


Abstract
MEK4 is an upstream kinase in MAPK signaling pathways where it phosphorylates p38 MAPK and JNK in response to mitogenic and cellular stress queues. MEK4 is overexpressed and induces metastasis in advanced prostate cancer lesions. However, the value of MEK4 as an oncology target has not been pharmacologically validated because selective chemical probes targeting MEK4 have not been developed. Despite a high level of sequence homology in the ATP-binding site, most reported MEK inhibitors are selective for MEK1/2 and display reduced potency toward other MEKs. Here, we present the first functional and binding selectivity-profiling platform of the MEK family. We applied the platform to profile a set of known kinase inhibitors and used the results to develop an in silico approach for small molecule docking against MEK proteins. The docking studies identified molecular features of the ligands and corresponding amino acids in MEK proteins responsible for high affinity binding versus those driving selectivity. WaterLOGSY and saturation transfer difference (STD) NMR spectroscopy techniques were utilized to understand the binding modes of active compounds. Further minor synthetic manipulations provide a proof of concept by showing how information gained through this platform can be utilized to perturb selectivity across the MEK family. This inhibitor-based approach pinpoints key features governing MEK family selectivity and clarifies empirical selectivity profiles for a set of kinase inhibitors. Going forward, the platform provides a rationale for facilitating the development of MEK-selective inhibitors, particularly MEK4 selective inhibitors, and repurposing of kinase inhibitors for probing the structural selectivity of isoforms.
Graphical abstract
Mitogen-activated protein kinase (MAPK) signaling pathways are conserved across eukaryotes from yeast to humans where they play a central role regulating cellular activities from survival and proliferation to stress response, differentiation, motility, and angiogenesis.1 MAPK pathways couple diverse extracellular signals (growth factors, hormones, cytokines, and environmental stresses) to distinct intracellular gene programs via a series of activating phosphorylation events. MAPK/Erk kinase (MEK) family enzymes, also called mitogen-activated protein kinase kinases (MAP2K or MKKs), are dual specificity kinases that phosphorylate serine/threonine and tyrosine residues within the activation loops of downstream MAP kinase effector proteins. Four distinct MAPK cascades have been identified and are referred to by the downstreammost MAPK protein: extracellular signal-regulated kinase (ERK1/2), c-Jun N-terminal kinase (JNK), p38 MAPK, and ERK5 (Figure 1).2
Figure 1.

(A) MEK proteins and their signaling pathways. (B) Integrated approaches to profile selectivity of MEK isoforms.
MAPK signaling cascades are dysregulated in human cancer and inflammatory diseases, and small molecule inhibitors targeting MAPK signaling components are under intense investigation in the clinic.3,4 A large number of MAPK inhibitors target MEK1/2.5,6 MEK1 and MEK2 exhibit nearly 90% sequence homology including a unique allosteric pocket that has been pharmacologically targeted to lock unphosphorylated MEK1/2 in a catalytically inactive state. Molecules targeting this allosteric pocket, such as the FDA-approved trametinib, exhibit unparalleled selectivity because they do not bind the conserved ATP-binding site.7 Most clinically relevant MEK inhibitors today target the MEK1/2 allosteric site and therefore show little activity against MEK3, MEK4, MEK5, MEK6, or MEK7. Indeed there is a dearth of chemical matter directed at these other MEK family members, which is surprising given their roles in a host of biological processes, and as a result their value as therapeutic targets has not been thoroughly investigated.
Compelling rationale exists for therapeutically targeting MEK family members beyond MEK1/2. Dysregulation of the p38 MAPK pathway has been implicated in a range of diseases including rheumatoid arthritis and other inflammatory disorders, cancer, cardiovascular disease, and neurodegeneration.8,9 Over 20 different p38 inhibitors have been tested in clinical trials, and none have progressed to phase III, prompting new strategies such as the inhibition of upstream activators MEK3 and MEK6.10,11 Aberrant JNK signaling has been attributed to a similarly wide-ranging list of human diseases, and therefore inhibitors targeting the upstream activators MEK4 and MEK7 could prove valuable.12 In addition to the p38 and JNK pathways, overexpression of MEK5 has been reported in a variety of cancers.13
One reason to target the MEKs is to cast a wider net of pharmacological activity compared to targeting downstream MAPKs. For example, existing p38 inhibitors target the four isoforms (α, β, γ, and δ) to varying degrees, whereas disrupting an upstream kinase would block all isoforms equally. Further, MEKs can activate alternative pathways, such as NF-κB, that provide overlapping functions with downstream MAPKs; inhibiting MEKs might thereby limit “escape routes” to therapeutic intervention. A second reason to target upsteam kinases is to take advantage of context-dependent activities including cell-type- and stimuli-specific signaling patterns. For example, although both MEK4 and MEK7 activate JNK, only MEK7 appears to mediate JNK-dependent inflammatory responses in immune cells. Thus, targeting MEK7 could alleviate harmful inflammation without broadly disrupting JNK function.
Our group has become interested in MEK4 as a target for the therapeutic inhibition of prostate cancer (PCa) metastasis.14 Although often described only as an activator of JNK, MEK4 also activates p38α and p38β. MEK4 is overexpressed in advanced PCa lesions and induces metastasis in PCa.15–19 MEK4 also appears to have a similar pro-invasion/prometastatic role in several other cancer types, including breast and pancreatic cancer.20 Through genetic and chemical approaches, MEK4 was shown to increase the invasive potential of PCa cells in vitro by upregulating the production of several matrix metalloproteinases (MMP’s) in response to TGF-β treatment.21 Overexpressing MEK4 increased the number of metastatic deposits observed in a PCa mouse model. These findings identify MEK4 as a clinically important therapeutic target and underscore the need to develop selective MEK4 probes for in vivo target validation in advanced cancer model systems.
Selective chemical tools targeting MEK4 and other family members apart from MEK1/2 could uncover new roles for these proteins in diverse human disesases. However, developing selective kinase inhibitors is met with the usual challenges of achieving specificity when targeting a conserved ATP-binding pocket. Over the past decade, new strategies for engineering selectivity have emerged including targeting inactive enzyme conformations (type II inhibitors), allosteric pockets (type III), and other distal sites involved in binding regulatory proteins (type IV).22 Alternatively a small “gatekeeper” close to the hinge region can expose a hydrophobic region to be exploited in some kinases.23 Targeting phosphate-binding loop (P-loop) conformations has emerged as another method for imparting selectivity to small molecules.24 A major limitation in these approaches is the requirement for detailed structural data for the target. As an alternative approach, Shokat et al. is widely known for utilizing engineered analog sensitive protein kinases as a tool for dissecting phospho-signaling pathways for elucidating the cellular function of individual kinases. 25,26 Recently, Heinzlmeir et al.27 utilized chemical proteomics and crystal structures along with an NMR experiment to facilitate the development of receptor tyrosine kinase EPHA2 inhibitors. Their approach is widely applicable to a variety of kinases. While their platform focuses on the application of a large set of clinical kinase drugs, the research presented here focuses on a highly focused set of kinase inhibitors. The selectivity profiling used in this study for the MEK family initiates with functional testing since binding and functional inhibition have been shown to not always correlate. Ultimately with regard to the development of selective inhibitors, a “map” of the structural features that could be exploited for a given target compared to closely related counter-targets would prove valuable in developing selective probes.
