanger (Letm1), and other mitochondrial proteins, relative to GAPDH mRNA (n = 5). B. Summary of Western blot quantitation expressed as normalized ratio of protein to GAPDH loading control (n = 8). C. Representative samples from Western blot data. D. For the subset of animals that contributed both protein and mRNA expression data, the graph plots the protein: GAPDH ratio on Western against the relative expression level on qPCR for each animal. Grey circles, data from non-uniporter genes across genotypes, and uniporter genes for WT and HET animals. Black squares, data from core uniporter genes for Tfam KO animals.
Further analyses confirmed that the cardiomyopathy in P10–P15 animals had not produced end-stage tissue remodeling. A prior study demonstrated enhanced oxidative stress and apoptotic cell death, but not extensive fibrosis, in 2–3 week old Tfam KO animals[8]. For oxidative stress, we measured H2O2 production using an Amplex Red assay (Fig. S2A–C). These showed a slight increase in ADP-stimulated H2O2 production in Tfam KO hearts (1.23±0.03), whereas maximal antimycin A-induced H2O2 production was reduced (0.32±0.07), consistent with diminished complex III activity. For cell death (Fig. S2D–E), we saw an increase in terminal deoxynucleotidyl transferase dUTP nick end labeled (TUNEL) nuclei in Tfam KO hearts. Similarly, we found that in 3 of 4 Tfam KO, but no control animals, we could detect cardiomyocytes staining for activated caspase 3 (Fig. S2F). Despite these indicators of cell death, Masson’s trichrome staining did not reveal any evidence of increased fibrosis in this age range (Fig. S2G–H). These results are congruent with the prior report [8].
As dilated cardiomyopathies progress, cardiac mitochondria fission and become more abundant, with increased mitochondrial mass seen in both human disease and animal models[6, 9, 11]. We thus examined mitochondrial abundance in P10–15 Tfam KO mice using several independent methods. First, complex II activity, which does not depend on TFAM, was preserved. Second, Tfam KO mitochondrial ultrastructure under electron microscopy remained comparable to that of controls, as were mitochondrial size distributions and volume density (Fig. 3A–C). Similarly, the mitochondrial yield from each heart was not different between Tfam KO and controls (Fig. 3D). Finally, we found no difference in citrate synthase activity, a marker of mitochondrial mass (Fig. 3E).
Figure 3. Mitochondrial ultrastructure and ΔΨ are preserved.
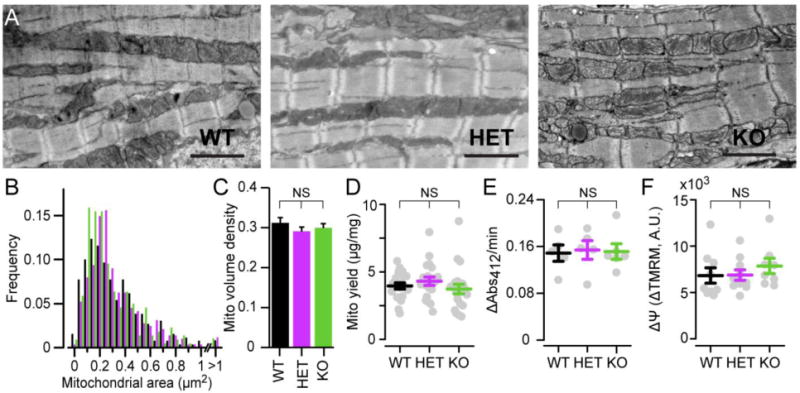
A. Exemplar transmission electron photomicrographs showing cardiac mitochondrial ultrastructure. Scale, 2 μm. B. Distribution of mitochondrial cross-sectional area in electron microscopy images across genotypes (means: WT, 0.34 ± 0.01 μm2; HET, 0.35 ± 0.01 μm2; KO, 0.31 ± 0.02 μm2, for n > 220 mitochondria per condition, with no difference between genotypes). C. Mitochondrial volume density in electron microscopy images (n = 13–24 images per condition). D. Mitochondrial yield measured as ratio of total protein in mitochondrial fraction (μg) to heart weight (mg, n = 19–21). E. Citrate synthase activity measured as increase in absorbance at 412 nm (ΔAbs412/min) produced by formation of 5,5′-dithiobis-(2-nitrobenzoic acid) (n = 6–7). F. ΔΨ assessment via FCCP-induced depolarization, measured as the total change in fluorescence in isolated cardiac mitochondria incubated in 20 μM TMRM (ΔTMRM, arbitrary units, A.U., n = 8–10).
We then examined ΔΨ, the voltage gradient driving ATP synthesis. We were unable to enzymatically dissociate high-quality cardiomyocytes for single-cell imaging from animals <P17. In P17 Tfam KO animals, single cardiomyocytes loaded with the voltage-sensitive dye TMRM (20 nM) fluoresced less intensely than controls (0.74 ± 0.02), suggesting an abnormal, depolarized ΔΨ (Fig. S3A–B). However, since the rest of our analyses were performed on mice aged P10–P15, we sought alternative means to test ΔΨ at younger ages. After mechanically disrupting cells, mitochondrial fractions were enriched by differential centrifugation and incubated in TMRM at high concentration (20 μM), which quenched fluorescence. Adding 5 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) to this suspension uncouples mitochondria, and the total TMRM fluorescence increase associated with dequenching is a very sensitive measure of ΔΨ [31]. Using this technique, Tfam KO mitochondria had ΔΨ indistinguishable from controls at younger ages (Fig. 3F). Together, these data reveal that the P10-P15 period features disease caused by energetic failure, but has not progressed to a late phase characterized by extensive tissue remodeling, impaired mitochondrial ultrastructure, and abnormal enzymatic function.
