

Abstract
Patient: Female, 41
Final Diagnosis: CML with myelodysplasia
Symptoms: Fatigue
Medication: Dasatinib • Azacitidine
Clinical Procedure: Haploidentical stem cell transplantation
Specialty: Hematology
Objective:
Rare co-existance of disease or pathology
Background:
CML presenting with a variant Philadelphia translocation, atypical BCR-ABL transcript, additional chromosomal aberrations, and evolving MDS is uncommon and therapeutically challenging. The prognostic significance of these genetic findings is uncertain, even as singular aberrations, with nearly no data on management and outcome when they coexist. MDS evolving during the course of CML may be either treatment-associated or an independently coexisting disease, and is generally considered to have an inferior prognosis. Tyrosine kinase inhibitors (TKI) directed against BCR-ABL are the mainstay of treatment for CML, whereas treatment modalities that may be utilized for MDS and CML include allogeneic stem cell transplant and – at least conceptually – hypomethylating agents.
Case Report:
Here, we describe the clinical course of such a patient, demonstrating that long-term combined treatment with dasatinib and azacitidine for coexisting CML and MDS is feasible and well tolerated, and may be capable of slowing disease progression. This combination therapy had no deleterious effect on subsequent potentially curative haploidentical bone marrow transplantation.
Conclusions:
The different prognostic implications of this unusual case and new therapeutic options in CML are discussed, together with a review of the current literature on CML presenting with different types of genomic aberrations and the coincident development of MDS. Additionally, this case gives an example of long-term combined treatment of tyrosine kinase inhibitors and hypomethylating agents, which could be pioneering in CML treatment.
MeSH Keywords: Azacitidine; Fusion Proteins, BCR-ABL; Leukemia, Myelogenous, Chronic, BCR-ABL Positive; Philadelphia Chromosome; Protein Kinase Inhibitors
Background
Chronic myelogenous leukemia (CML) is a chronic myeloproliferative disorder that is driven by the BCR-ABL1 oncogene. The reciprocal BCR-ABL1 translocation involves chromosomes 22 and 9 and leads to a fusion gene that encodes a constitutively active oncogenic kinase, typically referred to as p210BCR-ABL1. Details of the molecular pathogenesis of CML have been described in numerous seminal papers and excellent reviews [1]. Tyrosine kinase inhibitors (TKI) that inhibit BCR/ABL1 signaling have become the criterion standard of CML treatment, with 5 TKIs presently approved for clinical use: imatinib was followed by dasatinib, nilotinib and bosutinib as second-generation TKIs, and ponatinib as third-generation TKI. Together with rigorous cytogenetic and molecular monitoring of treatment response, this armamentarium has transformed CML from a mostly fatal leukemia to a disease with an excellent prognosis in the vast majority of patients, the goal of a normal life expectancy, and even prospect for cure in a subset of patients. The nearly invariable transition from an initial chronic phase (CP) to accelerated (AP) and ultimately blast phase (BP) in the pre-TKI era has become exceedingly rare. Importantly, the prognosis of patients that do experience such progression remains very poor despite all currently available treatment options. Consequently, patients destined to do poorly should be identified at an early stage. This relies on 2 complementary strategies: i) evaluation of the prognosis at diagnosis using a variety of scoring systems, such as the EUTOS, Sokal or Hasford scores [2–4], and ii) assessment of the speed of hematologic, cytogenetic and molecular responses during first-line or second-line therapy. The European Leukemia Net (ELN) provides distinct recommendations for CML treatment based on classification of a patient’s response as optimal or failure [5]. Additional warning signs that warrant close supervision, but for which no unequivocal treatment guidelines have been defined, include additional chromosomal aberrations (ACAs), either in the Ph-positive clone or in Ph-negative cells as evidence of clonal evolution, and atypical BCR-ABL1 transcripts. These aberrations, which may be identified at diagnosis or during therapy, have been variably associated with an inferior or uncertain prognosis. By themselves, none of these findings are considered an unequivocal trigger for changing therapy, although cytogenetic findings consistent with the presence or development of a myelodysplastic syndrome, e.g., monosomy 5 or monosomy 7, are considered ominous signs.
Myelodysplastic syndromes (MDS) are a group of diseases of the hematopoietic stem cell characterized by peripheral cytopenias that variably effect erythro-, thrombo-, and granulopoiesis and an increasing proportion of bone marrow blasts. As in CML, prognosis and treatment are based on several clinical scoring systems. Treatment of MDS is stage-dependent and includes supportive care (transfusions and antibiotic prophylaxis) and disease-modifying hypomethylating agents (azacitidine and/or decitabine) to stabilize the course of the disorder and delay acceleration into an acute myelogenous leukemia [6], or allogeneic stem cell transplantation in the small subset of patients deemed fit enough to undergo this procedure. In rare cases, MDS develops during treatment for CML [7]; no standard therapy has to date been established for patients in whom both diseases coexist.
In this report, we describe the case of a 41-year-old woman diagnosed with CML, whose clinical course was characterized by several of the above-mentioned features: an atypical transcript, ACAs, and an evolving MDS (Table 1).
Table 1.
Uncommon prognostic aspects of CML in this case.
| Feature | Frequency | Prognostic role in CML | Caveats | References |
|---|---|---|---|---|
| Variant BCR-ABL translocations | 6% | Inferior in pre-TKI era/unclear in TKI era | Speculated to be a marker of genomic instability | [9–11,13] |
| Atypical BCR-ABL transcripts | Sporadically | Uncertain | BCR-ABL1 transcript monitoring difficult | [15–17] |
| Additional chromosomal aberrations (ACAs) in Ph-positive clones | 5% (more common in AP and BP: 30–80%) | Negative predictor if present at initial diagnosis | Prognostic role unclear if developed under TKI, but considered as warning sing | [5,20–22] |
| ACAs in independent Ph-negative clones | Rare | Uncertain | Possibly TKI therapy induced | [23,27] |
| Myelodysplasia in CML | Rare | If associated with monosomy 7 poor | TKI side effects or MDS/MPN overlap syndrome | [7,28–30] |
| KRAS mutation | Very rare | Controversial prognostic role | Association with imatinib resistance reported | [34–36] |
| ASXL1 mutation | Frequent | May contribute to disease progression | Poor prognosis in MDS and MPNs | [37] |
| ETV6 mutation | Occasional | No data | Occurs in high risk MDS | [39,40] |
Prognostically relevant features illustrated in this case are generally of low frequency (except of ASXL1 mutation). Nevertheless, their influence on overall prognosis, while variable ranging from worsening prognosis to an uncertain role, have to be considered and should prompt rigorous BCR-ABL1 monitoring even when this is technically difficult such as in case of atypical transcripts.