A major goal at the onset was to leverage an established, small set of compounds from the literature shown to have off-target effects on MEK4 to probe for selectivity trends among the MEK kinase family. Here, we present a platform for mapping the pharmacological relatedness of all seven MEK family members to one another. We utilize the platform to identify structural features responsible for small molecule selectivity within the MEK family by correlating empirical activity data from a universal kinase assay with structural studies performed by in silico modeling. We validate our findings using Waterlogsy and saturation transfer difference (STD) NMR spectroscopy techniques to confirm the binding modes for select active compounds. The structural features identified here were validated using proof of concept synthetic analogs, and this knowledge will be leveraged in our future design of selective chemical probes. In addition, the general approach presented here can serve as a template for developing selective inhibitors against other closely related protein kinase families.
RESULTS AND DISCUSSION
Functional Profiling of Known Kinase Inhibitors against MEK Isoforms
A functional in vitro profiling platform was developed with active recombinant protein for each MEK isoform to measure the ability of a compound to inhibit phosphorylation of the downstream substrate. Because most established MEK inhibitors target MEK1/2 and our focus is to develop probes against other MEK proteins, specifically MEK4, we sought other means to identify candidate inhibitors for testing in our assay platform. ATP-competitive kinase inhibitors frequently interact with a spectrum of kinases, and the extent of cross-reactivity has become more apparent with the commercialization of kinome-wide profiling panels. 28–30 We searched two published kinome profiling data sets31,32 and selected seven compounds based on their binding affinity (Kd) to MEK4 and global kinome selectivity score (S[3 µM]; Figure S1). We first validated the binding of these compounds toward MEK4 with a fluorescence thermal shift (FTS) assay using previously described methods (Figure S2).33 This data was further compared to previously published binding affinities, showing a high thermal shift (ΔTm) with high binding affinity (Kd), e.g., PLX-4720 with Kd of <200 nM and ΔTm > 10 °C (Figure S3A). Nevertheless, binding affinity does not differentiate between agonist and antagonist activity or reveal a compound’s ability to functionally inhibit downstream substrate phosphorylation. Thus, we were motivated to develop a set of high throughput assays to rapidly characterize the functional activities for a set of compounds across the entire MEK family.
To profile inhibitors against all seven MEK isoforms, we optimized the universal ADP-Glo biochemical kinase assay to each MEK/substrate system (Figure 1). We used active MEK proteins incubated with ATP and full-length recombinant downstream substrates containing point mutations to render them catalytically inactive. In this assay, the luminescence signal is proportional to amount of ADP produced during the kinase reaction, which in turn is proportional to MEK kinase activity. By detecting ADP production, the assay measures universal MEK activity without substrate-specific antibodies. To ensure the profiling accurately determines the intrinsic affinities of the inhibitors for each MEK protein, the Km values for ATP were determined in every MEK/substrate system. The Km values were consistent with previous reports.34–36 The concentrations of enzyme and substrate were optimized for a sufficient signal/ background, and the enzyme reaction time was tested to determine initial velocity conditions (Figures S4 and S5).
The inhibitors selected from published binding data were tested against each MEK protein at an ATP concentration below the KM. The promiscuous inhibitor staurosporine was titrated against the enzyme system as a positive control (Figure 2A).36 Five of the seven compounds displayed potent inhibition against MEK4, and each compound potently inhibited at least one MEK protein (Figure 2B). By comparing the inhibitory profiles of the full set of compounds across the various MEKs, we can begin to determine the degree of “pharmacological similarity” between the kinases. For example, MEK1 and MEK2 appeared very similar with nearly identical compound activity profiles. MEK3 was unique because it was not inhibited by any of the inhibitors other than staurosporine. MEK5 was strongly inhibited by all compounds except GSK1838705A. The activity profiles of MEK4 and MEK6 were correlated. Although we hypothesized MEK7 would have a similar profile as MEK4 due to moderately high structural homology and sharing of a common substrate (JNK), this compound set revealed a unique profile for MEK7 with inhibition only by AST-487 and LY-333531. Interestingly, the published binding affinity data showed relatively weak correlation with functional inhibition for this set of compounds across the MEK family (Figures S3B and S7 for detailed comparison). Although the rationale for this is unclear, it underscores the importance of measuring functional inhibition, at least for the MEK family, when evaluating kinase inhibitor selectivity patterns.
Figure 2.

MEK isoform selectivity panel screen. (A) Staurosporine control data for each MEK isoform, assay validation. (B) Heat map of collected IC50 data on all MEK isoforms against selected inhibitors. Cells contain IC50 values in micromolarity. (C) Structural homology of MEK isoforms as a percentage of similarity (%). MEK3 and MEK5 missing in structural comparison due to lack of crystal structures. Red, blue, and green highlight the three most structurally homologous isoforms.
Isoform Active Site Comparison
As a first step in understanding the structural basis for the inhibitory patterns shown in Figure 2B, we asked if compound activities were correlated to the overall structural homology of the MEK proteins. In other words, did the enzyme with the highest degree of structural homology show similar inhibitory profiles? To answer this, we calculated structural homology between each pair of MEK enzymes by overlaying the tertiary structures of the enzymes for comparison using the protein structure alignment panel implemented in the Biopolymer module of the Tripos software. Considering one isoform as the fixed reference structure and the others as the movable structures with the atoms to fit option as C-α, the alignments were carried out. This process was repeated keeping each isoform as the reference structure and moving the others to generate an orthogonal table as shown in Figure 2C, note MEK3 and MEK5 currently lack the appropiate crystallographic data to perform the analysis. We observed a correlation between the inhibitory profiles against MEK1 and MEK2 (89% homology) and also against MEK4 and MEK6 (60% homology). However, the next most related protein pair, MEK4 and MEK7 (54% homology), exhibited little similarity across this compound set. We also performed a sequence homology of the MEK isoforms (being able to include MEK3 and MEK5), and overall the trends correlate between the sequence and the structural homology analysis with only slight differences in the percentages. Thus again, we were not able to isolate a specific trend across the compound set and isoforms (see Figure S8). Even with the sequence homology, we still lack critical information necessary to explain the unique resistance seen by MEK3 isoform. We hypothesized that more specific structural features than the overall three-dimensional structural and sequence homology would be needed to explain the pharmacological similarity patterns observed across the MEK family for these inhibitors. We therefore characterized the active sites for MEK1, MEK2, MEK4, MEK6, and MEK7 in silico to generate metrics describing the shape of the ligand binding pockets, the chemical properties of residues found in the active sites, and the physical properties of the activation loop residues.
We used high resolution ATP bound cocrystal structures of MEK1 (PDB: 3EQC), MEK2 (PDB: 1S9I), and MEK6 (PDB: 3VN9) in the analysis. No ATP-bound crystal structure for MEK7 is currently available, so we used an unbound structure (PDB: 3WZU), and using the Glide-XP docking module37 followed by a 10 ns molecular dynamics simulations, we identified the docked conformation of ATP. For MEK4, a low resolution ATP-bound structure is available (PDB: 3ALN) with missing or uncertain (high B value) side chain atoms for some active site residues. To address this, we carried out a protein refinement with tools of the Prime module available in the Schrodinger suite, which added the missing atoms and corrected the torsions of the active site residues Lys131, Leu236, and Lue180. After the corrections, the MEK4 structure was subjected to energy minimization in OPLS-2005 force field to make the structure suitable for further in silico studies. We then took the centroid location of ATP in the binding sites and used a 5 Å radius to determine all binding pocket residues (Table S1). To compute live protomols38 of the active site residues, we computationally sampled the pocket with three different probes, hydrophobic, hydrogen bond donor and acceptor (CH4, −NH, and −C═O), to identify all interacting pharmacophoric elements for the seven MEK isoforms (Table S2). We found the hydrophobic and hydrogen bond donor/ acceptor calculations to be similar for the MEK1–MEK2 and MEK4–MEK6 pairs. MEK7 was unique with more hydrophobic residues and fewer hydrogen bond donors and acceptors than the other MEK proteins.