3.3 Tfam KO cardiac mitochondria feature higher baseline Ca2+, increased Ca2+ influx, and diminished Ca2+ efflux
We next turned to an analysis of mitochondrial Ca2+. First, total mitochondrial Ca2+ content was measured by applying ICP-MS to mitochondrial pellets[32]. Tfam KO hearts had a 2.2 ± 0.5-fold increase over controls in mitochondrial Ca2+ content (Fig. 4A). This elevation was specific to Ca2+, as there was no difference in mitochondrial potassium content (Fig. S4A). Next, we measured electrophoretic Ca2+ uptake in mitochondrial fractions isolated by differential centrifugation. Mitochondrial suspensions were incubated in a Ca2+-sensitive dye restricted to the media[33]. A Ca2+ pulse produces a large fluorescence increase that declines as mitochondria take up the Ca2+ bolus (Fig. 4B). Notably, with this assay Tfam KO cardiac mitochondria sequestered Ca2+ at twice the rate as littermate controls (Fig. 4C, 2.0 ± 0.1), suggesting enhanced activity of the uniporter. Importantly, the total Ca2+ pulse amplitude was comparable across trials (Fig. S4B), so this faster rate was due to increased uptake by Tfam KO mitochondria. Next, we examined the predominant efflux pathway for Ca2+, the mitochondrial Na+-Ca2+ exchanger[34]. Following a pulse to load mitochondria with Ca2+ in Na+-free media, an injection of Ru360 and Na+ respectively blocks further transport through the uniporter and activates Ca2+ efflux. A final injection of the Na+-Ca2+ exchange blocker CGP-37157 largely abolishes further efflux, and allows measurement of the Na+-Ca2+ exchanger-specific rate (Fig. 4D). We found that efflux rates in Tfam KO mitochondria were reduced to 0.3 ± 0.1 of control rates (Fig. 4E).
Figure 4. Mitochondrial Ca2+ is increased in Tfam KO hearts.
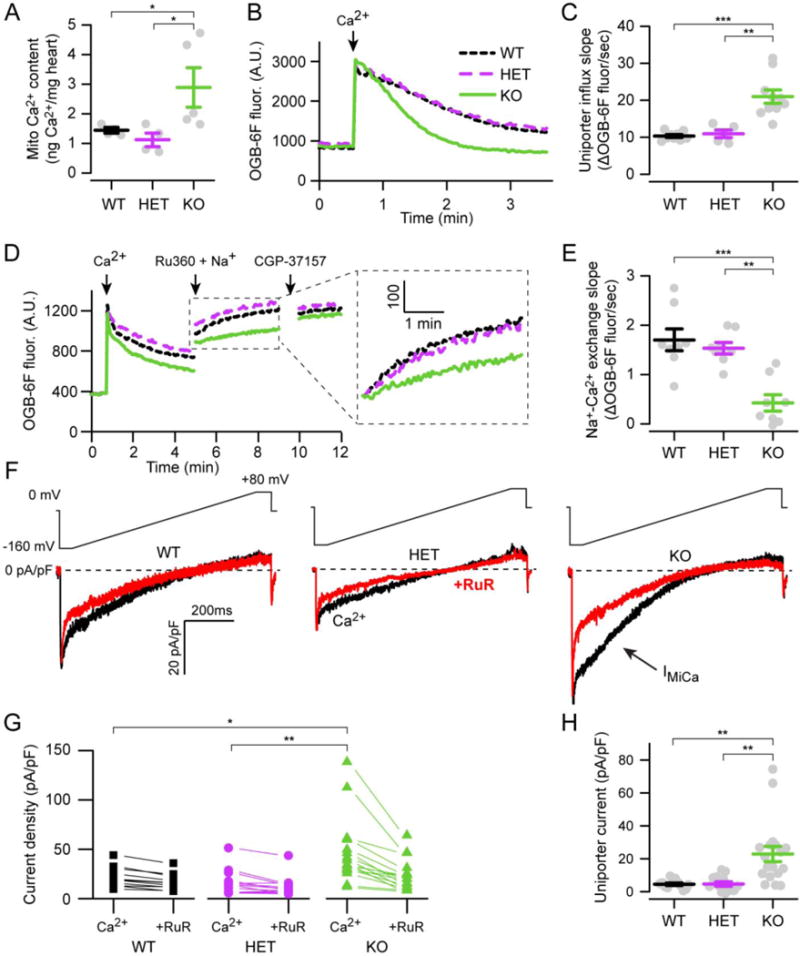
A. ICP-MS analysis of mitochondrial Ca2+ content (n = 4–5). B. Exemplar traces display Ca2+ uptake of cardiac mitochondria following a 50 μM pulse (arrow). C. Summary of Ca2+ uptake measured as linear slope fit to 15–75 sec after Ca2+ pulse (n = 5–10) D. Exemplar traces display protocol for measuring Ca2+ efflux rates. Cardiac mitochondria are pulsed with 15 μM Ca2+ in Na+-free media, followed by addition of 5 mM Na+ and 1 μM uniporter blocker (Ru360), followed by addition of a blocker of Na+-Ca2+ exchange (CGP-37157, 5 μM) as indicated. Inset shows Ca2+ efflux traces shifted vertically to start from the same point. E. Summary of Ca2+ efflux measured as linear slope fit following Na+ addition minus residual slope after CGP-37157 addition (n = 9). F. Exemplar whole-mitoplast current density traces in 100 mM Ca2+, with or without ruthenium red (RuR, 0.1 μM). The inwards mitochondrial Ca2+ current is indicated (IMiCa). Voltage ramp protocol is shown above the traces. G. Ca2+ current density at −160 mV with or without RuR. H. Summary data shows uniporter-specific Ca2+ current density at −160 mV, calculated by subtracting the +RuR from Ca2+ values (G, H: n = 12–17 mitoplasts per condition).