Case Report
A 41-year-old woman presented in 01/2001 with bone pain, leukocytosis, and thrombocytosis. Her WBC was 19 000/µl, ANC was 14 000/µl, Hb was 13.4 g/dl, and platelets were 517 000/mm3 (Figure 1A–1C). Cytogenetic analysis revealed a variant BCR-ABL1 translocation (46,XX,t(9;22;17)(q34;q11;q24)) (9 of 9 metaphases) (Table 2). Molecular genetic analysis by direct sequencing identified an atypical BCR-ABL1 transcript (p190Bcr-Abl (b2a3)), also referred to as (p190Bcr-Abl (e1a3)) according to revised nomenclature [8]. A diagnosis of Ph+ CML in chronic phase was established. Treatment with hydroxyurea was initiated in 02/2001. This resulted in control of WBC but no molecular response. Interferon-α was contraindicated due to clinical depression. Two years and 3 months after diagnosis (05/2003), imatinib was started at an initial dose of 400 mg/day. Peripheral edema necessitated dose reduction to 300 mg/day. The BCR-ABL1 ratio decreased to 18% after 1 year (04/2004) (detected with a p210-transcript assay via RT-PCR, with G6PD as housekeeping gene) and imatinib was continued at 300 mg/day. After 2 more years, a non-variant translocation (46,XX,t(9;22)[19/25]) was observed by cytogenetic analysis, accompanied by an increasing BCR-ABL1/ABL1 ratio (62%; detected with an p210 transcript assay via RT-PCR). The imatinib dose was increased to 300/400 mg alternating per day (Figure 2). Neither the variant translocation nor the atypical BCR-ABL1 transcript were detectable at that time using a p210 RT-PCR assay and nested PCR approach. Despite failure of TKI treatment, the patient refused an allogeneic hematopoietic stem cell transplantation (HSCT).
Figure 1.

Blood count. (A) Hemoglobin levels. The hemoglobin levels over time represent the course of the disease showing a transfusion-independent anemia (grade 1–2). Hb levels are presented in g/dl. (B) Thrombocyte count. Thrombocyte count also over time reflects disease progression with mild thrombocytopenia (grade 1–2), not resulting in any bleeding complications. Thrombocyte counts are shown in thrombocytes/nl. (C) Absolute neutrophil count. The absolute neutrophil count is the most sensitive parameter in the course of the disease of this patient. The progression results in a severe grade 4 granulocytopenia requiring antibiotic prophylaxis. ANC is shown in neutrophils/nl. Severe granulocytopenia did not change under dasatinib/azacitidine treatment.
Table 2.
Results of cytogenetic analysis (in all cases with viable cells, a female karyotype 45 resp. 46 XX was detected additionally).
| Date | Results | Therapy | Response |
|---|---|---|---|
| 17.02.2001 | t(9: 22: 17) [9/9] | Hydroxyurea | Primary diagnosis |
| 09.09.2004 | No viable cells | Imatinib | n.a. |
| 16.03.2006 | t(9: 22) [19/25] | Nilotinib | Partial cytogenetic remission (PCyR) |
| 09.08.2006 | t(9: 22: 17) [4/20], −7 [16/20] | Nilotinib | PCyR |
| 24.11.2006 | t(9: 22: 17) [6/20], −7 [14/20] | Nilotinib | PCyR |
| 06.02.2007 | t(9: 22: 17) [2/21], −7 [19/21] | Nilotinib | PCyR |
| 22.05.2007 | −7 [19/19] | Nilotinib | First complete cytogenetic remission (CCyR) |
| 04.09.2007 | −7 [20/20] | Nilotinib | CCyR |
| 08.01.2008 | −7 [21/21] | Nilotinib | CCyR |
| 30.04.2008 | t(9: 22: 17) [3/21], −7 [18/21] | Dasatinib | First cytogenetic relapse |
| 20.08.2008 | −7 [20/20] | Dasatinib | Second complete cytogenetic remission (CCyR) |
| 08.12.2008 | −7 [14/16] | Dasatinib | CCyR |
| 15.12.2009 | −7 [2/2] | Dasatinib + Azacitidine | CCyR |
| 15.10.2010 | −7 [20/20] | Dasatinib + Azacitidine | CCyR |
| 03.05.2011 | −7 [13/21], −7 der(22)t(2;22) [6/21], t(9;22;17) [2/21] | Dasatinib + Azacitidine | Second cytogenetic relapse |
The results of the continuous cytogenetic analysis are shown and illustrate clonal evolution and development of additional chromosomal aberrations and monosomy 7 under different subsequent therapies in this case including azacitidine and dasatinib combination. The numbers of detected cytogenetic abnormal cells are indicated in [/].
Figure 2.
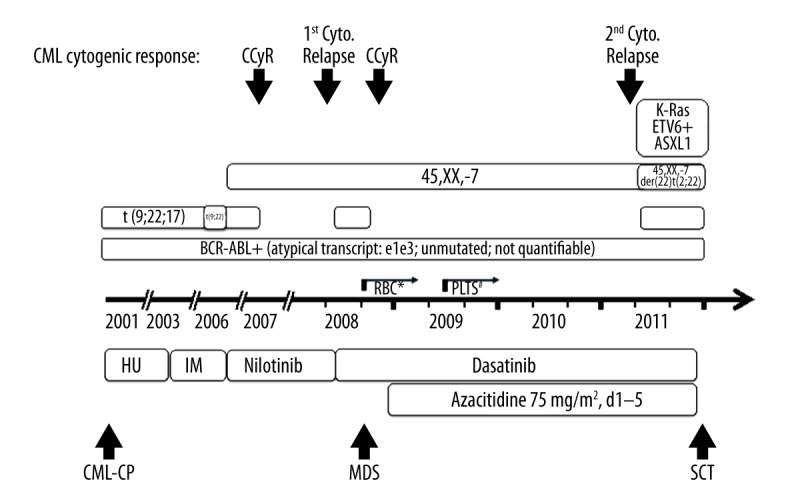
Disease and treatment history. The emergence of different clones and molecular aberrations correlates with the development of MDS and the loss of cytogenetic response. Notably, appearance of monosomy 7 predates manifestation of MDS by 2 years. The corresponding therapeutic regimens are shown (HU – hydroxyurea, IM – imatinib), demonstrating prolonged disease stabilization by combined dasatinib and azacitidine treatment for 4 years. The atypical BCR-ABL transcript was detectable continuously prior to SCT. Worsening of red blood count (RBC) and platelet count (Plts) are indicated by * and #, respectively.