Our analysis of active site residues provides an initial means to organize the MEK family into subsets likely to exhibit similar activity profiles in response to a given inhibitor, which binds to the active site. To expand our analysis, we calculated the size and shape of the active site for each MEK in terms of cavity depth (Å), area (Å2), and volume (Å3) (Table 1, top rows). Although all five active site pockets have a similar surface area, MEK1 and MEK2 have a larger volume and MEK7 has a significantly smaller cavity depth (p < 0.001).
Table 1.
MEK Isoform ATP Binding Pocket Comparisona
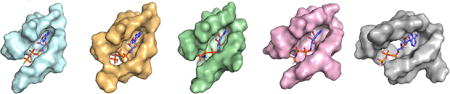
|
|||||
|---|---|---|---|---|---|
| MEK1 | MEK2 | MEK4 | MEK6 | MEK7 | |
| Cavity depth (Å) | 7.23 | 7.15 | 7.00 | 6.96 | 5.95 |
| Area (Å2) | 2354 | 2363 | 2276 | 2267 | 2291 |
| Volume (Å3) | 3638 | 3598 | 3222 | 3282 | 3212 |
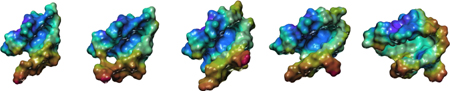
|
|||||
| Hydrophobicity (%) | 34.2 | 33.3 | 31.0 | 32.1 | 37.9 |
| Acidic (%) | 17.2 | 17.0 | 13.8 | 13.9 | 10.3 |
| Basic (%) | 10.3 | 10.0 | 13.8 | 14.3 | 10.3 |
| Neutral (%) | 37.9 | 38.0 | 41.4 | 41.0 | 41.4 |
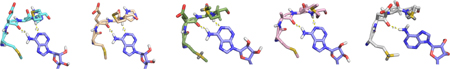
|
|||||
| Residues | M143EH | M147EH | M178EL | M129EL | M212EL |
| Local Curvature (Å−1) | 3.98 | 3.96 | 3.69 | 3.64 | 4.28 |
| Phi angle, φ (°) | 177.7 | 178.6 | 179.9 | 181.9 | 187.5 |
| Psi angle, ψ (°) | 112.2 | 111.0 | 115.0 | 114.6 | 136.3 |
Electrostatic potential map of MEK isoforms: red (positive); blue (negative).
We further analyzed the properties of the binding pockets to compare the chemical properties of residues from the active sites of the MEK isoforms (Table 1, middle rows). We found MEK1 and MEK2 exhibited very similar characteristics with a higher proportion of acidic residues than the others MEKs. MEK4 and MEK6 were also highly similar to each other. MEK7 has a significantly different binding pocket compared to the other isoforms with more hydrophobic residues and less charged residues than the other active sites we analyzed. These features provide another parameter leading to varied inhibitor selectivity over MEK4 and MEK6. To generate an electrostatic potential map of the active site binding cavities we assigned Kollman charges to the residues.39 This revealed that while MEK1, MEK2, MEK4, and MEK6 cavities are negatively charged, the MEK7 cavity shows patches of partial positive and negative charges. A lipophilic analysis of the binding pocket correlates strongly with the electrostatic calculations as can be seen in Figure S9, with MEK7 having a significantly less hydrophilic pocket. Taken together, the chemical and electrostatic analysis of active site residues from the five MEK proteins supports our earlier observations that MEK1-MEK2, MEK4-MEK6, and MEK7 represent somewhat distinct subsets within the MEK family and they are likely to respond in a similar fashion to a panel of inhibitors.
Since the inhibitors used in this study have previously been shown to act in an ATP-competitive manner, we analyzed the hinge region of the ATP-binding pocket in more detail (Table 1, bottom rows). Local curvature was analyzed to understand how inhibitors are interacting in the ligand binding cavities of the different MEK proteins. The topological local curvature can be quantified by assigning concave, convex, or saddle-type curvatures to each inside surface point of the cavities.40 This analysis revealed that MEK1 and MEK2 have nearly identical curvatures (3.98 Å−1 and 3.96 Å−1, respectively). MEK4 and MEK6 are also very similar to one another (3.69 Å−1 and 3.64 Å−1, respectively) while MEK7 exhibits a larger local curvature than all other MEK proteins (4.28 Å−1). The primary requirement for the type 1 kinase inhibitors is to have at least 1–3 hydrogen bonds with the hinge residues. Hence, a donor and an acceptor atom must be present in the inhibitors to form hydrogen bonds with the hinge residues. The hydrogen bond strength depends on the two torsional planes containing the donor–acceptor atoms in the hinge residues. We calculated the two torsional angles (φ and ψ) of the hinge regions which describe the backbone of the amino acids containing the carbonyl and amine groups in the pocket responsible for forming putative hydrogen bonds with the inhibitors. Once again, MEK1 and MEK2 appeared highly similar to both torsional angles appearing within about 1° of each other. The MEK4 and MEK6 torsional angles were within about 2° of each other. MEK7 was distinct with a φ angle of 187.5° and ψ angle of 136.3°, which we anticipate would significantly alter the “approach angle” at which an inhibitor will bind within the active site. Corresponding to this expectation, we noted a distinct orientation at which ATP is predicted to bind to the hinge region of MEK7 relative to those of the other MEK proteins (Table 1, bottom graphic). We expect the geometrical requirements caused by the rigid nature of the active state binding site may be exploited to develop compounds with excellent shape complementarity to MEK7.
In order to present an overall picture for comparing MEK family active site architecture, we implemented a principal component analysis (PCA) in a 148 × 148 × 148 descriptor space. Considering the active site residues of all the isoforms, we computed 135 EZ descriptors and added the 13 descriptors as mentioned above (Table 2S and Table 1).41 PCA is a multivariate statistical technique used for dimensionality reduction to facilitate visualization and interpretation of multidimensional data. In our analysis, the first three principle components captured 97.4% of the total variance from among the 148 active site descriptors. The 3-dimensional PCA plot further supports our classification of MEK family proteins into the following subsets: MEK1-MEK2, MEK4-MEK6, and MEK7 (Figure S10). Thus, the inhibitor selectivity profiles we empirically observed in our functional assays (Figure 1B) appear highly correlated with a number of distinct physicochemical features in the active site structures of the MEK proteins.
Moreover a clear separation can be seen when analyzing the binding pockets of these isoforms. MEK1–MEK2 have held true to their 89% overall structural homology with having nearly identical cavity shape and electrostatics. MEK4–MEK6, though at a lower level of similarity compared to MEK1– MEK2, still possesses relatively high similarity in parameters despite having a 60% in overall structural homology. The noticeably surprising difference is found in MEK7, whose structural homology to MEK4, at 54%, is only slightly lower than MEK6 to MEK4 but whose overall compound selectively and binding pocket analysis is very distinctive.
