

Abstract
Brown adipose tissues (BAT) burn lipids to generate heat through uncoupled respiration, thus representing a powerful target to counteract lipid accumulation and obesity. The tumor suppressor liver kinase b1 (Lkb1) is a key regulator of cellular energy metabolism; and adipocyte-specific knockout of Lkb1 (Ad-Lkb1 KO) leads to the expansion of BAT, improvements in systemic metabolism and resistance to obesity in young mice. Here we report the unexpected finding that the Ad-Lkb1 KO mice develop hindlimb paralysis at mid-age. Gene expression analyses indicate that Lkb1 KO upregulates the expression of inflammatory cytokines in interscapular BAT and epineurial brown adipocytes surrounding the sciatic nerve. This is followed by peripheral neuropathy characterized by infiltration of macrophages into the sciatic nerve, axon degeneration, reduced nerve conductance, and hindlimb paralysis. Mechanistically, Lkb1 KO reduces AMPK phosphorylation and amplifies mammalian target-of-rapamycin (mTOR)-dependent inflammatory signaling specifically in BAT but not WAT. Importantly, pharmacological or genetic inhibition of mTOR ameliorates inflammation and prevents paralysis. These results demonstrate that BAT inflammation is linked to peripheral neuropathy.
Keywords: Brown adipocyte, Liver kinase b1 (serine/threonine kinase 11), Inflammation, Sciatic nerve, mTOR, Paralysis
Highlights
-
•
Adipocyte specific knockout Lkb1 mice develop late onset hindlimb paralysis despite better metabolism in young animals.
-
•
Lkb1 KO in brown adipocytes promotes expression of proinflammatory cytokines that are linked to sciatic nerve neuropathy.
-
•
Rapamycin or genetic inhibition of mTOR ameliorates BAT inflammation and prevents paralysis.
Brown adipose tissues (BAT) burn lipids to dissipate heat, expansion of BAT is thus considered as an attractive strategy to counteract obesity. Here, we use a mouse model in which the Lkb1 gene is knockout in adipocytes to show that the knockout mice develop enlarged BAT, resulting in better systemic metabolism. However, the knockout mice later develop peripheral neuropathy and hindlimb paralysis due to inflammation of the enlarged BAT. Inhibition of a signaling cascade known as mTOR effectively blunted the inflammation of BAT and prevented hindlimb paralysis. Therefore, even though BAT is known to promote metabolic health in the context of obesity, chronic inflammation of BAT is linked to devastating peripheral neuropathy. This study further suggests that blockage of mTOR is an effective strategy to treat inflammation-induced peripheral neuropathy.
1. Introduction
Adipose tissues play key roles in regulating lipid metabolism, glucose homeostasis and inflammation (Baumgartner et al., 1986; Cohen and Spiegelman, 2016). White adipose tissues (WAT) mainly store energy surplus in form of triglyceride, and also function as a major endocrine organ through secreting adipokines (Peirce et al., 2014). Levels of WAT adipokines, such as resistin and leptin, were correlated with risks of dementia and neurodegenerative diseases such as Alzheimer's disease (AD), as well as neurological and neurovascular disorders (Arnoldussen et al., 2014; Bednarska-Makaruk et al., 2017; Pedditizi et al., 2016). Brown adipose tissues (BAT) are specialized in breaking down lipids to generate heat mediated by Ucp1 (Uncoupling protein 1) to defend against cold (Schulz and Tseng, 2013). BAT have also been reported to secrete endocrine factors to modulate systemic energy metabolism (Pfeifer, 2015; Wang et al., 2014). The thermogenic function of brown adipocytes makes BAT expansion a promising strategy to offset energy surplus and counteract obesity, but whether BAT expansion comes with side effects remains unexplored.
Peripheral neuropathy is a class of disease affecting peripheral nerves, often involves axonal abnormality or demyelination (Suter, 2006; Suter, 2007). Axons are normally enclosed by a myelin sheath with multilayered membrane, which functions as an insulator to greatly increase the velocity of axonal impulse conduction (Lemke and Axel, 1985). Myelination of axons is performed by oligodendrocytes in the central nervous system (CNS) and by Schwann cells in the peripheral nervous system (PNS). Neuro-inflammation is a common factor accounting for the pathogenesis of multiple neurodegenerative diseases (Qin et al., 2007), including Alzheimer's Disease (Rubio-Perez and Morillas-Ruiz, 2012), Parkinson's Disease (Barnum and Tansey, 2012), and peripheral neuropathies (Qin et al., 2007; Rubio-Perez and Morillas-Ruiz, 2012). However, what causes inflammation of the peripheral nerve is incompletely understood.
Lkb1 (Liver kinase b1) is a serine/threonine kinase involved in regulating cellular energy metabolism and immune response (Shaw, 2008; Filippov et al., 2013). Lkb1 directly phosphorylates and activates AMP-activated protein kinase (AMPK), and subsequently regulates the mammalian target-of-rapamycin (mTOR) pathway (Woods et al., 2003; Shaw et al., 2004). Recent studies have begun to uncover the function of Lkb1 in adipocytes. Specifically, aP2-Cre induced knockout of Lkb1 in preadipocytes results in lipodystrophy and premature death of the KO mice (Zhang et al., 2013). However, adiponectin-Cre (adipoq-Cre) mediated deletion of Lkb1 in mature adipocytes (Ad-Lkb1 mice) leads to expansion of BAT without significant alterations of WAT mass (Shan et al., 2016). The young Ad-Lkb1 mice exhibit elevated energy expenditure due to increased thermogenic function of BAT (Shan et al., 2016). Hence, Lkb1 plays distinct roles in different adipose tissues, as well as at different stages of adipogenesis.
Recent research has established an association between WAT expansion and inflammation (Bayer et al., 2014; Nov et al., 2013). Interestingly, Wernstedt et al. reported that proinflammatory signaling in the adipocyte is required for proper remodeling and expansion of WAT (Asterholm et al., 2014). However, whether inflammation is also associated with BAT expansion in Ad-Lkb1 KO mice during postnatal growth, and whether inflammation of BAT exerts any side effects remains unclear. Here, we performed microarray analysis to pinpoint inflammation as a key feature of hypertrophic BAT in the Ad-Lkb1 mouse model. Strikingly, we show that the Ad-Lkb1 mice develop hindlimb paralysis starting at 8-month old (middle age). As Adipoq-Cre only drives deletion of Lkb1 in adipocytes and induces hypertrophy of BAT but not WAT, development of hindlimb paralysis in the Ad-Lkb1 mouse makes it a good model to study the long-term consequences of BAT inflammation.
2. Materials and Methods
2.1. Animals
All procedures involving mice were approved by Institutional Animal Care and Use Committee. The Adipoq-Cre (stock #010803, RRID: IMSR_JAX:010803), Lkb1flox/flox (stock #014143, RRID: IMSR_JAX:014143) and mTOR flox/flox (stock #011009, RRID: IMSR_JAX:011009) mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed in the animal facility with free access to water and standard rodent chow food (TD.06414 Harlan). Male and female mice > 6-month-old were used unless otherwise indicated. PCR genotyping was done using protocols described by the supplier. For macrophage deletion treatment, Clodrosome or Vehicle was purchased from Encapsula NanoSciences company, which was delivered by intraperitoneal (IP) injection with 200uL (5 mg/ml) per mouse (8-months old, female) for initial injection and 100 μl each subsequent injection once weekly as described (Dace et al., 2009). Rapamycin (Calbiochem and LC Laboratories) was administered by IP injection at 4 mg/kg (Yilmaz et al., 2006). It was reconstituted in absolute ethanol at 10 mg/ml or 1 diluted in 5% Tween-80 (Sigma) before injection.