Shortly thereafter (06/2006), hematologic CR was lost with left-shift in the peripheral blood smear and mild-to-moderate cytopenia (WBC 2,61/nl, ANC 0.9/nl, plts. 138/nl and Hb 12.7 g/dl; Figure 1A–1C). Cytogenetic analysis (08/2006) revealed the same variant BCR-ABL1 translocation that had been observed at initial diagnosis, (46,XX,t(9;22;17)[4/20]) in conjunction with a newly occurring monosomy 7 (45,XX,-7 [16/20]) (Table 2). At that timepoint, no BCR-ABL mutation was detectable in Sanger sequencing. Imatinib was switched to nilotinib 400 mg/BD within a clinical trial, with hematologic remission within 2 months and a complete cytogenetic remission of the Ph-positive CML within 14 months, but persistence of the monosomy 7 in all metaphases (45,XX,-7 [19/19]) (Figure 2, Table 2). Accordingly, monosomy 7 was present in the Ph-negative clone.
Cytogenetic remission with respect to the t(9;22) lasted for an additional year until cytogenetic relapse occurred in 04/2008 (46,XX,t(9;22;17)[3/21]; 45,XX,-7 [18/21]) and treatment was switched to dasatinib (50 mg/day). Ph-negativity was regained within 3 months, with persistence of monosomy 7 in bone marrow cytogenetics (45,XX,-7 [20/20]) in 08/2008.
This coincided with worsening of cytopenias, appearance of profoundly dysmorphic megakaryopoiesis and erythropoiesis, and severely reduced granulopoiesis by bone marrow examination, without an increase of blasts; the additional diagnosis of an MDS was established (Figures 1A–1C, 2). The IPSS and WPSS scores were intermediate-1 and high, respectively. Dasatinib was continued to maintain control of CML and azacitidine was added in 12/2008 at a dosage of 75 mg/m2 s.c. for 5 consecutive days (days 1 to 5 of a 4-week treatment schedule, reduced dosage due to present cytopenia). Azacitidine administration was changed to i.v. infusion after severe skin irritation with s.c. administration. Combined dasatinib and azacitidine was continued for another 3 years, with a sustained complete cytogenetic response of the Philadelphia-positive clone, whereas monosomy 7 continued to be detected in 90% to 100% of metaphases on bone marrow analysis (Table 2). Peripheral blood counts showed transfusion-independent anemia (grade 1–2), mild thrombocytopenia (grade 1–2), and grade 4 (severe) granulocytopenia (Figure 1).
Cytogenetic relapse of the CML with reappearance of the t(9;22;17) in 2 of 21 metaphases and clonal evolution with a new distinct clone with t(2;22) in 6 of 21 metaphases was observed in 05/2011 (10 years after initial diagnosis and 2.5 years of combination therapy), with no additionally detectable BCR-ABL mutation upon Sanger sequencing. All remaining metaphases demonstrated monosomy 7 (13 of 21 metaphases) (Table 2). Molecular genetic analysis revealed KRAS, ASXL1, and ETV6 mutations (Figure 2), which by backtracking analysis of prior diagnostic bone marrow samples had not been present at initial diagnosis of CML and also not at development of myelodysplasia.
In view of persisting MDS with clonal evolution and resistant CML by cytogenetics, the patient agreed to undergo an allogeneic HSCT. As no matched related or unrelated donor could be identified, haplo-identical bone marrow transplantation (BMT) was performed in 09/2011 with one of her daughters as stem cell donor. Dasatinib and azacitidine were discontinued prior to start of the conditioning which included thiotepa, i.v. busulfan and fludarabine. GvHD prophylaxis was conducted with post-transplant cyclophosphamide and continuous treatment with mycophenolate-mofetil (MMF) and cyclosporine A (CSA). The patient engrafted and hematopiesis recovered adequately with complete donor chimerism. The BMT resulted in complete recovery of peripheral blood counts after 19 days (granulocytes >0.5/nl) resp. 30 days (thrombocytes >50/nl) (Figure 1A–1C), and complete cytogenetic and molecular remission.
The patient developed a mild acute grade 1 graft versus host disease (GvHD) of the oral cavity, which was treated with prednisolone. One year after BMT, she developed a steroid-dependent, moderate, chronic GvHD.
Complete cytogenetic and molecular remission persisted until 2.5 years after BMT, when atypical BCR-ABL1 transcripts were again detected by nested PCR, but a quantitative RT-PCR assay could not be performed due to the atypical transcript. Cytogenetics were normal and full donor chimerism persisted in the bone marrow. Treatment of molecular relapse with nilotinib led to a disappearance of BCR-ABL1 transcript in nested PCR analysis, even though nilotinib was discontinued after 28 days because of gastrointestinal and musculoskeletal side effects and elevated liver function tests (transaminases). Except for 2 analyses revealing low level atypical BCR-ABL1 transcripts on day +1036 and day +1477 by nested PCR, which disappeared without treatment at both timepoints, the patient has to date remained in complete cytogenetic and molecular remission with respect to both CML and MDS-associated aberrations.
In summary this disease and treatment course of CML with atypical BCR-ABL1 transcript, MDS and clonal evolution demonstrates the initial coexistence of 2 distinct diseases with development of TKI-refractory CML in the absence of a BCR-ABL kinase domain mutation.
Moreover, we to our knowledge for the first time demonstrate that long-term combined treatment with dasatinib and azacitidine is feasible and well tolerated, and may be capable of slowing disease progression. This combination may also be warranted for treatment of CML patients responding poorly to standard therapy.
Discussion
The above case provides several interesting insights into the relation of evolving cytogenetic and molecular findings during the development of CML-associated myelodysplasia, the kinetics of clonal evolution, and therapeutic options in the face of TKI failure. We reviewed these different aspects and the current understanding of their impact on prognosis, and discuss them in the context of the individual patient described above.
