

Abstract
High-fat diet (HFD)-induced obesity is reaching worldwide proportions. In addition to causing obesity, HFDs also induce a variety of health disorders, which includes cognitive decline. Hippocampal function may be particularly vulnerable to the negative consequences of HFD, and it is suspected that ‘primed’ neuroinflammatory processes may mediate this response. To examine the link between diet, hippocampal function and neuroinflammation, male Wistar rats were fed a medium or HFD. Hippocampal memory function was measured using contextual pre-exposure fear conditioning (CPE-FC). Rats fed a HFD demonstrated impaired memory, an effect that was augmented with longer duration of HFD consumption. HFD-induced memory impairments were linked to potentiated levels of interleukin-1 beta (IL-1β) protein in the hippocampus 2 h after the foot-shock that occurs during CPE-FC. Central IL-1 receptor antagonism, with intracisterna magna (ICM) administration of hIL-1RA prior to the foot-shock prevented the diet-induced memory disruption, suggesting a critical role for IL-1β in this phenomenon. Additionally, obese animals whose diet regimen was reversed from HFD back to standard chow recovered memory function and did not demonstrate a foot-shock-induced hippocampal IL-1β increase. Interestingly, dietary reversal neutralized the negative impact of HFD on memory and IL-1β, yet animals maintained physiological evidence of obesity (increased body mass and serum leptin), indicating that dietary components, not body mass, may mediate the negative effects on memory.
Keywords: High-fat diet, Hippocampus, Interleukin-1β, Fear conditioning, hIL-1RA, Dietary reversal, Memory
1. Introduction
There is a well-established link between human obesity and cognitive decline (Sellbom and Gunstad, 2012). Specifically, hippocampally-dependent functions may be particularly vulnerable (Kanoski and Davidson, 2011; Francis and Stevenson, 2013), as many studies have linked high-caloric diets with decreased contextual and spatial memory (Winocur and Greenwood, 2005; Valladolid-Acebes et al., 2011; Kosari et al., 2012; Ross et al., 2012; Yamada-Goto et al., 2012). This is most likely the result of dietary-induced alterations in hippocampal synaptic plasticity (Molteni et al., 2002; Wu et al., 2004; Stranahan et al., 2008; Hwang et al., 2010). However, the mechanisms underlying how high-caloric diets cause hippocampal dysfunction are largely unknown.
It is possible that diet and obesity may induce cognitive disruption by impacting neural inflammatory processes. Obesity is often characterized by increased and altered peripheral inflammatory processes (Mito et al., 2000; Das, 2010; Donath and Shoelson, 2011) and peripheral inflammation can induce neuroinflammation (Maier and Watkins, 1998), leading to the notion that central inflammatory processes may also be altered with obesity. The pro-inflammatory cytokine interleukin-1 beta (IL-1β) is required for proper hippocampal memory function (Schneider et al., 1998; Yirmiya et al., 2002; Avital et al., 2003). However, elevated or exaggerated central levels of IL-1β are detrimental to cognitive processing (Gibertini et al., 1995; Barrientos et al., 2002; Yirmiya and Goshen, 2011) as the duration of hippocampal-based cognitive impairment mirrors the duration of elevated hippocampal IL-1β (Barrientos et al., 2009a) and inflammation-induced cognitive decline is prevented when IL-1β action is blocked with IL-1 receptor antagonist (IL-1RA) (Pugh et al., 1998; Palin et al., 2004; Frank et al., 2010; Barrientos et al., 2012). Furthermore, there is growing evidence that increased IL-1β levels in the hippocampus are linked to deficits in hippocampal cognitive function in genetic mouse models of obesity (Dinel et al., 2011; Erion et al., 2014) and following HFD consumption (Pistell et al., 2010; Boitard et al., 2014).
In addition, there is growing evidence that certain conditions, such as stress (Johnson et al., 2003; Li et al., 2008; Frank et al., 2011; Loram et al., 2011), peripheral inflammation (Perry et al., 2007; Dantzer et al., 2008; Williamson et al., 2011), and advanced age (Barrientos et al., 2006; Chen et al., 2008) sensitize or ‘prime’ central inflammatory responses to a secondary challenge, leading to exaggerated central IL-1β levels. It is possible that obesity may induce neural inflammation by activating a similar ‘priming’ mechanism. Obesity, or exposure to HFD, may not induce neural inflammation directly, but rather could induce structural or phenotypic alterations in central innate immune cells (e.g., microglia) such that the occurrence of a secondary challenge, such as shock, would induce an amplified pro-inflammatory response that is greater than that which would be observed without the prior priming experience.
The present series of studies aimed to examine the impact of high-fat diet (HFD) on hippocampal-dependent memory function, and to evaluate whether altered hippocampal IL-1β responses might be involved in mediating cognition. hIL-1RA was used to determine whether blocking the action of IL-1β during a learning task would prevent the negative impact of HFD on memory. Furthermore, there is a growing body of evidence to suggest that short-term dietary reversal is able to reverse negative consequences of prior high-fat diet consumption (Hydock et al., 2013; Reeves et al., 2013). Therefore, we also sought to evaluate whether a switch from HFD to regular chow would induce recovery of cognitive functioning and linked neuroinflammation.
2. Materials and methods
2.1. Animals
Male Wistar rats (Harlan Laboratories) were used. All animals were approximately 2 months of age and weighed between 250 and 275 g at time of arrival. Subjects were pair housed in standard large cages (52 cm × 30 cm × 21 cm; L × W × H) with food and water administered ad libitum. The colony room was maintained at 22 °C on a 12-h light/dark cycle (lights on at 07:00 h). All experiments were conducted in accordance with protocols approved by the University of Colorado Animal Care and Use Committee.
2.2. Dietary manipulation
Animals were randomly assigned on date of arrival to receive one of three diet types (Harlan Laboratories). Control animals received standard rat chow (Reg; TD.8640, energy density of 3.0 kcal/g; 29% calories from protein, 54% from carbohydrates and 17% from fat (fat source: soybean oil). Animals selected for diet-induced obesity received one of two types of adjusted calorie diets that are considered to be medium-fat (Med; TD.88137; energy density of 4.5 kcal/g; 15.2% calories from protein, 42.7% from carbohydrates and 42% from fat (fat source: milk fat)) or high-fat (HFD; TD.06414, energy density of 5.1 kcal/g; 18.4% calories from protein, 21.3% from carbohydrates and 60.3% from fat (fat source: approximately 10:1 lard: soybean oil). The standard chow does not contain added sucrose, while the medium-fat diet is approximately 34.5% sucrose by weight, and the HFD is 12.1% sucrose by weight. Following Experiment 1, use of the medium-fat diet was discontinued. Upon arrival, animals were given free access to the assigned diet and remained on that diet thereafter, unless otherwise noted. The duration of dietary consumption prior to either testing or sample collection varied across experiments, with feeding time ranging from 12 to 24 weeks. The specifics of dietary duration for each experiment are outlined in Section 2.8. Some animals received a dietary reversal procedure that transitioned animals from the HFD to a regular diet. For this, as the removal of palatable food is a known stressor (South et al., 2012), animals were given a one-week period of mixed HFD and regular food to ease the stress of transition before complete reversal to the regular diet. All animals were weighed on date of arrival and re-weighed bi-weekly.
2.3. Contextual pre-exposure fear conditioning paradigm
A contextual pre-exposure fear-conditioning (CPE-FC) paradigm was chosen to assess hippocampal based memory deficits following diet-induced obesity. In this paradigm, subjects are placed in a conditioning environment, receive an immediate foot-shock, and are then removed. The immediate shock does not lead to fear conditioning to the context, putatively because the foot-shock occurs before subjects have formed a representation (hippocampal) of the context. Indeed, if the subjects are allowed to explore the environment prior to the immediate shock session (the pre-exposure experience), the immediate shock event then does condition fear to the contextual cues (Fanselow, 1990).
The CPE-FC paradigm was chosen for a number of reasons. First, studies suggest that hippocampal-based learning and memory function is particularly vulnerable to dietary influence (Kanoski and Davidson, 2011). Importantly, CPE-FC requires intact hippocampal functioning during all three phases (pre-exposure, conditioning with shock, and retrieval memory testing; described in more detail below) of the procedure (Rudy et al., 2002). Second, it was initially unclear whether diet-based cognitive deficits would be readily apparent, so it was anticipated that the relatively long duration between conditioning and testing that occurs with CPE-FC would accentuate any dietary-induced differences.
Additionally, behavioral tests of memory that require movement could be confounded by alterations in body mass. Prior research suggests that increased body mass from high-fat dietary manipulation does not impact basal activity compared to controls (Hill-Pryor and Dunbar, 2006) but other groups have observed reduced locomotor activity following HFD (Lavin et al., 2011). CPE-FC is a conservative test since memory is assessed by the presence or absence of freezing behavior (see below for further details), with more movement (less freezing behavior) indicating reduced memory for the context. If HFD animals have reduced memory for the conditioned context, this would be exhibited as increased movement compared to controls.