Ca2+ influx depends not only on the activity of the uniporter but other factors such as the amount of intramitochondrial buffering and the magnitude of ΔΨ driving uptake. It is unlikely that a change in ΔΨ caused the enhanced uptake, as this would require a larger ΔΨ than controls. Yet, we observed no difference at this age (Fig. 3F) and a decline in ΔΨ with disease progression (Fig. S3). Thus, we hypothesized that the increase in uptake was due to the uniporter. To confirm this, we turned to whole-mitoplast electrophysiology, the most accurate method to directly measure uniporter current[24, 35].
To measure uniporter current, we isolated cardiac mitoplasts, which are mitochondria with outer membranes stripped away. After accessing the matrix, ΔΨ was clamped across the entire inner membrane (whole-mitoplast mode). As in prior reports, we found that mouse cardiac mitochondrial currents were minuscule (Fig. 4F)[36]. To isolate the uniporter-mediated current, the total Ca2+ current was subtracted from the fraction left after uniporter inhibition with ruthenium red (RuR, Fig. 4G). In fact, the uniporter-mediated current was 5.0 ± 1.0-fold larger in mitochondria from Tfam KO mice compared to controls (Fig. 4H). The slight disparity in magnitude of Ca2+ enhancement between this measure and other assays may be due to a few outlier values present in the voltage clamp data. Without these, our results remained significant but the fold change was 3.7 ± 0.5. Thus, direct measurement of uniporter currents confirms that the enhanced mitochondrial Ca2+ uptake seen in these animals is mediated by increased transport through the uniporter.
Next, we examined several parameters to establish whether the change in Ca2+ uptake was specific or might reflect generalized mitochondrial dysfunction not captured in our prior assays (Fig. 3). First, we measured another transport pathway present in cardiac mitochondria, chloride (Cl−) flux through the inner membrane anion channel (IMAC)[37]. This redox-sensitive channel becomes active during periods of stress, depolarizing cardiac mitochondria[38]. Under our recording conditions, IMAC current becomes maximal at depolarized potentials, and its magnitude was unchanged between Tfam KO and control mitoplasts (Fig. S5A–B). In addition, mitoplast sizes, measured indirectly via capacitance values during electrophysiology, were comparable between Tfam KO and controls (Fig. S5C). Next, we examined if Tfam KO animals had higher levels of fibroblast mitochondria in our preparations compared to controls. Though an increased contribution of fibroblast mitochondria may partly explain differences in Ca2+ uptake, they are unlikely to be the main reason. Fibroblast numbers are lowest in young hearts, and the mitochondrial mass in cardiomyocytes far exceeds that of fibroblasts, demonstrated by the trivial amount of TFAM left in the KO animals (Fig. 1A–C). Nevertheless, to examine this in more detail, we quantified the levels of a fibroblast marker, vimentin, in Tfam KO versus control hearts, and found no difference (Fig. S5D). Finally, we examined whether the change in Ca2+ transport altered sensitivity to permeability transition, measured by pulsing mitochondria repeatedly with 10 μM Ca2+ boluses. In this assay, permeability transition becomes visible as a rise in the extra-mitochondrial Ca2+ fluorescence, which is blocked by incubating mitochondria with cyclosporin A (Fig. S6A). Tfam KO mitochondria required a similar total Ca2+ load to trigger permeability transition compared to controls (Fig. S6B). Thus, the enhanced Ca2+ transport seen in Tfam KO hearts appears to be a specific regulatory mechanism active in cardiomyocytes during energetic failure.
3.4 Altered Ca2+ transport in Tfam KO cardiac mitochondria is caused by changes in protein expression
Having established that Tfam KO cardiac mitochondria take up Ca2+ more avidly and release it more slowly than controls, we sought potential mechanisms for this effect. The most straightforward explanation for altered Ca2+ transport rates would be correlated changes in the expression of the uniporter and exchanger. To test this, we analyzed mRNA expression of genes encoding the uniporter, mitochondrial Na+-Ca2+ exchanger, and other mitochondrial genes (Fig. 5A). The exchanger is encoded by the Nclx gene[39]. The mitochondrial Ca2+ uniporter is a multi-subunit channel composed of a main pore-forming subunit (Mcu)[24, 33, 40, 41], core accessory subunits that target the channel and confer Ca2+ sensitivity and gating (Micu1–3, Emre, Mcub)[42–45], and other associated proteins that alter mitochondrial Ca2+ handling (Mcur1)[26, 46, 47].