In Silico Analysis of Inhibitors
Having observed this correlation between the functional activities of inhibitors and features of the enzyme binding pockets, we performed additional in silico analysis to determine the binding mode of select inhibitors and pinpoint key residues for inhibitor interaction between MEK family members. Inhibitors, PLX4720, AST487, and pazopanib, were docked to MEK4, MEK6, and MEK7 using the Glide docking engine implemented in the Schrodinger suite.42 Extra precision protocols along with the mutation based induced fit docking module of the Glide engine was used to determine the putative binding poses of each ligand with the docking results summarized in Table S3.33
When analyzing the predicted binding poses of PLX4720 to understand the 12-fold decrease in activity between MEK4 (5 µM) and MEK6 (60 µM; Figure 3A), we could see critical interactions that could be necessary for increased potency. We compared the Glide–XP docking score (a function of the binding energy),37 hydrogen bond energy, lipophilic potential energy, and the solvation penalty function in both the cases. It was observed that the ligand in MEK4, when compared to MEK6, had a 3.6 kcal greater docking score, 3.9 kcal in lipophilic score and 4.6 kcal in hydrogen bonding energy with a significantly lower penalty score of 2.6 kcal. The additional hydrogen bonding energy is due to the classical hydrogen bond that is formed in between the sulfonamide and backbone amide of Arg110 in MEK4. In MEK6, the corresponding residue is Arg61, and the backbone amide of this residue is 4.9 Å away from the sulfonamide of PLX4720. In addition, halogen bonding might also play a role in the potency of PLX4720 in MEK4 as one of the aromatic fluorine atoms is within 2.4 Å of the residue Lys131, but it must be noted that the angle in the docking pose is 131.5°, which is not the most conducive for halogen bonding. Docking analysis of PLX4720 pin-pointed subtle differences in MEK4 and MEK6, which were two isoforms with significant similarities.
Figure 3.

In silico analysis of inhibitors. General standard kinase active site representations for MEK4 and MEK6/7. (A) PLX4720 (cyano, stick) docked poses in MEK4 (green, cartoon) and MEK6 (pink, cartoon). (B) AST487 (magenta, stick) docked poses MEK4 (green) and MEK7 (gray). (C) Pazopanib (purple, stick) docked binding modes in MEK4 (green, cartoon) and MEK6 (pink, cartoon).
AST-487 has previously been predicted to bind in a type II manner, binding of an inhibitor to the inactive or open form of a kinase.43 On the basis of the functional profiling assay it was found that MEK7 and MEK5 were strongly inhibited by AST-487. AST-487 was found to be nearly 100-fold more active at inhibiting downstream phosphorylation for MEK7 (200 nM) than MEK4 (40 µM). Upon docking AST-487 into the active site of MEK7 and MEK4, we observed that the methylamine substituted pyrimidine ring aligns perfectly in the hinge region, forming two strong hydrogen bonds. In MEK4, the pyrimidine ring is not able to align properly in the hinge region and thus can only form 1 hydrogen bond. Also, the urea linkage in AST-487 interacts with the carbonyl backbone of the glycine residue in MEK7, while in MEK4 the urea does not appear to make such interactions. Between proper hinge alignment and supplemental interactions with the urea, AST-487 inhibits MEK7 significantly stronger then MEK4. Comparing the docking scores of AST-487 in both the above isoforms, we found that the Glide-XP docking score in MEK7 was 5.8 kcal more than that in MEK4. Again, the compound went into the binding site with a negligible penalty score of 0.02 kcal but had gained a 4.8 kcal lipophilic potential energy and 3.3 kcal more in hydrogen bond energy than MEK4.
Pazopanib is an FDA approved drug for renal cell carcinoma and is a multikinase inhibitor shown to inhibit c-KIT, FGFR, PDGFR, and VEGFR, with 140 nM, 74 nM, 84 nM, and 10–47 nM cell-free assay, respectively.44 Previous crystal structures have shown the binding mode of pazopanib analogs to VEGFR2, but to date no information exists regarding MEKs.45 As pazopanib was one of the most potent inhibitors screened against MEK4, we wanted to focus on its mode of binding. When comparing pazopanib’s mode of binding to MEK4 and MEK6 (Figure 3C), it can be seen that in both isoforms the compound binds to the hinge region via the aminopyrimidine. Through further analysis it can be predicted that the methyl-indazole plays a key role in the potency of this compound as in both docked structures this moiety is occupying a rather large hydrophobic pocket. Analysis of these subtle differences in conjunction with the distinct trends from the functional assay and the binding pocket comparison provide further evidence that can be used to develop selective inhibitors toward MEK4 and the MEK isoform family.
NMR Binding Experiments
With this efficient platform for pharmacological comparison of the MEKs in hand, we focused on the development of MEK4 inhibitors by validating our in silico binding models of MEK4 with NMR experiments designed to detect and characterize ligand–protein interactions. Specifically, binding confirmations of pazopanib and AST-487 were further investigated and confirmed through the use of recombinant MEK4 (MEK4-kinase domain, KD) in combination with ligand-based NMR techniques to detect and map the binding. WaterLOGSY has previously been shown to be useful for the detection of small molecule interactions with large proteins and their complexes.46 Accordingly, we first tested whether AST-487 and pazopanib (note: two previously published synthetic analogs of Pazopanib were used to overcome solubility issues,45 pazopanib-01 and pazopanib-02 with IC50 against MEK4 of 2 and 3.5 µM, respectively) bind to recombinant MEK4 using the WaterLOGSY experiment. In Figure 4A, the top spectrum corresponds to the 1D–NMR 1H spectrum of the downfield region of AST-487. The second spectrum corresponds to the WaterLOGSY spectrum observed for MEK4 in the absence of AST-487 (i.e., a control experiment), and the third spectrum corresponds to the WaterLOGSY spectrum observed for AST-487 in the presence of MEK4. The relatively strong positively phased resonances of AST-487 in the presence of MEK4 indicate that it is binding to MEK4. The presence of positively phased compound resonances can also be seen in pazopanib analogs (data not shown), and thus binding of pazopanib to MEK4 is also detected.
Figure 4.

NMR analysis of the interactions between MEK4 and AST-487/pazopanib. (A) WaterLOGSY NMR spectra of the AST-487 interaction with recombinant MEK4. For reference, the 1D NMR spectrum of the downfield region of AST-487 is shown. (B) WaterLOGSY NMR competition assay to demonstrate competitve binding between inhibitor and ATP. Schematic diagrams showing relative STD percentages (C) AST-487 and (D) Pazopanib-01 bound to recombinant MEK4.
Consequently, we tested the binding mode of AST-487 and pazopanib analogs to the MEK4 ATP binding pocket using a WaterLOGSY-based competition assay in the presence of ATP. As shown in Figure 4B, the addition of ATP significantly decreases the observed binding of AST-487 (the average reduction to resonance intensities was 43% ± 2%), pazopanib-01 (the average reduction to resonance intensities was 55% ± 7%), and pazopanib-02 (the average reduction to resonance intensities was 60% ± 4%) suggesting that ATP is displacing the compounds, strengthening the confidence in our in silico binding predictions.
We further characterized the compound interactions with MEK4 using saturation transfer difference (STD) NMR experiments, which give insight into the small molecule nonpolar protons in closest contact with the protein surface.47 A summary of the relative intensities of the AST-487 and pazopanib-01 STD spectrum are shown in Figure 4C and D, respectively, for their interactions with MEK4. On the basis of this analysis, AST-487 binds to MEK4 through the pyrimidine ethereal linked phenyl with limited interactions through the urea moiety and solvent exposed piperdine. This correlates directly with induced-fit docking simulations of AST-487 and MEK4 (Figure 3B). The piperdine moiety of AST-487 exhibits significantly less STD intensity, suggesting that it is more distant from the protein surface and thus may present an attractive site for modifications designed to increase affinity. Pazopanib interacts with MEK4 mainly with the indazole and pyrimidine rings, as seen by the higher STD intensities. The interactions calculated by STD confirmed the predicted binding confirmations through induced fit docking, which had originally predicted two energetically similar but different confirmations of pazopanib binding to MEK4. Taken together, the NMR experiments indicate that AST-487 and pazopanib bind to the ATP pocket of MEK4 in a competitive manner and complement and validate computational predictions as to the mode of binding.