2.2. Foot Print (Paralysis) Measurement
To measure the locomotion of WT and KO mice, both fore-paws were immersed in red ink and hind limbs were immersed in blue ink, then the mice were allowed to walk freely on white filter paper (Whatman).
2.3. Treadmill Measurement
Mouse ran treadmill test performed as described by Yue et al. (2016). In brief, mouse was trained on treadmill (Eco 3/6 treadmill; Columbus Instruments, Columbus, OH, USA) with a fixed 10% slope, at constant 10 m/min speed for 5 min daily for consecutively 3 days before test. On the exercise testing day, animals ran on the treadmill at 10 m/min for 5 min and the speed was increased by 2 m/min every 2 min until they were exhausted or a maximal speed of 30 m/min was achieved. The exhaustion was defined as the inability of the animal to run on the treadmill for 10 s despite mechanical prodding. Running time and maximum speed achieved was measured whereas running distance was calculated.
2.4. Hematoxylin-Eosin (H&E) and Immunostaining
Spinal cord, sciatic nerve, TA, Gas, EDL and SOL muscle tissues from the WT and Ad-Lkb1 KO mice were dissected and frozen immediately in OCT compound. Frozen samples were cross-sectioned (8–12 μm) using a Leica CM1850 cryostat. For H&E staining, the sections were stained in hematoxylin (30 min), rinsed in running tap water and stained in eosin (1 min). Immunostaining was performed as previously described (Shan et al., 2014). Briefly, sections were fixed in 4% PFA (except for fiber type detection), incubated in blocking buffer (5% goat serum, 2% BSA, 0.2% triton X-100 and 0.1% sodium azide in PBS) for 1 h, followed by incubation with the primary antibodies diluted in blocking buffer overnight. After washing with PBS, the samples were incubated with secondary antibodies and DAPI (diluted in PBS) for 45 min at room temperature. Fluorescent images were captured as single channel grayscale images using a Leica DM 6000B fluorescent microscope with a 20 × objective (NA 0.70) and assembled in Photoshop. Images for WT and KO samples were captured using identical parameters and both control and KO images were adjusted identically in Photoshop.
2.5. Transmission Electron Microscopy (TEM)
The sciatic nerve and spinal cord were fixed in 2.5% glutaraldehyde for 30 min and cut into 1 × 2-mm blocks. The blocks continued fixation in 2.5% glutaraldehyde for 1 h, followed by fixation in 2% osmium tetroxide for 1 h. All the fixatives were made with 0.1 M cacodylate buffer. After washing, the blocks were dehydrated in a graded ethanol series and then embedded in Epon Generic Resin. The sections with a thickness of about 90 nm were prepared with uranyl acetate and lead citrate stain and examined with a transmission electron microscope (Gatan Digital microscopy). The myelinated axons were counted by Image J (National Institutes of Health) and abnormal myelinated axons were characterized by de-compaction of myelin sheath, irregular shapes with myelin infoldings, or axonal swelling. The g-ratio and axon diameter were also analyzed by image J (Tao et al., 2009). In brief, g-ratio of each axon was calculated by the perimeter of axons (inner) divided by the perimeter of corresponding fibers (outer) to eliminate the bias on circularity. Axonal diameters were normalized by perimeters through equation: diameter = perimeter/π. For each mouse, three random images were counted 20–30 axons randomly. This procedure allows for inclusion of irregularly shaped axons and fibers and helps to eliminate biased measurement of diameters based on circularity.
2.6. Electrophysiological Recordings and Analysis
Sciatic nerves rapidly excised from the body were immediately placed in Krebs' solution (mM): 124 NaCl, 2 KCl, 1.2 KH2PO4, 1.3 MgSO4, 2 CaCl2, 10 dextrose, and 10 sodium ascorbate, bubbled continuously with 95% O2–5% CO2 to maintain pH of 7.2–7.4. After an hour of recovery, sciatic nerves were placed in a recording chamber for electrophysiological recordings, as detailed in past publications (Shi and Whitebone, 2006; Yan et al., 2016; Shi and Blight, 1996). In short, the chamber is composed of a central compartment circulating oxygenated Krebs' solution (2 ml/min) at 37 °C, two neighboring ‘sucrose gap’ compartments circulating isotonic sucrose (320 mM) (1 ml/min) and two outer compartments containing isotonic KCl (120 mM). Extracellular stimulating and recording electrodes were positioned such that action potentials were conducted and measured across the central compartment. Compound action potentials (CAPs), the spatio-temporal summation of all single axon action potentials, were generated by applying a supramaximal stimulus (110% of the maximal stimulus) in the form of 0.1 ms constant current unipolar pulses every 3 s and were recorded in real time. A bridge amplifier (Neurodata Instruments) was used for data acquisition and further analysis was accomplished using custom Labview software (National Instruments) installed on a Dell PC.
2.7. Microarray Analysis
RNA was extracted from 12-wk.-old WT and Ad-Lkb1 mice, three mice for each genotype. Gene expression was analyzed by microarray with Agilent SurePrint G3 Mouse GE 8 × 60 K chip as previously described (Bi et al., 2016). Genes with an average fold change ≥ 1.5folds were selected for pathway analysis through the DAVID Bioinformatics Resources 6.8 as instructed (Huang et al., 2009a, Huang et al., 2009b).
2.8. Real-time PCR
Total RNA was extracted from tissues using Trizol Reagent according to the manufacturer's instructions. RNA was treated with RNase-free DNase l to remove genomic DNA. The purity and concentration of total RNA were measured by a spectrophotometer (Nanodrop 3000, Thermo Fisher) at 260 nm and 280 nm. Ratios of absorption (260/280 nm) of all samples were between 1.8 and 2.0. Then 3 μg of total RNA were reversed transcribed using random primers and MMLV reverse transcriptase. Real-time PCR was carried out with a Roche Lightcycler 480 PCR System using SYBR Green Master Mix and gene-specific primers. The 2−∆∆CT method was used to analyze the relative changes of gene expression normalized against 18S rRNA as internal control.
2.9. Protein Extraction and Western Blot Analysis
Total protein was isolated from tissues using RIPA buffer contains 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS. Protein concentrations were measured using BCA Protein Assay Reagent (Thermo scientific). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore Corporation), blocked in 5% fat-free milk for 1 h at RT, then incubated with first antibodies (diluted in 5% milk) overnight at 4 °C. Lkb1 (sc-32245, 1:1000, RRID: AB_6278) and GAPDH (sc-32233, 1:1000, RRID: AB_627679) antibodies are from Santa Cruz biotechnology. mTOR (#2972, 1:1000, RRID: AB_330978) antibodies are from Cell signaling technology. The horseradish peroxidase (HRP)-conjugated secondary antibody (anti-rabbit IgG, 111-035-003, RRID: AB_10015289 or anti-mouse IgG, 115-035-003, RRID: AB_10015289, Jackson ImmunoResearch) were diluted 1: 10,000. Immunodetection was performed using enhanced chemiluminescence western blotting substrate (Santa Cruz biotechnology) and detected by FluorChem R System (ProteinSimple). Results shown in the figures are representative results from at least three independent experiments.
2.10. Data Analysis
All data are presented as means ± S.E.M. Comparisons were made by two-tailed Student's t-tests. Effects were considered significant at P < 0.05.
2.11. Data and Materials Availability
The microarray reported in this paper has been deposited in the Gene Expression Omnibus database (GEO: GSE89348).