2.3.1. Context pre-exposure fear conditioning (CPE-FC) behavioral procedures
The CPE-FC paradigm consists of three separate and distinct components: pre-exposure, immediate shock, and testing for freezing behavior (testing occurred both within the conditioned context A and in an alternate control context B). The CPE-FC procedure used here was adapted from Barrientos et al. (2002). The standard procedure spanned a one-week period and consisted of 4 active days.
2.3.1.1. Contextual pre-exposure and immediate shock sessions
For context pre-exposure (day 1), rats were taken two at a time from their home cage and transported to the conditioning context (context A) in a blue ice bucket with a lid and were placed into one of two identical contextual conditioning chambers. Ice buckets were used in order to establish an association between the contextual representation and the transport cues preceding placement of the rat in the context. Animals were allowed to freely explore the conditioning chamber for 5 min, after which animals were transported back to their home cage where they remained for approximately 30 s before initiation of the next transport and pre-exposure experience. This procedure was repeated 5 more times, with each additional exposure to the context lasting only 30 s before animals were placed in ice buckets and transported back to their home cage. The total duration of pre-exposure to the context lasted approximately 20 min per animal pair, and was designed for animals to intimately learn and link the transport and contextual cues of the overall pre-exposure experience. Immediate shock (day 2) in the context occurred 24 h following pre-exposure. Animals were transported via blue ice buckets to the conditioning chambers, immediately given one 2 s 1.5 mA shock, then quickly removed and returned to their home cage.
2.3.1.2. Contextual fear testing
Contextual fear was assessed during two 5-min testing sessions. These occurred on day 5 in the control context B, which was used to measure generalized freezing behavior, and on day 7 in the conditioned context A. Following conditioning, the CPE-FC procedure did not involve further shock so it was likely that testing sessions could facilitate extinction of any observable freezing behavior. Therefore, testing in the control context B occurred prior to testing in the conditioned context A. This diminished the possibility that lack of freezing in context B could be attributed to extinguished fear acquired during the context A test, and also supported the notion that any freezing observed in Context A (but not in context B) could be attributed to a contextually-specific fear memory for the conditioned context.
Fear behavior was assessed by placing each rat in context A or B and observing freezing behavior using a time sampling procedure in which, every 10 s, each rat was judged as either freezing or active at the instant the sample was taken. Freezing is the dominant defensive fear response for a rat and was defined as the absence of all visible movement, except for respiration. Scoring began approximately 10 s after the animal was placed into the chamber and continued for 5 min.
Whenever possible, blinded behavioral scoring was achieved by randomizing animals and altering identifying markings, which were later decoded. For some tests, it was not possible to maintain true blind scoring due to the noticeable nature of increased weight. To compensate for this possible bias, testing sessions were video recorded and a subsequent scorer later confirmed freezing behavior. Freezing scores were averaged between scorers and converted to percentages by dividing the number of positive freezing scores over the total number of sampling blocks. For context B, scores below 10 percent (%) total freezing were considered acceptable indicators of low baseline freezing behavior.
2.3.2. Apparatus
2.3.2.1. Context A (conditioning context)
The conditioning chambers consisted of one of two identical Igloo ice chests (54 L × 30 W × 27 H, cm) with white interiors, which were located in a room with overhead lights on. An activated 24-V DC light bulb and mini vent fan were mounted on the ceiling of each chest. The conditioning chambers (26 L × 21 W × 24 H, cm), placed inside each chest, were made of clear plastic. A 2 s 1.5 mA shock was delivered through a removable floor of stainless steel rods 1.5 mm in diameter, spaced 1.2 cm, center to center. Each rod was wired to a shock generator and scrambler (Coulbourn Instruments, Allentown, PA). Chambers were cleaned with a diluted Mr. Clean mixture before each animal was pre-exposed, conditioned or tested.
2.3.2.2. Context B (control context)
The control context B differed substantially from the conditioning context A with respect to transport, light, tactile and smell cues. Context B consisted of identical hollow clear plastic cylinders (approximately 25 cm in diameter, 35 cm high), which were placed inside white wooden boxes (30 L × 35 W × 50 H, cm). Boxes were illuminated with a small visible red light (7.5 watt red incandescent), with overhead room lighting off. A removable plastic tray and a small amount of fresh bedding (approx 1 cup) were placed on the floor of the context. Each chamber was cleaned with a diluted mixture of alcohol and water prior to animal testing. Animals remained in their home cage during transportation to the context B testing room, then moved to the context B chambers by hand.
2.4. Tissue collection
A rapid decapitation procedure was used for tissue collection in an effort to capture the true nature of inflammatory marker levels at the time of sacrifice. Trunk blood was collected in 10 ml glass tubes and placed on ice. Brains were rapidly extracted and the bilateral hippocampal formations dissected atop a glass plate resting on ice. Each hippocampal half was placed in a labeled 1.5 ml Eppendorf tube and flash frozen in liquid nitrogen. After all blood samples were collected, tubes containing blood were centrifuged at 4 °C at 4000g for 10 min and serum was collected and placed in a clean tube. All samples were stored at −80 °C until further processed or used for ELISA measurement.
2.5. Tissue processing for ELISA protein measurement
Levels of IL-1β in hippocampus and serum as well as serum leptin proteins were determined using commercially available rat-specific enzyme linked immunosorbent assay (ELISA) IL-1β and leptin protein kits (R&D Systems, Minneapolis, MN). The assays were performed according to the manufacturer's instructions. IL-1β was determined and is presented as picograms per 100 μg of total protein (pg/100 μg) for hippocampal samples and IL-1β and leptin are presented as picograms per milliliter (pg/ml) in serum.
Following dissection and tissue collection, hippocampal half samples were sonicated in 0.3 ml of a sonication buffer containing 50 mM Tris base and a mixture enzyme inhibitor (100 mM amino-n-caproic acid, 10 mM EDTA, 5 mM benzamidine HCl, and 0.2 mM phenylmethyl sulfonyl fluoride).
Tissues were mechanically homogenized using an ultrasonic cell disrupter (Thermo Fisher Scientific, Pittsburgh, PA). Sonication consisted of 20 s of cell disruption at 52% amplitude. Sonicated samples were centrifuged at 10,000g at 4 °C for 10 min. Supernatants were removed and stored at 4 °C until ELISA was performed. Bradford protein assays were also performed on hippocampal samples to determine total protein concentrations.
2.6. Intracisternal magnal (ICM) injection of human IL-1RA
To determine the impact of blocking central action of IL-1β at the time of contextual pre-exposure fear-conditioned shock, rats received an infusion of human recombinant IL-1 receptor antagonist (hIL-1RA; Amgen) into the cisterna magna (ICM) 2 h prior to the CPE-FC shock event, as described previously (Barrientos et al., 2012). Rats were briefly anesthetized with halothane and the dorsal aspect of the skull was shaved and swabbed with 70% EtOH. A 27-gauge needle attached via PE50 tubing to a 25 μl Hamilton syringe was inserted into the cisternal magna. To verify entry into the cerebral spinal fluid (CSF), approximately 2 μl of clear CSF was drawn and gently pushed back in and a 3 (μl total volume of hIL-1RA (dose of 112 μg) was slowly administered and allowed to absorb for 1 min before the needle was removed. Following injection, halothane was immediately discontinued and subjects were placed in a recovery box, and typically were awake, alert and mobile within 3 min. The same procedure was used and an equal volume of sterile saline was injected ICM for vehicle control animals. ICM injections were used because they do not require surgery or cannulae implantation, which are themselves inflammatory manipulations (Holguin et al., 2007) and because substances injected ICM spread readily throughout the central nervous system (CNS) (Proescholdt et al., 2000). As IL-1RA binds to but does not activate the IL-1 type-1 receptor, thereby preventing IL-1β signal transduction (Dinarello, 1998), it was anticipated that ICM IL-1RA would effectively block action of IL-1β within the CNS, including the hippocampus.
2.7. Data analysis
Statistical analyses were conducted using StatView version 5.0 software and Prism Graphpad version 5.0d. One-way and two-way ANOVAs were used, as appropriate. CPE-FC freezing scores (across time for the 5-min test sessions) data were analyzed using repeated measures ANOVAs. However, for simplicity, these data are graphically presented as summed total percentage for the entire test session. Where appropriate, Tukey's post-hoc comparisons were conducted to reveal pairwise differences between groups. All data are presented as mean ± SEM with sample sizes provided. Threshold for statistical significance was set at α = .05.