Simple Selectivity Perturbations
As an application of the outlined platform and to show proof of concept of how one can utilize this new information about the MEK family to gain selectivity when developing MEK-isoform specific inhibitors, we applied simple synthetic modifications to analogs, listed in the Supporting Information Table S4. Focusing initially on the JNK/p38 modules of the MAPk pathway, based of the selectivity profile Figure 2B, we were interested in determing if we could perturb the selectivity/potency of AST-487 from MEK7 toward MEK4 by targeting a “back” pocket of MEK4. The binding pocket analysis demonstrated that MEK7 has a much narrower binding pocket, and we know from the NMR studies that the methyl amine has fewer NOE interactions, meaning there is room for modifications. Taken all together, we choose to make a simple modification to the alkyl amine of AST-487, shown in Figure 5A. We can see a minimal but clear increase in potency toward MEK4 as the size of the alkyl group increases and a drastic decrease in MEK7 potency. These simple synthetic modifications accurately justify the information gained through this platform. In particular, since we have already shown MEK7 to be extremely unique among the MEK isoforms, this proves that slight alterations probing the hydrophobic pocket would be critical for improving MEK4 selectivity.
Figure 5.

Synthetic analogs to modulate selectivity along the p38/JNK modules. (A) Analogs of AST-487 with selective alterations of the alkyl amine group to selectively target MEK4 and MEK7. (B) Analogs of pazopanib focused on trying to target back pocket uniquely between MEK4 and MEK6. (C) Analogs of PLX-4720 focused of providing simple information on the selectivity difference seen between MEK4 and MEK6. Tables contain IC50 values in micromolarity determined by optimized selectivity ADPGlo assay. Blue arrows indicate a decrease, while the red arrows indicate an increase in IC50 values. A comprehensive list of analogs can be found in Supporting Information, Table S4.
With regard to pazopanib, which is an inhibitor with almost identical potencies toward MEK4 and MEK6, the structural alterations were designed to probe a back pocket thought to be present in both isoforms. Again, these alterations were perceived by NMR studies where fewer NOE interactions were observed on the methyl groups of the heterocycle. As can be seen by selected analogs shown in Figure 5B, when modifying the heterocycle that fits into the back pocket, a gradual increase in potency is seen in parallel for both MEK4 and MEK6, representing similar binding as previously discussed. This provides further evidence against this scaffold as a suitable starting point for gaining selectivity between MEK4 and MEK6.
As demonstrated earlier, PLX4720 has unique 10-fold selectivity over MEK6 for MEK4. We found this to be an ample starting point to explore some minor synthetic manipulations to confirm our selectivity hypothesis. Critical interactions observed using in silico experiments placed emphasis on the sulfonamide and dihalide moieties. To assess the importance of these features, simple analogs were made (Figure 5C). Simply eliminating the sulfonamide resulted in retention of potency and selectivity (compound 3a: 6.9 µM, MEK4; 75.1 µM, MEK6) while altering the backbone halide from a chloro to a bromo completely destroyed the selectivity toward MEK4 (compound 3c: 1.4 µM, MEK4; 2.5 µM, MEK6). When moving the difluoro from the 2,6 position to the 3,5 position, the selectivity returned by ~4 fold but the potency toward MEK4 decreased (compound 3d: 27 µM, MEK4; >100 µM, MEK6). Ultimately the halide at the 2-position on the phenyl ring was found to be critical for potency, and the chloro of the backbone is crucial for selectivity.
CONCLUSION
Despite the prevalence of aberrant MAPK signaling in human cancers and stress pathways and the crucial location of MAP2Ks in this pathway, limited research has progressed to target the entire MEK kinase family. This underscores the need to expand the scope of chemical probes and drug development outside of just MEK1/2 to the entirety of the MEK isoform family, which possess unique positions in the MAPK signaling pathway but with unexplored chemical and biological space. The MEK family is usually categorized based on their downstream substrate. Herein, we have shown that a simple and robust inhibitor-based profiling platform of the MEK isoforms provides a foundation for designing selective inhibitors and also leads to new insights into the relationships within MEK kinase isoforms. This information can be exploited to categorize and prioritize based on pharmacological inhibitor similarity. We utilized published inhibitors with “off-target” binding affinities toward MEK4 as a set of molecules that were further profiled in a functional biochemical assay across the entire MEK family. The empirical binding affinities and functional potencies, along with sequential in silico docking studies, were used to predict the molecular features of the ligands, and the corresponding amino acids in the MEK kinases, responsible for activity and selectivity across this kinase family.
We have confirmed that MEK1 and MEK2 are highly similar isoforms as seen by their compound selectivity screen and binding pocket analysis. Shown herein are significant similarities among MEK4 and MEK6 as well as subtle differences, which will be critical to understanding how to incorporate selectivity into future probe development. It is hypothesized that in future MEK probe development, MEK4 and MEK6 should be considered together, as both their inhibitor selectivity screen and binding pocket analysis were similar. On the contrary, the data presented herein support MEK7, though having a similar downstream substrate of JNK to MEK4 and a relatively high sequence homology to MEK4, having very dif ferent inhibitor selectivity. MEK5 being the most recently discovered MEK kinase isoform appears to relate most closely to MEK7 on compound selectivity, which stresses the need for more structural information to better understand MEK5–MEK7 structural similarities. Moreover, based purely on the functional profiling and previous binding affinity data, MEK3 clearly has unique binding modes, which further emphasizes the need for additional structural information to better pursue inhibitor development. In the absence of crystallographic data, validation with NMR spectroscopy techniques informed the binding modes of active compounds in MEK4, which is further progressing the development of selective MEK4 inhibitors.
Analysis of these subtle differences in conjunction with the distinct trends from the functional assay and the binding pocket comparison of a focused set of compounds rationally simplifies the complex structure–activity relationships of protein–small molecule interactions among the MEKs and therefore provides guidance for further probe development. With the abundance of available data of human kinases and the activity of kinase inhibitors, we anticipate that this platform may be broadly implemented in the future to facilitate the development of isoform selective inhibitors and define a SAR in the absence of crystallagraphic costructures.
METHODS
General Information
Compounds (staurosporine, pazopanib, AST-487, GSK-1838705A, flavopiridol, PLX-4720, ruxolitinib) were purchased from MedChemExpress (Monmouth Junction, NJ), and LY333531 was purchased from Cayman Chemical (Ann Arbor, MI). Pazopanib analogs with increased solubility were synthesized by following a previously published procedure.45
Recombinant Protein Expression
Human MAP2K4-EE (residues 37–399; Figure S2B), containing mutations that mimic a constitutively active/phosphorylated state, S257E and T261E, and human MAP2K4-KD (residues 80–399; Figure S2B) were expressed as previously described.33 Constructs were expressed in KRX Single Step cells and purified using affinity chromatography. A 50 mL fraction of Lurie-Bertani (LB) medium was inoculated with a single colony from a plate streaked with KRX cells and grown overnight at 220 rpm and 37 °C with ampicillin and chloramphenicol antibiotics. A total of 10 mL of this overnight culture was used to inoculate 1 L of Terrific Broth (TB) medium (with the same antibiotics). 0.5% l-arabinose was added to KRX cell cultures for growth. Cultures were grown at 37 °C until the OD600 reached 0.6–0.8. At an appropriate OD600, the KRX cells were induced by adding 0.1 mM isopropyl-1-thio-d-galactopyr-anoside (IPTG) and 0.25% l-rhamnose, followed by 4 h of 37 °C growth. After 4 h, the temperature was reduced to 25 °C overnight. Cells were collected by centrifugation at 8000 rpm for 10 min and the pellet resuspended in lysis buffer. Cells were thawed and sonicated at 90% amplitude, 2 s on/10 s off for 30 min. The lysate was centrifuged at 18 000 rpm for 40 min and the supernatant collected for purification. A Cobalt Affinity chromatography column was employed to purify constructs.