3. Results
3.1. Ad-Lkb1 Mice Develop Hindlimb Paralysis Starting From 8-month Old
We generated Adipoq-Cre/Lkb1flox/flox (Ad-Lkb1) mice to specifically knock out (KO) Lkb1 in adipocytes (Fig. 1a). Young Ad-Lkb1 mice exhibited BAT (but not WAT) hypertrophy, resulting in better glucose tolerance, insulin sensitivity and resistance to high fat diet induced obesity (Shan et al., 2016). Interestingly, the Ad-Lkb1 mice developed dysfunction of both hind limbs and became paralyzed starting at 8 months old. This was characterized by clenching of both hind limbs to the body when suspended by the tail (Fig. 1b), dragging, clumsy and autonomous twitching of both hind limbs, and inability to maintain body balance (Fig. 1c and movie S1). Furthermore, footprint assay showed that the forelimbs of Ad-Lkb1 mice maintained normal mobility but the hindlimbs could not move forward normally (Fig. 1d). On average, 18% of the male Ad-Lkb1 mice exhibited hindlimb paralysis at 9 months old, reaching 100% at 11-month-old (Fig. 1e). The Ad-Lkb1 females developed paralysis about one month earlier than did the males, with about 22% mice paralyzed at 8 months old, and 100% at 10-month-old (Fig. 1f). These data demonstrate that Ad-Lkb1 mice develop the chronic and late onset hindlimb paralysis.
Fig. 1.

Ad-Lkb1 mice develop hindlimb paralysis starting at 8-month-old. (a) Targeting strategy for adipocyte-specific deletion of Lkb1. Black box represents deleted exons and triangles represent LoxP sites. (b and c) Representative images of WT and paralyzed KO mice (8-month-old). KO mice clench the hind limbs to the body when suspended by the tail (b), drag hind limbs and are unable to keep the body balance (c). (d) Foot-prints of a paralyzed KO and a normal WT mouse (9-month-old), fore-paws were immersed in red ink and hind limbs were immersed in blue ink. (e and f) Occurrence rate of paralysis in WT and KO male (n = 11 pairs) and female (n = 14 pairs) mice. The number of samples are biological replicates.
3.2. Ad-Lkb1 KO Leads to Axon Degeneration in the Sciatic Nerve
We further examined the etiology of paralysis in Ad-Lkb1 mice. Axon abnormality and subsequent degeneration of sciatic nerve is a common cause of hindlimb paralysis (Douglas et al., 2009). Semi-thin transverse sections of sciatic nerve showed lower fiber density and fewer myelinated axons in the KO compared to WT mice (Fig. 2a and b). Transmission electron microscopy (TEM) revealed that axons of KO mice exhibited abnormal morphology characterized by demyelination, myelin sheath de-compaction, myelin inflodings, axon swelling and increased endoneurial space (Fig. 2c). Measurement of g-ratio, the ratio of inner axonal diameter to outer diameter of myelin sheath, revealed a trend of demyelination and decompaction in KO sciatic nerves, manifested by wider dispersal of g-ratios (Fig. 2d). In addition, axon diameter distribution showed a shift from medium-large axons to smaller axons in KO mice (Fig. 2e). Quantitatively, the Ad-Lkb1 sciatic nerves had significantly fewer normally myelinated axons and more abnormally myelinated axons compared to those of WT mice (Fig. 2f). Consistently, KO mice had higher percentage of abnormal axons than WT (Fig. 2g). At the molecular level, the mRNA levels of myelination related genes, including apolipoprotein E (Apoe), myelin protein zero (Mpz) and peripheral myelin protein 22 (Pmp22) were downregulated in the sciatic nerves of Ad-Lkb1 mice compared to the WT counterparts (Fig. 2h). At the functional level, axon demyelination in the Ad-Lkb1 sciatic nerve significantly reduced the amplitude of compound action potentials (CAP) (Fig. 2i). These results suggest that Lkb1 KO in adipocytes induces degeneration and functional loss of axons in the sciatic nerve.
Fig. 2.

Ad-Lkb1 KO leads to axon abnormality in sciatic nerves. (a) Semi-thin transverse sections of sciatic nerve showing lower fiber density in KO mice than WT mice. Scale bar: 100 μm. (b) Total number of myelinated axon per sciatic nerve in WT and KO mice (N = 4 pairs). (c) Ultrastructure transmission electron microscopy (TEM) images showing compact and completely myelinated fibers in WT mice. KO sciatic nerves had increased endoneurial space (red asterisk) and axonal fibers with myelin disruption, decompaction (red arrow) and folding (blue arrow). (d and e) G-ratio (d) and axon size distribution (e) of myelinated axons in WT and KO sciatic nerve were analyzed as described in method. N = 4 pairs. WT = 333 axons, KO = 250 axons. (f and g) Numbers of normal, abnormal myelinated axons per area (f) and percentages of abnormal axons (g). Three random pictures of each sample were counted (N = 4 pairs). (h) Relative mRNA levels of myelination related genes (Apoe, Mpz, Krox24 and Pmp22) in WT (N = 4) and paralyzed KO mice (N = 3). (i) CAP (compound action potential) amplitude of WT (N = 4) and paralyzed KO (N = 3) mice. All the mice are 8–12 months old and the number of samples are biological replicates. Error bars: S.E.M., *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t-test).
We also examined if sciatic nerve degeneration affects hind limbs muscles. KO mice had significantly lower muscle weights than did the control mice (Fig. S1a). H&E staining revealed that muscle fiber size appeared smaller in muscles of paralyzed Ad-Lkb1 mice (Fig. S1b). In addition, the complexity of neuromuscular junction (NMJ) was reduced in Lkb1 KO mice compared with that of WT mice (Fig. S1c). Furthermore, fiber type immunostaining in gastrocnemius (GAS) and tibialis anterior (TA) muscles showed that the Lkb1 KO contained more type IIA muscle fibers in GAS (blue box region) and TA muscles, compared to those of WT mice (Fig. S1d). Similarly, the extensor digitorum longus (EDL) and soleus (SOL) muscles of the Lkb1 KO mice contained more type IIA fibers, but fewer type IIX/IIB fibers compared with the corresponding muscles of WT mice (Fig. S1e). These results are consistent with previous observations in human peripheral neuropathies in which demyelination and abnormality in sciatic nerve lead to muscle atrophy, accompanied by myofiber type switching toward slower types and reduced complexity of NMJ (Lamonte et al., 2002).
3.3. Ad-Lkb1 KO Induces Expression of Pro-inflammatory Genes in Brown Adipocyte and Macrophage Infiltration in Sciatic Nerves
Schwann cells constitute the major cell type in the sciatic nerve, and form the myelin sheath that insulates and functionally supports the peripheral nerve fibers (Jessen and Mirsky, 2016). The severe sciatic nerve abnormality in the Ad-Lkb1 mice is similar to the phenotype of mice with Schwann cell specific deletion of Lkb1 (Pooya et al., 2014; Beirowski et al., 2014), raising the possibility that the Adipoq-Cre may induce ectopic KO of Lkb1 in Schwann cells. To examine the possibility, we performed Cre/LoxP lineage tracing using the Adipoq-Cre and Rosa26Td-Tomato (Adipoq-Td) reporter mice. Td (a red fluorescent protein) signals were found in classical WAT and BAT, but not in the spinal cord, liver, spleen, muscle, heart, or brain (Fig. S2a). Although Td signals were observed in sciatic nerves along the periphery (Fig. S2b), they are limited to epineurial adipocytes that were marked by Bodipy staining (Fig. S2c and d). Direct labeling the Schwann cells in cross and longitudinal sections of sciatic nerve with the S100b antibody revealed that the Td positive cells were never positive for S100b (Fig. S2e). These data indicate that Adipoq-cre specifically marks adipocytes but not Schwann cells, and exclude the possibility that the paralysis phenotype of Ad-Lkb1 mice is due to ectopic deletion of Lkb1 in Schwann cells.