Nclx mRNA expression was 0.4 ± 0.1 of control levels (Fig. 5A). Surprisingly, and contrary to the functional data showing enhanced Ca2+ uptake, transcripts of subunits forming the mitochondrial Ca2+ uniporter were also reduced (range: 0.49–0.65 ± 0.04–0.07). To examine if the qPCR data reflected protein levels, we measured band intensity after Western blotting, a semi-quantitative means to gauge candidate protein expression (Fig. 5B–C). We used the same loading control, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), as in our qPCR analysis. Consistent with the reduced Nclx transcripts, protein levels for the Na+-Ca2+ exchanger were also reduced in Tfam KO mitochondria (0.65 ± 0.05). For core uniporter subunits, (MCU, MICU1, EMRE, and MCUB), a 1.5–1.9 fold increase in protein levels was detected (range: 1.5–1.9 ± 0.1–0.2). The only uniporter-associated protein showing a decrease, MCUR1, appears to be a regulator rather than a core channel component[26, 46–48]. Taken together, our data suggest that the enhanced Ca2+ uptake reflects a post-transcriptional mechanism increasing or stabilizing the amount of uniporter present in mitochondria. This effect can be visualized in Fig. 5D, where we map cardiac protein and qPCR levels for all the genes tested across controls and Tfam KO animals. For all genes except for the uniporter subunits from Tfam KO animals, transcript versus protein levels are directly correlated (gray circles, Fig. 5D). The Tfam KO uniporter subunits (black squares, Fig. 5D) clearly form an outlier group with excess protein expression compared to their transcript levels. Thus, the enhancement in mitochondrial Ca2+ appears to result from coincident changes in protein levels of the mitochondrial Ca2+ uniporter and Na+-Ca2+ exchanger.
3.5 Ca2+ stimulates respiration in Tfam KO mitochondria more than control
Next, we examined the effects of mitochondrial Ca2+ on OXPHOS. Physiological Ca2+ concentrations appear not to affect ETC complex I-IV activity directly but can alter OXPHOS in two important ways[3]. Ca2+ entry mildly uncouples mitochondria, increasing the stimulus for electron flow through complex I–IV. Ca2+ also stimulates pyruvate dehydrogenase phosphatase, several dehydrogenases of the citric acid cycle, and the F1-FO-ATP synthase, all ultimately enhancing NADH production and ATP synthesis [3, 49].
We tested these two effects separately. First, we subjected mitochondria incubated in 20 μM TMRM (dequenching mode) to a Ca2+ pulse to measure changes in ΔΨ. This pulse should depolarize mitochondria as Ca2+ enters the matrix, visible as a rise in TMRM fluorescence with dequenching (Fig. 6A). Subsequently, enhanced ETC function should repolarize ΔΨ, visible as a reduction in fluorescence. We measured these parameters as the maximal deflection in TMRM fluorescence (Fig. 6B), an index for Ca2+-induced uncoupling, and the slope of TMRM fluorescence recovery (Fig. 6C), an index for the balance between ETC activity and depolarization associated with continued Ca2+ entry. In this test, Ca2+ pulses produced greater depolarization in Tfam KO mitochondria (Fig. 6B), reflecting enhanced Ca2+ uptake in these samples. Subsequently, Tfam KO mitochondria also repolarized faster than their control counterparts (Fig. 6C). Thus, the direct uncoupling effects of Ca2+, though larger in the Tfam KO mice, were largely complete within a few minutes, consistent with prior reports[49].
Figure 6. Increasing Ca2+ boosts mitochondrial respiration in Tfam KO hearts more than controls.

A. Exemplar trace of ΔΨ response to a 15 μM Ca2+ bolus, measured via TMRM dequenching as in Fig. 3F. B. Maximal change in TMRM signal following Ca2+ bolus (n = 8–14). C. Kinetics of ΔΨ measured as linear slope fit to 30–150 sec after Ca2+ pulse (n = 8–14). D. Exemplar raw traces of mitochondrial O2 consumption stimulated by 0.5 μM FCCP, visible as an increase in τFL of an O2-sensing fluorophore. After maximal consumption of O2 in the sealed chamber, the τFL values plateau. E. Peak relative oxygen consumption rates (OCR) stimulated by incubation with 0.5 μM FCCP, calculated from the rising portion of τFL curves as in (D) (n = 9–13). F. Exemplar raw traces showing inhibition of O2 consumption, stimulated by 0.5 mM ADP, in mitochondria incubated with 1 μM oligomycin. G. Oligomycin summary data (n = 5–6). H. Exemplar raw traces of ADP-stimulated mitochondrial O2 consumption in the presence or absence of Ca2+. I. Summary of Ca2+ effects on ADP-stimulated OCR (n = 9–13). J. Fold change in OCR stimulated by Ca2+ (n = 9–13).
Next, we studied the effect of Ca2+ on ATP synthesis. Directly comparing synthesis rates in the presence or absence of Ca2+ is difficult[49], as divalent binding alters the kinetics of the luciferase reaction[50]. Therefore, as with our analysis in the absence of Ca2+, we normalized ATP synthesis rates to the WT level for each set of samples run in parallel. Remarkably, we found that the deficiency in Tfam KO ATP synthesis noted in Fig. S1G–H was rescued by incubating mitochondria in Ca2+ (Fig. S7A–C). ATP synthesis rates in Tfam KO cardiac mitochondria were comparable to WT, despite the larger transient uncoupling noted previously.