2.8. Experimental design
2.8.1. Experiment 1: Impact of diet type on weight gain, cognitive performance and obesity development, following 12 weeks of dietary intervention
The current diet-induced obesity animal literature varies greatly with respect to use of specific components and duration of dietary manipulation. Subtle variations in diet can greatly influence resulting weight gain, as well as the linked physiological and cognitive outcomes (Winocur and Greenwood, 2005; Dziedzic et al., 2007). Therefore, the primary aim of Experiment 1 was to establish a paradigm for the purposes of studying the impact of high-fat diet on inducing hippocampal-based cognitive function.
To establish this paradigm, Wistar rats were assigned to receive regular rat chow, the med-fat diet or HFD. After 12 weeks, weight gain progression and CPE-FC performance were assessed and compared across diet types. Lastly, serum leptin was measured and compared to final body mass as an added physiological measure of the impact of increased fat in the diet. The presence of elevated serum leptin levels is a well established component of obesity and associated metabolic alterations (Zimmet et al., 1999). Therefore, leptin measures were used to confirm that dietary manipulations produced physiological alterations (in addition to weight gain) in a manner that is consistent with obesity, but leptin levels were not used as a determinant of obesity, nor to make claims on the metabolic status of the animals.
2.8.2. Experiment 2: Further characterization of the relationship between dietary manipulation, duration of HFD exposure and body mass with performance on CPE-FC, following 20 weeks of dietary intervention
The primary goal of Experiment 2 was to further characterize the nature of HFD-induced weight gain and the resulting impact on memory performance. Specifically, we sought to determine whether a longer duration of feeding on a HFD (20 weeks as opposed to the 12-weeks used in Experiment 1) would amplify memory disruption, as well as to determine whether the influence of HFD on memory function would persist following a switch from HFD to a normal diet. Therefore, memory function was assessed in three groups of Wistar animals that had been give one of the following dietary interventions: the regular diet (20 weeks), the HFD (20 weeks) or following a ‘dietary reversal’ which consisted of switching animals from the HFD to regular chow (20 weeks on HFD, 4 weeks on regular diet). After the allotted time, animal groups were tested for behavior in CPE-FC.
The second portion of Experiment 2 served as a control to validate the CPE-FC paradigm for use with animals given a HFD. It is already established that the immediate shock event alone, without benefit of the prior pre-exposure, is not sufficient to associate the context with the shock and initiate a later freezing response (Fanselow, 1990). To confirm that the pre-exposure and the shock events are both required to induce later freezing behavior under the current conditions, rats were fed regular or HFD for 24 weeks and then given CPE-FC. Half of the animals from each diet type did not receive the pre-exposure experience (day 1) but did get the immediate shock on day 2. The remaining animals were given the established pre-exposure experience of day 1, but on day 2, animals were placed in the conditioning context and immediately removed without receiving a shock. All animals were tested for behavioral freezing in context A on day 7.
2.8.3. Experiment 3: Dietary influence on central and peripheral inflammatory response to CPE-FC shock
The association between learning and memory deficits with elevations in hippocampal IL-1β led to the speculation that alterations in central cytokines could be mediating the observed memory decline found here. Furthermore, data from our laboratory, as well as others, suggests that inflammatory events occurring in the periphery often result in de novo production of pro-inflammatory cytokines in the brain (van Dam et al., 1992; Nguyen et al., 1998; Barrientos et al., 2009b) further indicating that HFD, or possibly the resulting increase in inflammatory adipose tissue in the periphery, could induce neuroinflammation. However, preliminary studies did not reveal diet-induced differences in CNS basal IL-1β or other markers of inflammation. As previously discussed, there is ample evidence to suggest that central innate immune processes are capable of being ‘primed’ such that constitutive levels of cytokines appear normal, yet a secondary challenge induces an amplified inflammatory response. It is believed that functional alterations in the central innate immune cell type, microglia, mediate the priming process (Frank et al., 2007). Furthermore, microglia are not evenly distributed within the brain but rather are found at higher concentrations in select brain areas such as the hippocampus (Lawson et al., 1990), which indicates hippocampal function may be particularly vulnerable to negative consequences of inflammatory priming.
It was initially thought that the brief shock that occurs during CPE-FC would be too mild a stressor to induce inflammation in the current model for HFD-induced obesity. However, a recent study demonstrated that a brief foot shock was enough to induce elevations of IL-1β in rats that had received early life infection, an event that appears to prime microglia (Williamson et al., 2011). The purpose of Experiment 3, then, was to test the hypothesis that HFD leads to IL-1β increases in the hippocampus in response to a fear-conditioning shock. To test this hypothesis, animals were given contextual pre-exposure on day 1 and sacrificed on day 2, 2 h after receiving either a fear-conditioning shock or being placed in the context without shock. To determine the dietary impact of immediate shock on hippocampal IL-1β levels, comparisons were made between animals that had received a regular diet (20 weeks), the HFD (20 weeks) or were given dietary reversal (20 weeks on HFD then 4 weeks regular chow). In addition, serum was analyzed to determine dietary and shock-induced alterations on IL-1β and leptin.
2.8.4. Experiment 4: The effect of ICM hIL-1RA injection 2 h prior to CPE-FC shock on later memory performance
The established link between elevated IL-1β and declines in memory function, as well as the results of Experiment 3, suggested increased IL-1β as a primary factor mediating the memory disruption observed in HFD animals. However, it was also possible that elevated IL-1β was correlated with, but unrelated to, diet-induced memory decline. Experiment 4 aimed to determine whether blocking the central action of IL-1β protein at the time of CPE-FC shock would prevent diet-induced decreased fear conditioning/memory. To do this, animals were conditioned using the CPE-FC paradigm after they had been consuming a regular or HFD for 20 weeks. An ICM injection of hIL-1RA or vehicle was administered 2 h prior to the immediate shock event of day 2. Animals were later scored (day 7) for freezing behavior within the conditioned context A.
3. Results
3.1. Experiment 1: Establishment and characterization of diet type on diet-induced obesity and CPE-FC performance following 12 weeks of dietary manipulation
3.1.1. Impact of diet type on weight gain
Overall, Wistar rats gained most weight from eating an increased-fat diet as compared to the standard rat chow (n = 6). Additionally, there was no difference in weight gain induced by the medium (n = 7) and high-fat diets (n = 6), such that both the medium and HFD induced similar weight gain (Fig. 1A). A repeated measures 1-way ANOVA shows that diet type had a significant impact on weight gain (F(2) = 6.402, p = .0325), with post-hoc evaluations showing that HFD (but not medium) produced significantly elevated weight gain compared to the regular diet.
Fig. 1.
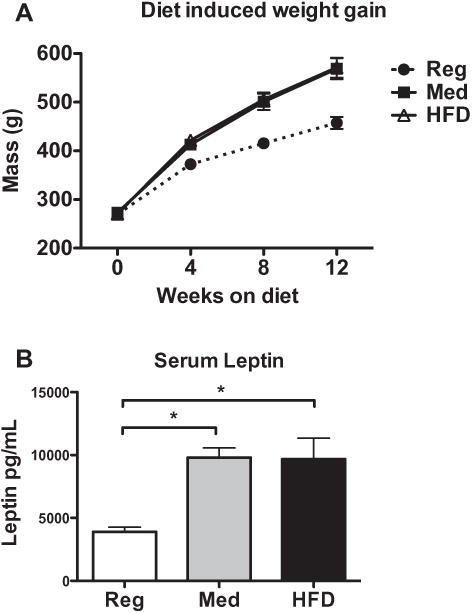
(A) Rats given the medium and high-fat diets gained significantly more body weight than regular-diet controls. There was no difference in weight gain between the medium and high-fat diet groups. (B) Serum leptin was elevated in animals fed either a medium or high-fat diet compared to regular-diet controls. *p < .05.
3.1.2. Impact of diet type on serum leptin levels
All animals on a Med or HFD demonstrated elevated serum leptin levels compared to regular diet controls (Fig. 1B), and increased body mass significantly predicted increased serum leptin. A 1-way ANOVA revealed a significant effect of diet on serum leptin levels (F(2) = 9.752, p = .0019), such that consumption of high-caloric diets induced higher leptin levels. Post-hoc tests demonstrated that animals fed either a medium or HFD had elevated leptin compared to controls.
In addition, there was a very strong correlation between body mass and leptin levels (R(16) = .7068, p < .0001), such that increased raw body mass is an adequate predictor of elevated leptin and is thus a strong indicator of a physiological state associated with obesity (data not presented graphically).
3.1.3. Impact of dietary manipulation on CPE-FC performance
3.1.3.1. Conditioned context A
Rats fed a HFD demonstrated memory declines as measured by decreased freezing behavior during exposure to context A in CPE-FC as compared to regular-diet controls (Fig. 2A). Merged total freezing scores for both test sessions are presented in Fig. 2. A 1-way repeated measures ANOVA comparing the impact of diet on freezing scores (across the 5-min test session) revealed a significant effect of diet (F(2) = 6.449, p = .0215) such that HFD animals (n = 6) froze significantly less than regular-diet animals (n = 6), as indicated by post-hoc evaluations.