A main phenotype of the Ad-Lkb1 mice is brown fat hypertrophy (Shan et al., 2016), which may boost adipokine secretion. We therefore hypothesized that Lkb1 null adipocytes induce sciatic nerve dysfunction by a paracrine/endocrine mechanism. To test this hypothesis, we first sought to understand how BAT hypertrophy alters gene expression, through microarray analysis of BAT from 3-month-old WT and Ad-Lkb1 mice, before development of hindlimb paralysis (Fig. 3a and b). Pathway analysis of the microarray data revealed that Lkb1 KO upregulated several inflammation-related pathways, including “Chemokine signaling”, “Toll-like receptor signaling”, “Jak-STAT signaling”, “Cytokine-cytokine receptor interaction” and “ECM-receptor interaction” pathways (Fig. 3a and b). Further qPCR analysis confirmed the higher mRNA levels of many inflammatory cytokines (Tumor necrosis factor a, Tnfa; C-C motif chemokine ligand 2, Ccl2, also called Mcp1; C-C motif chemokine ligand 7, Ccl7, also called Mcp3) in BAT from 2, 6, 8-month-old KO mice, prior to onset of hindlimb paralysis (Fig. 3c). To verify if Lkb1 KO cell-autonomously upregulates the expression of pro-inflammatory cytokines in brown adipocytes, we cultured brown adipocytes from WT and Ad-Lkb1 mice and found that Tnfa, Ccl2 and Ccl7 were expressed at higher levels in Lkb1 KO compared to WT adipocytes (Fig. 3d). Interestingly, mRNA levels of these inflammatory genes were not affected by Lkb1 KO in white adipose tissues (inguinal WAT, iWAT and epididymal WAT, eWAT) that did not undergo hypertrophy (Fig. S3a and b), suggesting that hypertrophy is the main trigger of adipocyte inflammation. Consistent with previous observation that epineurial adipocytes exhibit brown adipocytes characteristics (Salisbury et al., 2012), we found that epineurial adipocytes express Ucp1 (Fig. S4a), a specific marker of brown adipocytes. To determine if epineurial adipocytes are inflamed in the Ad-Lkb1 mice, we examined mRNA levels of these inflammatory cytokines in the sciatic nerve. Indeed, mRNA levels of Tnfa, interleukin 1b (Il1b) tended to be higher in Ad-Lkb1 than in WT sciatic nerves (Fig. S4b). The mRNA levels of Ccl2 were ~ 4 folds higher in Ad-Lkb1 than in WT sciatic nerves (Fig. S4b). Collectively, these results indicated that Lkb1 KO specifically induced expression of pro-inflammatory genes in brown adipocytes including epineurial adipocytes, but not in white adipocytes.
Fig. 3.

Ad-Lkb1 KO induces inflammation in brown adipocyte and macrophage recruitment into sciatic nerve. (a) Microarray volcano plot of genes significantly up- or down-regulated in Ad-Lkb1 (N = 3) versus WT BAT (N = 3). Colored dots or triangles denote genes in the indicated pathways. (b) Heat map showing relative levels (fold changes = KO signal/WT signal) of the representative genes from pathways listed in A. (c and d) Relative mRNA levels of Lkb1, Tnfa, Ccl2 and Ccl7 genes in BAT (c) from 2 month (N = 4 pairs), 6 month (N = 4 pairs), 8 month (N = 5 pairs) old mice and mature brown adipocyte (d, N = 4 repeats), determined by qPCR. (e and f) Relative mRNA levels of CD-68 (e, WT, N = 5, KO, N = 6) and immunofluorescence labeling of CD68-expressing cells (f, N = 3 pairs, three random pictures of each sample were counted) in sciatic nerve from WT and paralyzed KO mice. Error bars: S.E.M., *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar: 100 μm. The number of samples are biological replicates.
We next asked how BAT inflammation induces sciatic nerve degeneration and dysfunction. Chemokines, including Ccl2 and Ccl7, provide directional cues and induce infiltration of inflammatory macrophages (Luster, 1998). Notably, the macrophage marker CD68 was also expressed at higher levels in Ad-Lkb1 than in WT sciatic nerves (Fig. 3e), suggesting macrophage infiltration. Indeed, there were more CD68 positive cells (macrophages) in cross section of sciatic nerves of Lkb1 KO mice than in those of WT mice (Fig. 3f). In addition, the expression of inflammatory genes was elevated in spinal cords of Ad-Lkb1 mice only after, but not prior to, the development of paralysis (Fig. S5a and b). This observation is consistent with the notion that sciatic nerve injury can lead to inflammation in spinal cord (Echeverry et al., 2011). These results indicate that pro-inflammatory cytokines from Lkb1 null brown adipocytes promote macrophage infiltration into the sciatic nerve, leading to nerve degeneration.
3.4. Anti-Inflammation Treatment Delays the Occurrence of Paralysis
To confirm if inflammation and macrophage infiltration leads to hindlimb paralysis in Ad-Lkb1 KO mice, we treated pre-symptomatic mice with the macrophage depletion agent clodrosome (Fig. 4a). Immunostaining confirmed that clodrosome reduced the abundance of macrophages in the sciatic nerve (Fig. 4b). Clodrosome also reduced the mRNA levels of CD68 and several pro-inflammatory chemokines (Fig. 4c). These data indicate that clodrosome effectively depleted macrophages and reduced inflammation in the sciatic nerve.
Fig. 4.

Clodrosome-mediated depletion of macrophages delays the occurrence and ameliorates the severity of paralysis. (a) Experimental design for clodrosome treatment. IP: intraperitoneal. (b and c) CD68 immunostaining (b, N = 3 pairs, three random pictures were counted from each mice) and relative mRNA levels of pro-inflammatory genes (c, N = 5 pairs) of sciatic nerve from 3 treatment groups: WT, KO + Vehicle and KO + Clodros. Clodros: Clodrosome. (d) Mice behavioral scores before (upper) and after (bottom) treatment in WT, KO + Vehicle and KO + Clodros groups, N = 5 pairs of age/sex-matched littermates (L1-L5). Mild paralysis: clenching of hind limbs to the body when suspended by the tail, dragging hind limbs and moving slowly. Severe paralysis: hind limbs can't move and the mice are unable to keep body balance. (e) Distance (upper) and speed (bottom) of mice ran on a treadmill after clodrosome treatment for 4 weeks. N = 5 pairs. (f) CAP amplitude for WT (N = 6), KO + Vehicle (N = 4) and KO + Clodros (N = 5) groups. The number of samples are biological replicates. Error bars: S.E.M., *P < 0.05, **P < 0.01, ***P < 0.001, WT compared with KO + Vehicle groups. #P < 0.05, ##P < 0.01, KO + Vehicle compared with KO + Clodros groups (Student's t-test). Scale bar: 100 μm.
Next, we assessed the histology of sciatic nerve and motor function of mice before and after clodrosome treatment in both WT and Ad-Lkb1 littermates (5 L labeled as L1-L5). Strikingly, clodrosome prevented the development or ameliorated the severity of paralysis in the Ad-Lkb1 mice (Fig. 4d). Clodrosome also improved the exercise capacity (running distance and speed on treadmill) of the Ad-Lkb1 mice (Fig. 4e). At the structural level, clodrosome partially rescued the abnormal morphology of axon in Ad-Lkb1 sciatic nerves (Fig. S6a). Both the number and percentage of normal myelinated axons were increased by clodrosome treatment (Fig. S6b). Compared with vehicle control treatment, clodrosome treatment also induced a shift of axon size from small-medium axons to larger axons (Fig. S6c). Furthermore, clodrosome significantly improved the CAP amplitude of sciatic nerve (Fig. 4f). Together, these results demonstrate that inflammation and macrophage infiltration result in sciatic motor axonal abnormality and development of hindlimb paralysis in Ad-Lkb1 mice.