Variant Bcr-Abl translocations and atypical transcripts
Variant BCR-ABL translocations involve more or other chromosomes than chromosomes 9 and 22. Their frequency in CML patients is approximately 6% [9] and in the pre-imatinib era the prognosis was suggested to be inferior [10]. With TKI-based therapy, conflicting results have been reported: an inferior prognosis was suggested by Stagno et al. based in a small cohort of 10 CML patients treated with imatinib or nilotinib as first-line therapy (7 suboptimal response, 1 TKI failure, and 2 optimal responses) [11]. In contrast, El-Zimarty and Marzochchi et al. reported that response and outcome of 30 patients treated with imatinib was identical to that of 44 patients harboring the common t(9;22) translocation in terms of complete cytogenetic remission (CCyR) and major molecular remission (MMR) [12,13]. Variant translocations have been speculated to be markers of genomic instability [10], [11] with a consequently inferior prognosis, but they do not constitute a warning sign according to ELN criteria [5] (Table 1).
Atypical BCR-ABL1 transcripts have different sizes and breakpoints compared to the usual p210 or p190 transcript and have been reported in approximately 1–2% of BCR-ABL1-positive ALL patients [14] and more sporadically in CML [15]. Atypical transcripts are usually noticed on polyacrylamide gel in conventional PCR because of their different size, but may be missed in some cases. Failure to identify atypical transcripts can have a negative impact on treatment outcome due to inadequate disease monitoring (Table 1). As in our case, quantification in RT-PCR assays can be difficult, particularly when transcript numbers are low. Therefore, qualitative detection in a nested PCR approach can be helpful. The p190Bcr-Abl b2a3 (or e1a3) transcript detected in our patient has so far been reported only in rare cases of CML [16] and ALL [17]. It has been proposed that atypical transcripts lacking exon a2 should have a more benign course of the disease [16]. In line with this, several publications described a benign course under TKI treatment in CML patients with these transcripts [18,19] (Table 1).
Contrary to these reports, our patient displayed an unfavorable course of disease, with primary resistance to imatinib, an only brief CCyR of less than one year duration on nilotinib, and a temporary cytogenetic response to dasatinib, leading to the indication for allogeneic SCT as discussed below.
Additional chromosomal aberrations (ACAs) in Ph-positive clones
Additional chromosomal aberrations (ACAs) can occur in the Ph-positive and Ph-negative clones. ACAs in the Ph-positive clone occur in approximately 5% of patients overall, and increase in frequency in late chronic, accelerated, and blast phase CML (30–80%) [20]. Chromosomes Y, 7, 8, and 19 are involved most frequently [20,21]. Presence of ACAs already at diagnosis has been suggested by Luatti et al. to have a negative impact on prognosis with imatinib treatment, based on delayed achievement of CCyR and MMR. These authors propose closer monitoring, especially with major route chromosomal aberrations [20]. ACAs in a Ph-positive clone developing during TKI are considered hallmarks of clonal evolution, and are variably associated with imatinib resistance [21]. While some studies showed no adverse impact of ACAs on the probability of achieving a MMR, other reports have linked ACAs with adverse prognosis with TKI treatment (imatinib) as well as in the pre-TKI era [22]. Despite the uncertain clinical relevance of ACAs representing clonal evolution, current ELN guidelines consider ACAs as a “warning sign“ [5].
Additional chromosomal aberrations (ACAs) in Ph-negative clones
The appearance of ACAs in Ph-negative cells is a rare occurrence, has been observed under treatment with interferon-α and imatinib [23], and most frequently involves chromosomes 8, 7, and Y. Unmasking of preexisting ACAs by treatment appears to be the most common cause [24], but the possibility that imatinib itself could induce ACAs by impairing DNA damage repair has been raised [25]. The overall role of imatinib in promoting ACA development remains unclear, however the clinical relevance of ACAs in a Ph-negative clone is also uncertain [24]. Most ACAs are typical of those seen in AML and MDS, but very few CML patients with ACAs actually developed clinically overt MDS (11%), and progression to AML appears to be even less frequent [23]. The clinical course following emergence of ACAs is highly variable: Some studies report an incidence of ACAs of 3.4% to 8.7% under imatinib treatment, a median time to appearance of 13.3 months and no association with MDS or CML progression [26] or a negative impact on outcome [9,27] and in some cases even an only transient appearance is described [26]. The presence of ACAs in Ph negative cells seems to have no impact on the median time to CCyR with imatinib treatment, or on overall and progression free survival [27] (Table 1).
In monosomy 7 in particular, results are variable: Kovitz et al. identified 17 patients treated with imatinib who developed MDS or AML. Ten of these patients had chromosome 7 abnormalities, and in 5 cases a monosomy 7, suggesting that monosomy 7 denotes a higher probability for appearance of an MDS [28] and this coinciding with ACAs suggest a poor prognosis of patients with monosomy 7 [28]. In 2011, Groves et al. reported that patients with monosomy 7 or del(7q) in Ph-negative clones in CML have a significant risk of a second myeloid malignancy, with 15 of 50 patients developing MDS or AML within 6 months of ACA detection [29]. However, benign disease course has also been described [30], without the appearance of MDS despite of the presence of monosomy 7 [26].
In summary, detection of ACAs warrants continued cytogenetic analyses rather than reliance on monitoring only of BCR-ABL1 transcripts. Any therapeutic interventions have to be considered on the basis of the individual ACA. For example, appearance of monosomy 7 alone, without clinical signs of myelodysplasia, is a warning sign but does not constitute an indication for MDS-directed therapy [27]. These patients should be monitored closely in order not to miss development of MDS, but for individual patients, clinical decisions need to consider the heterogeneity in outcome among patients with monosomy 7 and dysplasia, as both benign courses as well as rapid progression to AML have been described. Further studies are needed to elucidate the factors underlying the variable prognosis of CML patients with ACAs and myelodysplasia.
Myelodysplasia in CML
Myelodysplasia in CML patients can been observed as a TKI-related side effect and as development of an MDS/MPN overlap syndrome. An MDS may be suspected in case of unexplained cytopenia, which must be distinguished from the initial and usually transient cytopenia that may occur during the early period of TKI therapy, which, if prolonged, is an adverse risk feature in CP-CML [15]. The frequency of severe neutro- and thrombopenias and anemia are reported in the range of 4–21%, and 2–12% thrombopenia and 0–10%, respectively, and are comparable with nilotinib and dasatinib treatment [31]. Overall, MDS is a rare cause of cytopenia in patients with CML [7,28]; a causal relationship with TKI treatment has been postulated partly because MDS has not been observed during interferon-alpha treatment [32]. Coexistence of dysplasia and myeloproliferative features resembling an MPN/MDS overlap syndrome constitutes a specific entity classified as atypical CML/MDS [33]. It is defined as a BCR-ABL1-negative disease with less than 20% blasts in the bone marrow and hypercellular granulocytic expansion with dysplasia by WHO criteria; no certain therapy has been established to date. The association between ACAs typical of MDS, e.g., monosomy 7 and myelodysplasia, is described above and in Table 1.