Fig. 2.
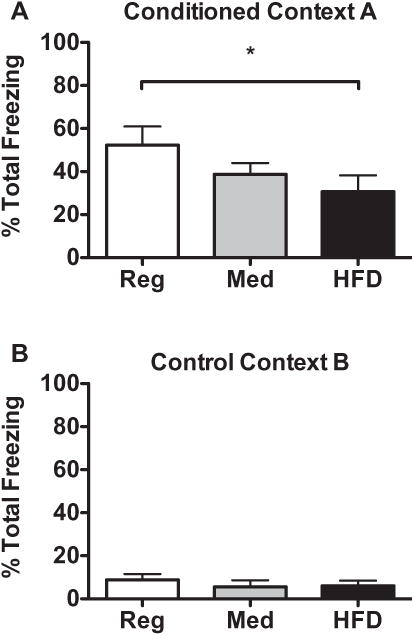
(A) Animals fed HFD had reduced memory as measured by total freezing behavior within conditioned context A compared to regular-diet controls. *p < .05. (B) There was very low freezing behavior observed in control context B, with no differences between diet groups, indicating lack of generalized fear behavior to a non-conditioned context.
3.1.3.2. Control context B
As anticipated, there was very little overall freezing behavior in the control context B (Fig. 2B), for all diet groups observed, indicating that the substantial freezing observed in Context A could not be attributed to generalized freezing. A 1-way repeated measures ANOVA demonstrated no diet-induced differences in freezing between groups (F(2) = 1.108, p = .3762). Overall group averages were below the pre-established baseline level of 10% for freezing in Context B, indicating that no groups expressed generalized freezing behavior to a non-fear conditioned context. Therefore, data for context B are not presented hereafter.
A primary purpose of Experiment 1 was to establish the diet that would most robustly produce obesity as well as demonstrate a hippocampal-specific cognitive deficit. Based on the results of Experiment 1, use of the medium-fat diet was discontinued for all subsequent studies. As Experiment 1 established that 12 weeks of consuming HFD was sufficient time for animals to demonstrate reduced conditioned fear memory, Experiment 2 aimed to determine whether prolonged HFD consumption beyond 12 weeks, and the resulting additional weight gain, would further amplify the deficit in memory function.
3.2. Experiment 2: Impact of duration of diet, type of dietary intervention, and overall body mass with performance on CPE-FC
3.2.1. CPE-FC performance following 20 weeks of HFD
As anticipated, animals that consumed a HFD for 20 weeks (N = 6) demonstrated significantly lower freezing behavior in context A of CPE-FC (Fig. 3) compared to the average freezing of regular diet controls (N = 6; t(10) = 2.431, p = .0354). This demonstrates that the memory decline that occurs from HFD persists with prolonged or long-term consumption.
Fig. 3.
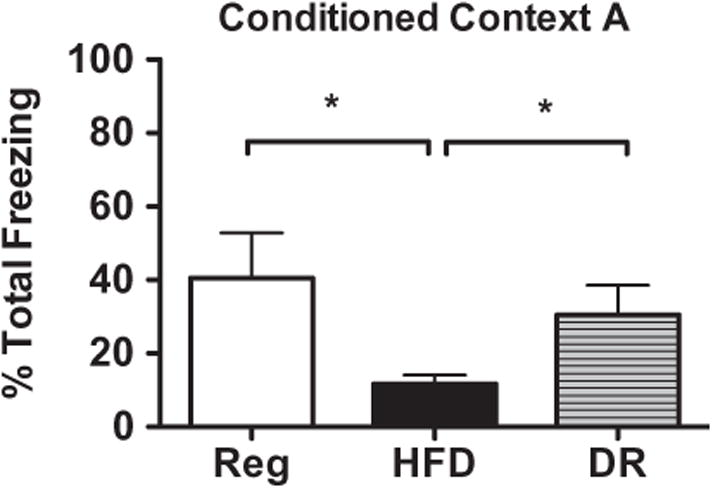
Animals fed HFD for 20 weeks froze significantly less overall compared to regular diet controls when tested in context A of CPE-FC. Animals that experienced the dietary reversal paradigm (DR) demonstrated freezing comparable to regular diet animals, indicating that discontinuing HFD consumption enabled recovery of memory function. *p < .05.
3.2.2. Impact of dietary reversal on CPE-FC performance
Animals previously fed HFD then given a short-term dietary reversal (DR) paradigm completely recovered hippocampal memory function as measured by CPE-FC (Fig. 3). A repeated measures 1-way ANOVA comparing DR animals to regular and HFD groups on freezing scores across the 5-min testing session confirmed that diet impacted freezing behavior (F(2) = 17.33, p = .0012). Post-hoc tests revealed that HFD animals froze significantly less compared to both regular diet and DR animal groups, which were not significantly different from each other. Therefore, DR enabled a complete recovery of cognitive function equivalent with regular diet controls.
The HFD and DR animals were from the same feeding cohort and were allowed to consume the HFD for the same duration. After 20 weeks, animals were randomly assigned to receive immediate CPE-FC or else were given DR and switched to the standard chow (4 additional weeks) before being tested on CPE-FC. Therefore, it is safe to presume that the DR animals, if also tested after 20 weeks of HFD, would have demonstrated memory deficits similar to that observed in the HFD group. This indicates that, despite having received HFD for 20 weeks, the relatively brief switch from HFD to regular chow was able to completely reverse memory deficits, thereby negating the damaging impact of the prior high-fat diet consumption.
3.2.3. Comparison of 12 and 20 weeks of HFD duration on CPE-FC performance
In order to determine whether a longer duration of HFD feeding augmented the reduction of freezing behavior in CPE-FC, the freezing scores of animals given HFD for 12 weeks (Experiment 1) were compared with freezing scores from rats given HFD for 20 weeks (Experiment 2; comparison not shown graphically). An evaluation of average total freezing times revealed that animals on the HFD for 20 weeks froze significantly less compared to animals that consumed HFD for only 12 weeks (t(10) = 2.743, p = .0207).
The overall freezing scores of control regular-diet animals at 12 weeks (52.2% ± 8.592, n = 6) were not statistically different from values observed at 20 weeks (39.4% ± 12.31, n = 6; t(10) = .8510, p = .4147). However, there was a trend for a longer feeding duration (on regular diet) to induce lower overall freezing behavior. In an effort to be conservative and account for this trend, raw freezing scores of HFD animals at 12 and 20 weeks were transformed to percent of same-duration regular-diet controls and again compared. Percent of control values from animals on the HFD for 12 weeks (58.5% ± 14.5) are significantly higher than percent of control values of HFD animals at 20 weeks (23.2% ± 4.72; t(10) = 2.313, p = .0433). Therefore, prolonged HFD consumption further reduced freezing behavior on CPE-FC, such that the longer the duration of HFD feeding, the worse was the resulting hippocampal-dependent memory deficit.
These results indicate that the most pronounced hippocampal-dependent cognitive decline occurs after a prolonged duration (20+ weeks) of high-fat dietary manipulation. Therefore, for all subsequent experiments, duration of dietary intervention lasted in the range of 20–24 weeks.
3.2.4. Comparison of dietary intervention with body mass and performance on CPE-FC
Based on the results from Experiment 1, it was unclear whether reduced freezing in CPE-FC was due to components of the diet or if it was a side effect of increased body mass. Therefore, an additional aim of Experiment 2 was to address this uncertainty. Animals that had been fed HFD and were designated for dietary reversal were the same weight (737.3 g ± 41.5, n = 6) as were animals that remained on the HFD (735.3 g ± 52.9, 6) at the time of the diet switch (20 weeks). As anticipated, the dietary reversal caused animals to stop gaining weight and instead facilitated weight loss (Fig. 4), such that those animals had slightly lower overall average body mass at the time of CPE-FC at 24 weeks (678.3 g ± 37.8, 6). However, the body mass of the DR group at the time of CPE-FC was not significantly lower when compared to the same animals at 20 weeks or when compared to the HFD animals tested on CPE-FC at 20 weeks. Therefore, the DR animals demonstrated improved memory performance while still exhibiting increased body mass. This suggests that dietary component, not reduction in body mass, was likely the primary cause for the recovered memory function observed in the DR animals.
Fig. 4.