To more robustly quantify the stimulation exerted by matrix Ca2+ in Tfam KO hearts, we analyzed mitochondrial respiration. To avoid the effects of Ca2+ uncoupling, respiration was monitored over a longer period, as ΔΨ largely repolarized within 5 minutes after a single Ca2+ pulse (Fig. 6A) [49]. This assay minimizes the direct effects associated with increased Ca2+ accumulation in Tfam KO mitochondria, allowing us to focus on the steady-state effects of such uptake. To measure respiration, we incubated mitochondria in media containing an oxygen-sensitive fluorophore (MitoXpress Xtra dye), which allowed assays on smaller mitochondrial quantities (20 μg) for prolonged periods. Oxygen quenching of the dye shortens its fluorescence lifetime (τFL), allowing measurement of mitochondrial oxygen consumption in the sealed chamber as an increase in τFL. Before studying the effects of Ca2+, we performed several quality control assays. Maximal oxygen consumption, induced by uncoupling mitochondria in 0.5 μM FCCP, was indistinguishable between Tfam KO and controls (Fig. 6D–E). Thus, despite compromised ETC activity, cardiac mitochondria from KO animals have substantial reserve capacity. Furthermore, oxygen consumption stimulated by 0.5 mM ADP was mitochondrial, as it ceases with inhibition of the ATP synthase in 1 μM oligomycin (Fig. 6F–G).
Next, we measured ADP-stimulated respiration in mitochondrial suspensions containing either the Ca2+ chelator EGTA (1 mM, Ca2+ depleted) or 10 μM EGTA + 12 μM Ca2+. As expected, for WT or HET cardiac mitochondria, Ca2+ enhanced respiration, visible as a faster rise in τFL (Fig. 6H). For these controls, Ca2+ increased oxygen consumption 1.7 ± 0.1 fold (Fig. 6I–J), consistent with prior reports[49]. Tfam KO mitochondria had a markedly different response. After depletion of mitochondrial Ca2+, Tfam KO mitochondria respired at half the control rates (Fig. 6H–I, S5A; 0.45 ± 0.12), and in some instances, was almost absent (Fig. 6I, far right-most exemplar). When incubated with Ca2+, however, mitochondrial respiration increased 2.5 ± 0.2 fold, approaching control rates (Fig. 6I–J). Thus, the increased Ca2+ in Tfam KO mitochondria produces a robust activation of respiration.
4. DISCUSSION
4.1 Conclusion
We show here that a mouse model of mitochondrial cardiomyopathy features increased mitochondrial Ca2+ levels, using multiple independent techniques: analysis of mitochondrial Ca2+ content, imaging of Ca2+ transport, direct electrophysiological measurement of uniporter currents, and assays of Ca2+ transporter protein expression. Such increases can improve deficits in OXPHOS in in vitro assays. Whether such increased matrix Ca2+ compensates for deficient energy synthesis or ultimately leads to mitochondrial dysfunction in vivo remains to be established, and will require further studies where the genes responsible for mitochondrial Ca2+ transport, such as Mcu, or overload can be selectively deleted in Tfam KO animals.
4.2 Regulation of mitochondrial Ca2+ transport in cardiac or skeletal myopathies
Besides describing the Ca2+ phenotype present in these animals, we show that the cardiomyopathy features an increase in the mitochondrial Ca2+ uniporter. Although several prior studies have shown increased mitochondrial Ca2+ content in the setting of disease, typically in muscle or nervous system, such increases have not implicated enhanced uniporter expression. Much of this prior evidence comes from skeletal muscle. The prevalence of mitochondrial Ca2+ overload in skeletal muscle disorders may reflect the larger magnitude of uniporter-mediated transport. Uniporter current density in skeletal muscle is 30 times larger than in heart[36], and becomes active at lower cytoplasmic Ca2+ levels[51]. Thus, given the already-large flux, skeletal muscle mitochondria may be far more prone to pathological Ca2+ overload, and not achieve any benefit by enhancing the flux further. Indeed, in the analysis of skeletal muscle myopathy caused by muscle-specific Tfam deletion[52], mitochondrial Ca2+ overload during repetitive stimulation was attributed to the permeability transition pore. Similarly, the tendency to deleterious mitochondrial Ca2+ overload in skeletal muscle is seen in malignant hypothermia[32], and myopathies caused by MICU1 mutations[53].
Mitochondrial Ca2+ overload also appears to cause cardiac injury in heart failure[54, 55]. However, there has been a lack of evidence for regulation of the uniporter, and investigators have yet to establish if mitochondrial Ca2+ transport is regulated in other forms of cardiac energetic failure. In another model of cardiac mitochondrial disease, mice with complex I deficiency have normal lifespan and cardiac function, but develop accelerated heart failure with stress[56]. Mitochondria from these animals were more sensitive to Ca2+ overload, but the investigators did not assess whether Ca2+ uptake rates changed during heart failure progression. Here, we found no sensitization to Ca2+-induced permeability transition in the Tfam KO mice. Nevertheless, Tfam KO may still be more likely to undergo permeability transition, as enhanced baseline Ca2+ and Ca2+ influx, and diminished Ca2+ efflux all may be driving mitochondrial Ca2+ closer to activating the permeability transition.