In our patient, MDS was initially classified as intermediate-1 by IPSS and high by WPSS. Clinical evidence for myelodysplasia was first noted 7.5 years after CML was diagnosed and 5.5 years after detection of monosomy 7, which coincided with start of TKI therapy, illustrating the long latency period until the myelodysplasia became clinically apparent.
Recurrent molecular aberrations
During the course of the disease, our patient developed 3 additional mutations: KRAS, ASXL1, and ETV6. Backtracking by molecular analysis of cryopreserved probes demonstrated that these genomic aberrations had not been present when MDS was clinically first diagnosed (Figure 2). KRAS belongs to the RAS superfamily of signaling proteins. Aberrant RAS function is associated with hyperproliferative developmental disorders and cancers [34]. KRAS mutations occur with a frequency below 5% in different subtypes of MDS and its prognostic relevance remains uncertain [35]. In CML, RAS mutations are very rare and their precise role in disease development and prognostic relevance is controversial [34], although they have been associated with imatinib resistance in individual patients with CML [36]. ASXL1 is a histone-modifying enzyme; therefore, it is a part of the epigenetic regulatory machinery. Loss-of-function mutations of ASXL1 are found in 11–21% of MDS patients and in 10–15% of patients with myeloproliferative syndromes, and are associated with a poor prognosis [37]. In CML, ASXL1 mutations have been reported in CP and BP and are relatively frequent [37]. It is not clear whether they are late or early events during disease development, but they seem to contribute to disease progression [38]. ETV6 encodes a transcription factor and is frequently involved in translocations and deletions in hematologic malignancies [39]. ETV6-PDGFRB translocations, for example, have been described in AML secondary to MDS and in MDS patients with high-risk features [40]. A large study by Haferlach et al. revealed that ETV6 rearrangements occur rarely in MDS (0.2%) [39], whereas data on ETV6 mutations in CML are extremely limited, with few case reports and an apparent association with atypical BCR-ABL1 negative CML [41]. Therefore its prognostic impact is unknown to date (Table 1).
In our patient, the appearance of KRAS, ASXL1, and ETV6 mutations were markers of disease progression and considered to portend an unfavorable prognosis (Figure 2), which, in conjunction with appearance of myelodysplasia, prompted addition of azacitidine. To date, no other data on prolonged combined treatment with TKI and a hypomethylating agent in the setting of CML chronic phase and myelodysplasia have been reported.
Treatment options for TKI failure in CML
The criteria for TKI failure are regularly updated and are described in the ELN guidelines [5]. They include a lack of hematologic, cytogenetic, and molecular responses at specified timepoints (3, 6, and 12 months after TKI start). Therapeutic options include a switch to other second- or third-generation TKIs, considering kinase domain mutational status and risk of side effects, HSCT (extensively reviewed e.g. by [42]), or experimental treatment in a clinical trial [5]. Novel agents in current clinical testing include allosteric BCR-ABL inhibitors (ABL001) [43], autophagy inhibitors (hydrochloroquine) [44], JAK2 inhibitors (Ruxolitinib) [45] and modulation of immune checkpoints by antibodies, e.g. nivolumomab [46]. Notably, there are no reports to date on the outcome of transplantation in CML-associated myelodysplasia as described in this report.
TKI treatment after allogeneic HSCT
As HSCT is performed today mainly in high-risk CML patients in case of TKI failure [42], a post-transplant TKI strategy becomes more and more important. Current recommendations clearly suggest a continuation of TKI treatment after HSCT if performed due to BC [47]. If HSCT was performed in AP or CPCML, there are in principle a preemptive and a MRD-triggered approach, like in Ph+ ALL [48]. To date, there are no clear recommendations on this, but strict MRD monitoring is essential after HSCT and, in case of TKI treatment, one has to consider the following caveats: data are available mainly for imatinib, but most patients who underwent transplant showed initial resistance to imatinib, and data about second- and third-generation TKIs are limited. If TKIs are administered prophylactically, TKI treatment can be started within the first month after HSCT and is in general well tolerated, although one has to be aware of drug-to-drug interactions (immunosuppressive treatment) and a potential weak hematopoiesis. In case of relapse upon routine BCR-ABL measurement, molecular, cytogenetic, and hematologic relapse after HSCT can often successfully be treated with TKIs [49] and there is also an option of donor lymphocyte infusion in combination with TKI treatment [50].
Rationale for Azacitidine and TKI combination therapy
Clinical experience with hypomethylating agents in CML is limited. High-dose decitabine was reported in a study with CML patients in accelerated or blast phase in the pre-TKI era [51]. Decitabine at a dose of 750–1000 mg/m2 per course for 5 days was administered to 20 patients in blast phase and 17 patients in accelerated phase. Objective response rate was 25% and 53% in blast phase and accelerated phase, respectively. Patients in blast phase reached complete hematologic remission (CHR) in 10%, PCyR in 5%, and 15% had bone marrow CR without platelet recovery. Of the patients treated, 35% returned to second chronic phase (with 2 patients showing PCyR) and 18% showed hematologic improvement or partial hematologic response. Low-dose decitabine was administered to 5 CML patients at a dose of 15–20 mg/m2 intravenously for 10-, 15-, or 20-day cycles (1 chronic, 1 accelerated and 3 blast phase patients). Two patients achieved a partial response and two a complete hematologic response [52]. The same group reported a phase II study of low-dose decitabine in CML patients resistant to imatinib. Twelve patients in chronic, 17 patients in accelerated, and 6 patients in blast phase were treated with 10–15 mg/m2 for 10 days every 6 weeks. A CHR was reached by 17–50%, PHR in 33–17%, a major CyR in 17–25%, and a minor CyR in 17–33% of patients [53].
Azacitidine treatment in combination with immunosuppressive drugs have been reported to be beneficial in rare cases of MDS [54]. The combination of a hypomethylating agent and a TKI was tested in 2 interesting studies: in a phase II study combining low-dose decitabine (15 mg/m2 for 5 days) with imatinib (600 mg/day) in patients with accelerated (n=18) and blast phase (n=10), the CHR rate was 20% and 39%, respectively, and the major CyR rate 17% and 20% [55]. In another study, reported by Ghez et al., 5 patients in myeloid blast crisis were treated with the combination of 5-azacitidine for 7 days in 28-day cycles in combination with a second-generation TKI. All patients achieved a CHR and 2 showed a CyR and a MMR after 3 to 10 months of treatment [56]. The feasibility and efficacy of long-term combination of a TKI for MDS secondary to CML has not yet been explored.