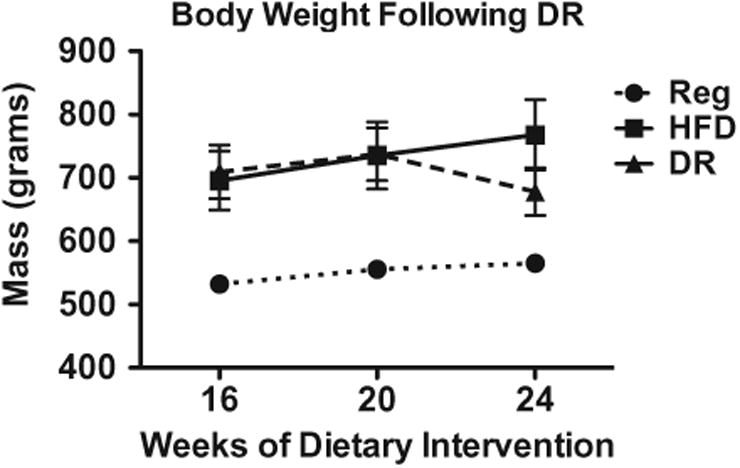
Animals that experienced the dietary reversal (DR) feeding paradigm lost weight as a result of the diet switch, however average body mass for the DR group was not significantly less than the HFD animals at 24 weeks. Also, DR animals still weighed more than regular-diet controls, indicating that the memory function recovery in the DR group was not due to obesity reversal.
3.2.5. Evaluation and establishment of CPE-FC parameters for use with HFD-induced obese rats
Within the CPE-FC paradigm, the immediate shock event, without benefit of the prior exposure to the context, is not typically an adequate experience on its own to condition fear to the context. It has been argued that the failure to conditioning occurs because when the shock happens immediately, there has not been sufficient time to form a hippocampal-based representation of the context that could be associated with the shock (Fanselow, 1990). To verify that this is true with the current model, a control group of regular and HFD animals (24 weeks feeding duration) were given the standard CPE-FC procedure with either the pre-exposure session or shock event eliminated. All animals (both regular diet and HFD, n = 4 per group), that had pre-exposure but did not receive an immediate shock, showed no freezing behavior at all during the later test in context A (0%; data not shown graphically). For animals that had received immediate shock (with no context pre-exposure), very low freezing behavior was observed for HFD (4.2% ± 3.3) and regular diet (7.5 ± 3.4; data not shown). These levels were all below 10% freezing, which was the pre-set level to indicate lack of generalized freezing behavior. These data confirm that both the pre-exposure experience and shock event, separately, are required to form a contextual representation of the conditioning context and then to associate that representation with the foot shock.
3.3. Experiment 3: Dietary influence on central and peripheral inflammatory response to CPE-FC shock
3.3.1. Hippocampal IL-1β protein
An initial comparison was done examining samples collected on day 2 of CPE-FC, 2 h after exposure where regular and HFD animals were given either no shock (baseline) or the shock that typically occurs during conditioning. HFD did not increase basal (non-shock) levels of IL-1β in hippocampus (Fig. 5A) compared to regular diet animals, nor did the shock event alone elevate IL-1β within regular diet animals. However, consumption of HFD combined with the foot-shock lead to an increase in IL-1β protein levels. A 2-way ANOVA comparing HFD-induced alterations in hippocampal IL-1β protein levels following shock or no shock revealed a significant effect of diet (F(1,23) = 7.923, p = .0098, n = 7), shock (F(1,23) = 8.326, p = .0082) and an interaction of diet and shock (F(1,23) = 9.434, p = .0054) such that only the HFD animals that also received a shock displayed elevated IL-1β protein levels above the other groups. Post-hoc tests revealed there were no baseline (non-shock) dietary differences in levels of hippocampal IL-1β protein between regular and HFD animal groups (p > .05) and regular diet animals did not demonstrate altered IL-1β levels in response to a shock compared to non-shock controls (p > .05).
Fig. 5.
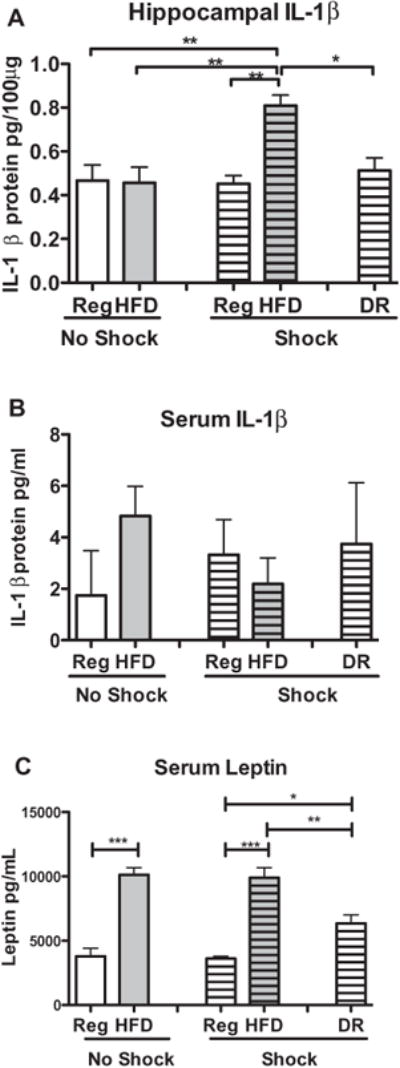
Samples collected 2 h after CPE-FC with exposure to either a shock or no shock revealed: (A) IL-1β protein levels in hippocampus were unaffected by diet when animals are not exposed to shock, however HFD combined with a shock event produced an IL-1β protein increase. DR animals do not demonstrate this priming as they had IL-1β protein levels comparable to regular-diet controls. **p < .01;*p < .05. (B) Serum IL-1β protein levels were not affected by diet nor shock, with no group differences in serum IL-1β protein, thereby discounting the likelihood that the HFD-induced priming seen in HC is a result of peripheral contamination. (C) Serum leptin levels were altered by diet, but not shock, with animals on HFD demonstrating significantly elevated leptin levels. Leptin levels in DR animals were reduced compared to HFD, but also still significantly elevated compared to the regular-diet group indicating physiological evidence of obesity had not resolved. ***p < .001; **p < .01;*p < .05.
The shock event was required to see evidence of diet-induced alterations on hippocampal IL-1β. Therefore, in an effort to conserve animal use, a dietary reversal (DR) non-shock control group was not included. The impact of shock on the DR group demonstrates that a return to the control diet clearly reversed any potentiating effects of the shock event on IL-1β production. This conclusion was confirmed by a 1-way ANOVA comparing post-shock IL-1β protein levels from regular-diet, HFD and DR groups (F(2) = 16.453, p = .0001), such that only HFD animals demonstrated elevated IL-1β protein. Post-hoc analyses show that IL-1β protein levels in the hippocampus are significantly higher in HFD animals compared to both regular diet and dietary reversal groups (p < .05). In addition, dietary reversal animals demonstrated IL-1β protein levels that were not significantly different from regular diet animals (p > .05). Thus, the DR procedure was able to reverse the impact of prior consumption of HFD and prevent the shock-induced pro-inflammatory response in the hippocampus.
3.3.2. Serum IL-1β protein
A potential difficulty with regard to the hippocampal IL-1β measurements is that animals were sacrificed using a rapid decapitation method to reduce stress- and pentobarbital-induced increases in the pro-inflammatory cytokine IL-1β. This prevented the use of intra cardiac saline perfusion to remove peripheral immune cells from the central nervous system vasculature. Therefore, the elevation of observed hippocampal IL-1β protein could have been partly, or completely, caused by differences in circulating IL-1β. If this were true, the pattern of group differences in hippocampal IL-1β would be mirrored in circulating IL-1β. To determine if similar pro-inflammatory expression would be observed peripherally, serum samples were analyzed to determine IL-1β protein levels (Fig. 5B). Clearly, neither the shock nor the diet altered peripheral IL-1β. A 1-way ANOVA comparing regular, HFD and DR serum samples following both shock or no shock revealed no significant differences between any groups (F(4) = 1.052, p = .3877). These results indicate that the observed levels of IL-1β in the hippocampal tissues were not produced by peripheral contamination.
3.3.3. Serum leptin
Serum leptin was also examined for any diet or shock-induced alterations (Fig. 5C). As can be observed, diet, but not foot-shock, altered leptin levels. A 2-way ANOVA comparing regular and HFD animals post-shock or no-shock (DR post-shock group excluded) revealed only a significant effect of diet (F(1,24) = 111.2, p < .0001), such that all HFD animals, regardless of shock, had elevated serum leptin levels. There were no effects of shock, or an interaction of diet and shock (p > .05). A 1-way ANOVA comparing regular diet, HFD and DR on serum leptin levels post-shock demonstrated a highly significant effect of diet (F(2) = 28.4, p < .0001). As expected, HFD animals had significantly elevated leptin compared to regular diet animals (p < .001). In addition, DR leptin levels were higher than regular diet animals (p < .05), but were also significantly reduced compared to the HFD group (p < .01). Additionally, across groups, body mass was significantly correlated with serum leptin (R(20) = .7653, p < .0001). It is important to note that DR animals sustained physiological evidence of an obese state (as indicated by persistent increased body mass and elevated leptin levels), yet no longer showed a primed central immunological response to the shock.