3.5. Lkb1 Ablation Induces Inflammation through Activation of mTOR Signaling
Deletion of Lkb1 in adipose tissues has been reported to activate the mTOR pathway in BAT through inhibition of AMPK activity (Shan et al., 2016). Consistently, Ad-Lkb1 KO in BAT but not in WAT reduced the phosphorylation of AMPK, but increased the level of pS6/S6, an indicator of mTOR signaling (Fig. S7a-b). We hypothesized that activation of mTOR in brown adipocytes promote their production of inflammatory cytokines, as previously reported (Xie et al., 2014). To test this hypothesis, we inhibited the mTOR pathway by rapamycin (Fig. 5a) and found that rapamycin treatment protected KO mice from development of hindlimb paralysis (Fig. 5b). In contrast, all the KO mice treated with vehicle control developed mild or sever hindlimb paralysis (Fig. 5b). This was further confirmed by exercise performance test, showing improved motor functions after rapamycin treatment (Fig. 5c). Moreover, rapamycin normalized the expression of inflammation-related genes and nerve conduction in sciatic nerve of KO mice to levels similar to those of WT mice (Fig. 5d and e). These data indicate that Lkb1 knockout induces adipocyte inflammation through activation of mTOR pathway.
Fig. 5.

Rapamycin rescues the Ad-Lkb1 induced hindlimb paralysis. (a) Experimental design for rapamycin treatment. 7–8 months old mice were injected with Rapamycin once a day for one week and subsequently injected every other day for 4 weeks, then collecting samples. (b) Mice behavioral scores before (upper) and after (bottom) treatment in WT, KO + Con (Control) and KO + Rapa (Rapamycin) groups, N = 3 pairs of age/sex-matched littermates (L1-L3). Mild paralysis: clenching of hind limbs to the body when suspended by the tail, dragging hind limbs and moving slowly. Severe paralysis: hind limbs can't move and the mice are unable to keep body balance. (c) Distance (left) and speed (right) of mice ran on a treadmill before treatment (upper) and after Rapamycin treatment (bottom) for 5 weeks. N = 3 pairs, 7–8 months old. (d) Relative mRNA levels of pro-inflammatory factors and chemokines of BAT and sciatic nerve in WT (N = 3), KO + Con (N = 3) and KO + Rapa (N = 3) groups. (e) CAP amplitude for WT (N = 3), KO + Con (N = 3) and KO + Rapa (N = 3) groups. *P < 0.05, **P < 0.01, WT compared with KO + Con mice; #P < 0.05, ##P < 0.01, KO + Con compared with KO + Rapa mice (Student's t-test). The number of samples are biological replicates. Error bars: S.E.M.
To further validate if activation of mTOR pathway is responsible for adipocyte inflammation in Ad-Lkb1 mice, we generated Adipoq-Cre/Lkb1f/f/mTOR f/f double knockout (DK) mice to concomitantly delete Lkb1 and mTOR in adipocytes (Fig. 6a and b). Strikingly, DK mice were behaviorally normal and completely free of paralysis through the age of 2-year-old (Fig. 6c and movie S2). DK also normalized the expression of proinflammatory genes in BAT to levels similar to those of WT (Fig. 6d). Furthermore, DK mice had reduced infiltration of CD68 positive macrophages in the sciatic nerve (Fig. 6e). At the ultrastructural level, sciatic nerve axons in DK mice had completely normal morphology with myelin sheath remained compact (Fig. 6f) and had normal size distribution of axons (Fig. 6g). Quantitatively, the number and percentage of abnormally myelinated axons were comparable in DK and WT mice, much lower than those of KO mice (Fig. 6h and i). DK mice also had significant higher nerve conductance compared to KO mice, though it remained lower than that of the WT (Fig. 6j). These results indicate that Lkb1 deletion induced adipocyte inflammation through activation of the mTOR pathway.
Fig. 6.

Ad-Lkb1 induces hindlimb paralysis through activation of mTOR signaling. (a and b) The mRNA and protein expression of Lkb1 and mTOR in BAT from WT (N = 4) and DK (N = 5) mice (8-month-old), **P < 0.01. (c) Representative images and foot-print of WT, Lkb1 KO and Lkb1/mTOR DK mouse (10-month-old). Fore-paws were immersed in red ink and hind limbs were immersed in blue ink. (d) Relative mRNA levels of pro-inflammatory factors and chemokines of BAT in WT (N = 3), KO (N = 3) and DK (N = 3) groups. (e) CD68 immunostaining of sciatic nerve from WT, KO and DK mice, N = 3 pairs (three random pictures were counted from each mice), scale bar: 100 μm. (f and g) sciatic nerve TEM images (f) and axon size distribution (g) of WT, KO and DK mice (male, 9-month-old), N = 3 pairs. (h and i) Numbers of normal, abnormal myelinated axons per area (h) and percentages of abnormal axons (i), N = 3 pairs. (j) CAP amplitudes of WT (N = 4), KO (N = 7) and DK (N = 6) from 8–10-month old mice. The number of samples are biological replicates. Error bars: S.E.M., *P < 0.05, **P < 0.01, KO compared with WT mice; #P < 0.05, ##P < 0.01, DK compared with KO mice (Student's t-test).
4. Discussion
In this study, we found that ablation of Lkb1 in adipocytes (including epineural adipocytes) induced inflammation and macrophage invasion in sciatic nerves, leading to severe sciatic axon abnormality and hindlimb paralysis. Accordingly, macrophage depletion in Ad-Lkb1 mice delays the development of hindlimb paralysis. We further provide genetic evidence that Lkb1 KO induces inflammation through activation of mTOR pathway. These results establish a role of brown adipocytes in regulating the function of peripheral nerve (Fig. 7) and their inflammation may lead to peripheral neuropathies.
Fig. 7.
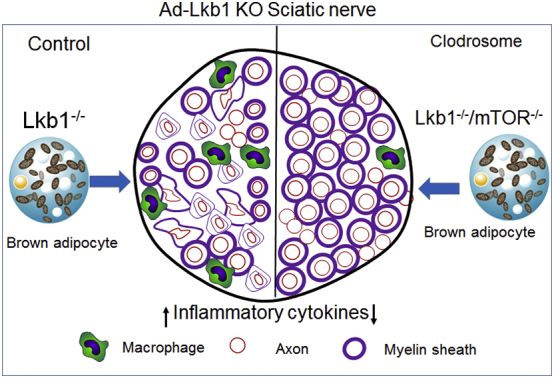
Schematic summary of Ad-Lkb1 induced inflammation and axonal abnormality in sciatic nerves through mTOR pathway.
Hindlimb paralysis is often a consequence of peripheral nerve neuropathy. Earlier clinical observations reported that the most common cause of peripheral neuropathy is diabetes and obesity (Thurston et al., 1995). However, Ad-Lkb1 mice are neither obese nor diabetic (Shan et al., 2016). On the contrary, Ad-Lkb1 mice have higher energy expenditure, better glucose tolerance and insulin sensitivity, and are more resistant to high fat diet (HFD)-induced obesity prior to development of paralysis (Shan et al., 2016). In addition, clinical data suggest that male diabetic patients are more vulnerable to develop peripheral neuropathies than females (Aaberg et al., 2008). In contrast, Ad-Lkb1 females had earlier onset of paralysis than males, suggesting that continuous chronic inflammation induces more severe tissue damages in females than in males (Casimir et al., 2013). Therefore, we exclude the possibility that hindlimb paralysis in the Ad-Lkb1 mice is a result of diabetes or systemic metabolic dysfunction.