Our patient had an indication for treatment with azacitidine on the basis of her worsening risk score (evolving to intermediate-2 at 6 months after diagnosis), with persistent grade 3–4 neutropenia, transfusion dependency, and increasing bone marrow fibrosis. Initially, it was difficult to distinguish whether the dysplasia with cytopenia was therapy-associated or reflected progression of CML. Allogeneic HSCT was indicated based on the course of her CML, but no matched sibling or unrelated donor was available, and no third-generation TKI was approved at that time. The decision to combine azacitidine and dasatinib was made with the following caveats: lack of data on combined dasatinib and azacitidine, the risk of aggravating cytopenia, and the potential for drug-drug interactions. The subsequent clinical course was characterized by sustained CCyR, but with persistence of detectable BCR-ABL1 transcripts. The MDS remained clinically and cytogenetically stable for 3 years, with appearance of a KRAS mutation as a possible negative prognostic factor for acceleration of MDS into AML [57]. Despite these adverse genetic findings, the patient’s clinical course remained stable for additional several months with continued combination therapy.
Our rationale for combining a TKI with a hypomethylating agent was supported by preclinical data: DNA methylation stimulates carcinogenesis by modification of DNA expression and consecutive silencing of tumor suppressors [58]. It has been shown that DNA methylation increases in progressive disease in CML [59] and that hypomethylating agents have single-agent activity in CML, even in imatinib-resistant cases [53]. Moreover, synergistic effects of imatinib and decitabine have previously been shown in vitro in CML [60]. Accordingly, combined administration of hypomethylating agents and TKIs had the potential for enhanced and possibly synergistic activity compared with single-agent treatment.
Conclusions
This case demonstrates an unusual course of CML, in which a variant translocation (t(9;22;17)) and an aberrant BCR-ABL transcript (e1a3) were detected at initial diagnosis, the latter being apparent not by routine RT-PCR but in nested PCR analysis. Primary treatment failure in response to imatinib according to ELN guidelines [5] prompted switching to nilotinib but was complicated by acquisition of additional chromosomal abnormalities (monosomy 7) in a Ph-negative clone. Nilotinib treatment resulted in a transient CCyR but no major molecular response (MMR). Cytogenetic relapse accompanied by pancytopenia posed a diagnostic challenge, with a differential diagnosis of acceleration of the CML or emergence of MDS. This cytogenetic relapse was treated with a switch to dasatinib. Based on cytologic features during the further disease course, with pronounced dysplasia of the megakaryocyte and erythroid lineages, severe granulocytopenia but normal blast cell content, and cytogenetic detection of monosomy 7, a diagnosis of MDS was established. This prompted addition of azacitidine to dasatinib treatment, which was well tolerated and achieved prolonged clinical stabilization. Subsequent evidence of clonal evolution was development of a KRAS mutation and loss of cytogenetic remission after 4 years under combination treatment.
Haploidentical BMT was performed as potentially curative therapy, resulting in a sustained complete cytogenetic remission, full donor chimerism, and undetectable BCR-ABL1 (checking for both typical and atypical transcripts) except for one inter-current molecular relapse 2.5 years after transplant, which was successfully treated with nilotinib, and two further detections revealing low level atypical BCR-ABL1 transcripts on day +1036 and day +1477, which disappeared without treatment.
This case demonstrates the feasibility of long-term combined therapy with a hypomethylating agent and a TKI in patients with CML coincident with MDS, but also highlights the continued importance of allogeneic HSCT, including alternative donor transplant, as a definite curative treatment option. The pivotal role of appropriate molecular monitoring, including atypical BCR-ABL1 transcripts and awareness of additional aberrations unrelated to CML but diagnostic of a second hematologic malignancy such as MDS, is also emphasized.
Footnotes
Conflict of interest
None.
Source of support: Fabian Lang received support from the Frankfurter Förderung “Nachwuchswissenschaftler” and the EUTOS funding program. Fabian Lang and Oliver G. Ottmann had advisory roles at Novartis, Ariad, Sanofi Aventis, and Bristol-Myers Squibb. Fabian Lang received funding from Novartis. Oliver G. Ottmann was funded by Novartis, Bristol-Myers Squibb, and the Deutsche José Carreras Leukämie Stiftung
References:
- 1.Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol. 2015;94:107–21. doi: 10.1007/s00277-015-2325-z. [DOI] [PubMed] [Google Scholar]
- 2.Hasford J, Baccarani M, Hoffmann V, et al. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: The EUTOS score. Blood. 2011;118:686–92. doi: 10.1182/blood-2010-12-319038. [DOI] [PubMed] [Google Scholar]
- 3.Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in ‘good-risk’ chronic granulocytic leukemia. Blood. 1984;63:789–99. [PubMed] [Google Scholar]
- 4.Hasford J, Pfirrmann M, Hehlmann R, et al. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. Writing Committee for the Collaborative CML Prognostic Factors Project Group. J Natl Cancer Inst. 1998;90:850–58. doi: 10.1093/jnci/90.11.850. [DOI] [PubMed] [Google Scholar]
- 5.Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108:1809–20. doi: 10.1182/blood-2006-02-005686. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen-Bjergaard J, Andersen MK, et al. Genetic pathways in therapy-related myelodysplasia and acute myeloid leukemia. Blood. 2002;99:1909–12. doi: 10.1182/blood.v99.6.1909. [DOI] [PubMed] [Google Scholar]
- 7.Schmitt-Graeff A, Hochhaus A. Hematological side effects of tyrosine kinase inhibition using imatinib. Pathologe. 2006;27:40–46. doi: 10.1007/s00292-005-0806-x. [DOI] [PubMed] [Google Scholar]