3.4. Experiment 4: Impact of hIL-1RA blockade of IL-1β at time of CPE-FC shock on diet-induced cognitive function
As expected, HFD strongly interfered with expression of memory for contextual fear. However, this diet-induced memory deficit was completely blocked by preventing the action of IL-1β at the time of CPE-FC shock with an ICM injection of hIL-1RA (Fig. 6). A 2-way repeated measures ANOVA comparing freezing levels (across the 5-min test session) of animals fed regular or HFD and given either saline or hIL-1RA 2 h prior to CPE-FC shock revealed neither a main effect of drug (F(1,32) = 2.74, p = .1076) nor a main effect of diet (F(1,32) = 3.383, p = .0751) but did illustrate a significant interaction of diet type with drug treatment (F(1,32) = 5.003, p = .0324), such that only HFD animals that were given saline had significantly decreased freezing scores compared to the other three groups. As expected, there was also a significant main effect of time (F(4,128) = 26.632, p < .0001) such that all animal groups demonstrated a reduction in freezing behavior as the testing sessions progressed. In addition, there was an interaction of time and drug (F(4,128) = 2.471, p = .0478) indicating that hIL-1RA facilitated fear extinction during the test session in both the regular and HFD animals compared to their saline-treated same-diet controls. There was not a significant 3-way interaction between diet, drug and time (p > .05), nor a 2-way interaction of diet and time (p > .05). Post-hoc analyses of overall freezing scores by group show that HFD animals given saline had reduced freezing compared to all regular diet animals (saline: p = .0083; hIL-1RA: p = .0167) and HFD animals given hIL-1RA (p = .0041). Also, HFD animals given hIL-1RA were not significantly different from either of the regular diet animals, regardless of drug treatment (p > .05), indicating that the presence of hIL-1RA at the time of the shock eliminated the detrimental effect of HFD.
Fig. 6.
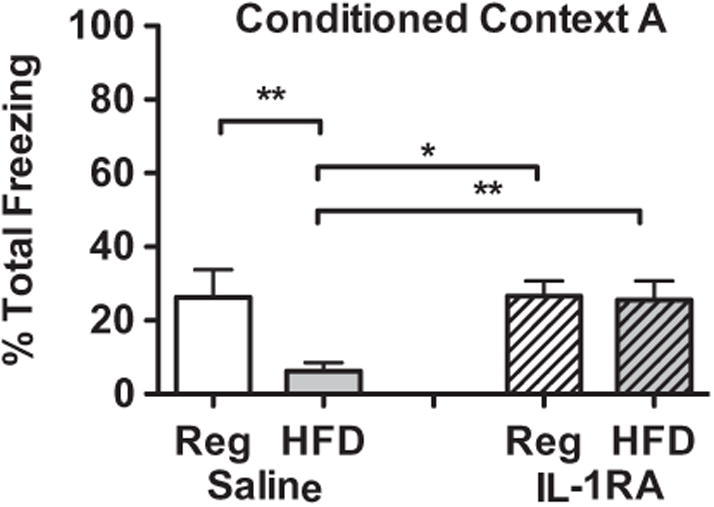
Animals that consumed HFD had decreased freezing behavior in context A of CPE-FC compared to regular-diet controls. Antagonism of IL-1β action with an ICM injection of hIL-1RA prior to CPE-FC shock eliminated the HFD-induced decrease in behavior indicating the memory deficit is mediated by action of IL-1β at the time of CPE-FC shock. **p < .01; *p < .05.
These data demonstrate a number of important outcomes. First, blockade of IL-1 receptors at the time of shock does not inhibit memory formation in regular diet animals, yet allowing the full action of elevated IL-1β in the HFD animals does. This suggests that the action of excess IL-1β during the conditioning experience leads to memory disruption. Furthermore, the pre-exposure experience is as important for fear conditioning as is the shock event and there was no drug manipulation until 24 h after the pre-exposure experience in these animals. The observation that HFD animals given hIL-1RA learned equally well compared to regular-diet controls suggests that HFD does not alter how animals learn from or respond to the pre-exposure experience.
4. Discussion
In sum, the present series of experiments clearly show that HFD increased body mass and disrupted hippocampal memory function in Wistar rats. The data suggest that the diet produced memory deficits by priming a shock-induced increase in hippocampal IL-1β protein, which at elevated levels disrupted later behavioral memory expression. The negative impact of diet was prevented by either blocking action of IL-1β with hIL-1RA or by preventing the primed stress-induced increase in IL-1β using a short-term dietary reversal prior to conditioning. Furthermore, the data suggest that the primary factor mediating cognitive disruption was the continued intake of HFD, not increased body mass from prior HFD consumption.
The diets used in the present study are routinely labeled ‘medium-fat’ and ‘high-fat’, however, it should be noted that they differ from the standard diet in many other ways. Indeed, they might also be considered high-sugar diets. The medium-fat diet contains 341.46 g/kg of sucrose (34.5% sucrose by total weight) while the high-fat diet has 90 g/kg sucrose and 160 g/kg maltodextrin (12.1% sucrose by weight) as sweeteners. Therefore, while the fat content is elevated above standard chow, these diets are also more palatable and the overall caloric content is denser. It is possible that any number of alterations in the macronutrient composition of the diets could have led to the observed negative cognitive consequences. It should also be noted that 60% calories from fat may not be considered by some to be ‘high-fat’, as ketogenic diets can contain up to 85–95% fat, and may actually be beneficial for health (Ruskin et al., 2009; Krikorian et al., 2012; Mobbs et al., 2013).
Animals fed the high-fat diet demonstrated elevated IL-1β protein levels in the hippocampus following a brief foot-shock that occurred during CPE-FC. The regular-diet animals showed no shock-induced changes in IL-1β protein. This suggests that HFD produces an altered response to a stressor by inducing neural inflammatory processes. This notion is supported by observations that waist circumference in humans is linked to augmented physiological stress responses (Brydon, 2011) and that obesity induces increased vulnerability of the CNS to the negative impacts of stress and injury (Yehuda et al., 2005; Bruce-Keller et al., 2009).
Dietary reversal was able to eliminate the negative impacts of prior HFD consumption on memory disruption and elevated hippocampal IL-1β in response to foot shock. However, while the DR animals lost weight and had reduced leptin levels, these animals continued to exhibit increased body mass and increased leptin levels compared to regular diet controls. Therefore, the benefits of dietary reversal occurred prior to the complete resolution of either increased body mass or leptin.
It should be noted that no conclusions are being made about obesity or the metabolic status of the animals in the current studies. Rather, the goal is to distinguish between HFD consumption and the resulting physiological impacts of such consumption with respect to cognitive and neural inflammatory processing. Following experiment 1, we discontinued use of the medium-fat diet because it did not induce measurable deficits in hippocampal cognitive function, despite inducing elevated weight gain comparable to HFD. Therefore, we did not examine what impact the medium-fat diet would have had on shock-induced levels of hippocampal IL-1β. In the future, this would be interesting to examine as a means to further distinguish diet and increased body mass with cognitive function and neural inflammatory processes. Due to the well-established inflammatory nature of enlarged adipose stores, it is possible that increased peripheral adiposity must occur simultaneously with continued consumption of HFD in order for the cognitive effects and neural inflammatory priming to be observed. While leptin may induce IL-1β release from microglia cells in culture (Pinteaux et al., 2007), the present data indicate that the presence of elevated body mass and leptin are not sufficient alone (in absence of HFD consumption) to produce similar inflammatory effects. What is not known, and is beyond the scope of the current paper, is how quickly the benefits of DR can be observed following diet switch, or if any amount of weight loss, even minimally, must occur in order to observe those effects.
The effects of DR, in the present study, are mirrored in a short-term diet and exercise intervention study in humans, in which significant improvements in inflammatory markers of metabolic health were observed prior to obesity reversal (Izadpanah et al., 2012). In addition, Kosari et al. (2012) found that a 60% fat diet altered spatial memory, but the effect was unrelated to body weight, while (Badman et al., 2009) found improved health markers, but not altered body weight, of genetically-induced obese mice fed a ketogenic diet. In addition, very short-term consumption of a high-fat diet (4 days) is sufficient to induce pro-inflammatory changes in adipose tissue (Ji et al., 2012) prior to inducing significant weight gain. The evidence suggests that, perhaps, health and cognitive consequences are mediated less in the long-term by adipose stores but rather in the short-term by macronutrient composition of the diet. Although ‘high-fat diet consumption’ and ‘obesity’ are typically viewed as synonymous, as they typically occur simultaneously, distinguishing between the two may be important for future mechanistic research in this area. Although it is interesting and important to note that diet alteration can improve the negative consequences of HFD on memory function, it does not address what, mechanistically, may mediate diet-induced memory dysfunction, nor does it determine what specific memory processes are altered by HFD consumption.