By studying Ca2+ transport via whole-mitoplast voltage clamping relatively early in the cardiomyopathy, we bypassed several confounding factors associated with analyses of end-stage failure, such as an increased mass of fissioned mitochondria and reduced ΔΨ. In the late stages of heart failure, enhanced mitochondrial Ca2+ uptake may be obscured, especially if such feedback regulation ultimately leads to mitochondrial Ca2+ overload and selective injury to those very cardiomyocytes that took up the most Ca2+. Indeed, the effect of deleting Mcu variably affects cardiac injury depending on the exact mouse and injury model[57–59]. Moreover, in end-stage failing human hearts, mitochondrial Ca2+ uptake appears to be blunted[60]. Given this complexity in mitochondrial Ca2+ transport in cardiac injury, mitochondrial cardiomyopathies present a unique opportunity to study regulation of this pathway and these diseases may best respond to therapies targeting mitochondrial Ca2+ transport.
4.3 Mechanism regulating mitochondrial Ca2+ transport in Tfam KO hearts
The regulation of mitochondrial Ca2+ described here is unlikely to be due to direct transcriptional control by TFAM, as it is a transcription factor for mtDNA and not nuclear-encoded genes, such as the uniporter or exchanger subunits. In fact, recent genome-wide studies show that while TFAM coats mitochondrial DNA, it fails to bind nuclear DNA, and is essentially absent in the nucleus[61]. In addition, Tfam HET animals at this age had no observable change in mitochondrial Ca2+ transport despite reduced transcription of mtDNA-encoded ETC subunits, effects mediated directly by TFAM. Thus, increased mitochondrial Ca2+ in Tfam KO animals is not due to a direct alteration in uniporter or exchanger transcription caused by TFAM, but rather to the heart failure that develops in its absence.
The mechanism for enhanced mitochondrial Ca2+ in Tfam KO hearts appears to vary based on the transport pathway. The reduction in Ca2+ efflux appears mediated by a reduction in Nclx transcription, which leads to consequent reduction in Na+-Ca2+ exchanger protein levels. Such a reduction may ultimately predispose to sudden death via mitochondrial Ca2+ overload, as seen in other forms of heart failure [62]. The situation for the mitochondrial Ca2+ uniporter is more complicated, since we find decreased transcripts but increased protein expression of channel subunits. Such discrepancy between transcript and protein levels can occur for up to 20% of genes assayed in heart failure versus control animals[20]. However, the changes we see here likely reflect a regulatory pathway rather than heart failure epiphenomena, as we found similar magnitudes and concordant changes across all the core uniporter subunits assayed. Potential mechanisms for regulating the uniporter include altering transcript expression[63], changing channel conductance[64, 65], changing channel oligomerization state[66–68], or changing channel sensitivity to cytoplasmic Ca2+ [51]. Future studies will need to establish the regulatory mechanism active in Tfam KO hearts and whether it stabilizes the channel or modifies its underlying behavior.
Supplementary Material
Highlights.
KO of mitochondrial transcription factor Tfam models mitochondrial cardiomyopathy
Cardiac mitochondria from Tfam KO mice take up Ca2+ at twice the rate as controls
Cardiac mitochondria from Tfam KO mice release Ca2+ at half the rate as controls
These changes reflect increased MCU complexes and decreased NCLX protein
Mitochondrial Ca2+ maintains respiratory rates despite compromised OXPHOS
Acknowledgments
We thank Nils-Göran Larsson and C. Ronald Kahn for providing Tfamfl/fl mice, Kenneth Spitzer for assistance with cardiomyocyte imaging, Diego Fernandez and Christopher Anderson for ICP-MS core services (Geology and Geophysics, University of Utah), Maria Ericsson for electron microscopy core services (Harvard Medical School), and staff at the Research Histology core (Huntsman Cancer Institute, University of Utah and ARUP Research institute) for tissue processing.