- 8.Liu LG, Tanaka H, Ito K, et al. Chronic myelogenous leukemia with e13a3 (b2a3) type of BCR-ABL transcript having a DNA breakpoint between ABL exons a2 and a3. Am J Hematol. 2003;74:268–72. doi: 10.1002/ajh.10429. [DOI] [PubMed] [Google Scholar]
- 9.Fabarius A, Leitner A, Hochhaus A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: Long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118:6760–68. doi: 10.1182/blood-2011-08-373902. [DOI] [PubMed] [Google Scholar]
- 10.Faderl S, Moshe T, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;16:164–72. doi: 10.1056/NEJM199907153410306. [DOI] [PubMed] [Google Scholar]
- 11.Stagno F, Vigneri P, Del Fabro V, et al. Influence of complex variant chromosomal translocations in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Acta Oncol. 2010;49:506–8. doi: 10.3109/02841861003660031. [DOI] [PubMed] [Google Scholar]
- 12.Marzocchi G, Castagnetti F, Luatti S, et al. Variant Philadelphia translocations: Molecular-cytogenetic characterization and prognostic influence on frontline imatinib therapy, a GIMEMA Working Party on CML analysis. Blood. 2011;117:6793–800. doi: 10.1182/blood-2011-01-328294. [DOI] [PubMed] [Google Scholar]
- 13.El-Zimaity MMT, Kantarjian H, Talpaz M, et al. Results of imatinib mesylate therapy in chronic myelogenous leukaemia with variant Philadelphia chromosome. Br J Haematol. 2004;125:187–95. doi: 10.1111/j.1365-2141.2004.04899.x. [DOI] [PubMed] [Google Scholar]
- 14.Burmeister T, Schwartz S, Taubald A, et al. Atypical BCR-ABL mRNA transcripts in adult acute lymphoblastic leukemia. Haematologica. 2007;92:1699–702. doi: 10.3324/haematol.11737. [DOI] [PubMed] [Google Scholar]
- 15.Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–84. [PubMed] [Google Scholar]
- 16.Al-Ali HK, Leiblein S, Kovacs I, et al. CML with an e1a3 BCR-ABL fusion: Rare, benign, and a potential diagnostic pitfall. Blood. 2002;100:1092–93. doi: 10.1182/blood-2002-03-0930. [DOI] [PubMed] [Google Scholar]
- 17.Langabeer SE. The e1a3 BCR-ABL1 fusion transcript in philadelphia chromosome-positive acute lymphoblastic leukemia. Ann Lab Med. 2015;35:540–41. doi: 10.3343/alm.2015.35.5.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder DS, McMahon R, Cohen SR, Slovak ML. Chronic myeloid leukemia with an e13a3 BCR-ABL fusion: Benign course responsive to imatinib with an RT-PCR advisory. Am J Hematol. 2004;75:92–95. doi: 10.1002/ajh.10456. [DOI] [PubMed] [Google Scholar]
- 19.McCarron SL, Langabeer SE, Bolger K, et al. Molecular response to imatinib in chronic myeloid leukaemia with a variant e13a3 BCR-ABL1 fusion. Med Oncol. 2015;32:452. doi: 10.1007/s12032-014-0452-3. [DOI] [PubMed] [Google Scholar]
- 20.Luatti S, Castagnetti F, Marzocchi G, et al. Additional chromosomal abnormalities in Philadelphia-positive clone: Adverse prognostic influence on frontline imatinib therapy: A GIMEMA Working Party on CML analysis. Blood. 2012;120:761–67. doi: 10.1182/blood-2011-10-384651. [DOI] [PubMed] [Google Scholar]
- 21.Falchi L, Rege-Cambrin G, Fava C, et al. Sustained molecular remissions are achievable with tyrosine kinase inhibitor therapy in patients with chronic myeloid leukemia and additional cytogenetic clonal evolution. Cancer Genet Cytogenet. 2010;199:139–42. doi: 10.1016/j.cancergencyto.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Jabbour E, Kantarjian H. Introduction: Chronic myelogenous leukemia (CML) Semin Hematol. 2007;44:S1–S3. doi: 10.1053/j.seminhematol.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Loriaux M, Deininger M. Clonal cytogenetic abnormalities in Philadelphia chromosome negative cells in chronic myeloid leukemia patients treated with imatinib. Leuk Lymphoma. 2004;45:2197–203. doi: 10.1080/10428190410001723278. [DOI] [PubMed] [Google Scholar]
- 24.Bumm T, Mueller C, Al-Ali HK, et al. Emergence of clonal cytogenetic abnormalities in Ph-cells in some CML patients in cytogenetic remission to imatinib but restoration of polyclonal hematopoiesis in the majority. Blood. 2003;101:1941–49. doi: 10.1182/blood-2002-07-2053. [DOI] [PubMed] [Google Scholar]
- 25.Shafman T, Khanna KK, Kedar P, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387:520–23. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 26.Medina J, Kantarjian H, Talpaz M, et al. Chromosomal abnormalities in Philadelphia chromosome-negative metaphases appearing during imatinib mesylate therapy in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase. Cancer. 2003;98:1905–11. doi: 10.1002/cncr.11729. [DOI] [PubMed] [Google Scholar]
- 27.Deininger MWN, Cortes J, Paquette R, et al. The prognosis for patients with chronic myeloid leukemia who have clonal cytogenetic abnormalities in philadelphia chromosome-negative cells. Cancer. 2007;110:1509–19. doi: 10.1002/cncr.22936. [DOI] [PubMed] [Google Scholar]
- 28.Kovitz C, Kantarjian H, Garcia-Manero G, et al. Myelodysplastic syndromes and acute leukemia developing after imatinib mesylate therapy for chronic myeloid leukemia. Blood. 2006;108:2811–13. doi: 10.1182/blood-2006-04-017400. [DOI] [PubMed] [Google Scholar]
- 29.Groves MJ, Sales M, Baker L, et al. Factors influencing a second myeloid malignancy in patients with Philadelphia-negative -7 or del(7q) clones during tyrosine kinase inhibitor therapy for chronic myeloid leukemia. Cancer Genet. 2011;204:39–44. doi: 10.1016/j.cancergencyto.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 30.Navarro JT, Feliu E, Grau J, et al. Monosomy 7 with severe myelodysplasia developing during imatinib treatment of Philadelphia-positive chronic myeloid leukemia: 2 cases with a different outcome. Am J Hematol. 2007;82:849–51. doi: 10.1002/ajh.20859. [DOI] [PubMed] [Google Scholar]
- 31.Wei G, Rafiyath S, Liu D. First-line treatment for chronic myeloid leukemia: Dasatinib, nilotinib, or imatinib. J Hematol Oncol. 2010;3:47. doi: 10.1186/1756-8722-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talpaz M, Hehlmann R, Quintás-Cardama A, et al. Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia. 2013;27:803–12. doi: 10.1038/leu.2012.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foucar K. Myelodysplastic/myeloproliferative neoplasms. Am J Clin Pathol. 2009;132:281–89. doi: 10.1309/AJCPJ71PTVIKGEVT. [DOI] [PubMed] [Google Scholar]