An injection of hIL-1RA prior to the shock experience of CPE-FC prevented diet-induced memory disruption, strongly suggesting that action of IL-1β at the time of shock mediates the effect of HFD on memory performance. Also, all animals in Experiment 4 showed relatively reduced freezing compared to other cohorts run through CPE-FC for other experiments. However, the dietary impact on freezing behavior within the saline animals proportionately remained clear, as did the recovery of function in HFD animals given hIL-1RA. It is possible that the brief anesthetization during the saline/hIL-1RA ICM injections may have had a later impact on freezing behavior expression. Furthermore, all groups of rats were un-treated during the pre-exposure phase, with the only intervention at that point being diet, as the injection procedure did not occur until just prior to the immediate shock event on day 2. Because the hIL-1RA injection did not occur until after pre-exposure, it is clear that HFD animals learned normally during the pre-exposure session as HFD animals treated with hIL-1RA demonstrated memory equivalent with controls. The data suggest that HFD animals may learn during the pre-exposure experience normally, and HFD animals are able to adequately demonstrate expression of the fear memory once it has been acquired (HFD + IL-1RA animals) so it might be that the memory process of consolidation is most vulnerable to HFD. However, we did not conduct a short-term memory test for the contextual fear after shock exposure, so it is not possible to implicate any particular aspect of memory function as being disrupted by HFD. The only group that demonstrated impaired memory in Experiment 4 was the HFD/saline group. Therefore, it was the action of potentiated levels of IL-1β, as induced in the HFD animals by the immediate shock, which acted as the primary facilitating factor in the ensuing memory impairment. Thus, HFD does not appear to interfere with all facets of memory function, or even hippocampal-dependent memory in all contexts. Rather, it seems that in order to observe the impact of a HFD on memory disruption, the learning experience may need to coincide with a secondary challenge, such as stress or infection. It is important to note that there is no claim that the dietary impact on cognition exclusively targets hippocampal function. While there is some evidence that hippocampal function may be particularly vulnerable to the influence of HFD (Heyward et al., 2012), amygdala-dependent cognition may also be impacted by HFD (Yamada-Goto et al., 2012).
The single foot-shock did not increase levels of IL-1β in serum, thus diet-induced elevations in IL-1β protein in hippocampus after HFD must have originated centrally. Therefore, there is reason to believe that the elevated IL-1β after the shock exposure is the result of diet-induced changes to cells within the CNS. It is well established that functional changes in microglia can mediate primed central inflammatory processes (Combrinck et al., 2002; Frank et al., 2007; Ransohoff and Perry, 2009). Furthermore, the primed neural inflammation that occurs with aging is mediated by microglia (Barrientos et al., 2010) and there are many similarities between obesity and age-induced cognitive and health impairments (Cohen, 2010; Uranga et al., 2010), suggesting that obesity may induce neural inflammatory priming via similar mechanisms.
It is generally understood that IL-1β protein production is complex and requires multiple steps, and it is not yet clear how a HFD alters central innate immune function such that a sensitized overproduction of IL-1β occurs upon stimulation. Interestingly, saturated fatty acids are able to stimulate microglia to induce NFkB and pro-inflammatory cytokine expression (Lee et al., 2003; Milanski et al., 2009), and levels of free fatty acids are known to be elevated centrally with obesity (Greenwood and Winocur, 1996). Taken together, this evidence suggests that consumption of a HFD may induce ‘primed’ CNS innate immune cells, and this process may occur via fatty acid signaling. However, this has not yet been demonstrated.
References
- Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, Yirmiya R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. 2003;13:826–834. doi: 10.1002/hipo.10135. [DOI] [PubMed] [Google Scholar]
- Badman MK, Kennedy AR, Adams AC, Pissios P, Maratos-Flier E. A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am J Physiol Endocrinol Metab. 2009;297:E1197–E1204. doi: 10.1152/ajpendo.00357.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun. 2009;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Watkins LR, Maier SF. Memory impairments in healthy aging: role of aging-induced microglial sensitization. Aging Dis. 2010;1:212–231. [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Hein AM, Frank MG, Watkins LR, Maier SF. Intracisternal interleukin-1 receptor antagonist prevents postoperative cognitive decline and neuroinflammatory response in aged rats. J Neurosci. 2012;32:14641–14648. doi: 10.1523/JNEUROSCI.2173-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27:723–732. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res. 2002;134:291–298. doi: 10.1016/s0166-4328(02)00043-8. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Watkins LR, Rudy JW, Maier SF. Characterization of the sickness response in young and aging rats following E. coli infection. Brain Behav Immun. 2009b;23:450–454. doi: 10.1016/j.bbi.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitard C, Cavaroc A, Sauvant J, Aubert A, Castanon N, Laye S, Ferreira G. Impairment of hippocampal-dependent memory induced by juvenile high-fat diet intake is associated with enhanced hippocampal inflammation in rats. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Keller JN, Morrison CD. Obesity and vulnerability of the CNS. Biochim. Biophys. Acta. 2009;1792:395–400. doi: 10.1016/j.bbadis.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brydon L. Adiposity, leptin and stress reactivity in humans. Biol Psychol. 2011;86:114–120. doi: 10.1016/j.biopsycho.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RA. Obesity-associated cognitive decline: excess weight affects more than the waistline. Neuroepidemiology. 2010;34:230–231. doi: 10.1159/000297745. [DOI] [PubMed] [Google Scholar]
- Combrinck MI, Perry VH, Cunningham C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience. 2002;112:7–11. doi: 10.1016/s0306-4522(02)00030-1. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das UN. Obesity: genes, brain, gut, and environment. Nutrition. 2010;26:459–473. doi: 10.1016/j.nut.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16:457–499. doi: 10.3109/08830189809043005. [DOI] [PubMed] [Google Scholar]
- Dinel AL, Andre C, Aubert A, Ferreira G, Laye S, Castanon N. Cognitive and emotional alterations are related to hippocampal inflammation in a mouse model of metabolic syndrome. PLoS ONE. 2011;6:e24325. doi: 10.1371/journal.pone.0024325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Dziedzic B, Szemraj J, Bartkowiak J, Walczewska A. Various dietary fats differentially change the gene expression of neuropeptides involved in body weight regulation in rats. J Neuroendocrinol. 2007;19:364–373. doi: 10.1111/j.1365-2826.2007.01541.x. [DOI] [PubMed] [Google Scholar]
- Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci. 2014;34:2618–2631. doi: 10.1523/JNEUROSCI.4200-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS. Factors governing one-trial contextual conditioning. Anim Learn Behav. 1990;18:264–270. [Google Scholar]
- Francis H, Stevenson R. The longer-term impacts of Western diet on human cognition and the brain. Appetite. 2013;63:119–128. doi: 10.1016/j.appet.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2007;21:47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Hein AM, Biedenkapp JC, Watkins LR, Maier SF. IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344xBN F1 rats. Brain Behav Immun. 2010;24:254–262. doi: 10.1016/j.bbi.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Watkins LR, Maier SF. Stress- and glucocorticoid-induced priming of neuroinflammatory responses: potential mechanisms of stress-induced vulnerability to drugs of abuse. Brain Behav Immun. 2011;25(Suppl 1):S21–S28. doi: 10.1016/j.bbi.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibertini M, Newton C, Friedman H, Klein TW. Spatial learning impairment in mice infected with Legionella pneumophila or administered exogenous interleukin-1-beta. Brain Behav Immun. 1995;9:113–128. doi: 10.1006/brbi.1995.1012. [DOI] [PubMed] [Google Scholar]
- Greenwood CE, Winocur G. Cognitive impairment in rats fed high-fat diets: a specific effect of saturated fatty-acid intake. Behav Neurosci. 1996;110:451–459. doi: 10.1037//0735-7044.110.3.451. [DOI] [PubMed] [Google Scholar]
- Heyward FD, Walton RG, Carle MS, Coleman MA, Garvey WT, Sweatt JD. Adult mice maintained on a high-fat diet exhibit object location memory deficits and reduced hippocampal SIRT1 gene expression. Neurobiol Learn Mem. 2012;98:25–32. doi: 10.1016/j.nlm.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill-Pryor C, Dunbar JC. The effect of high fat-induced obesity on cardiovascular and physical activity and opioid responsiveness in conscious rats. Clin Exp Hypertens. 2006;28:133–145. doi: 10.1080/10641960500468326. [DOI] [PubMed] [Google Scholar]