The late onset of paralysis suggests that the magnitude of BAT hypertrophy-induced inflammation is low grade, which may not reach a threshold level to cause systemic inflammatory response and nerve degeneration in young animals. Chronic low-grade inflammation is similarly associated with white fat expansion and obesity, leading to neuropathy (Miscio et al., 2005; Pop-Busui et al., 2016). Interestingly, low levels of adipocyte inflammation are essential for the healthy expansion of adipose tissues (Asterholm et al., 2014). However, sustained chronic inflammation could slowly accumulate and lead to side effects in the long run. In the Ad-Lkb1 mice, the sustained inflammation of BAT and production of inflammatory cytokines may have reached a threshold to signal macrophage infiltration and sciatic nerve degeneration by middle ages.
Degeneration of sciatic nerve in Ad-Lkb1 KO mice is manifested by reduced number of axons, abnormal axonal morphology, increased number of small axons and loss of CAP. These changes of sciatic nerve are sufficient to compromise its function. Although the average g-ratio in KO mice is not higher than WT, the g-ratios are more widely scattered (dispersed), suggesting de-compaction (which leads to a smaller g-ratio) and demyelination (which leads to larger g-ratio). Although the severe sciatic nerve abnormality in the Ad-Lkb1 mice is similar to the phenotype of mice with Schwann cell specific deletion of Lkb1 (Beirowski et al., 2014; Pooya et al., 2014), the etiology in the Ad-Lkb1 model is not due to ectopic deletion of Lkb1 in Schwann cells. First, lineage tracing study fails to detect any ectopic Adipoq-cre activity in Schwann cells. Second, the late onset of paralysis in Lkb1 KO mice indicates that initial myelination is normal in the KO mice, and further suggests that the peripheral neuropathy is not primarily caused by defects in Schwann cells.
The lineage tracing analysis indicates that Ad-Lkb1mediates the deletion of Lkb1 in both white and brown adipocytes. Interestingly, disruption of Lkb1 only leads to upregulation of proinflammatory cytokines in brown, but not in white, adipocytes. This may be due to hypertrophy of BAT, but not WAT, in Ad-Lkb1 mice. Alternatively, BAT-specific inhibition of AMPK and activation of mTOR (independent of BAT hypertrophy) could also explain the upregulation of inflammatory cytokines in BAT but not WAT. Our Ucp1 expression analysis and a previous study (Salisbury et al., 2012) suggest that epineurial adipocytes are similar to classic brown adipocytes, which explains the elevated expression of inflammatory genes in sciatic nerve. In theory, proinflammatory cytokines from both classical and epineurial brown adipocytes may have contributed to the development of hind limb paralysis. Proinflammatory factors from classic BAT may be secreted into the circulation and delivered to the sciatic nerve. Supporting this notion, adipokines, such as leptin (Bonda et al., 2014), resistin (Bokarewa et al., 2005) and adiponectin (Fry et al., 2006) from WAT, are reported to regulate neuroinflammation through the blood circulation (Parimisetty et al., 2016). On the other hand, local inflammation medicated by epineurial brown adipocytes may directly trigger macrophage infiltration and sciatic nerve degeneration. Supporting this notion, previous research reported that leptin secreted from epineurial adipocytes of the sciatic nerve is critical for the development of tactile allodynia through macrophage activation in mice after partial sciatic nerve ligation (Maeda et al., 2009). Future studies using epineurial adipocyte-specific or classical brown adipocyte-specific Cre to delete Lkb1 will directly address the relative contribution of BAT and epineurial fat inflammation to peripheral nerve disease. Given lack of the specific molecular makers to distinguish classical intrascapular and epineurial brown adipocytes, this remains a challenging task at present time.
Ccl2 and Ccl7 cytokines are reported to mediate recruitment of monocytes and macrophages (Shi and Pamer, 2011). Consistently, increased Ccl2/7 expression and macrophage infiltration was observed in sciatic nerves. Macrophages can be further divided into early proinflammatory M1 and late anti-inflammatory M2 macrophages (Italiani and Boraschi, 2014). M1 efficiently produces toxic effector molecules (ROS and NO) and inflammatory cytokines (Il-1β, Tnfa, Il-6) (Gordon and Taylor, 2005). Our qPCR results identified an elevated expression of M1 markers and thus pointed infiltrated macrophage to M1 macrophages. Clodrosome is widely used to specifically deplete macrophages, including macrophages in sciatic nerves (Zhu et al., 2015). We show that clodrosome treatment normalized the expression of pro-inflammatory genes and delayed the occurrence of paralysis. These results demonstrate that inflammation of Ad-Lkb1 KO leads to hindlimb paralysis.
Interestingly, conditional KO of Lkb1 driven by rat insulin 2 promoter (RIP2)-Cre (RIP2-Cre/Lkb1 flox/flox mice) also results in paralysis (Sun et al., 2011). However, the etiology of hindlimb paralysis in the RIP2-Cre/Lkb1 flox/flox mice was due to ectopic deletion of Lkb1 in the spinal cord and brain that affected neuronal polarity (Sun et al., 2011). In contrast, our adipoq-cre lineage tracing excluded the possibilities of Cre activity or adipocyte presence in the spinal cord. Other studies also support the notion that adipocytes are only found in the epineurium of peripheral nerves, but not in the spinal cord (Schlaepfer, 1969; Kato et al., 1990; Verheijen et al., 2003). It was reported that sciatic nerve injury could disrupt blood–spinal cord barrier, leading to secondary inflammation in the spinal cord (Echeverry et al., 2011). Consistent with this notion, we observed spinal cord inflammation only after, but not before, the development of paralysis. Thus, the inflammatory responses in the spinal cord are secondary to sciatic nerve lesions.
The rescue of hindlimb paralysis by rapamycin treatment or concomitant KO of mTOR in the Ad-Lkb1 mice suggests that Lkb1 KO activates mTOR to elicit inflammatory responses. It has been reported that deletion of Lkb1 in adipose tissues reduces phosphorylation of AMPK and augments mTOR signaling (Shan et al., 2016), which plays a pivotal role in regulating immune responses (Weichhart et al., 2015). This is also supported by the observation that mice treated with rapamycin (mTOR inhibitor) have enhanced anti-inflammation activity (Xie et al., 2014). Furthermore, several groups have reported that mTORC1 inhibition reduces Ccl2 secretion by immune cells, including monocytes, macrophages and dendritic cells (Weichhart et al., 2015). Consistently, we found that Lkb1 KO-induced activation of mTOR pathway upregulates Ccl2 expression in brown adipocyte. Alternatively, deletion of Lkb1 may have led to inflammation through suppression of autophagy in brown adipocytes. It has been reported that inhibition of AMPK activity results in loss of autophagy through activation of mTOR signaling to phosphorylate Ulk1 Ser 757 and disrupt the interaction between AMPK and Ulk1, a key regulator for autophagy (Ren et al., 2016; Kim et al., 2011). Also, perturbations in autophagy-protein-dependent functions in immunity contribute to chronic inflammatory diseases and autoimmune diseases (Levine et al., 2011; Zhong et al., 2016). Although our data confirm that deletion of Lkb1 induces inflammation through mTOR pathway in adipocytes, the specific involvement of autophagy in this process warrants future investigation.
The following are the supplementary data related to this article.
Movement of WT mouse (upper) and initial paralyzed Ad-Lkb1 mouse (bottom) in the new cages.
{kind=link}
Movement of WT (left), initial paralyzed Ad-Lkb1 (middle) and DK (right) mice in the new cages.
{kind=link}
Supplementary figures
Author Contributions
Y.X. and S.K. conceived the project; Y.X., T.S., J.P., N.N. and Z.J. performed experiments; Y.X., C.W., J.P., M.D., F.Y., R.S., W.J., K·H., X.S., G.Y. and S.K. analyzed and interpreted data; Y.X. and S.K. wrote the paper.