- 34.Ouerhani S, Bougatef K, Soltani I, et al. The prevalence and prognostic significance of KRAS mutation in bladder cancer, chronic myeloid leukemia and colorectal cancer. Mol Biol Rep. 2013;40:4109–14. doi: 10.1007/s11033-013-2512-8. [DOI] [PubMed] [Google Scholar]
- 35.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122:4021–34. doi: 10.1182/blood-2013-09-381665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwal A, Eide CA, Harlow A, et al. An activating KRAS mutation in imatinib-resistant chronic myeloid leukemia. Leukemia. 2008;22:2269–72. doi: 10.1038/leu.2008.124. [DOI] [PubMed] [Google Scholar]
- 37.Boultwood J, Perry J, Pellagatti A, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24:1062–65. doi: 10.1038/leu.2010.20. [DOI] [PubMed] [Google Scholar]
- 38.Makishima H, Visconte V, Sakaguchi H. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–10. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haferlach C, Bacher U, Schnittger S, et al. ETV6 rearrangements are recurrent in myeloid malignancies and are frequently associated with other genetic events. Genes Chromosom Cancer. 2012;51:328–37. doi: 10.1002/gcc.21918. [DOI] [PubMed] [Google Scholar]
- 40.Raynaud DS, Baens M, Grosgeorge J, et al. Fluorescence in situ hybridization analysis of t(3; 12)(q26; p13): A recurring chromosomal abnormality involving the TEL gene (ETV6) in myelodysplastic syndromes. Blood. 1996;88:682–89. [PubMed] [Google Scholar]
- 41.Keung YK, Beaty M, Steward W, et al. Chronic myelocytic leukemia with eosinophilia, t(9;12)(q34;p13), and ETV6-ABL gene rearrangement: Case report and review of the literature. Cancer Genet Cytogenet. 2002;138:139–42. doi: 10.1016/s0165-4608(02)00609-x. [DOI] [PubMed] [Google Scholar]
- 42.Jabbour E, Cortes J, Kantarjian HM, et al. Allogeneic stem cell transplantation for patients with chronic myeloid leukemia and acute lymphocytic leukemia after Bcr-Abl kinase mutation-related imatinib failure. Blood. 2006;108:1421–23. doi: 10.1182/blood-2006-02-001933. [DOI] [PubMed] [Google Scholar]
- 43.Hantschel O. Allosteric BCR-ABL inhibitors in Philadelphia chromosome-positive acute lymphoblastic leukemia: Novel opportunities for drug combinations to overcome resistance. Haematologica. 2012;97:157–59. doi: 10.3324/haematol.2012.061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–23. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plosker GL, Robinson DM. Nilotinib. Drugs. 2008;68:449–59. doi: 10.2165/00003495-200868040-00005. [DOI] [PubMed] [Google Scholar]
- 46.Topalian SL, Hodi FS, Brahmer JR. Safety, activity, and immune correlates of anti-PD-1 Antibody in Cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saussele S, Silver RT. Management of chronic myeloid leukemia in blast crisis. Ann. Hematol. 2015;94:S159–65. doi: 10.1007/s00277-015-2324-0. [DOI] [PubMed] [Google Scholar]
- 48.Pfeifer H, Wassmann B, Bethge W, et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR-ABL1-positive acute lymphoblastic leukemia. Leukemia. 2013;27:1254–62. doi: 10.1038/leu.2012.352. [DOI] [PubMed] [Google Scholar]
- 49.Klyuchnikov E, Kröger N, Brummendorf TH, et al. Current status and perspectives of tyrosine kinase inhibitor treatment in the post-transplant period in patients with chronic myelogenous leukemia (CML) Biol Blood Marrow Transplant. 2010;16:301–10. doi: 10.1016/j.bbmt.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 50.Savani BN, Montero A, Kurlander R, et al. Imatinib synergizes with donor lymphocyte infusions to achieve rapid molecular remission of CML relapsing after allogeneic stem cell transplantation. Bone Marrow Transplant. 2005;36:1009–15. doi: 10.1038/sj.bmt.1705167. [DOI] [PubMed] [Google Scholar]
- 51.Kantarjian HM, O’Brien SM, Keating M, et al. Results of decitabine therapy in the accelerated and blastic phases of chronic myelogenous leukemia. Leukemia. 1997;11:1617–20. doi: 10.1038/sj.leu.2400796. [DOI] [PubMed] [Google Scholar]
- 52.Issa JPJ, Garcia-Manero G, Giles FJ, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–40. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 53.Issa JPJ, Gharibyan V, Cortes J, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23:3948–56. doi: 10.1200/JCO.2005.11.981. [DOI] [PubMed] [Google Scholar]
- 54.Endo M, Sekikawa A, Tsumura T, et al. A case of myelodysplastic syndrome with intestinal behçet’s disease-like symptoms treated by prednisolone and azacitidine. Am J Case Rep. 2015;16:827–31. doi: 10.12659/AJCR.895431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oki Y, Kantarjian HM, Gharibyan V, et al. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer. 2007;109:899–906. doi: 10.1002/cncr.22470. [DOI] [PubMed] [Google Scholar]
- 56.Ghez D, Micol JB, Pasquier F, et al. Clinical efficacy of second generation tyrosine kinase inhibitor and 5-azacytidine combination in chronic myelogenous leukaemia in myeloid blast crisis. Eur J Cancer. 2013;49:3666–70. doi: 10.1016/j.ejca.2013.07.147. [DOI] [PubMed] [Google Scholar]
- 57.Stephenson J, Lizhen H, Mufti GJ. Possible co-existence of RAS activation and monosomy 7 in the leukaemic transformation of myelodysplastic syndromes. Leuk Res. 1995;19:741–48. doi: 10.1016/0145-2126(95)00056-t. [DOI] [PubMed] [Google Scholar]
- 58.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–67. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen TT, Mohrbacher AF, Tsai YC, et al. Quantitative measure of c-abl and p15 methylation in chronic myelogenous leukemia: Biological implications. Blood. 2000;95:2990–92. [PubMed] [Google Scholar]
- 60.La Rosée P, Johnson K, Corbin AS, et al. In vitro efficacy of combined treatment depends on the underlying mechanism of resistance in imatinib-resistant Bcr-Abl-positive cell lines. Blood. 2004;103:208–15. doi: 10.1182/blood-2003-04-1074. [DOI] [PubMed] [Google Scholar]