SOURCES OF FUNDING
This work was supported by the Heart, Lung, and Blood Institute and Institute of Neurological Disorders and Stroke of the NIH under awards R00HL124070 (D.C.), T32HL007572 (P.R.H.), and T32NS007473 (P.R.H.), American Heart Association under award 13FTF16890003 (D.C.), and the Nora Eccles Treadwell Foundation (D.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
ABBREVIATIONS
- AA
antimycin A
- CSA
cyclosporin A
- ETC
Electron transport chain
- IMAC
Inner membrane anion channel
- OXPHOS
Oxidative phosphorylation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None
References
- 1.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circulation research. 2013;113(6):709–24. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circulation research. 2004;95(2):135–45. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 3.Balaban RS. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochimica et biophysica acta. 2009;1787(11):1334–41. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Hattab AW, Scaglia F. Mitochondrial Cardiomyopathies. Front Cardiovasc Med. 2016;3:25. doi: 10.3389/fcvm.2016.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmgren D, Wahlander H, Eriksson BO, Oldfors A, Holme E, Tulinius M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J. 2003;24(3):280. doi: 10.1016/s0195-668x(02)00387-1. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nature genetics. 1999;21(1):133–7. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 7.Stiles AR, Simon MT, Stover A, Eftekharian S, Khanlou N, Wang HL, Magaki S, Lee H, Partynski K, Dorrani N, Chang R, Martinez-Agosto JA, Abdenur JE. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol Genet Metab. 2016;119(1–2):91–9. doi: 10.1016/j.ymgme.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(7):4038–43. doi: 10.1073/pnas.061038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):3136–41. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavi P, Hansson A, Zhang SJ, Larsson NG, Westerblad H. Abnormal Ca(2+) release and catecholamine-induced arrhythmias in mitochondrial cardiomyopathy. Human molecular genetics. 2005;14(8):1069–76. doi: 10.1093/hmg/ddi119. [DOI] [PubMed] [Google Scholar]
- 11.Ahuja P, Wanagat J, Wang Z, Wang Y, Liem DA, Ping P, Antoshechkin IA, Margulies KB, Maclellan WR. Divergent mitochondrial biogenesis responses in human cardiomyopathy. Circulation. 2013;127(19):1957–67. doi: 10.1161/CIRCULATIONAHA.112.001219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A, Magrini G, Campana C, Fortina P, Gavazzi A, Narula J, Vigano M. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. The American journal of pathology. 1998;153(5):1501–10. doi: 10.1016/S0002-9440(10)65738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diakos NA, Navankasattusas S, Abel ED, Rutter J, McCreath L, Ferrin P, McKellar SH, Miller DV, Park SY, Richardson RS, Deberardinis R, Cox JE, Kfoury AG, Selzman CH, Stehlik J, Fang JC, Li DY, Drakos SG. Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch During Mechanical Unloading of the Failing Human Heart: Implications for Cardiac Reloading and Conditioning. JACC Basic to translational science. 2016;1(6):432–444. doi: 10.1016/j.jacbts.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96(7):2190–6. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 15.Yue R, Xia X, Jiang J, Yang D, Han Y, Chen X, Cai Y, Li L, Wang WE, Zeng C. Mitochondrial DNA oxidative damage contributes to cardiomyocyte ischemia/reperfusion-injury in rats: cardioprotective role of lycopene. Journal of cellular physiology. 2015;230(9):2128–41. doi: 10.1002/jcp.24941. [DOI] [PubMed] [Google Scholar]
- 16.Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, Walker UA. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation. 2003;108(19):2423–9. doi: 10.1161/01.CIR.0000093196.59829.DF. [DOI] [PubMed] [Google Scholar]
- 17.Kanazawa A, Nishio Y, Kashiwagi A, Inagaki H, Kikkawa R, Horiike K. Reduced activity of mtTFA decreases the transcription in mitochondria isolated from diabetic rat heart, American journal of physiology. Endocrinology and metabolism. 2002;282(4):E778–85. doi: 10.1152/ajpendo.00255.2001. [DOI] [PubMed] [Google Scholar]
- 18.Gao Z, Barth AS, DiSilvestre D, Akar FG, Tian Y, Tanskanen A, Kass DA, Winslow RL, Tomaselli GF. Key pathways associated with heart failure development revealed by gene networks correlated with cardiac remodeling. Physiological genomics. 2008;35(3):222–30. doi: 10.1152/physiolgenomics.00100.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. The Journal of physiology. 2003;551(Pt 2):491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster DB, Liu T, Kammers K, O’Meally R, Yang N, Papanicolaou KN, Talbot CC, Jr, Cole RN, O’Rourke B. Integrated Omic Analysis of a Guinea Pig Model of Heart Failure and Sudden Cardiac Death. Journal of proteome research. 2016;15(9):3009–28. doi: 10.1021/acs.jproteome.6b00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, Tsutsui H. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005;112(5):683–90. doi: 10.1161/CIRCULATIONAHA.104.524835. [DOI] [PubMed] [Google Scholar]
- 22.Kunkel GH, Chaturvedi P, Tyagi SC. Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail Rev. 2016;21(5):499–517. doi: 10.1007/s10741-016-9561-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. The Journal of clinical investigation. 1997;100(1):169–79. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhuri D, Sancak Y, Mootha VK, Clapham DE. MCU encodes the pore conducting mitochondrial calcium currents. eLife. 2013;2:e00704. doi: 10.7554/eLife.00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nature protocols. 2012;7(6):1235–46. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 26.Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1602264113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Will Y, Hynes J, Ogurtsov VI, Papkovsky DB. Analysis of mitochondrial function using phosphorescent oxygen-sensitive probes. Nature protocols. 2006;1(6):2563–72. doi: 10.1038/nprot.2006.351. [DOI] [PubMed] [Google Scholar]
- 28.Gaussin V, Van de Putte T, Mishina Y, Hanks MC, Zwijsen A, Huylebroeck D, Behringer RR, Schneider MD. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):2878–83. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3467–72. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long Q, Yang K, Yang Q. Regulation of mitochondrial ATP synthase in cardiac pathophysiology. Am J Cardiovasc Dis. 2015;5(1):19–32. [PMC free article] [PubMed] [Google Scholar]