Secondary Binding Assay
Small molecule screening by fluorescence-based thermal shift (FTS) assay was performed as described previously using an Echo550 (Labcyte) for compound transfer and a Mantis (Formulatrix, Bedford, MA) to dispense protein into 384-well PCR plates.33 Purified MAP2K4-EE was appropriately diluted in a buffer containing 100 mM Hepes at pH 7.5 and 150 mM NaCl. All assay experiments used 2 µg of protein per well and 5 nL of 5000X Sypro Orange (Invitrogen) up to a total volume of 10 µL, with a resultant protein concentration of 4.65 µM. Stock solutions (40 mM in DMSO) of the inhibitors were prepared, and serial dilutions were performed in a 384-well Echo Source plate. The PCR plates were sealed with an optical seal, shaken, and centrifuged after protein and compounds were added. Thermal scanning (10 to 95 °C at 1.5 °C/ min) was performed using a real-time PCR setup (CFX384 - Biorad Laboratories), and fluorescence intensity was measured after every 10 s. Curve fitting, melting temperature calculation, and report generation on the raw FTS data were performed using Excel. Figure S2A shows ΔTm for the inhibitors against the two constructs. Figure S3A shows a comparison of previously published high binding affinity correlating a high thermal shift (ΔTm) with high binding affinity (Kd) to validate binding assays, constructs, and compounds.
General Biochemical Assay Procedure
All MEK substrates used in the ADP-Glo enzyme assays were produced recombinantly in E. coli. ERK2 (Uniprot P28482-1) containing the enzyme-inactivating point mutation K54R was purchased from Proqinase (Freiburg, Germany). p38alpha (Uniprot Q16539-1) with inactivating mutation K53A, ERK5 (Uniprot Q13164-1) with inactivating mutation K84R, and JNK1beta (Uniprot P45983-3) with inactivating mutation K55M were synthesized and cloned into the pMCSV7 expression vector. All substrates were fused to a 6xHis tag on the N-terminus and lacked the initiator methionine. ERK5 was truncated after Q398. Proteins were purified first by nickel affinity chromatography followed by ion exchange and size exclusion chromatography. All full length activated MEK kinase isoforms were purchased from Carna Bioscience: MAP2K1 (07–141), MAP2K2 (07–142), MAP2K3 (07–143), MAP2K4 (07–144), MAP2K5 (07–145), MAP2K6 (07–146), and MAP2K7 (07–148).
ADP-Glo reagents were purchased from Promega Corporation (Madison, WI) and 10× kinase buffer was purchased from Cell Signaling Technology (Cat# 9802, Danvers, MA). White 384-well Proxiplates (PerkinElmer) were used for the assay. A Mantis liquid handler (Formulatrix, Bedford, MA) was used to dispense all liquids into the desired wells, except for the transfer of compounds. Similar to binding assay, stock solutions (40 mM in DMSO) of the inhibitors were prepared, and serial dilutions made in an 384-well Echo Source plate and using an Echo550 (Labcyte) compounds were transferred to the Proxiplate. Protein mixture (MEK and substrate, 3 µL) was added to Proxiplate, and compounds were transferred and allowed to incubate for 30 min at RT. ATP (1 µL) was then added and reacted for the desired time at RT. After the necessary time, the first ADP-Glo reagent (4 µL) was added and allowed to sit for 45 min at RT. Then, kinase detection reagent (8 µL) was added, and after an incubation time of 45 min, the luminescence was read on an EnSpire Plate Reader (PerkinElmer). Optimized conditions for each MEK isoform were used for screening, see Table S4. Staurosporine was used as a positive control each time the assay was performed. To plot, analyze the data, and calculate all kinase reaction biochemical values, Prism from GraphPad Software (La Jolla, CA) was used.
Binding Pocket Analysis
Using the MOLCAD suite42,48,49 implemented in Tripos software, we computed the electrostatic potential maps and binding cavities and characterized the hydro-phobicity and acidic, basic, and neutral residues percentage of the binding pockets.
Protein Preparation
The high-resolution crystal structures of MEK1, MEK2, MEK6, and MEK7 isoforms with the protein databank accession codes 3EQC, 1S9I, 3VN9, and 3WZU, respectively, were considered for the analysis of the binding sites and for docking experiments. The protein preparation tools implemented in Surflex docking panel was used to prepare the receptors. The hydrogens were added in the hydrogen bonding orientations. Gasteiger charges were assigned, replacing the “b” values (uncertainty values), and irrelevant torsions were eliminated. All these structures were subjected to Ramachandran’s map,50 and 92–95% of the residues of these structures were found to be in a favorable region. The rest of the residues were in the generously allowed domains. When we analyzed the MEK4 crystal structure (3ALO.pdb) in MolProbity,51 we observed that less than 85% of residues were in the favored region. Again, we noticed that the structure had 79% favored rotamers with bad angles and bonds. The MolProbity score of this structure is only 63%. The MolProbity score of a quality crystal structure is usually more than 90%. More importantly, some of the side chain atoms of the ATP binding sites were either missing or have high “b” values. On the basis of this analysis, this structure is not a quality structure to carry out in silico studies. In order to fix this structure, we used the Prime module of the Schrodinger software, which corrected the irrelevant side chains; fixed the missing atoms; corrected undesired orientations of Asn, Gln, or His residues; and replaced the “b-values” with optimized potential for liquid simulations (OPLS) charges.
Ligand Preparation
Using the ligand preparation panels implemented in Sybyl (Tripos) and Schrodinger, all the ligands (pazopanib, AST-487, GSK-1838705A, flavopiridol, PLX-4720, LY333531, ruxolitinib) were prepared at physiological pH = 7.4 ± 1.
Docking of the Ligands
For docking of the ligands, we used the Surflex docking tools implemented in the Sybyl platform of Tripos software.52 The ATP binding sites of the earlier prepared isoform structures of MEK1, 2, 6, and 7 were identified, and the ligand-based protomols were generated for all the different isoforms. The protomols are computational representations of the intended binding sites to which putative ligands would be aligned. In the Surflex docking, the “GeomX” option was used where multistating initial conformations of the ligands were allowed for docking. The predock and postdock minimization conditions were also carried out before and after the docking simulations.