Conflict of Interest Statement
The authors declare that they have no competing interests.
Acknowledgments and Funding Sources
This work was supported by a grant from the US National Institutes of Health (R01CA212609 to S.K.), Innovation Fund for Medical Sciences (CIFMS) from Chinese Academy of Medical Sciences (2016-I2M-1-012), and the National Science and Technology Major Project of China (2016ZX08006003 to G.Y.). Y.X. was supported by a pre-doctoral fellowship from the Chinese Scholarship Council (20140630014). The funders had no role in the study design, data collection, data analysis, interpretation, or writing of the report.
We thank Phillip Reid for assistance with video editing. Jun Wu and Mary Larimore for mouse colony maintenance; Qing Jiang and Chao Yang for technical support, and members of the Kuang Laboratory for comments.
Contributor Information
Gongshe Yang, Email: gsyang@nwsuaf.edu.cn.
Shihuan Kuang, Email: skuang@purdue.edu.
References
- Aaberg M.L., Burch D.M., Hud Z.R., Zacharias M.P. Gender differences in the onset of diabetic neuropathy. J. Diabetes Complicat. 2008;22:83–87. doi: 10.1016/j.jdiacomp.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Arnoldussen I.A., Kiliaan A.J., Gustafson D.R. Obesity and dementia: adipokines interact with the brain. Eur. Neuropsychopharmacol. 2014;24:1982–1999. doi: 10.1016/j.euroneuro.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asterholm I.W., Tao C., Morley T.S., Wang Q.A., Delgado-Lopez F., Wang Z.V., Scherer P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20:103–118. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnum C.J., Tansey M.G. Neuroinflammation and non-motor symptoms: the dark passenger of Parkinson's disease? Curr. Neurol. Neurosci. Rep. 2012;12:350–358. doi: 10.1007/s11910-012-0283-6. [DOI] [PubMed] [Google Scholar]
- Baumgartner R.N., Roche A.F., Guo S., Lohman T., Boileau R.A., Slaughter M.H. Adipose-tissue distribution - the stability of principal components by sex, ethnicity and maturation stage. Hum. Biol. 1986;58:719–735. [PubMed] [Google Scholar]
- Bayer J.K., Grandoch M., Fender A.C., Ruther U., Fischer J.W. Role of HA matrix in adipose tissue: Possible implications for adipose tissue expansion, inflammation and insulin resistance. Naunyn Schmiedeberg's Arch. Pharmacol. 2014;387:S29. [Google Scholar]
- Bednarska-Makaruk M., Graban A., Wisniewska A., Lojkowska W., Bochynska A., Gugala-Iwaniuk M., Slawinska K., Lugowska A., Ryglewicz D., Wehr H. Association of adiponectin, leptin and resistin with inflammatory markers and obesity in dementia. Biogerontology. 2017;18:561–580. doi: 10.1007/s10522-017-9701-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B., Babetto E., Golden J.P., Chen Y., Yang K., Gross R.W., Patti G.J., Milbrandt J. Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat. Neurosci. 2014;17:1351–1361. doi: 10.1038/nn.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi P., Yue F., Karki A., Castro B., Wirbisky S.E., Wang C., Durkes A., Elzey B.D., Andrisani O.M., Bidwell C.A., Freeman J.L., Konieczny S.F., Kuang S. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J. Exp. Med. 2016;213:2019–2037. doi: 10.1084/jem.20160157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokarewa M., Nagaev I., Dahlberg L., Smith U., Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 2005;174:5789–5795. doi: 10.4049/jimmunol.174.9.5789. [DOI] [PubMed] [Google Scholar]
- Bonda D.J., Stone J.G., Torres S.L., Siedlak S.L., Perry G., Kryscio R., Jicha G., Casadesus G., Smith M.A., Zhu X.W., Lee H.G. Dysregulation of leptin signaling in Alzheimer disease: evidence for neuronal leptin resistance. J. Neurochem. 2014;128:162–172. doi: 10.1111/jnc.12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimir G.J., Lefevre N., Corazza F., Duchateau J. Sex and inflammation in respiratory diseases: a clinical viewpoint. Biol. Sex Differ. 2013;4:16. doi: 10.1186/2042-6410-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P., Spiegelman B.M. Cell biology of fat storage. Mol. Biol. Cell. 2016;27:2523–2527. doi: 10.1091/mbc.E15-10-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dace D.S., Khan A.A., Stark J.L., Kelly J., Cross A.H., Apte R.S. Interleukin-10 overexpression promotes Fas-ligand-dependent chronic macrophage-mediated demyelinating polyneuropathy. PLoS One. 2009;4:e7121. doi: 10.1371/journal.pone.0007121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas D.S., Moran J.L., Bermingham J.R., Chen X.J., Brindley D.N., Soliven B., Beier D.R., Popko B. Concurrent Lpin1 and Nrcam Mouse Mutations Result in Severe Peripheral Neuropathy with Transitory Hindlimb Paralysis. J. Neurosci. 2009;29:12089–12100. doi: 10.1523/JNEUROSCI.2029-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverry S., Shi X.Q., Rivest S., Zhang J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J. Neurosci. 2011;31:10819–10828. doi: 10.1523/JNEUROSCI.1642-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov S., Pinkosky S.L., Lister R.J., Pawloski C., Hanselman J.C., Cramer C.T., Srivastava R.A., Hurley T.R., Bradshaw C.D., Spahr M.A., Newton R.S. ETC-1002 regulates immune response, leukocyte homing, and adipose tissue inflammation via LKB1-dependent activation of macrophage AMPK. J. Lipid Res. 2013;54:2095–2108. doi: 10.1194/jlr.M035212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M., Smith P.M., Hoyda T.D., Duncan M., Ahima R.S., Sharkey K.A., Ferguson A.V. Area postrema neurons are modulated by the adipocyte hormone adiponectin. J. Neurosci. 2006;26:9695–9702. doi: 10.1523/JNEUROSCI.2014-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S., Taylor P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Italiani P., Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen K.R., Mirsky R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016;594:3521–3531. doi: 10.1113/JP270874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H., Yamamoto T., Yamamoto H., Ohi R., So N., Iwasaki Y. Immunocytochemical characterization of supporting cells in the enteric nervous system in Hirschsprung's disease. J. Pediatr. Surg. 1990;25:514–519. doi: 10.1016/0022-3468(90)90563-o. [DOI] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamonte B.H., Wallace K.E., Holloway B.A., Shelly S.S., Ascano J., Tokito M., Van Winkle T., Howland D.S., Holzbaur E.L. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lemke G., Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. 1985;40:501–508. doi: 10.1016/0092-8674(85)90198-9. [DOI] [PubMed] [Google Scholar]
- Levine B., Mizushima N., Virgin H.W. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster A.D. Chemokines - Chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- Maeda T., Kiguchi N., Kobayashi Y., Ikuta T., Ozaki M., Kishioka S. Leptin derived from adipocytes in injured peripheral nerves facilitates development of neuropathic pain via macrophage stimulation. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13076–13081. doi: 10.1073/pnas.0903524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miscio G., Guastamacchia G., Brunani A., Priano L., Baudo S., Mauro A. Obesity and peripheral neuropathy risk: a dangerous liaison. J. Peripher. Nerv. Syst. 2005;10:354–358. doi: 10.1111/j.1085-9489.2005.00047.x. [DOI] [PubMed] [Google Scholar]