- 31.Voronina SG, Barrow SL, Gerasimenko OV, Petersen OH, Tepikin AV. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. The Journal of biological chemistry. 2004;279(26):27327–38. doi: 10.1074/jbc.M311698200. [DOI] [PubMed] [Google Scholar]
- 32.Giulivi C, Ross-Inta C, Omanska-Klusek A, Napoli E, Sakaguchi D, Barrientos G, Allen PD, Pessah IN. Basal bioenergetic abnormalities in skeletal muscle from ryanodine receptor malignant hyperthermia-susceptible R163C knock-in mice. The Journal of biological chemistry. 2011;286(1):99–113. doi: 10.1074/jbc.M110.153247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–5. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cardenas C, Shanmughapriya S, Rajan S, Vallem S, Chen X, Foskett JK, Cheung JY, Houser SR, Madesh M. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28(11):4936–49. doi: 10.1096/fj.14-256453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–4. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 36.Fieni F, Bae Lee S, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nature communications. 2012;3:1317. doi: 10.1038/ncomms2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borecky J, Jezek P, Siemen D. 108-pS channel in brown fat mitochondria might Be identical to the inner membrane anion channel. The Journal of biological chemistry. 1997;272(31):19282–9. [PubMed] [Google Scholar]
- 38.Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. The Journal of biological chemistry. 2007;282(30):21889–900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):436–41. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–40. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol. 2015;16(9):545–53. doi: 10.1038/nrm4039. [DOI] [PubMed] [Google Scholar]
- 42.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467(7313):291–6. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PloS one. 2013;8(2):e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342(6164):1379–82. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. The EMBO journal. 2013;32(17):2362–76. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca(2+) uptake that regulates cellular metabolism. Nature cell biology. 2012;14(12):1336–43. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S, Madesh M. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell reports. 2016;15(8):1673–85. doi: 10.1016/j.celrep.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell metabolism. 2015;21(1):109–16. doi: 10.1016/j.cmet.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 49.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase, American journal of physiology. Cell physiology. 2000;278(2):C423–35. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 50.Lee RT, Denburg JL, McElroy WD. Substrate-binding properties of firefly luciferase. II. ATP-binding site. Archives of biochemistry and biophysics. 1970;141(1):38–52. doi: 10.1016/0003-9861(70)90103-7. [DOI] [PubMed] [Google Scholar]
- 51.Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, Szabo I, Zanotti G, Rizzuto R, Raffaello A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Molecular cell. 2016;64(4):760–773. doi: 10.1016/j.molcel.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 52.Aydin J, Andersson DC, Hanninen SL, Wredenberg A, Tavi P, Park CB, Larsson NG, Bruton JD, Westerblad H. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Human molecular genetics. 2009;18(2):278–88. doi: 10.1093/hmg/ddn355. [DOI] [PubMed] [Google Scholar]
- 53.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE, Roper H, Santen GW, Niks EH, van der Pol WL, Lindhout D, Raffaello A, De Stefani D, den Dunnen JT, Sun Y, Ginjaar I, Sewry CA, Hurles M, Rizzuto R, U.K. Consortium. Duchen MR, Muntoni F, Sheridan E. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nature genetics. 2014;46(2):188–93. doi: 10.1038/ng.2851. [DOI] [PubMed] [Google Scholar]
- 54.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. The Journal of clinical investigation. 2007;117(9):2431–44. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(36):11389–94. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell metabolism. 2013;18(2):239–50. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell reports. 2015;12(1):15–22. doi: 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell reports. 2015;12(1):23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nature cell biology. 2013;15(12):1464–72. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michels G, Khan IF, Endres-Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T, Hoppe UC. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation. 2009;119(18):2435–43. doi: 10.1161/CIRCULATIONAHA.108.835389. [DOI] [PubMed] [Google Scholar]
- 61.Wang YE, Marinov GK, Wold BJ, Chan DC. Genome-wide analysis reveals coating of the mitochondrial genome by TFAM. PloS one. 2013;8(8):e74513. doi: 10.1371/journal.pone.0074513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017;545(7652):93–97. doi: 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan L, Huang BJ, Ma XE, Wang SY, Feng J, Lv F, Liu Y, Liu Y, Li CM, Liang DD, Li J, Xu L, Chen YH. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int J Mol Sci. 2015;16(3):5420–33. doi: 10.3390/ijms16035420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491(7423):269–73. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fieni F, Johnson DE, Hudmon A, Kirichok Y. Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature. 2014;513(7519):E1–2. doi: 10.1038/nature13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O-Uchi J, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, Yi B, Zhang H, Gross P, Mishra J, Ainbinder A, Kettlewell S, Smith GL, Dirksen RT, Wang W, Rizzuto R, Sheu SS. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxidants & redox signaling. 2014;21(6):863–79. doi: 10.1089/ars.2013.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, Riitano MF, Worth AM, Seelam A, Carvalho E, Subbiah R, Jana F, Soboloff J, Peng Y, Cheung JY, Joseph SK, Caplan J, Rajan S, Stathopulos PB, Madesh M. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Molecular cell. 2017 doi: 10.1016/j.molcel.2017.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Konig T, Troder SE, Bakka K, Korwitz A, Richter-Dennerlein R, Lampe PA, Patron M, Muhlmeister M, Guerrero-Castillo S, Brandt U, Decker T, Lauria I, Paggio A, Rizzuto R, Rugarli EI, De Stefani D, Langer T. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Molecular cell. 2016;64(1):148–162. doi: 10.1016/j.molcel.2016.08.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.