For MEK4, the docking of different ligands was carried out using Schrodinger software as the protein structure was corrected using this platform. The Glide docking engine implemented in the Schrödinger software suite was utilized. Following the protocols of the Glide docking engine, which is built upon a grid-based algorithm, a 12 Å3 grid was generated in the active site of the target protein. Again, using this corrected structure of MEK4 and defining the ligand binding site, ligand flexible and receptor fixed docking was carried out for all the ligands. We failed to dock the compounds with this docking experiment. It was observed that the active site residues Lys131, Asp247, Leu236, Lys187, Leu180, and Il2108 were preventing the entry of the ligands. Hence, we carried out mutation based induced fit docking53 of the compounds, as these are known inhibitors of MEK4 isoforms. These residues were mutated to alanine temporarily, and the ligands were prepositioned into the active site pocket with a softer Van der Waals potential and were energy minimized using OPLS2005 force field. After prepositioning the ligands, the residues were than mutated back, and applying the most stringent Van der Waals and electrostatic constraints, the complex structures (ligand–protein) were again minimized in OPLS2003 force field. During this process, the side chains of the aforementioned residues were perturbed without changing the backbone (Cα). This induced fit docking aims to improve the docking of the ligands, in which the receptor adjusts in the presence of the ligands.
Principle Component Analysis (PCA)
PCA (Biovia, Discovery Studio, Version 4.1, 2014) is an orthogonal linear transformation technique that transforms the data set into a new coordinate system such that the variance of the data is maximized in the first few principal components called PCA1, PCA2, and PCA3. Hence, this technique is used to reduce the dimension of the data by keeping only the first few principal components. A 3-D plot of the dependent variables (MEK1, MEK2, MEK4, MEK6, and MEK7) can show the distribution of the variables in a 3D space.
NMR Binding Studies General Procedure
All nuclear magnetic resonance (NMR) experiments were performed on a Bruker 800-MHz spectrometer. Experimental conditions were a 0.5 µM MEK4 construct (EE or KD) and 50–100 µM inhibitor in 20 mM PO4/pH 7.4, 100 mM NaCl in 90% H2O, and 10% D2O (WaterLOGSY) or 100% D2O (STD) at 25 °C in 3 mm NMR tubes. The WaterLOGSY experiments were performed as previously described.54,55 Water was selectively saturated using a 2 ms square shaped pulse with a mixing time of 2 s and a relaxation delay of 2.5 s. STD experiments were performed as previously described. 56,57 In these experiments, protein 1H was saturated with a train of 50 ms Gaussian-shaped pulses at 100 Hz power for 1 s with “on” resonance saturation at −1 ppm and “off” resonance saturation at 30 ppm (the relaxation delay was 2.5 s before the saturating pulses). Spectra were processed by NMRPipe with a 5 Hz line broadening function and analyzed by NMRDraw.58 Relative % STD was defined as 100 × STDobs/STDmax where STD = ΔI/Ioff, ΔI = Ioff − Ion, and Ioff and Ion are the intensities observed for the various resonances after the “off” and “on” presaturation of MEK-4. Errors in the WaterLOGSY and STD were estimated as ΔI/Iref((NΔI/ΔI)2 + (NIoff/Iref)2)0.5, where NΔI and NIref are the noise calculated by NMRDraw in the appropriate spectrum (no protein in WaterLOGSY and Ioff in the STD).
ATP Competition Experiments
In these experiments, we tested the binding of 100 µM inhibitor (AST487, pazopanib-01, pazopanib-02) to 5uM MEK4-KD using WaterLOGSY. We then repeated the experiments with a 10-fold excess of ATP (1 mM). NMRDraw was used to measure the difference in resonance intensities between two measurements. Sample conditions: 100 µM AST487 + 5 µM MEK4-KD ± 1 mM ATP (in 3 mm NMR tubes). Then a decrease in line intensity for each resonance was measured.
Synthesis of Analogs
Synthetic procedures and characterization data can be found in the Supporting Information.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the National Cancer Institute (NCI; R01 CA188015). K.D. thanks the National Science Foundation (NSF) for graduate research fellowship (DGE-1324585).
Footnotes
ASSOCIATED CONTENT
- Figures S1-S10, Tables S1-S4, general procedure for synthesis of analogs, and NMR spectra (PDF)
The authors declare no competing financial interest.
References
- 1.Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J. Hematol. Oncol. 2013;6:27. doi: 10.1186/1756-8722-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 3.Hsueh C-T, Liu D, Wang H. Novel biomarkers for diagnosis, prognosis, targeted therapy and clinical trials. Biomark. Res. 2013;1:1. doi: 10.1186/2050-7771-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr. Opin. Pharmacol. 2005;5:350–356. doi: 10.1016/j.coph.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Micel LN, Tentler JJ, Tan A-C, Selby HM, Brunkow KL, Robertson KM, Davis SL, Klauck PJ, Pitts TM, Gangolli E, et al. Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol. Cancer Ther. 2015;14:317–325. doi: 10.1158/1535-7163.MCT-13-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poplin E, Feng Y, Berlin J, Rothenberg ML, Hochster H, Mitchell E, Alberts S, O’Dwyer P, Haller D, Catalano P, Cella D, Benson AB. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2009;27:3778–3785. doi: 10.1200/JCO.2008.20.9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guma M, Hammaker D, Topolewski K, Corr M, Boyle DL, Karin M, Firestein GS. Antiinflammatory functions of p38 in mouse models of rheumatoid arthritis: Advantages of targeting upstream kinases MKK-3 or MKK-6. Arthritis Rheum. 2012;64:2887–2895. doi: 10.1002/art.34489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 10.Fisk M, Gajendragadkar PR, Maki-Petaja KM, Wilkinson IB, Cheriyan J. Therapeutic potential of p38 MAP kinase inhibition in the management of cardiovascular disease. Am. J. Cardiovasc. Drugs. 2014;14:155–165. doi: 10.1007/s40256-014-0063-6. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discovery. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 12.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Drew BA, Burow ME, Beckman BS. MEK5/ ERK5 pathway: the first fifteen years. Biochim. Biophys. Acta, Rev. Cancer. 2012;1825:37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu L, Ding Y, Catalona WJ, Yang XJ, Anderson WF, Jovanovic B, Wellman K, Killmer J, Huang X, Scheidt KA, Montgomery RB, Bergan RC. MEK4 Function, Genistein Treatment, and Invasion of Human Prostate Cancer Cells. J. Natl. Cancer Inst. 2009;101:1141–1155. doi: 10.1093/jnci/djp227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 16.Huang X, Chen S, Xu L, Liu Y, Deb DK, Platanias LC, Bergan RC. Genistein inhibits p38 map kinase activation, matrix metalloproteinase type 2, and cell invasion in human prostate epithelial cells. Cancer Res. 2005;65:3470–3478. doi: 10.1158/0008-5472.CAN-04-2807. [DOI] [PubMed] [Google Scholar]
- 17.Lotan TL, Lyon M, Huo D, Taxy JB, Brendler C, Foster BA, Stadler W, Rinker-Schaeffer CW. Up-regulation of MKK4, MKK6 and MKK7 during prostate cancer progression: an important role for SAPK signalling in prostatic neoplasia. J. Pathol. 2007;212:386–394. doi: 10.1002/path.2194. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Pan Y, Dai JL. Evidence of MKK4 pro-oncogenic activity in breast and pancreatic tumors. Oncogene. 2004;23:5978–5985. doi: 10.1038/sj.onc.1207802. [DOI] [PubMed] [Google Scholar]