- Holguin A, Frank MG, Biedenkapp JC, Nelson K, Lippert D, Watkins LR, Rudy JW, Maier SF. Characterization of the temporo-spatial effects of chronic bilateral intrahippocampal cannulae on interleukin-1beta. J Neurosci Methods. 2007;161:265–272. doi: 10.1016/j.jneumeth.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, Chen CT, Liang KC, Ho IK, Yang WS, Chiou LC. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring) 2010;18:463–469. doi: 10.1038/oby.2009.273. [DOI] [PubMed] [Google Scholar]
- Hydock DS, Lien CY, Jensen BT, Schneider CM, Hayward R. Switching to a low-fat diet attenuates the intensified doxorubicin cardiotoxicity associated with high-fat feeding. Cancer Chemother Pharmacol. 2013;71:1551–1560. doi: 10.1007/s00280-013-2154-5. [DOI] [PubMed] [Google Scholar]
- Izadpanah A, Barnard RJ, Almeda AJ, Baldwin GC, Bridges SA, Shellman ER, Burant CF, Roberts CK. A short-term diet and exercise intervention ameliorates inflammation and markers of metabolic health in overweight/obese children. Am J Physiol Endocrinol Metab. 2012;303:E542–E550. doi: 10.1152/ajpendo.00190.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Sun S, Xia S, Yang L, Li X, Qi L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem. 2012;287:24378–24386. doi: 10.1074/jbc.M112.371807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Hansen MK, Watkins LR, Maier SF. Effects of prior stress on LPS-induced cytokine and sickness responses. Am J Physiol Regul Integr Comp Physiol. 2003;284:R422–R432. doi: 10.1152/ajpregu.00230.2002. [DOI] [PubMed] [Google Scholar]
- Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103:59–68. doi: 10.1016/j.physbeh.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosari S, Badoer E, Nguyen JC, Killcross AS, Jenkins TA. Effect of western and high fat diets on memory and cholinergic measures in the rat. Behav Brain Res. 2012;235:98–103. doi: 10.1016/j.bbr.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Krikorian R, Shidler MD, Dangelo K, Couch SC, Benoit SC, Clegg DJ. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol Aging. 2012;33(425):e419–e427. doi: 10.1016/j.neurobiolaging.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin DN, Joesting JJ, Chiu GS, Moon ML, Meng J, Dilger RN, Freund GG. Fasting induces an anti-inflammatory effect on the neuroimmune system which a high-fat diet prevents. Obesity (Silver Spring) 2011;19:1586–1594. doi: 10.1038/oby.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–37051. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- Li S, Wang C, Wang W, Dong H, Hou P, Tang Y. Chronic mild stress impairs cognition in mice: from brain homeostasis to behavior. Life Sci. 2008;82:934–942. doi: 10.1016/j.lfs.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Loram LC, Taylor FR, Strand KA, Frank MG, Sholar P, Harrison JA, Maier SF, Watkins LR. Prior exposure to glucocorticoids potentiates lipopolysaccharide induced mechanical allodynia and spinal neuroinflammation. Brain Behav Immun. 2011;25:1408–1415. doi: 10.1016/j.bbi.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF, Watkins LR. Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol Rev. 1998;105:83–107. doi: 10.1037/0033-295x.105.1.83. [DOI] [PubMed] [Google Scholar]
- Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DM, Anhe G, Amaral ME, Takahashi HK, Curi R, Oliveira HC, Carvalheira JB, Bordin S, Saad MJ, Velloso LA. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. 2009;29:359–370. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito N, Hosoda T, Kato C, Sato K. Change of cytokine balance in diet-induced obese mice. Metabolism. 2000;49:1295–1300. doi: 10.1053/meta.2000.9523. [DOI] [PubMed] [Google Scholar]
- Mobbs CV, Mastaitis J, Isoda F, Poplawski M. Treatment of diabetes and diabetic complications with a ketogenic diet. J Child Neurol. 2013 doi: 10.1177/0883073813487596. [DOI] [PubMed] [Google Scholar]
- Molteni R, Barnard RJ, Ying Z, Roberts CK, Gomez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–814. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J Neurosci. 1998;18:2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palin K, Bluthe RM, Verrier D, Tridon V, Dantzer R, Lestage J. Interleukin-1beta mediates the memory impairment associated with a delayed type hypersensitivity response to bacillus Calmette–Guerin in the rat hippocampus. Brain Behav Immun. 2004;18:223–230. doi: 10.1016/j.bbi.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Pinteaux E, Inoue W, Schmidt L, Molina-Holgado F, Rothwell NJ, Luheshi GN. Leptin induces interleukin-1beta release from rat microglial cells through a caspase 1 independent mechanism. J Neurochem. 2007;102:826–833. doi: 10.1111/j.1471-4159.2007.04559.x. [DOI] [PubMed] [Google Scholar]
- Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. 2010;219:25–32. doi: 10.1016/j.jneuroim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proescholdt MG, Hutto B, Brady LS, Herkenham M. Studies of cerebrospinal fluid flow and penetration into brain following lateral ventricle and cisterna magna injections of the tracer [14C]inulin in rat. Neuroscience. 2000;95:577–592. doi: 10.1016/s0306-4522(99)00417-0. [DOI] [PubMed] [Google Scholar]
- Pugh CR, Kumagawa K, Fleshner M, Watkins LR, Maier SF, Rudy JW. Selective effects of peripheral lipopolysaccharide administration on contextual and auditory-cue fear conditioning. Brain Behav Immun. 1998;12:212–229. doi: 10.1006/brbi.1998.0524. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Reeves JG, Suriawinata AA, Ng DP, Holubar SD, Mills JB, Barth RJ., Jr Short-term preoperative diet modification reduces steatosis and blood loss in patients undergoing liver resection. Surgery. 2013 doi: 10.1016/j.surg.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Ross AP, Bruggeman EC, Kasumu AW, Mielke JG, Parent MB. Nonalcoholic fatty liver disease impairs hippocampal-dependent memory in male rats. Physiol Behav. 2012;106:133–141. doi: 10.1016/j.physbeh.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Barrientos RM, O'Reilly RC. Hippocampal formation supports conditioning to memory of a context. Behav Neurosci. 2002;116:530–538. doi: 10.1037//0735-7044.116.4.530. [DOI] [PubMed] [Google Scholar]
- Ruskin DN, Kawamura M, Masino SA. Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS ONE. 2009;4:e8349. doi: 10.1371/journal.pone.0008349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci USA. 1998;95:7778–7783. doi: 10.1073/pnas.95.13.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellbom KS, Gunstad J. Cognitive function and decline in obesity. J Alzheimers Dis. 2012;30(Suppl. 2):S89–S95. doi: 10.3233/JAD-2011-111073. [DOI] [PubMed] [Google Scholar]
- South T, Westbrook F, Morris MJ. Neurological and stress related effects of shifting obese rats from a palatable diet to chow and lean rats from chow to a palatable diet. Physiol Behav. 2012;105:1052–1057. doi: 10.1016/j.physbeh.2011.11.019. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranga RM, Bruce-Keller AJ, Morrison CD, Fernandez-Kim SO, Ebenezer PJ, Zhang L, Dasuri K, Keller JN. Intersection between metabolic dysfunction, high fat diet consumption, and brain aging. J Neurochem. 2010;114:344–361. doi: 10.1111/j.1471-4159.2010.06803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valladolid-Acebes I, Stucchi P, Cano V, Fernandez-Alfonso MS, Merino B, Gil-Ortega M, Fole A, Morales L, Ruiz-Gayo M, Del Olmo N. High-fat diets impair spatial learning in the radial-arm maze in mice. Neurobiol Learn Mem. 2011;95:80–85. doi: 10.1016/j.nlm.2010.11.007. [DOI] [PubMed] [Google Scholar]
- van Dam AM, Brouns M, Louisse S, Berkenbosch F. Appearance of interleukin-1 in macrophages and in ramified microglia in the brain of endotoxin-treated rats: a pathway for the induction of non-specific symptoms of sickness? Brain Res. 1992;588:291–296. doi: 10.1016/0006-8993(92)91588-6. [DOI] [PubMed] [Google Scholar]
- Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early-life infection. J Neurosci. 2011;31:15511–15521. doi: 10.1523/JNEUROSCI.3688-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging. 2005;26(Suppl. 1):46–49. doi: 10.1016/j.neurobiolaging.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur J Neurosci. 2004;19:1699–1707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- Yamada-Goto N, Katsuura G, Ochi Y, Ebihara K, Kusakabe T, Hosoda K, Nakao K. Impairment of fear-conditioning responses and changes of brain neurotrophic factors in diet-induced obese mice. J Neuroendocrinol. 2012;24:1120–1125. doi: 10.1111/j.1365-2826.2012.02327.x. [DOI] [PubMed] [Google Scholar]
- Yehuda S, Rabinovitz S, Mostofsky DI. Mediation of cognitive function by high fat diet following stress and inflammation. Nutr Neurosci. 2005;8:309–315. doi: 10.1080/00268970500509972. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Winocur G, Goshen I. Brain interleukin-1 is involved in spatial memory and passive avoidance conditioning. Neurobiol Learn Mem. 2002;78:379–389. doi: 10.1006/nlme.2002.4072. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Boyko EJ, Collier GR, de Courten M. Etiology of the metabolic syndrome: potential role of insulin resistance, leptin resistance, and other players. Ann N Y Acad Sci. 1999;892:25–44. doi: 10.1111/j.1749-6632.1999.tb07783.x. [DOI] [PubMed] [Google Scholar]