- Nov O., Shapiro H., Ovadia H., Tarnovscki T., Dvir I., Shemesh E., Kovsan J., Shelef I., Carmi Y., Voronov E., Apte R.N., Lewis E., Haim Y., Konrad D., Bashan N., Rudich A. Interleukin-1beta regulates fat-liver crosstalk in obesity by auto-paracrine modulation of adipose tissue inflammation and expandability. PLoS One. 2013;8:e53626. doi: 10.1371/journal.pone.0053626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parimisetty A., Dorsemans A.C., Awada R., Ravanan P., Diotel N., Lefebvre D'hellencourt C. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J. Neuroinflammation. 2016;13:67. doi: 10.1186/s12974-016-0530-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedditizi E., Peters R., Beckett N. The risk of overweight/obesity in mid-life and late life for the development of dementia: a systematic review and meta-analysis of longitudinal studies. Age Ageing. 2016;45:14–21. doi: 10.1093/ageing/afv151. [DOI] [PubMed] [Google Scholar]
- Peirce V., Carobbio S., Vidal-Puig A. The different shades of fat. Nature. 2014;510:76–83. doi: 10.1038/nature13477. [DOI] [PubMed] [Google Scholar]
- Pfeifer A. NRG4: an endocrine link between brown adipose tissue and liver. Cell Metab. 2015;21:13–14. doi: 10.1016/j.cmet.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Pooya S., Liu X., Kumar V.B., Anderson J., Imai F., Zhang W., Ciraolo G., Ratner N., Setchell K.D., Yoshida Y., Jankowski M.P., Dasgupta B. The tumour suppressor LKB1 regulates myelination through mitochondrial metabolism. Nat. Commun. 2014;5:4993. doi: 10.1038/ncomms5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop-Busui R., Ang L., Holmes C., Gallagher K., Feldman E.L. Inflammation as a therapeutic target for diabetic neuropathies. Curr. Diab. Rep. 2016;16:29. doi: 10.1007/s11892-016-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Wu X., Block M.L., Liu Y., Breese G.R., Hong J.S., Knapp D.J., Crews F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Xu X., Wang Q., Ren S.Y., Dong M., Zhang Y. Permissive role of AMPK and autophagy in adiponectin deficiency-accentuated myocardial injury and inflammation in endotoxemia. J. Mol. Cell. Cardiol. 2016;93:18–31. doi: 10.1016/j.yjmcc.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Rubio-Perez J.M., Morillas-Ruiz J.M. A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal. 2012;2012 doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury E.A., Lazard Z.W., Ubogu E.E., Davis A.R., Olmsted-Davis E.A. Transient brown adipocyte-like cells derive from peripheral nerve progenitors in response to bone morphogenetic protein 2. Stem Cells Transl. Med. 2012;1:874–885. doi: 10.5966/sctm.2012-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer W.W. Experimental lead neuropathy: a disease of the supporting cells in the peripheral nervous system. J. Neuropathol. Exp. Neurol. 1969;28:401–418. [PubMed] [Google Scholar]
- Schulz T.J., Tseng Y.H. Brown adipose tissue: development, metabolism and beyond. Biochem. J. 2013;453:167–178. doi: 10.1042/BJ20130457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan T.Z., Zhang P.P., Liang X.R., Bi P.P., Yue F., Kuang S.H. Lkb1 is indispensable for skeletal muscle development, regeneration, and satellite cell homeostasis. Stem Cells. 2014;32:2893–2907. doi: 10.1002/stem.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan T., Xiong Y., Zhang P., Li Z., Jiang Q., Bi P., Yue F., Yang G., Wang Y., Liu X., Kuang S. Lkb1 controls brown adipose tissue growth and thermogenesis by regulating the intracellular localization of CRTC3. Nat. Commun. 2016;7 doi: 10.1038/ncomms12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R.J. LKB1: cancer, polarity, metabolism, and now fertility. Biochem. J. 2008;416:e1–3. doi: 10.1042/BJ20082023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R.J., Kosmatka M., Bardeesy N., Hurley R.L., Witters L.A., Depinho R.A., Cantley L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Blight A.R. Compression injury of mammalian spinal cord in vitro and the dynamics of action potential conduction failure. J. Neurophysiol. 1996;76:1572–1580. doi: 10.1152/jn.1996.76.3.1572. [DOI] [PubMed] [Google Scholar]
- Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Whitebone J. Conduction deficits and membrane disruption of spinal cord axons as a function of magnitude and rate of strain. J. Neurophysiol. 2006;95:3384–3390. doi: 10.1152/jn.00350.2005. [DOI] [PubMed] [Google Scholar]
- Sun G., Reynolds R., Leclerc I., Rutter G.A. RIP2-mediated LKB1 deletion causes axon degeneration in the spinal cord and hind-limb paralysis. Dis. Model. Mech. 2011;4:193–202. doi: 10.1242/dmm.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U. Myelin and axons: Lessons learned from inherited peripheral neuropathies. Neuromuscul. Disord. 2006;16:644–645. [Google Scholar]
- Suter U. Myelin and axons: lessons learned from inherited peripheral neuropathies. Neuron Glia Biol. 2007;2:S19. [Google Scholar]
- Tao Y., Dai P., Liu Y., Marchetto S., Xiong W.-C., Borg J.-P., Mei L. Erbin regulates NRG1 signaling and myelination. Proc. Natl. Acad. Sci. 2009;106:9477–9482. doi: 10.1073/pnas.0901844106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston J.H., Mcdougal D.B., Jr., Hauhart R.E., Schulz D.W. Effects of acute, subacute, and chronic diabetes on carbohydrate and energy metabolism in rat sciatic nerve. Relation to mechanisms of peripheral neuropathy. Diabetes. 1995;44:190–195. doi: 10.2337/diab.44.2.190. [DOI] [PubMed] [Google Scholar]
- Verheijen M.H., Chrast R., Burrola P., Lemke G. Local regulation of fat metabolism in peripheral nerves. Genes Dev. 2003;17:2450–2464. doi: 10.1101/gad.1116203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.X., Zhao X.Y., Meng Z.X., Kern M., Dietrich A., Chen Z., Cozacov Z., Zhou D., Okunade A.L., Su X., Li S., Bluher M., Lin J.D. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat. Med. 2014;20:1436–1443. doi: 10.1038/nm.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichhart T., Hengstschlager M., Linke M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015;15:599–614. doi: 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A., Johnstone S.R., Dickerson K., Leiper F.C., Fryer L.G., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Xie L., Sun F., Wang J., Mao X., Yang S.H., Su D.M., Simpkins J.W., Greenberg D.A., Jin K. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J. Immunol. 2014;192:6009–6019. doi: 10.4049/jimmunol.1303492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Page J.C., Shi R. Acrolein-mediated conduction loss is partially restored by K + channel blockers. J. Neurophysiol. 2016;115:701–710. doi: 10.1152/jn.00467.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O.H., Valdez R., Theisen B.K., Guo W., Ferguson D.O., Wu H., Morrison S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Yue F., Bi P., Wang C., Li J., Liu X., Kuang S. Conditional loss of pten in myogenic progenitors leads to postnatal skeletal muscle hypertrophy but age-dependent exhaustion of satellite cells. Cell Rep. 2016;17:2340–2353. doi: 10.1016/j.celrep.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Wang Q., Song P., Zou M.H. Liver kinase b1 is required for white adipose tissue growth and differentiation. Diabetes. 2013;62:2347–2358. doi: 10.2337/db12-1229. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhong Z., Sanchez-Lopez E., Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016;166:288–298. doi: 10.1016/j.cell.2016.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Soderblom C., Krishnan V., Ashbaugh J., Bethea J.R., Lee J.K. Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol. Dis. 2015;74:114–125. doi: 10.1016/j.nbd.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movement of WT mouse (upper) and initial paralyzed Ad-Lkb1 mouse (bottom) in the new cages.
Movement of WT (left), initial paralyzed Ad-Lkb1 (middle) and DK (right) mice in the new cages.
Supplementary figures