- 19.Pavese JM, Ogden IM, Voll EA, Huang X, Xu L, Jovanovic B, Bergan RC. Mitogen-activated protein kinase kinase 4 (MAP2K4) promotes human prostate cancer metastasis. PLoS One. 2014;9:e102289. doi: 10.1371/journal.pone.0102289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cunningham SC, Gallmeier E, Hucl T, Dezentje DA, Calhoun ES, Falco G, Abdelmohsen K, Gorospe M, Kern SE. Targeted deletion of MKK4 in cancer cells: a detrimental phenotype manifests as decreased experimental metastasis and suggests a counterweight to the evolution of tumor-suppressor loss. Cancer Res. 2006;66:5560–5564. doi: 10.1158/0008-5472.CAN-06-0555. [DOI] [PubMed] [Google Scholar]
- 21.Xu L, Bergan RC. Genistein inhibits matrix metalloproteinase type 2 activation and prostate cancer cell invasion by blocking the transforming growth factor beta-mediated activation of mitogen-activated protein kinase-activated protein kinase 2–27-kDa heat shock protein pathway. Mol. Pharmacol. 2006;70:869–877. doi: 10.1124/mol.106.023861. [DOI] [PubMed] [Google Scholar]
- 22.Muller S, Chaikuad A, Gray NS, Knapp S. The ins and outs of selective kinase inhibitor development. Nat. Chem. Biol. 2015;11:818–821. doi: 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- 23.Koeberle SC, Romir J, Fischer S, Koeberle A, Schattel V, Albrecht W, Grutter C, Werz O, Rauh D, Stehle T, Laufer SA. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat. Chem. Biol. 2012;8:141–143. doi: 10.1038/nchembio.761. [DOI] [PubMed] [Google Scholar]
- 24.Chaikuad A, Tacconi EMC, Zimmer J, Liang Y, Gray NS, Tarsounas M, Knapp S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014;10:853–860. doi: 10.1038/nchembio.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Lopez MS, Dar AC, LaDow E, Finkbeiner S, Yun C-H, Eck MJ, Shokat KM. Structure-guided inhibitor design expands the scope of analog-sensitive kinase technology. ACS Chem. Biol. 2013;8:1931–1938. doi: 10.1021/cb400376p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okuzumi T, Ducker GS, Zhang C, Aizenstein B, Hoffman R, Shokat KM. Synthesis and evaluation of indazole based analog sensitive Akt inhibitors. Mol. BioSyst. 2010;6:1389–1402. doi: 10.1039/c003917a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinzlmeir S, Kudlinzki D, Sreeramulu S, Klaeger S, Gande SL, Linhard V, Wilhelm M, Qiao H, Helm D, Ruprecht B, Saxena K, Medard G, Schwalbe H, Kuster B. Chemical proteomics and structural biology define EPHA2 inhibition by clinical kinase drugs. ACS Chem. Biol. 2016;11:3400–3411. doi: 10.1021/acschembio.6b00709. [DOI] [PubMed] [Google Scholar]
- 28.Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 29.Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat. Methods. 2005;2:17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- 30.Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signaling. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 32.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 33.Krishna SN, Luan C-H, Mishra RK, Xu L, Scheidt KA, Anderson WF, Bergan RC. A fluorescence-based thermal shift assay identifies inhibitors of mitogen activated protein kinase kinase 4. PLoS One. 2013;8:e81504. doi: 10.1371/journal.pone.0081504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.App. ATP Km values (µM) as measured and used at ProQinase for testing of Protein Kinases with a FlashPlate-based radiometric protein kinase assay. ProQinase, Breisgau; Germany: 2015. [Google Scholar]
- 35.Smyth LA, Collins I. Measuring and interpreting the selectivity of protein kinase inhibitors. J. Chem. Biol. 2009;2:131–151. doi: 10.1007/s12154-009-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kinase Profiling Book. CarnaBiosciences; Kobe, Japan: 2015. [Google Scholar]
- 37.Small-Molecule Drug Discovery Suite 2014-4. Glide, version 6.5. Schrödinger, LLC; New York: 2014. [Google Scholar]
- 38.Cleves AE, Jain AN. Knowledge-guided docking: accurate prospective prediction of bound configurations of novel ligands using Surflex-Dock. J. Comput.-Aided Mol. Des. 2015;29:485–509. doi: 10.1007/s10822-015-9846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 40.Zachmann CD, Brickmann J. Hausdorff dimension as a quantification of local roughness of protein surfaces. J. Chem. Inf. Model. 1992;32:120–122. doi: 10.1021/ci00005a020. [DOI] [PubMed] [Google Scholar]
- 41.Sandberg M, Eriksson L, Jonsson J, Sjostrom M, Wold S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998;41:2481–2491. doi: 10.1021/jm9700575. [DOI] [PubMed] [Google Scholar]
- 42.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 43.Wodicka LM, Ciceri P, Davis MI, Hunt JP, Floyd M, Salerno S, Hua XH, Ford JM, Armstrong RC, Zarrinkar PP, Treiber DK. Activation state-dependent binding of small molecule kinase inhibitors: structural insights from biochemistry. Chem. Biol. 2010;17:1241–1249. doi: 10.1016/j.chembiol.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Bukowski RM, Yasothan U, Kirkpatrick P. Pazopanib. Nat. Rev. Drug Discovery. 2010;9:17–18. doi: 10.1038/nrd3073. [DOI] [PubMed] [Google Scholar]
- 45.Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, Davis-Ward RG, Epperly AH, Hinkle KW, Hunter RN, III, Johnson JH, et al. Discovery of 5-[[4-[(2, 3-Dimethyl-2 H-indazol-6-yl) methylamino]-2-pyrimidinyl] amino]-2-methyl-benzene-sulfonamide (Pazopanib), a Novel and Potent Vascular Endothelial Growth Factor Receptor Inhibitor. J. Med. Chem. 2008;51:4632–4640. doi: 10.1021/jm800566m. [DOI] [PubMed] [Google Scholar]
- 46.Dalvit C, Pevarello P, Tato M, Veronesi M, Vulpetti A, Sundstrom M. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biomol. NMR. 2000;18:65–68. doi: 10.1023/a:1008354229396. [DOI] [PubMed] [Google Scholar]
- 47.Mayer M, Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem., Int. Ed. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 48.Brickmann J, Exner TE, Keil M, Marhofer RJ. Molecular graphics-Trends and perspectives. J. Mol. Model. 2000;6:328–340. [Google Scholar]
- 49.Heiden W, Moeckel G, Brickmann J. A new approach to analysis and display of local lipophilicity/hydrophilicity mapped on molecular surfaces. J. Comput.-Aided Mol. Des. 1993;7:503–514. doi: 10.1007/BF00124359. [DOI] [PubMed] [Google Scholar]
- 50.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 51.Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins: Struct, Funct, Genet. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 52.Jain AN. Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput.-Aided Mol. Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 53.Ansari FL, Wadood A, Ullah A, Iftikhar F, Ul-Haq Z. In silico studies of urease inhibitors to explore ligand-enzyme interactions. J. Enzyme Inhib. Med. Chem. 2009;24:151–156. doi: 10.1080/14756360801945598. [DOI] [PubMed] [Google Scholar]
- 54.Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B. WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR. 2001;21:349–359. doi: 10.1023/a:1013302231549. [DOI] [PubMed] [Google Scholar]
- 55.Ramirez BE, Antanasijevic A, Caffrey M. Ligand screening using NMR. In: Anderson WF, editor. Structural Genomics and Drug Discovery: Methods and Protocols. Springer; New York: 2014. pp. 305–313. [DOI] [PubMed] [Google Scholar]
- 56.Celigoy J, Ramirez B, Tao L, Rong L, Yan L, Feng Y-R, Quinnan GV, Broder CC, Caffrey M. Probing the HIV gp120 envelope glycoprotein conformation by NMR J. Biol. Chem. 2011;286:23975–23981. doi: 10.1074/jbc.M111.251025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem., Int. Ed. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 58.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.